

Abstract
Fibronectin (FN) may be involved in time‐ and stage‐dependent and inter‐related controlled processes of inflammation, coagulation, and wound healing accompanying peripheral arterial disease (PAD). In the present study, FN and FN‐containing extra‐domain A (EDA‐FN), macromolecular FN‐fibrin complexes, and FN monomer were analysed in the plasma of 142 PAD patients, including 37 patients with restenosis, for 37 months after revascularisation. FN concentration increased significantly in the plasma of PAD patients within 7 to 12 months after revascularisation, whereas the high concentration of EDA‐FN was maintained up to 24 months, significantly higher in the group 7 to 12 months after revascularisation with recurrence of stenosis and lower in the PAD groups 1 to 3 months and 4 to 6 months after revascularisation with comorbid diabetes and ulceration, respectively. The relative amounts of FN‐fibrin complexes up to 1600 kDa and FN monomer were significantly higher, within intervals of 4 to 24 months and 4 to 6 months after revascularisation, respectively. Moreover, the relative amounts of 750 to 1600 kDa FN‐fibrin complexes within 13 to 24 months after revascularisation were higher in comparison with those in the group without restenosis. In conclusion, high levels of EDA‐FN and FN‐fibrin complexes could have potential diagnostic value in the management of PAD patients after revascularisation, predicting restenosis risk.
Keywords: EDA‐fibronectin, fibronectin, FN‐fibrin complexes, peripheral arterial disease, restenosis
1. INTRODUCTION
Peripheral arterial disease (PAD) is a chronic, progressive disease occurring as a result of atherosclerosis in the arterial system of the extremities. Restriction of blood flow due to arterial stenosis or occlusion may cause intermittent claudication, and in more advanced stages of the disease, it may result in critical limb ischaemia with pain at rest or trophic lesions, and moreover, it results in an increased risk of loss of a limb if not treated.1, 2 Endovascular interventions—percutaneous transluminal angioplasty (PTA) or stent implantation—do not stop the disease but help to achieve a greater lumen diameter as well as compression and shifting of the atherosclerotic plaque, which in consequence may significantly reduce the risk of restenosis. Artery intervention is associated with endothelial denudation and endothelial impaired function and inevitably causes trauma to the blood vessel wall.3, 4 Endothelial injury triggers both local and systemic inflammation with a typical cellular response.5 Moreover, the rupture of the endothelial layer, preconditioned by atherosclerotic plaque, exposes sub‐endothelial extracellular matrix (ECM) proteins from the basement membrane to flowing blood. Some of them, for example fibronectin (FN), take part in possible thrombus formation, vessel occlusion, and the repair process.6, 7
FN is a mosaic, multi‐domain, and multifunctional glycoprotein found on cell surfaces, in blood plasma, and other biological fluids reviewed in.8, 9, 10 FN is a dimer (~500 kDa) of 2 identical or nearly identical 230‐ to 250‐kDa polypeptides composed of repeating types I, II, and III sequences assembled into several domains with binding sites for ECM proteins (eg, collagen, FN itself, or FN fragment/s), cell surface receptors (integrins, bacterial FN receptors), blood protein derivatives (fibrin), and glycosaminoglycans (heparin)8 and may or may not possess alternatively spliced type III extra‐domains A (EDA) and B (EDB) and the oncofetal IIICS region.10, 11
FN exists in 2 major forms, as plasma fibronectin (pFN) and cellular fibronectin (cFN). pFN is synthesised and secreted by hepatocytes to the blood stream at a relatively high concentration of about 280 mg/L, where it circulates as a soluble compact inactive globular molecule lacking EDA and EDB domains.8, 10, 12 cFN, containing variable proportions of the EDA (EDA‐FN) and EDB (EDB‐FN) domains, is released by various cell types (eg, endothelial cells, fibroblasts, macrophages, monocytes, lymphocytes, smooth muscle cells, platelets). EDA‐FN is reported to play the role of a “danger signal” for leukocytes to cause adverse endothelial remodelling after arterial damage.13 Its fibrillar form supports efficient platelet aggregation and pro‐coagulant activity.14 Expression of EDA‐FN is up‐regulated in a disease‐related tissue condition and stress signals by some ECM components, cytokines, and transforming growth factor beta (TGF‐β). In normal blood plasma, it remains in a relatively low concentration of 2 to 3 mg/L.10, 11, 15, 16 Plasma FN is thought to be inactive under normal conditions, being a reservoir for tissue needs.17 Inflammatory environmental, local transient hypoxia, and pH changes stimulate transfer of pFN from blood to tissue, where it participates in the modulation of haemostasis, fibrin clot formation, ECM remodelling, provisional matrix formation, and tissue repair.6, 8, 18, 19 Hou et al20 postulated that pFN is a participant in a FN‐mediated “protein wave” of haemostasis that precedes the classical first wave of haemostasis, that is, it prevents platelet aggregation and thrombus formation immediately after injury, before fibrinogen to fibrin conversion. During clot formation, pFN is accumulated in granules of platelet‐producing megakaryocytes, and upon platelet degranulation, FN is released from the granules and assembled on the surface of platelets, preventing them from aggregating via a fibrin‐dependent pathway and via a fibrin‐independent mechanism.8, 19
During an enhanced thrombotic process, pFN is covalently cross‐linked by factor XIIIa through an isopeptide bond to the carboxyl terminal compact and flexible αC‐domain of fibrin deposited on the endothelium after activation of a coagulation cascade and fibrinogen conversion to fibrin.18 Other molecules of fibrin together with additional FN molecules, including FN degradation products and the EDA isoform, as well as some components of the ECM and/or plasma, might be attached to that heterodimer during the formation of a fibrin clot or thrombus, or they can be further used for ECM remodelling. In the early phase of the wound‐healing process, the pFN incorporated into the fibrin matrix forms a provisional matrix in which FN adopts extended conformations within the FN‐fibrin matrix, with exposure of cryptic binding domains to facilitate migration and adhesion of cells in the area of thrombus formation.8 Plasma FN changes its function in the presence of fibrin: it shows prerequisite pro‐thrombotic activities and supports platelet aggregation and thrombus growth and stability6, 21, 22 while at the same time being an important self‐limiting regulator to prevent haemorrhage and excessive thrombus formation and vessel occlusion.23 In contrast, EDA‐FN is deposited at the site of injury in the late phase of the wound‐healing process.6, 10, 11 Multiple wound‐related signal transduction pathways control the deposition of EDA‐FN, and the EDA insert can, in turn, trigger pathways that induce inflammation; increase ECM molecule deposition, including other FN molecules and collagen; and activate fibroblasts, creating a vicious cycle that aggravates and delays wound healing.6, 8, 24
FN‐fibrin complexes were also found in human blood plasma and were demonstrated by agarose immunoblotting under semi‐denaturing, non‐reducing conditions as a series of FN‐fibrin bands with molecular masses from 750 to 2100 kDa.25 Such macromolecular complexes were demonstrated by Kątnik‐Prastowska's group in the plasma of patients with recurrent respiratory infections,25 atherosclerosis with advanced coronary artery disease,26 aging, and also multi‐morbidity, especially when coexisting with cardiovascular diseases and osteoarthrosis16 but rarely in healthy individuals.15, 25, 26, 27 It is postulated that their occurrence might be related to molecular events connected with interplaying processes of inflammation, immunity, and coagulation27 and/or may also be involved in fibrin clearance from the circulation in inflammatory conditions.28
Witkiewicz and coworkers have analysed some inflammatory and haemostatic parameters in the plasma of patients with advanced PAD of the lower extremities after endovascular revascularisation and in relation to new restenosis.29, 30 Continuing their work, and in light of the significant role of FN in interplaying processes of inflammation, coagulation, and wound healing, we determined the levels of FN molecular forms (FN, EDA‐FN, FN‐fibrin complexes and FN degradation products). In our study, we analysed the concentrations of FN and EDA‐FN and relative amounts of the soluble FN‐fibrin complexes and FN monomer in 142 plasma samples of PAD patients after revascularisation. The data obtained were related to disease status 37 months after revascularisation as well as to restenosis occurrence, comorbid diabetes, and ulceration. We believe that the determination of FN molecular status in the context of the disease may help in better understanding the multidirectional role of FN in human arterial pathophysiology, and it could have potential diagnostic value in the management of PAD patients after revascularisation, predicting restenosis risk.
2. MATERIALS AND METHODS
2.1. Patients
The study included 142 patients (87 men and 55 women) aged 45 to 88 years (66.0 ± 9.7 years) with lower limb PAD who visited the Angiology Unit of the Wrocław Regional Specialist Hospital up to 5 times after they had undergone successful peripheral endovascular revascularisation at the Angiology and Vascular Surgery Centers in Wrocław and Kraków. A revascularisation procedure, such as PTA or stent implantation, had been performed in patients with lower limb ischaemia (stages IIb, III, or IV according to the Fontaine classification) from 1 to 18 months before recruitment. The participants were recruited between August 2010 and June 2012 based on a medical interview, physical examination, and some haematological and biochemical tests. All PAD patients provided informed consent to participate in the study. The study was approved by the Bioethical Committee at the Regional Specialist Hospital in Wroclaw, approval no. KB/2/2008, and the study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki.
Lower limb ischaemia was diagnosed based on clinical symptoms, physical examination, and ankle‐brachial pressure index (ABPI) measurement. Before revascularisation and after restenosis formation, vascular ultrasound, computed tomography, and/or arteriography were performed; walking tests were conducted; and ABPI was measured. After revascularisation, the health status of patients was assessed every 3 months, 5 times during the period of the experiment, with vascular ultrasound and blood collection.
Of 142 PAD patients included in the study, 67 (47.2%) also had various forms of ischaemic heart disease (some with a history of myocardial infarction), 120 (84.5%) arterial hypertension, and 86 (60.6%) dyslipidaemia; 90 (63.4%) were ex‐smokers, 33 (23.2%) were current smokers, and 19 (13.4%) never smoked. Type 2 diabetes was observed in 87 (60.8%) and overweight and obesity in 106 (74.1%). Patients with diabetes maintained adequate glycaemic control on diet, oral hypoglycaemic agent treatment, insulin alone, or a combination of oral anti‐diabetic drugs and insulin. In 16 patients (8 diabetic, 8 non‐diabetic), superficial trophic changes in the foot (ulceration or necrosis) were found, which were classified as uninfected or mildly infected—grade 1 to 2 according to Infectious Diseases Society of America/International Working Group on the Diabetic Foot (IDSA/IWGDF). None of the patients presented signs of a systemic inflammatory response. As a result of the treatment, in 6 cases, the wounds were healed completely during the study period. In other patients, the wounds were stable or partially healed. Peripheral endovascular revascularisation was performed on the following arteries: iliac, femoral superficial, popliteal, and crural. In 21 PAD patients, a restenosis occurred within 6 months after revascularisation.
The PAD patients’ plasma samples were divided into groups in relation to the time period after revascularisation was performed: (1) from 1 to 3 months; n = 73, 49 men and 24 women, aged 48 to 86 (64.7 ± 9.0) years; (2) from 4 to 6 months; n = 85, 54 men and 31 women, aged 50 to 88 (66.2 ± 9.4) years; (3) from 7 to 12 months; n = 178, 117 men and 61 women, aged 45 to 88 (66.2 ± 9.7) years; (4) 13 to 24 months; n = 202, 121 men and 81 women, aged 5 to 89 (67.8 ± 9.5) years; and (5) from 25 to 37 months; n = 46, 29 men and 17 women, aged 45 to 83 (67.0 ± 8.8) years.
The PAD patients’ group was also divided into 2 subgroups with respect to restenosis occurrence: restenosis, n = 37, 19 men and 18 women, aged 48 to 81 (64.0 ± 8.6) years, and without restenosis, n = 105, 68 men and 37 women, aged 45 to 88 (66.7 ± 10.0) years; 2 subgroups with respect to comorbid diabetes: diabetes, n = 87, 53 men and 34 women, aged 45 to 88 (65.7 ± 9.1) years, and without diabetes, n = 55, 34 men and 21 women, aged 47 to 88 (66.5 ± 10.8) years; 2 subgroups with respect to ulcer: ulcer, n = 16, 10 men and 6 women, aged 55 to 86 (66.5 ± 8.9) years, and without ulcer, n = 126, 76 men and 50 women, aged 45 to 88 (66.0 ± 9.8) years. The normal reference group (N), consisting of 37 healthy adult volunteers, 19 men and 18 women, aged 40 to 90 (64.7 ± 12.7) years, included in the study after informed consent had been obtained, was recruited from the staff and their family members of Wrocław Medical University as well as individuals attending the Wroclaw Biobank (a joint project of Wroclaw Research Centre EIT Plus and Wroclaw Medical University), who were not under the care of a medical specialist due to any acute or chronic illness and for whom the results of the basic laboratory parameters included in the study were within normal limits.
2.2. Sampling
The venous blood (4.5 mL) was drawn from an antecubital vein after overnight fasting into whole blood tubes with 0.106 mol/L buffered trisodium citrate with a 9:1 blood to anticoagulant ratio. The plasma was obtained by centrifugation of blood samples at 2500g for 15 minutes. The samples were aliquoted to 0.2 mL portions and stored at −76°C until analysis. Before use, frozen samples were thawed for 1 hour at 20°C to allow solubilisation of FN from possible aggregates.
2.3. Quantification of FN and EDA‐FN
The levels of total FN and the EDA‐FN isoform were determined by 2 enzyme‐linked immunosorbent assays (ELISA)16, 31 based on the immunoreactivity of FN with the 2 specific, well‐defined monoclonal antibodies anti‐FN (FN30‐8; M010; TaKaRa Bio Inc., Shiga, Japan) and anti‐EDA‐FN (MAB 1940; Chemicon International Inc., Temecula, California), respectively. Briefly, anti‐FN diluted 1:10 000 or anti‐EDA‐FN diluted 1:3000 was used as a coating agent in the wells of a microtitre plate (Nalge Nunc International, Naperville, Illinois) as well as to bind FN or EDA‐FN from the samples. For total FN measurements, a human plasma FN standard (Sigma Chemical Co, St. Louis, Missouri) at a concentration of 3 to 50 ng/well and plasma samples that were 4000‐ and 8000‐fold diluted were used. For the EDA‐FN test, the cellular FN from human foreskin fibroblasts (Sigma Chemical Co) from 3 to 50 ng/well as standard and plasma samples that were 25‐, 50‐, and 100‐fold diluted were used. The amount of FN bound was quantified by primary rabbit anti‐FN antibodies (Sigma Chemical Co), diluted to 1:5000 for total FN and 1:3500 for EDA‐FN, and secondary goat anti‐rabbit peroxidase conjugated immunoglobulin (Sigma Chemical Co), diluted to 1:30 000 for total FN and 1:3000 for EDA‐FN. Then, the wells were assayed by a colorimetric reaction using the H2O2/ortho‐phenylenediamine dihydrochloride system and measured in absorbance units (AU) in a Stat Fax 2100 Microplate Reader (Awareness Technology Inc., Palm City, Florida) at 492 nm, with 630 nm as a reference filter. All ELISA immune binding and washing steps, except the coating step, were carried out in Tris‐buffered saline (TBS), pH 7.4, containing 0.1% Tween 20 (TBS‐T). For total FN, 0.2% bovine serum albumin in TBS‐T and 0.2% Tween 20 for the EDA‐FN test were used as blocking agents. Both the total FN and EDA‐FN concentrations are given in mg/l.
2.4. SDS‐polyacrylamide gel electrophoresis
Plasma samples, each containing a constant amount of 300 ng of FN, were subjected to sodium dodecyl sulfate (SDS)/7.5% polyacrylamide gel electrophoresis under reducing conditions according to Laemmli,32 subsequently blotted onto nitrocellulose (SERVA Electrophoresis GmbH, Heidelberg, Germany), blocked with 3% non‐fat milk in TBS, and developed with anti‐FN monoclonal antibody (FN30‐8; TaKaRa Shuzo Co. Ltd., diluted 1:5000) and with rabbit anti‐mouse immunoglobulins conjugated with horseradish peroxidase (Sigma Chemical Co., diluted 1: 10 000) as described previously.31 The colour reaction was carried out using the H2O2/3,3‐diaminobenzidine system. The bands were scanned and analysed by densitometry using GelScan V6.0 (BioSciTec GmbH, Frankfurt/Main, Germany). Human haptoglobin type 2‐1 preparation,33 which is known to form a stable, distinct series of polymers with the molecular masses of the first 5, 107, 162, 217, 274, and 331 kDa, was used as the molecular mass standard.34
2.5. Semi‐denaturing SDS‐agarose gel electrophoresis
Semi‐denaturing SDS‐agarose gel electrophoresis and subsequent capillary transfer for analysis of soluble plasma supramolecular FN‐fibrin complexes were carried out as described previously.25 The plasma sample containing 300 ng of FN was loaded on 1.5% agarose (Standard Low ‐mr, Bio‐Rad Laboratories, Hercules, California) prepared previously as a horizontal slab gel in Tris‐acetate‐EDTA (TAE) buffer (40 mM Tris, 20 mM acetic acid, 1 mM ethylenediaminetetraacetic acid (EDTA), pH 8.5) with 0.1% SDS and then separated in TAE buffer with 0.1% SDS at 60 V for 3 hours in the Sub‐Cell GT System (Bio‐Rad Laboratories), transferred to a nitrocellulose membrane (Serva Electrophoresis GmbH) with TBS buffer, and left overnight. The membrane was blocked with 3% non‐fat milk in TBS and subjected to immunoblotting with anti‐FN monoclonal antibody (FN30‐8; TaKaRa Shuzo Co. Ltd., diluted 1:10 000) and rabbit anti‐mouse immunoglobulins conjugated to horseradish peroxidase (Sigma, diluted 1:5000). The colour reaction was developed with hydrogen peroxide/3,3′‐diaminobenzidine. The immunoblots were scanned and analysed by densitometry using GelScan V6.0 (BioSciTec GmbH). The relative amounts of the FN molecular forms were expressed as the percentage of the total number of pixels in a lane. The von Willebrand factor (vWF, TaKaRa Shuzo Co. Ltd.) present in normal human plasma as a series of defined polymers in multiples of 450 kDa and a preparation of plasma FN (Sigma Chemical Co.) were used as standard molecular masses, which were detected using a rabbit anti‐human vWF (Dako, Glostrup, Denmark, 1:10 000) and goat anti‐rabbit peroxidase‐labelled IgG (Sigma Chemical Co.; 1:20 000).
2.6. Statistics
The variables associated with the occurrence of the FN monomer and FN‐fibrin complexes were non‐normally right skewed distributed, while those of FN concentration showed almost normal distribution, so non‐parametric statistical tests were used. Comparisons between 2 independent groups were performed by using the Mann‐Whitney U test, whereas Kruskal‐Wallis non‐parametric analysis of variance (ANOVA) and post‐hoc analysis (multiple comparison range test) tests were used to compare more than 2 groups. Frequency of occurrence is the ratio of the number of samples containing the FN form to the total number of samples. Data are presented as means ± SDs, median, and (25th and 75th) quartiles, and P values lower than .05 were regarded as significant in each statistical test. All the statistical analyses were performed using Statistica version 12 PL (StatSoft Inc., Tulsa, Oklahoma).
3. RESULTS
3.1. Plasma FN and EDA‐FN
3.1.1. Dynamics within post‐revascularisation period
The mean value of FN concentration in the plasma of PAD patients 1 to 3 months after revascularisation (Table 1: 261.7 ± 56.4 mg/L) was similar to that of the age‐matched normal group (270.1 ± 70.7 mg/L). For the samples taken at 4 to 6 months and 7 to 12 months, it was significantly higher (291.7 ± 71.0 mg/L; P < .01 and 295.4 ± 70.6 mg/L; P < .0003, respectively), and finally, after 1 year, it decreased to the value observed in the normal age‐matched group.
Table 1.
FN and EDA‐FN concentrations in plasma of PAD patients in relation to period after revascularisation
| Plasma FN | FN concentration (mg/L) | |||||
|---|---|---|---|---|---|---|
| Groups of plasma PAD samples after revascularisation | ||||||
| (A) 1‐3 months | (B) 4‐6 months | (C) 7‐12 months | (D) 13‐24 months | (E) 25‐37 months | (N) Normal age‐matched group | |
| FN |
n = 73 261.7 ± 56.4 256.0 220.8‐288.0 |
n = 85 291.7 ± 71.0 289.3 248.0‐333.0 (A) P < 0.01 |
n = 178 295.4 ± 70.6 295.3 254.3‐331.4 (N) P < 0.04 (A) P < 0.0003 (D) P < 0.04 |
n = 202 278.3 ± 64.8 271.3 231.0‐312.7 |
n = 46 274.2 ± 58.7 265.3 233.5‐317.3 |
n = 37 270.1 ± 70.7 264.1 207.3‐327.0 |
| EDA‐FN |
n = 73 5.3 ± 6.6 3.9 2.7‐4.8 (N) P < 0.0000 |
n = 82 3.9 ± 3.6 3.1 2.4‐4.1 (N) P < 0.0004 |
n = 135 4.1 ± 3.4 2.9 2.4‐4.7 (N) P < 0.0001 |
n = 117 4.2 ± 3.4 3.4 2.4‐4.8 (N) P < 0.0001 |
n = 27 3.4 ± 1.7 2.9 2.3‐4.3 (N) P < 0.03 |
n = 37 2.5 ± 0.9 2.3 2.0‐3.4 |
| % EDA‐FN to FN |
n = 73 2.1 ± 2.6 (N) P < 0.0000 |
n = 82 1.4 ± 1.4 (A) P < 0.02 |
n = 135 1.5 ± 1.2 (N) P < 0.02 (A) P < 0.006 |
n = 117 1.6 ± 1.5 (N) P < 0.0009 |
n = 27 1.3 ± 0.7 |
n = 37 1.0 ± 0.5 |
FN and EDA‐FN concentrations were determined by 2 independent ELISA using specific monoclonal antibodies anti‐FN (FN30‐8; M010; TaKaRa Bio Inc., Shiga, Japan)31 and anti‐EDA‐FN (MAB 1940; Chemicon International Inc., Temecula, California).16 For details, see section 2. Data are presented as mean values ± SD, median, and (25th and 75th) quartiles. Significantly different from groups: (N) normal age‐matched, (A) 1 to 3 months, and (D) 13 to 24 months after treatment. Statistical analysis was performed using the Mann‐Whitney U test to compare 2 independent patient and reference groups and Kruskal‐Wallis non‐parametric ANOVA and post‐hoc analysis for comparing among patient subgroups.
The high mean value of EDA‐FN concentration in the plasma of PAD patients after revascularisation found for the group sampled at 1 to 3 months (5.3 ± 6.6 mg/L) decreased gradually but negligibly in the subsequent time intervals of 4 to 6 months (3.9 ± 3.6 mg/L), 7 to 12 months (4.1 ± 3.4 mg/L), 13 to 24 months (4.2 ± 3.4 mg/L), and 25 to 37 months (3.4 ± 1.7 mg/L). All above mentioned EDA‐FN values were significantly higher than that of the age‐matched normal group (2.5 ± 0.9 mg/L).
3.1.2. Dependence related to restenosis occurrence
For 37 months after revascularisation, no differences were observed in the plasma FN concentration of PAD patients between the groups with and without the occurrence of restenosis (Figure 4A). In contrast, during the first year after revascularisation, the EDA‐FN concentration (Figure 4B) showed an ascending pattern in the restenosis group and descending in the group without restenosis. Moreover, the EDA‐FN concentration was significantly higher in the restenosis group sampled within 7 to 12 months after revascularisation (5.4 ± 2.5 mg/L; P < .0005) in comparison with that in the group without restenosis (3.9 ± 3.5 mg/L).
Figure 4.
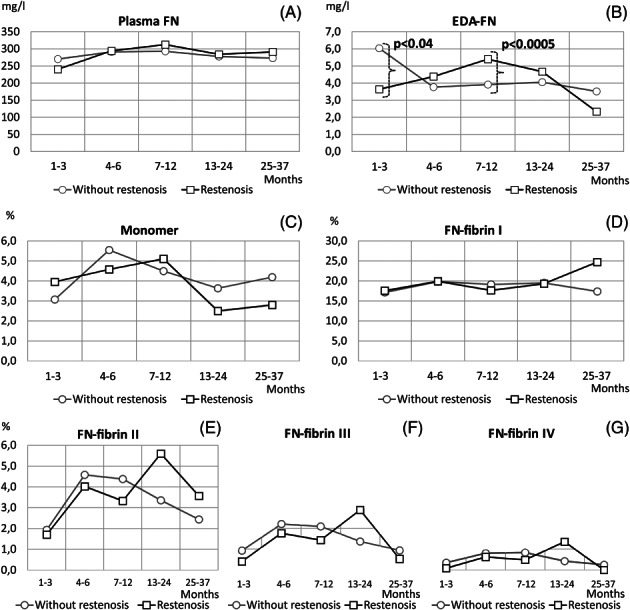
Levels of FN molecular forms in plasma of PAD patients with respect to restenosis occurrence. Mean values of data: concentrations (mg/l) of plasma FN (A) and EDA‐FN (B); relative amounts (%) of FN monomer (C) and FN‐fibrin complexes I (D), II (E), III (F), and IV (G) shown with respect to occurrence of restenosis (—□—) and without restenosis (—○—) after revascularisation: 1 to 3 months (without restenosis: n = 54, restenosis: n = 23), 4 to 6 months (without restenosis: n = 66, restenosis: n = 19), 7 to 12 months (without restenosis: n = 155, restenosis: n = 23), 13 to 24 months (without‐restenosis: n = 180, restenosis: n = 22), and 25 to 37 months (without restenosis: n = 43, restenosis: n = 3)
3.1.3. Dependence related to comorbid diabetes and ulcer
For 37 months after revascularisation, no differences were observed in the plasma FN concentration of PAD patients with comorbid diabetes (Figure 2A) and with ulceration (Figure 3A) when compared with the respective groups without these disorders. On the other hand, the EDA‐FN concentration of PAD patients with diabetes (Figure 2B) exclusively in the group sampled 1 to 3 months after revascularisation (3.9 ± 2.5 mg/L) and with ulceration (Figure 3B) in the group sampled 4 to 6 months after revascularisation (2.4 ± 0.5 mg/L) was significantly lower when compared with the corresponding groups without these disorders (7.4 ± 9.5 mg/L, P < .02 and 4.1 ± 3.7 mg/L, P < .009, respectively).
Figure 2.
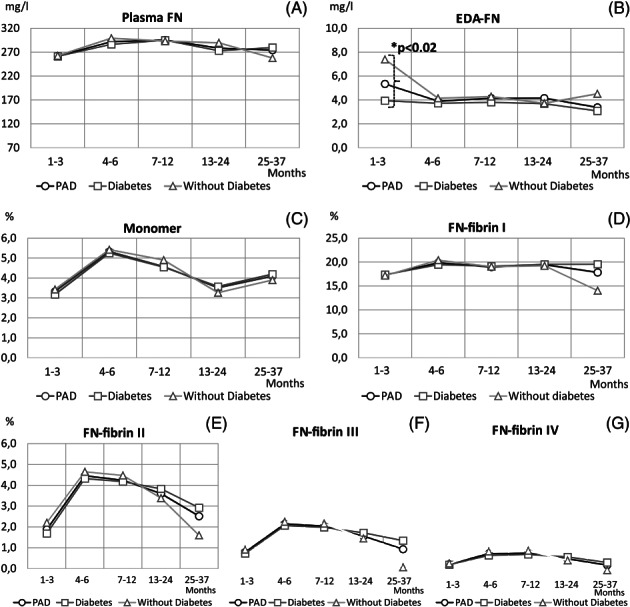
Levels of FN molecular forms in plasma samples of PAD patients with respect to comorbid diabetes. Mean values of data: concentrations (mg/l) of plasma FN (A) and EDA‐FN (B); relative amounts (%) of FN monomer (C) and FN‐fibrin complexes I (D), II (E), III (F), and IV (G) of all PAD patients (—○—), PAD patients with diabetes (—□—), and PAD patients without diabetes (—Δ—) shown in relation to period after revascularisation: 1 to 3 months (diabetes: n = 42, without diabetes: n = 30), 4 to 6 months (diabetes: n = 49, without diabetes: n = 36), 7 to 12 months (diabetes: n = 98, without diabetes: n = 66), 13 to 24 months (diabetes: n = 120, without diabetes: n = 66), and 25 to 37 months (diabetes: n = 30, without diabetes: n = 12). *Significantly different between groups of PAD with comorbid diabetes and PAD with diabetes excluded
Figure 3.
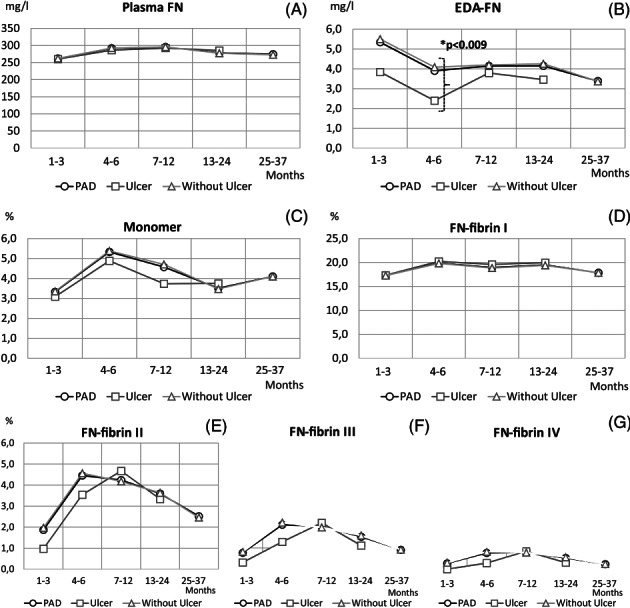
Levels of FN molecular forms in plasma samples of PAD patients with respect to ulceration. Mean values of data: concentrations (mg/l) of plasma FN (A) and EDA‐FN (B); relative amounts (%) of FN monomer (C) and FN‐fibrin complexes I (D), II (E), III (F), and IV (G) of all PAD patients (—○—), PAD patients with previous ulcer (—□—), and PAD patients without ulcer (—Δ—) shown in relation to period after revascularisation: 1 to 3 months (ulcer: n = 7, without ulcer: n = 66), 4 to 6 months (ulcer: n = 9, without ulcer: n = 76), 7 to 12 months (ulcer: n = 24, without ulcer: n = 154), 13 to 24 months (ulcer: n = 26, without ulcer: n = 176), and 25 to 37 months (ulcer: n = 0, without ulcer: n = 45). *Significantly different between groups of PAD with comorbid ulcer and PAD with ulcer excluded
3.2. Plasma FN‐fibrin complexes
SDS‐agarose gel electrophoresis of PAD plasma samples under non‐reducing conditions with subsequent FN blotting demonstrated the presence of up to 8 FN bands (Figure 1), with molecular masses corresponding to an FN dimer (500 kDa), FN monomer (~250 kDa), and FN‐fibrin complexes I to VI (750, 1000, 1300, 1600, 1900, and 2200 kDa, respectively). FN‐fibrin complex I occurred in nearly all PAD plasma samples, complexes I to II in 39% to 67%, I to IV in 10% to 28%, I to VI in up to 2%, and no FN‐fibrin complexes were detected in 1% of PAD samples (Table 2). Their presence and relative amount were related to the period of time after revascularisation (Table 2) and restenosis occurrence (Figure 4D‐G). In contrast, 75.7% of samples of the age‐matched normal group exclusively demonstrated the presence of the FN‐fibrin complex I (Figure 1, lanes 1 to 2; Table 2).
Figure 1.
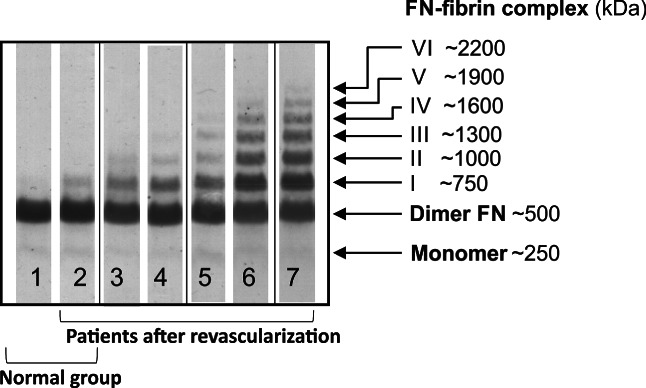
Representative immunopatterns of FN‐fibrin complexes in plasma of PAD patients after revascularisation of ischaemic lower limbs. Blood plasma samples of 142 PAD patients and 37 normal individuals were subjected to SDS‐agarose gel electrophoresis under non‐reducing conditions (25). The bands were transferred to a nitrocellulose membrane (Serva Electrophoresis GmbH, Heidelberg, Germany) and developed with anti‐FN monoclonal antibody (Clone FN30‐8, TaKaRa Shuzo Co. Ltd., Shiga, Japan). Lanes 1 to 2, normal plasma; lanes 2 to 7, PAD patients’ plasma
Table 2.
Occurrence and relative amount of FN monomer and FN‐fibrin complexes in plasma of PAD patients in relation to the period after revascularisation
| FN or FN‐fibrin complex (kDa) | Frequency of occurrence and relative amount (%) of FN bands showed by agarose immunoblotting (Figure 1) | ||||||
|---|---|---|---|---|---|---|---|
| Groups of plasma PAD samples after revascularisationNumber of samples (n) | |||||||
| (A) 1‐3 months (73) | (B) 4‐6 months (85) | (C) 7‐12 months (178) | (D) 13‐24 months (202) | (E) 25‐37 months (46) | (N) Normal age‐matched (37) | ||
| Monomer (230‐250) |
76.3% (56) 3.3 ± 3.0 |
91.8% (78) 5.3 ± 4.1 (N) P < 0.02 (A) P < 0.009 |
92.7% (165) 4.6 ± 3.2 |
89.6% (181) 3.5 ± 2.8 (B) P < 0.002 (C) P < 0.01 |
95.7% (44) 4.1 ± 2.5 |
89.2% (33) 3.5 ± 2.2 |
|
| Dimer (460‐500) |
100% (73) 76.3 ± 14.2 |
100% (85) 67.3 ± 15.1 |
100% (178) 69.1 ± 16.2 |
100% (202) 71.2 ± 14.2 |
100% (46) 74.3 ± 11.6 |
100% (37) 89.6 ± 6.3 |
|
| FN‐fibrin complexes | I (750) |
91.8% (67) 17.3 ± 9.1 (N) P < 0.0000 |
100% (85) 19.9 ± 7.1 (N) P < 0.0000 |
100% (178) 18.9 ± 7.4 (N) P < 0.0000 |
99.5% (201) 19.5 ± 7.0 (N) P < 0.0000 |
100% (46) 17.8 ± 7.5 (N) P < 0.0000 |
75.7% (28) 5.0 ± 4.0 |
| II (1000) |
38.4% (28) 1.9 ± 3.6 (N) P < 0.0009 |
65.9% (56) 4.5 ± 5.1 (N) P < 0.0000 (A) P < 0.0007 |
63.5% (113) 4.2 ± 5.3 (N) P < 0.0000 (A) P < 0.0005 |
67.3% (136) 3.6 ± 4.4 (N) P < 0.0000 (A) P < 0.001 |
58.7% (27) 2.5 ± 3.0 (N) P < 0.0000 |
0 | |
| III (1300) |
23.2% (17) 0.8 ± 2.1 (N) P < 0.047 |
45.9% (39) 2.1 ± 3.3 (N) P < 0.0000 (A) P < 0.04 |
44.9% (80) 2.0 ± 3.4 (N) P < 0.0000 (A) P < 0.03 |
40.6% (82) 1.5 ± 2.9 (N) P < 0.0000 |
34.8% (16) 0.9 ± 2.1 (N) P < 0.006 |
0 | |
| IV (1600) |
9.6% (7) 0.3 ± 1.2 |
28.2% (24) 0.8 ± 1.8 (N) P < 0.01 |
21.9% (39) 0.8 ± 2.0 (N) P < 0.002 |
16.8% (34) 0.5 ± 1.5 |
15.2% (7) 0.2 ± 0.9 |
0 | |
| V (1900) |
5.5% (4) 0.1 ± 0.6 |
9.4% (8) 0.2 ± 0.8 |
11.2% (20) 0.3 ± 1.1 |
7.9% (16) 0.1 ± 0.7 |
4.3% (2) 0.03 ± 0.1 |
0 | |
| VI (2200) |
2.7% (2) 0.04 ± 0.3 |
2.4% (2) 0.04 ± 0.3 |
1.7% (3) 0.03 ± 0.3 |
1.5% (3) 0.03 ± 0.3 |
0 | 0 | |
The bands of plasma FN forms corresponding to FN monomer, dimer, and FN‐fibrin complexes I to VI were showed as described previously25 and shown in Figure 1. The bands were scanned and analysed by densitometry using GelScan V6.0 (BioSciTec GmbH, Frankfurt/Main, Germany). Data are presented as the percentage of the occurrence frequency (number of samples containing the band) and relative amount (%) of FN band as mean values ± SD. For details, see section 2.
Statistical analysis was performed using the Mann‐Whitney U test to compare 2 independent patient and reference groups and Kruskal‐Wallis non‐parametric ANOVA and post‐hoc analysis for comparing among patient subgroups. Significantly different from (N) normal age‐matched group and patients’ group of (A) 1 to 3 months, (B) 4 to 6 months, and (C) 7 to 12 months after revascularisation.
3.2.1. Relative amount of FN‐fibrin complexes during post‐revascularisation period
The relative amounts of FN‐fibrin complexes I to III within 1 to 37 months after revascularisation (Table 2, P < .00001) and complex IV within 4 to 6 and 7 to 12 months after revascularisation (Table 2, P < .01 and P < .002) showed significantly higher mean values than those in the normal age‐matched group. FN‐fibrin complexes V and VI occurred in a few samples in relatively small amounts.
The relative amount of the 750‐kDa FN‐fibrin complex I (Table 2) was nearly at a stable level in the plasma in the periods 1 to 3 (17.3% ± 9.1%), 4 to 6 (19.9% ± 7.1%), 7 to 12, 13 to 24 (19.5% ± 7.0%), and 25 to 37 (17.8% ± 7.5%) months after revascularisation; however, those values were significantly higher (P < .00001) than in the age‐matched normal group (5.0% ± 4.0%).
The relative amounts of FN‐fibrin complexes II to IV in the PAD patients’ samples within 4 to 6 and 7 to 12 months after revascularisation (Table 2) increased more than 2‐fold compared with that of 1 to 3 months after revascularisation. From the 13th month after revascularisation, the relative amounts of FN‐fibrin complexes II to IV decreased gradually. Their relative amounts were significantly higher (Table 2) than in the age‐matched normal group.
3.2.2. FN‐fibrin complexes with respect to restenosis occurrence
No significant differences were observed between groups with and without restenosis in the relative amount of the 750‐kDa FN‐fibrin complex I within 37 months of observation. The relative amounts of FN‐fibrin complexes II to IV were higher in the restenosis group than in those without restenosis (complex II: 5.6% ± 6.4% and 3.4% ± 4.1%; complex III: 2.9% ± 4.2% and 1.4% ± 2.6%; complex IV: 1.3% ± 2.5% and 0.4% ± 1.3%) (Figure 4D‐G).
3.2.3. FN‐fibrin complexes with respect to comorbid diabetes and ulcer
During 24 months after revascularisation, no significant differences were observed in the relative amounts of FN‐fibrin complexes I to IV between PAD groups with and without comorbid diabetes (Figure 2D‐G) as well as ulceration (Figure 3D‐G). However, the mean values of the relative amounts of FN‐fibrin complexes I to III in the period 25 to 37 months after revascularisation were higher in the PAD group with comorbid diabetes (Figure 2D‐F).
3.3. FN monomer
The FN monomer band occurred in PAD patients’ plasma 1 to 3 months after revascularisation (76.3%) less frequently than in the next 4 to 37 months (89.6%‐95.7%) and in the age‐matched normal group (89.2%) (Table 2).
3.3.1. Relative amount of FN monomer during post‐revascularisation period
The relative amount of FN monomer (Table 2) in PAD patients’ plasma 1 to 3 months after revascularisation (3.3% ± 3.0%) was at the same level as in the age‐matched group (3.5% ± 2.2%); then, it increased significantly in the 4 to 6 months group (5.3% ± 4.1%, P < .009) and gradually decreased in the groups sampled at 7 to 12, 13 to 24, and 25 to 37 months after revascularisation (4.6% ± 3.2%, 3.5% ± 2.8%, and 4.1% ± 2.5%, respectively).
3.3.2. Relative amount of FN monomer with respect to restenosis occurrence
The differences in the relative amounts of FN monomer between groups with restenosis and without restenosis were non‐significant within 1 year after revascularisation (the 1‐3, 4‐6, and 7‐12 months groups), but in the later period, it became significantly lower in the restenosis group at 13 to 24 months (2.5% ± 2.2%, P < .0006) after revascularisation (Figure 4C).
3.3.3. Relative amount of FN monomer with respect to comorbid diabetes and ulcer
No significant difference was observed in the relative amount of the FN monomer between PAD groups with and without comorbid diabetes (Figure 2C) as well as ulceration (Figure 3C) within 37 months of observation.
4. DISCUSSION
Our results demonstrated that the FN molecular forms (FN and its EDA isoform, FN‐fibrin complexes, and FN monomer) present in the plasma of patients with PAD showed increased levels within the first year after the endovascular intervention, reaching values close to those in the normal age‐matched group up to 37 months. However, maintaining or further increasing high levels of plasma EDA‐FN at 7 to 12 months and FN‐fibrin complexes at 13 to 24 months after revascularisation in PAD patients’ plasma was found to be related to the occurrence of restenosis. Moreover, decreasing EDA‐FN concentration during the first 6 months after revascularisation was associated with changes influenced by comorbid diabetes and/or ulceration. In contrast, concentration of FN and levels of FN monomer and FN‐fibrin complexes in plasma of PAD patients were not directly associated with comorbid diabetes or tissue ulceration.
The analyses of the levels of FN and EDA‐FN (Table 1) and the showed bands of FN‐fibrin complexes and FN monomer/s (Figure 1 and Table 2) in the plasma of PAD patients after revascularisation allowed us to distinguish 3 main time‐dependent stages, arbitrarily named by us as: (1) early, up to 3 months after revascularisation, with twice as high concentration of EDA‐FN (5.3 ± 6.6 mg/L) as in the normal group (2.5 ± 0.9 mg/L, P < .00001) and with the occurrence of a series of supramolecular FN‐fibrin complexes; (2) intermediate, from 4 to 24 months, with a still high EDA‐FN concentration (3.9 ± 3.6 to 4.1 ± 3.4 mg/L) and a significant increase of the total FN concentration (295 ± 70.6 mg/L; P < .0003), relative amounts of supramolecular FN‐fibrin complexes (P < .00001), and FN monomer (P < .009) compared with the data for the first stage (FN: 261 ± 56.4 mg/L) and normal age‐matched (FN: 274 ± 58.7 mg/L) groups; and (3) late, 2 years after revascularisation and medical therapy, in which the mean values of the analysed parameters become lower although negligibly higher than in the normal age‐matched group.
The mean values of the FN molecular forms analysed with respect to the mentioned time periods showed high values of SD, which may result from the individual course of PAD, degree of atherosclerotic changes, therapies used, diseases accompanying aging (mean age of patients was higher than 60 years) as well as variable ability of the patients’ immunological system to respond to inflammatory agents. In spite of the above objections, the following scenario for FN molecular form engagement during the post‐revascularisation period can be postulated. The hepatic‐origin plasma FN, present in a significantly high concentration up to 12 months after medical intervention (Table 1, Figure 4), might opsonise and adhere to different cells, facilitating cell migration and, through phagocytosis, remove cellular debris.8 In addition, during tissue wound healing, the requirements for FN become greater, and the plasma FN can be used in the early phase of controlled repair of wounds. It influences the initial rate of fibrin structure matrix formation,35 and through binding with other ECM molecules (ie, fibrin, collagen, GAG), it forms a provisional matrix.6, 11, 36, 37
The vascular injuries provoked by the atherosclerotic lesions induce chronic, low‐grade inflammation. Accumulated leukocytes in the injured artery as well as endothelial and smooth muscle cells produce pro‐inflammatory molecules, including EDA‐FN.15 EDA‐FN with its known pro‐inflammatory and pro‐thrombotic activities15 is secreted at high concentrations into the plasma of PAD patients during the first and second stages after revascularisation (Table 1) and with disease progression and restenosis occurrence (Figure 4B). Some changes in EDA‐FN concentration have also been found in the plasma of patients with advanced coronary artery disease,27 thrombosis,15, 38 and diabetes.39, 40, 41
Diabetes and ulceration were reported to be associated with increased deposition of FN in ECM and linked with the occurrence of FN modified by glycation and/or hypoxia. Such structural modification may cause its reduced functionality.39, 41, 42, 43, 44 The lack of EDA‐FN as well as a low concentration of functional EDA‐FN may lead to exacerbation of vascular damage and delay in normal tissue remodelling.45 Thus, in our opinion, the low mean values of plasma EDA‐FN concentration in the groups of PAD patients with comorbid diabetes in the period of 1 to 3 months after revascularisation (3.9 ± 2.5 mg/L) (Figure 2B) and PAD complicated by ulceration within 3 to 6 months after revascularisation (2.4 ± 0.5 mg/L) (Figure 3B), compared with those of non‐diabetic (7.4 ± 3.5 mg/L; P < .02) and ulcer‐free (4.1 ± 3.7 mg/L; P < .009) groups, might be related to its insufficient local production by impaired endothelium and other cells involved in arterial remodelling and its deposition in the ECM. However, it cannot be excluded that modified EDA‐FN was also not efficiently recognised by the monoclonal antibody used in FN‐ELISA, and in consequence, we observed the decreased EDA‐FN concentration.
Simultaneously, in oxidoreductive conditions accompanying vessel wall inflammation and atherosclerosis, and vascular remodelling, some FN molecules undergo degradation, producing FN monomers (Figure 1, Table 2). FN degradation products and dimers of FN have a tendency to associate among themselves and to form complexes with fibrin, a product of fibrinogen conversion during coagulation cascade activation initiated by tissue factor released from damaged endothelium.46 The fibrils of FN‐fibrin are deposited rapidly in the site of arterial injury, where they may play a dual role. On one hand, they support platelet aggregation and clot formation, take part in normal haemostasis, and initiate tissue remodelling.6, 8 On the other hand, pFN, alone and through interactions with fibrin, is able to switch from supporting haemostasis to inhibiting thrombosis and vessel occlusion depending on the fibrin gradient and, thereby, is a self‐limiting regulator in thrombosis.21, 22 Moreover, it is reported that through the uptake of fibrin into FN‐fibrin complexes, plasma FN can protect against extravascular accumulation of fibrin. Moreover, the FN‐fibrin interaction may also be involved in fibrin clearance from the circulation in inflammatory conditions (28).
In most plasma samples, the relative amounts of FN‐fibrin complexes gradually decreased from 12 months after revascularisation, probably when the tissue remodelling and wound healing ended. In contrast, the recurrence of stenosis was associated with significant increasing levels of EDA‐FN (Figure 4B) and FN‐fibrin complexes (Figure 4D‐G) (particularly those of masses 1000 kDa and higher) in the plasma of PAD patients within 7 to 12 months after the medical intervention. The maintained high level of FN‐fibrin complexes in PAD patients’ plasma samples might reflect a hypercoagulable state. Such a situation might also be associated with delayed arterial healing,47 excessive deposition of matrix components in a vessel wall, destruction of normal tissue architecture, failure of transition to a mature matrix, and recurrence of stenosis.
The described patterns of increasing levels of liver‐derived plasma FN and EDA‐FN released from cells to the circulation, soluble FN‐fibrin complexes, and FN degradation products in plasma of PAD patients were dynamic within about 1 year after revascularisation and demonstrated a natural tendency to reach nearly normal values up to 2 years. However, the increasing concentration of EDA‐FN within the first year after revascularisation and high relative amounts of macromolecular FN‐fibrin complexes in PAD patients maintained up to 2 years after revascularisation may be molecular signs of a risk of restenosis occurrence.
5. CONCLUSION
Some molecular forms of FN are involved in time‐ and stage‐dependent and inter‐related processes of inflammation, coagulation, and wound healing accompanying PAD. The increasing levels of liver‐derived plasma FN and EDA‐FN released from cells to the circulation, soluble FN‐fibrin complexes, and FN degradation products in plasma of PAD patients were dynamic within about 1 year after revascularisation and showed a natural tendency to reach nearly normal values up to 2 years. However, the decrease of EDA‐FN concentration in plasma of PAD patients in the first months of recovery after revascularisation might be connected with coexisting diabetes and ulceration. Moreover, the increasing concentration of EDA‐FN within the first year after revascularisation and high relative amounts of macromolecular FN‐fibrin complexes in PAD patients maintained up to 2 years after revascularisation may be molecular signs of a risk of restenosis occurrence. The determination of FN molecular forms could have potential diagnostic value in the management of PAD patients after revascularisation.
ACKNOWLEDGEMENTS
The research was supported by the projects of WROVASC‐Integrated Centre of Cardiovascular Medicine cofunded by the European Regional Development Fund (POIG.01.01.02‐02‐001/08‐06), Innovative Economy Operational Program 2007 to 2013 1‐1, and Wroclaw Centre of Biotechnology, the Leading National Research Centre (KNOW) programme for the years 2014 to 2018.
Conflict of interest
The authors have no competing interests to declare.
Author contribution
M. P. designed study, performed research, analyzed data, wrote the paper. D. K.‐G. performed research, analyzed data. W. W. conceived of study. D. K. designed study. M. K. analyzed data, designed study. W. K. wrote the paper. I. K.‐P. conceived of and designed study, analyzed data, wrote the paper.
Pupek M, Krzyżanowska‐Gołąb D, Kotschy D, et al. Time‐dependent changes in extra‐domain A‐fibronectin concentration and relative amounts of fibronectin‐fibrin complexes in plasma of patients with peripheral arterial disease after endovascular revascularisation. Int Wound J. 2018;15:649–659. 10.1111/iwj.12909
REFERENCES
- 1. Jones WS, Schmit KM, Vemulapalli S, et al. Treatment Strategies for Patients With Peripheral Artery Disease. Vol 118. Rockville, MD: AHRQ Comparative Effectiveness Review; 2013. Report No.: 13‐EHC090‐EF. [Google Scholar]
- 2. Hiatt WR, Goldstone J, Smith SC Jr, et al. Atherosclerotic peripheral vascular disease symposium II: nomenclature for vascular diseases. Circulation. 2008;118:2826‐2829. [DOI] [PubMed] [Google Scholar]
- 3. Dangas GD, Claessen BE, Caixeta A, Sanidas EA, Mintz GS, Mehran R. In‐stent restenosis in the drug‐eluting stent era. J Am Coll Cardiol. 2010;56(10):1897‐1907. [DOI] [PubMed] [Google Scholar]
- 4. Belay T. Endothelial repair and regeneration following intimal injury. J Cardiovasc Transl Res. 2016;9(2):91‐101. [DOI] [PubMed] [Google Scholar]
- 5. Otsuka F, Byrne RA, Yahagi K, et al. Neoatherosclerosis: overview of histopathologic findings and implications for intravascular imaging assessment. Eur Heart J. 2015;36(32):2147‐2159. [DOI] [PubMed] [Google Scholar]
- 6. Lenselink EA. Role of fibronectin in normal wound healing. Int Wound J. 2015;12:313‐316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang Y, Gallant RC, Ni H. Extracellular matrix proteins in the regulation of thrombus formation. Curr Opin Hematol. 2016;23:280‐287. [DOI] [PubMed] [Google Scholar]
- 8. To WS, Midwood KS. Plasma and cellular fibronectin: distinct and independent functions during tissue repair. Fibrogenesis Tissue Repair. 2011;4:21. 10.1186/1755-1536-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zollinger AJ, Smith ML. Fibronectin, the extracellular glue. Matrix Biol. 2017;60‐61:27‐37. [DOI] [PubMed] [Google Scholar]
- 10. White ES, Baralle FE, Muro AF. New insights into form and function of fibronectin splice variants. J Pathol. 2008;216:1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. White ES, Muro AF. Fibronectin splice variants: understanding their multiple roles in health and disease using engineered mouse models. IUBMB Life. 2011;63:538‐546. [DOI] [PubMed] [Google Scholar]
- 12. Lemańska‐Perek A, Pupek M, Polańska B, Leszek J, Kątnik‐Prastowska I. Alterations in molecular status of plasma fibronectin associated with aging of normal human individuals. Clin Biochem. 2013;46:787‐794. [DOI] [PubMed] [Google Scholar]
- 13. De Haan J, Smeets M, Pasterkamp G, Arslan F. Danger signals in the initiation of the inflammatory response after myocardial infarction. Mediat Inflamm. 2013;2013:206039. 10.1155/2013/206039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Maurer E, Schaff M, Receveur N, et al. Fibrillar cellular fibronectin supports efficient platelet aggregation and procoagulant activity. Thromb Haemost. 2015;114(6):1175‐1188. [DOI] [PubMed] [Google Scholar]
- 15. Chauhan AK, Kisucka J, Cozzi MR, et al. Prothrombotic effects of fibronectin isoforms containing the EDA domain. Arterioscler Thromb Vasc Biol. 2008;28(2):296‐301. [DOI] [PubMed] [Google Scholar]
- 16. Pupek M, Pawłowicz R, Lindner K, et al. Occurrence of fibronectin‐fibrin complexes in plasma of patients with multimorbidity due to the inflamm‐aging phenomenon. Exp Gerontol. 2016;77:19‐28. [DOI] [PubMed] [Google Scholar]
- 17. Moretti FA, Chauhan AK, Iaconcig A, Porro F, Baralle FE, Muro AF. A major fraction of fibronectin present in the extracellular matrix of tissues is plasma‐derived. J Biol Chem. 2007;282:28057‐28062. [DOI] [PubMed] [Google Scholar]
- 18. Maurer LM, Tomasini‐Johansson BR, Mosher DF. Emerging roles of fibronectin in thrombosis. Thromb Res. 2010;125(4):287‐291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yi M, Sakai T, Fässler R, Ruoslahti E. Antiangiogenic proteins require plasma fibronectin or vitronectin for in vivo activity. PNAS. 2003;100:11435‐11438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hou Y, Carrim N, Wang Y, Gallant RC, Marshall A, Ni H. Platelets in hemostasis and thrombosis: Novel mechanisms of fibrinogen‐independent platelet aggregation and fibronectin‐mediated protein wave of hemostasis. J Biomed Res. 2015;29(6):437‐444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang Y, Reheman A, Spring CM, et al. Plasma fibronectin supports hemostasis and regulates thrombosis. J Clin Invest. 2014;124(10):4281‐4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang Y, Ni H. Fibronectin: extra domain brings extra risk? Blood. 2015;125:3043‐3044. [DOI] [PubMed] [Google Scholar]
- 23. Wang Y, Ni H. Fibronectin maintains the balance between hemostasis and thrombosis. Cell Mol Life Sci. 2016;73(17):3265‐3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Barker TH, Engler AJ. The provisional matrix: setting the stage for tissue repair outcomes. Matrix Biol. 2017;60–61:1‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Krzyżanowska‐Gołąb D, Lemańska‐Perek A, Pupek M, et al. Identification of soluble supramolecular FN‐fibrin complexes in human plasma. J Immunoassay Immunochem. 2014;35:412‐427. [DOI] [PubMed] [Google Scholar]
- 26. Lemańska‐Perek A, Polańska B, Krzyżanowska‐Gołąb D, Kątnik‐Prastowska I. Occurrence of soluble supra‐molecular FN‐fibrin complexes in the plasma of children with recurrent respiratory infection. Ann Clin Biochem. 2015;52:441‐447. [DOI] [PubMed] [Google Scholar]
- 27. Lemańska‐Perek A, Krzyżanowska‐Gołąb D, Pupek M, Klimeczek P, Witkiewicz W, Kątnik‐Prastowska I. Analysis of soluble molecular fibronectin‐fibrin complexes and EDA‐fibronectin concentration in plasma of patients with atherosclerosis. Inflammation. 2016;39:1059‐1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pankov R, Yamada KM. Fibronectin at a glance. J Cell Sci. 2002;115:3861‐3863. [DOI] [PubMed] [Google Scholar]
- 29. Kotschy D, Kotschy M, Masłowski L, et al. Inflammatory markers in peripheral arterial disease patients after endovascular revascularization with new restenosis. Acta Angiol. 2014;20(2):47‐59. [Google Scholar]
- 30. Kotschy D, Kotschy M, Socha P, et al. Witkiewicz. Tissue factor and other hemostatic parameters in patients with advanced peripheral artery disease after endovascular revascularization – search for hemostatic factors which indicate restenosis. Adv Clin Exp Med. 2015;24(1):93‐98. [DOI] [PubMed] [Google Scholar]
- 31. Pupek M, Krzyżanowska‐Gołąb D, Dyła T, Lemańska‐Perek A, Jankowska R, Kątnik‐Prastowska I. Presence of high‐molecular‐weight forms and domain alterations of fibronectin in pleural effusion of patients with lung cancer. Clin Biochem. 2009;42(7–8):654‐661. [DOI] [PubMed] [Google Scholar]
- 32. Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680‐685. [DOI] [PubMed] [Google Scholar]
- 33. Kątnik I, Jadach J. Immunoaffinity purification of human haptoglobin using monoclonal antibodies. Arch Immunol Ther Exp (Warsz). 1993;41(5–6):303‐308. [PubMed] [Google Scholar]
- 34. Hooper DC, Peacock AC. Determination of the subunit composition of haptoglobin 2‐1 polymers using quantitative densitometry of polyacrylamide gels. J Biol Chem. 1976;251(19):5845‐5851. [PubMed] [Google Scholar]
- 35. Ramanathan A, Karuri N. Fibronectin alters the rate of formation and structure of the fibrin matrix. Biochem Biophys Res Commun. 2014;443(2):395‐399. [DOI] [PubMed] [Google Scholar]
- 36. Yates CC, Bodnar R, Wells A. Matrix control of scarring. Cell Mol Life Sci. 2011;68(11):1871‐1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Labat‐Robert J. Cell‐matrix interactions, the role of fibronectin and integrins. A survey. Pathol Biol. 2012;60(1):15‐19. [DOI] [PubMed] [Google Scholar]
- 38. Prakash P, Kulkarni PP, Lentz SR, Chauhan AK. Cellular fibronectin containing extra domain A promotes arterial thrombosis in mice through platelet Toll‐like receptor 4. Blood. 2015;125:3164‐3172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Roy S, Cagliero E, Lorenzi M. Fibronectin overexpression in retinal microvessels of patients with diabetes. Invest Ophthalmol Vis Sci. 1996;37(2):258‐266. [PubMed] [Google Scholar]
- 40. Kanters SDJM, Banga JD, Algra A, Frijns RCJM, Beutler JJ, Fijnheer R. Plasma levels of cellular fibronectin in diabetes. Diabetes Care. 2001;24(2):323‐327. [DOI] [PubMed] [Google Scholar]
- 41. Pastino AK, Greco TM, Mathias RA, Cristea IM, Schwarzbauer JE. Stimulatory effects of advanced glycation endproducts (AGEs) on fibronectin matrix assembly. Matrix Biol. 2017;59:39‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kunkemoeller B, Kyriakides TR. Redox signaling in diabetic wound healing regulates extracellular matrix deposition. Antioxid Redox Signal. 2017;27(12):823‐838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Maione AG, Smith A, Kashpur O, et al. Altered ECM deposition by diabetic foot ulcer‐derived fibroblasts implicates fibronectin in chronic wound repair. Wound Repair Regen. 2016;24(4):630‐643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Widgerow AD. Chronic wounds – is cellular “reception” at fault? Examining integrins and intracellular signaling. Int Wound J. 2013;10(2):185‐192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cappellari GG, Barazzoni R, Cattin L, Muro AF, Zanetti M. Lack of fibronectin extra domain A alternative splicing exacerbates endothelial dysfunction in diabetes. Sci Rep. 2016;6:37965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xu XR, Zhang D, Oswald BE, et al. Platelets are versatile cells: New discoveries in hemostasis, thrombosis, immune responses, tumor metastasis and beyond. Crit Rev Clin Lab Sci. 2016;53(6):409‐430. [DOI] [PubMed] [Google Scholar]
- 47. Finn AV, Nakazawa G, Joner M, et al. Vascular responses to drug eluting stents: importance of delayed healing. Arterioscler Thromb Vasc Biol. 2007;27(7):1500‐1510. [DOI] [PubMed] [Google Scholar]