

Abstract
Connective tissue growth factor (CCN2/CTGF) and transforming growth factor β1 (TGF‐β1) are important regulators of skin wound healing, but controversy remains regarding their expression in epithelial cell lineages. Here, we investigate the expression of CCN2 in keratinocytes during reepithelialisation and its regulation by TGF‐β1. CCN2 was detected in the epidermis of healing full‐thickness porcine wounds. Human keratinocytes were incubated with or without 10 ng/ml TGF‐β1, and signalling pathways were blocked with 10‐μM SIS3 or 20‐μM PD98059. Semi‐quantitative real‐time PCR was used to study CCN2 mRNA expression, and western blot was used to measure CCN2, phosphorylated‐ERK1/2, ERK1/2, phosphorylated‐Smad3 and Smad2/3 proteins. CCN2 was transiently expressed in neoepidermis at the leading edge of the wound in vivo. In vitro, CCN2 expression was induced by TGF‐β1 at 2 hours (7·5 ± 1·9‐fold mRNA increase and 3·0 ± 0·6‐fold protein increase) and 12 hours (5·4 ± 1·9‐fold mRNA increase and 3·3 ± 0·6‐fold protein increase). Compared with inhibiting the SMAD pathway, inhibiting the mitogen‐activated protein kinase (MAPK) pathway was more effective in reducing TGF‐β1‐induced CCN2 mRNA and protein expression. Inhibition of the MAPK pathway had minimal impact on the activity of the SMAD pathway. CCN2 is expressed in keratinocytes in response to tissue injury or TGF‐β1. In addition, TGF‐β1 induces CCN2 expression in keratinocytes through the ras/MEK/ERK pathway. A complete understanding of CCN2 expression in keratinocytes is critical to developing novel therapies for wound healing and cutaneous malignancy.
Keywords: CCN2, Cell migration, Keratinocytes, Reepithelialisation, Wound healing
Introduction
The skin protects the body from the external environment 1. The protective barrier is provided by the keratinocytes of the epidermis and by the dermis, which consists of fibroblasts that produce extracellular matrix 2, 3. Upon injury, normal wound healing must occur in an organised fashion for tissue integrity to be restored. Cutaneous wound healing is a complex process involving the migration of inflammatory cells to the wound site, deposition of extracellular matrix by fibroblasts and the re‐establishment of an intact epithelial barrier. These processes are tightly regulated by a variety of growth factors and cytokines, and abnormalities in any of these processes may result in the clinical phenotype of chronic non‐healing wounds or hypertrophic scars 4, 5. Although novel therapies have been developed to augment normal wound healing, treatment of aberrant wounds remains a complex endeavour that costs the United States over 25 billion dollars annually 6.
The re‐establishment of an intact epithelial barrier is an essential feature to a healed wound and is accomplished through the directed migration of keratinocytes in response to molecular mediators. CCN2, previously named connective tissue growth factor, has emerged as a regulator of cell migration. CCN2 is the second member of the CCN family of six cysteine‐rich proteins named Cyr61, CCN2, Nov and WISP1, 2 and 3 7, 8, 9. Members of the CCN family of proteins contain four structurally conserved domains that regulate cellular activity by binding molecules such as growth factors, integrins and proteoglycans 10. In the context of normal wound healing, CCN2 is well established as a profibrotic factor that plays a major role in cell proliferation, migration, adhesion, angiogenesis and maintenance of the extracellular matrix 7, 11. Expression of CCN2 was initially thought to be confined to cells of mesenchymal origin, but a growing body of evidence indicates that CCN2 is also expressed in epithelial cells 12, 13, 14, 15, 16, 17. This is due in part to advances in cancer genomics, which has linked over‐expression of CCN2 to multiple carcinomas, particularly metastatic melanoma 18, 19, 20.
The potential role of CCN2 in the physiology of certain non‐healing wounds, fibrosis and cutaneous malignancy presents a unique opportunity for the development of novel therapeutics. CCN2 and its intracellular pathways in epidermal cells are thus clinically important as they may one day serve as both a biomarker and therapeutic for a variety of pathologies. The main obstacle to using CCN2 as a therapeutic target will be its cell type‐specific expression and widespread involvement in matricellular processes. Modulating expression of CCN2 in a single organ may have an unpredictable impact on many cell types as illustrated by the diverse effects of CCN2 on keratinocytes, fibroblasts and melanocytes. For instance, our previous finding that CCN2 promotes the migration of keratinocytes suggests that inhibiting dermal CCN2 expression for anti‐fibrotic therapy may prevent reepithelialisation 15. Conversely, up‐regulating CCN2 expression with the intent of achieving closure of a non‐healing wound may pose a risk of cancer secondary to keratinocytes or melanocytes exhibiting more invasive and migratory behaviours. Thus, while CCN2 holds great promise for the development of novel therapeutics, a better understanding of the cellular events and signalling pathways involved in its expression are needed.
Transforming growth factor β1 (TGF‐β1) is one of the most potent inducers of CCN2 9, 21, 22. TGF‐β1‐regulated gene expression is generally mediated through Smad signalling 23. TGF‐β1 binds to its primary (type II) receptor, allowing recruitment, transphosphorylation and activation of the signalling (type I) receptor. This receptor complex activates receptor‐activated Smads (R‐Smads), primarily Smad 2 and Smad 3, by phosphorylating serine residues at the C‐terminal. The activated R‐Smads form a complex with Smad 4 and subsequently translocate into the nucleus, where they bind to Smad‐binding elements (SBE) in promoters of TGF‐β1‐responsive genes. In addition to Smad signalling, TGF‐β1 is known to activate other signalling pathways, including the mitogen‐activated protein kinase (MAPK) pathway. Within the MAPK pathway, TGF‐β1 directly regulates CCN2 expression through effector proteins ERK, p38 and JNK in a variety of cell types to facilitate cellular activity 24. Regulation of CCN2 by TGF‐β1 may be cell type‐specific even within a single organ. For instance, TGF‐β1 induces CCN2 through the Smad signalling pathway in hepatocytes but has little effect on CCN2 expression in hepatic stellate cells 25. Conversely, the Jak‐Stat signalling pathway is intimately involved in TGF‐β1 induction of CCN2 in lung endothelial cells 26.
Numerous studies have identified cross‐talk between Smad and ERK signalling activated by TGF‐β1. ERK induced by TGF‐β1 can regulate Smad activity and function by direct phosphorylation of Smads or indirectly by modifying the activity of coactivators/corepressors that mediate Smad‐DNA binding 27, 28, 29. In addition, Smads have been shown to mediate activation of ERK, adding more complexity to the relationship between the Smad and ERK pathways 27, 30. It has been shown that both Smad and ERK signalling cascades are involved in TGF‐β1‐induced CCN2 expression in mesenchymal cells 31, 32, 33. However, in keratinocytes, the potential interactions between Smad and ERK in TGF‐β1‐induced CCN2 expression remain unexplored. Thus, we sought to investigate the localisation of CCN2 in epidermal wound healing and to characterise TGF‐β1‐induced CCN2 in the context of the Smad and ERK signalling pathways.
Methods
Ethics statement
The research protocol and consenting procedures were reviewed and approved by the institutional review boards (IRB) of Partners Human Research Committees at the Brigham and Women's Hospital (IRB protocol no. 2010P002947). The study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki. Human skin was obtained from patients undergoing elective plastic surgery. The IRB committee deemed full written consent unnecessary, and oral consent was considered appropriate as no patient identifiers were collected, and the tissue would have otherwise been routinely discarded. Oral consent was obtained after careful review of an informational sheet describing what researchers would do with the otherwise‐discarded tissue. After the surgery, medical staff contacted the researchers, and the de‐identified tissue was collected. This study was conducted according to the principles expressed in the Declaration of Helsinki.
All animal procedures were approved by the Harvard Medical Area Standing Committee on Animals and conformed with regulations related to animal use and other federal statutes (protocol number 693). All animals were handled in strict accordance with good animal practice, and the study was conducted adhering to the institution's guidelines for animal husbandry.
Reagents
Recombinant human TGF‐β1 was purchased from Calbiochem (Gibbstown, NJ, USA). MEK 1 inhibitor PD98050 was obtained from Cell Signaling Technology (Danvers, MA); Smad 3 inhibitor SIS3 was from Millipore (Bedford, MA); and actinomycin D was from Sigma (St. Louis, MO). Polyclonal antibodies against CCN2 (L‐20) came from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Rabbit‐anti‐phospho‐p44/42 (phosphorylated ERK), rabbit anti‐p44/42 (ERK), rabbit‐anti‐phospho‐Smad3 (Ser 423/425), rabbit‐anti‐Smad 2/3 and rabbit‐anti‐lamin A/C were from Cell Signaling Technology. HRP‐conjugated mouse‐anti‐GAPDH was purchased from Abcam (Cambridge, MA). Biotinylated secondary antibodies (rabbit‐anti‐goat and donkey‐anti‐rabbit) were from Vector Laboratories (Burlingame, CA). Alexa‐fluor‐488 donkey‐anti‐goat IgG and Alexa‐fluor‐488 donkey‐anti‐rabbit IgG were from Invitrogen (Carlsbad, CA). Reagents used for positive controls included rat thoracic aorta myoblast whole cell lysate (A10) from Santa Cruz Biotechnology. Dispase, trypsin and ethylenediaminetetraacetic acid (EDTA) were obtained from Invitrogen. EpiLife keratinocyte medium and keratinocyte supplement were from GIBCO (Grand Island, NY). Dulbecco's Modified Eagle's Medium (DMEM) was from Sigma Aldrich (Steinheim, Germany). High pure RNA isolation kits were purchased from Roche Diagnostics (Indianapolis, IN). Quantitative real‐time Polymerase Chain Reaction (qRT‐PCR) kits were obtained from Applied Biosystems (Foster City, CA). FAM™Taqman®MGB probes and Taqman® Universal PCR Master Mix as well as TaqMan probes and primers for 18S RNA were purchased from Applied Biosystems. Bicinchoninic Acid (BCA) protein assay reagent kit as well as all western blot reagents was purchased from Bio‐Rad (Hercules, CA). Chemiluminescence detection kit was from GE Health Care (Westborough, MA). NE‐PER™ nuclear and cytoplasmic extraction reagents were purchased from Thermo Scientific (Rockford, IL).
Porcine wound model
The in vivo wound model has been extensively described 34, 35. Briefly, full‐thickness wounds measuring 1·5 × 1·5 cm were created on the dorsum of three pigs and covered with sterile dry gauze. The pigs were returned to the pen and monitored during recovery from anaesthesia. Biopsies were taken on postoperative days 6, 10, 14, 18 and 22 with a 0·5‐cm margin from the tattoo marking the wound border, with a total of three samples per pig per biopsy day (n = 9). Tissue samples were embedded in an optimal cutting temperature compound (Tissue‐Tek O.C.T., Sakura, Fintec, CA) and snap‐frozen in liquid nitrogen.
Immunohistochemistry and immunofluorescence staining
Cryosections (6 µm) were incubated at 4°C overnight with primary antibody diluted 1:200. Slides were subsequently incubated with biotinylated rabbit anti‐goat followed by incubation with avidin‐biotin‐peroxidase complex. Aminoethylcarbazole was used for detection, and sections were counterstained with haematoxylin.
For immunofluorescence detection of CCN2 and phosphorylated Smad 3, keratinocytes were cultured on collagen I‐coated cover slips in a serum‐free medium for 24 hours. As indicated by the figure legends, some chambers were pre‐treated with 20‐μM MEK inhibitor PD98059 for 60 minutes prior to the addition of 10 ng/ml TGF‐β1 for 24 hours. Keratinocytes cultured without the addition of inhibitors or growth factors served as negative controls. Following treatment, cells were fixed in 4% paraformaldehyde for 20 minutes at room temperature. After 5 minutes permeabilisation with 0·1% Triton X‐100, non‐specific binding sites were blocked using 1% BSA in PBS. CCN2 was detected with primary antibodies diluted 1:300, followed by incubation with Alexa Fluor 488 donkey anti‐goat IgG, diluted 1:500. Phosphorylated Smad 3 was detected with primary antibodies diluted 1:500, followed by incubation with Alexa Fluor 568 donkey anti‐goat IgG, diluted 1:500. Slides were mounted with aqueous mounting medium containing 4′,6‐diamidino‐2‐phenylindole (DAPI) (ProLong Gold, Invitrogen) and assessed by fluorescence microscopy.
Cell culture
Primary human keratinocytes were isolated from the epidermis as previously described 34. Briefly, skin was incubated at 4°C with 1 U/ml dispase in DMEM overnight, and the epidermis was mechanically separated from the underlying dermis. The epithelial sheets were treated with 0·05% trypsin and 0·01% EDTA for 10 minutes. Keratinocytes were cultured in EpiLife keratinocyte medium supplemented with bovine insulin (5 µg/ml), hydrocortisone (0·5 µg/ml), human recombinant epidermal growth factor (0·1 µg/ml), 0·4% bovine pituitary extract and 60 μM calcium chloride. Cells were expanded using serial passage, and cells from passages 1–3 were used for the experiments.
RNA preparation
Primary human keratinocytes were grown in serum‐free conditions as described above. Keratinocytes from days 1–7 were harvested each day for RNA isolation. When indicated, keratinocytes were treated with 1, 5 or 10 ng/ml TGF‐β1 for 2 or 12 hours. For inhibition studies, keratinocytes were pre‐treated with 20‐μM MEK 1 inhibitor PD98050, 10 μM Smad 3 inhibitor SIS3 and/or 5 µg/ml actinomycin D for 1 hour prior to incubation with 10 ng/ml TGF‐β1, as indicated in the figure legends. Cells were harvested with 0·05% trypsin and 0·01% EDTA, washed with PBS, and pelleted via centrifugation at 200 g for 7 minutes at 4°C. The high pure RNA isolation kit was used to extract total RNA from the cells according to kit protocol.
Quantitative real‐time PCR
As per the thermal cycling protocol, cycling consisted of 2 minutes at 95°C and 10 minutes at 95°C, followed by 45 cycles at 95°C for 15 seconds and 60°C for 1 minute. The cycle threshold was set within the linear phase of the target gene amplification and above the non‐template control background in order to calculate the cycle number at which the transcription was detected as denoted by Ct. The instrument spectral compensations by the 7900 HT SDS software version 2.1 (Applied Biosystems) was used to collect data. Samples were run in triplicate, and expression values were normalised with respect to the housekeeping gene 18S RNA employing the ΔΔCt method 36.
Western blot
Keratinocytes were grown to 80% confluence in serum‐free conditions. Cells were collected with an ice‐cold plastic cell scraper at 10 minutes, 30 minutes, 60 minutes or 0–12 hours after adding 10 ng/ml TGF‐β1. In some treatment groups, keratinocytes were pre‐treated with 10‐μM Smad 3 inhibitor SIS3 and/or 20‐μM MEK inhibitor PD98059 for 1 hour prior to the addition of TGF‐β1. Total cell lysate was separated into cellular and nuclear proteins using the NE‐PER™ nuclear and cytoplasmic extraction reagents according to manufacturer's instructions 37. Cytosolic proteins were first extracted by disrupting cell membranes, followed by centrifugation. Intact nuclei were washed with cold PBS and then lysed with high‐salt NE‐PER™ buffer. Total protein was obtained by lysing cells with radioimmunoprecipitation assay buffer supplemented with protease and phosphatase inhibitor cocktails. Protein concentration was determined using the BCA protein assay reagent kit according to the manufacturer's instructions, and the cell lysates were stored at –80°C. Protein from the cell lysate (40 µg) was boiled in sample buffer and electrophoresed through a 4–20% gradient Tris‐glycine ready gel and subsequently transferred to nitrocellulose membranes by immunoblotting. After 1 hour blocking in 5% BSA at room temperature, membranes were incubated with 1:1000 dilutions of goat‐anti‐CCN2, horseradish peroxidase–conjugated mouse‐anti‐GAPDH, rabbit‐anti‐phospho‐p44/42 (phospho ERK), rabbit anti‐p44/42 (ERK), rabbit‐anti‐phospho‐Smad 3 (Ser 423/425) or rabbit‐anti‐Smad 2/3 at 4°C overnight, followed by incubation with HRP‐conjugated rabbit‐anti‐goat or goat‐anti‐rabbit secondary antibodies (dilution 1:5000) for 1 hour at room temperature. Bands were visualised using an enhanced chemiluminescence detection system with dyed molecular‐weight markers. Densitometric analysis of the bands was carried out with ImageJ Software (IH‐Image – http://rsbweb.nih.gov/ij). The relative phosphorylated‐ERK and phosphorylated‐Smad 3 values were obtained from normalisation against the total ERK 1/2 or total Smad 2/3 values, respectively, or normalisation of CCN2 against GAPDH values.
Statistical analysis
Values are given as mean ± SD. Student's t‐test was used when comparing two groups. Statistical comparison of more than two groups was performed using one‐way analysis of variance (ANOVA) with Dunn's post hoc test. Statistical analysis was performed using GraphPad Prism 5.0 (GraphPad, LaJolla, CA), and P values of <0·05 were considered significant.
Results
CCN2 expression in vivo and in vitro
We have previously shown that CCN2 is expressed by the in‐growing neoepidermis during wound healing 15. Full‐thickness porcine wounds were allowed to heal, and the temporospatial expression of CCN2 was studied over a period of 22 days. The in‐growing neoepidermis stained positive for CCN2 (Figure 1A). The expression of CCN2 was confined to the suprabasal layers of keratinocytes (Figure 1B). To assess the percentage of the wounds expressing CCN2, the ratio of CCN2‐expressing neoepidermis to negative neoepidermis was calculated. On day 6, the wounds were 29 ± 7% reepithelialised, and the entire neoepidermis stained positive for CCN2 (Figure 2A and B). On day 10, the wounds were 51 ± 6% reepithelialised, and CCN2 expression started to diminish around the original border. On day 14, the wounds were fully resurfaced, and CCN2 was expressed in the central parts of the wounds in a 0·6:1 ratio. The central parts of the wounds remained positive throughout day 18, and there was no detectable CCN2 expression 22 days after wounding, indicating that CCN2 protein expression in epidermis is transient.
Figure 1.
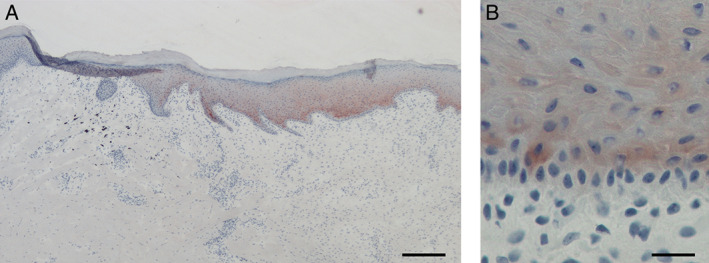
Epidermal connective tissue growth factor (CCN2) expression in vivo. (A) CCN2 protein expression in vivo was detected by immunohistochemistry 10 days after wounding. CCN2 was transiently expressed in the leading edge of the neoepidermis. Red colour represents AEC‐positive immunoreactivity, and the blue colour is the haematoxylin counter‐stain. (B) Magnified image of the dermal–epidermal junction showing that CCN2 expression was confined to the suprabasal keratinocytes. Scale bar equals 200 µm in (A) and 25 µm in (B).
Figure 2.
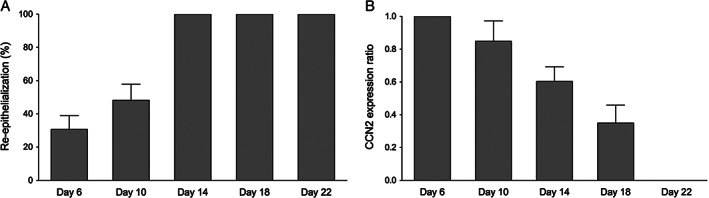
Connective tissue growth factor (CCN2) is transiently expressed during reepithelialisation. (A) The full‐thickness porcine wounds were fully reepithelialised by day 14. (B) The linear ratio of CCN2‐expressing neoepidermis to total epidermis showed a transient expression of CCN2 during reepithelialisation. Graphs show mean ± SD from four independent experiments, n = 8.
TGF‐β1 induces CCN2 mRNA expression in keratinocytes
One of the most potent inducers of CCN2 is TGF‐β1, which was recently shown to up‐regulate the expression of CCN2 in corneal epithelial cells 17. To determine whether TGF‐β1 induces CCN2 expression in human keratinocytes, cells were incubated with or without the addition of 1, 5 or 10 ng/ml TGF‐β1 for 2 or 12 hours (Figure 3A). All concentrations of TGF‐β1 led to an increase in CCN2 mRNA expression at 2 and 12 hours. Incubation with 1 ng/ml of TGF‐β1 led to a 7·6 ± 1·9‐fold increase in CCN2 mRNA expression at 2 hours (P < 0·001) and a 5·5 ± 2·2‐fold increase in CCN2 mRNA expression at 12 hours (P < 0·001) compared to the untreated control. Incubation with 5 ng/ml of TGF‐β1 led to a 8·1 ± 1·4‐fold increase in CCN2 mRNA expression at 2 hours (P < 0·001) and a 6·1 ± 1·7‐fold increase in CCN2 mRNA expression at 12 hours (P < 0·001) compared to untreated control. Incubation with 10 ng/ml of TGF‐β1 led to an 8·4 ± 1·9‐fold increase in CCN2 mRNA expression at 2 hours (P < 0·001) and a 6·7 ± 1·8‐fold increase in CCN2 mRNA expression at 12 hours (P < 0·001) compared to untreated control. As maximal increase of CCN2 expression was observed after the addition of 10 ng/ml TGF‐β1, this concentration was chosen for subsequent experiments.
Figure 3.
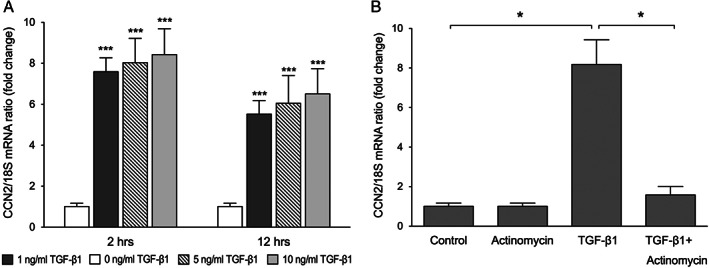
Transforming growth factor β1 (TGF‐β1) induces connective tissue growth factor (CCN2) mRNA expression in keratinocytes. (A) Keratinocytes were incubated with or without the addition of 1, 5 or 10 ng/ml TGF‐β1 for 2 and 12 hours, and CCN2 mRNA expression was analysed by quantitative real‐time PCR. Graphs show mean ± SD from three independent experiments, n = 6. (B) To verify that the increased expression of CCN2 by TGF‐β1 is because of active transcription, keratinocytes were pre‐treated with 5 µg/ml actinomycin D for 60 minutes prior to the addition of TGF‐β1. CCN2 mRNA expression was analysed by quantitative real‐time PCR after 24 hours incubation. Graphs show mean ± SD from three independent experiments, n = 6. *P <0·05 vs. 0 ng/ml TGF‐β1 (control), ***P < 0·001 vs. control.
To verify that increased expression of CCN2 by TGF‐β1 is because of active transcription and not release of pre‐existing CCN2 mRNA, keratinocytes were pre‐treated with 5 µg/ml actinomycin D for 60 minutes prior to incubation with 10 ng/ml TGF‐β1 for 2 hours (Figure 3B). Actinomycin D treatment blocked TGF‐β1‐induced CCN2 mRNA expression, suggesting transcriptional regulation of CCN2 by TGF‐β1.
TGF‐β1 enhances CCN2 protein production in keratinocytes
To determine whether TGF‐β1‐induced CCN2 mRNA expression was reflected by elevated synthesis of CCN2 protein, keratinocytes were cultured in the presence or absence of 10 ng/ml TGF‐β1 for 24 hours, and CCN2 protein was detected by immunofluorescence. The results indicate that CCN2 protein levels were relatively low in the absence of TGF‐β1 (Figure 4A). The addition of TGF‐β1 led to increased CCN2 production. The expression of CCN2 was primarily perinuclear, as described previously (Figure 4B) 38. Time course analysis of CCN2 expression showed CCN2 expression as early as 30 minutes after TGF‐β1 addition (Figure 4C). Maximum levels were reached after 6 hours' stimulation with 10 ng/ml TGF‐β1 (3·9 ± 0·5‐fold increase compared to 1·0 ± 0·2‐fold with untreated control, P < 0·05) (Figure 4D).
Figure 4.
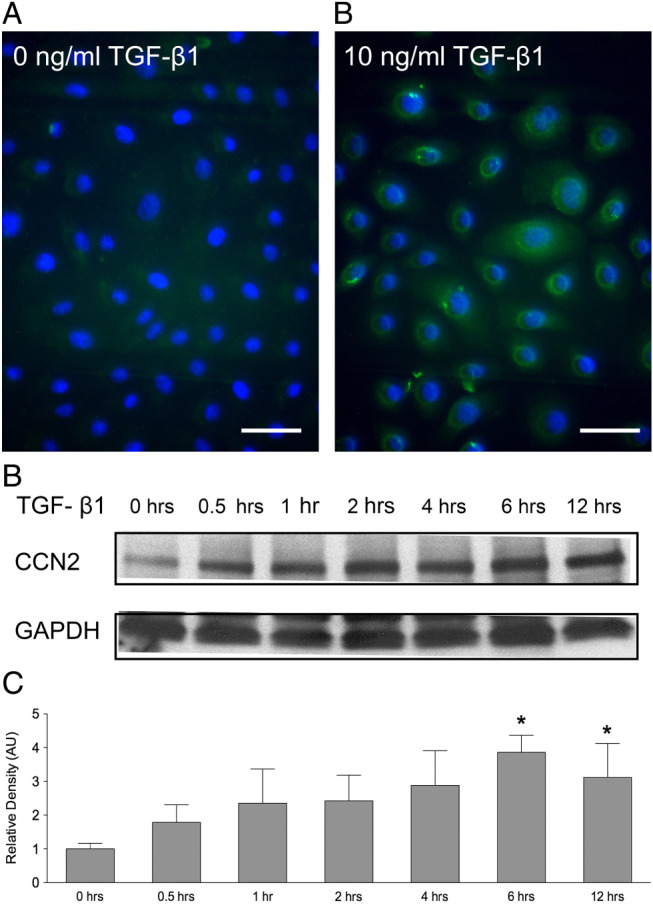
Transforming growth factor β1 (TGF‐β1) increases connective tissue growth factor (CCN2) protein production in keratinocytes. CCN2 protein production in vitro was detected by immunohistochemistry. Green staining indicates CCN2, and blue staining indicates nuclei. (A) After 24 hours' culture in a serum‐free control culture media, keratinocytes expressed low levels of CCN2. (B) The addition of 10 ng/ml TGF‐β1 increased CCN2 protein production. The experiments were repeated three times with similar results. Scale bar equals 20 µm. (C) TGF‐β1‐induced CCN2 protein production was detected with western blot. An increased production of CCN2 was observed after the addition of TGF‐β1, and significantly increased levels of CCN2 were detected after 6 and 12 hours. One representative western blot out of three independently performed experiments is shown. (D) The relative density of the bands was normalised to GAPDH. Graphs show mean ± SD from three independent experiments, n = 6. *P < 0·05 versus untreated control.
TGF‐β1 activates the SMAD signalling pathway in human keratinocytes
To identify intracellular signalling pathways involved in the regulation of CCN2 by TGF‐β1, keratinocytes were incubated with 10 ng/ml TGF‐β1 for 0–60 minutes. Immunoblotting was performed on cell lysates, and phosphorylated Smad 3 was measured (Figure 5A). Smad 3 was chosen for this study as it has previously been shown that TGF‐β1 induction of CCN2 does not occur in Smad 3 knockout cells 39. Phosphorylation of Smad 3 was detected after 10 minutes and by 30 minutes had reached a maximum (Figure 5A and B). Therefore, the 30‐minute post‐treatment time point was selected for subsequent analysis of Smad activation. Total Smad 2/3 remained constant at all time points. Next, we tested the effect of Smad 3‐specific inhibitor SIS3 on TGF‐β1‐induced Smad 3 phosphorylation in keratinocytes. SIS3 is a selective inhibitor of TGF‐β1‐induced Smad 3 phosphorylation and Smad 3‐mediated cellular signalling but has no effect on Smad 2 or ERK activation 40. Pre‐treatment of keratinocytes with 10 μM SIS3 for 60 minutes blocked TGF‐β1‐induced Smad phosphorylation at 30 minutes (Figure 5C and D). Therefore, this time point was chosen for all subsequent studies of the involvement of Smad signalling in the induction of CCN2 expression.
Figure 5.
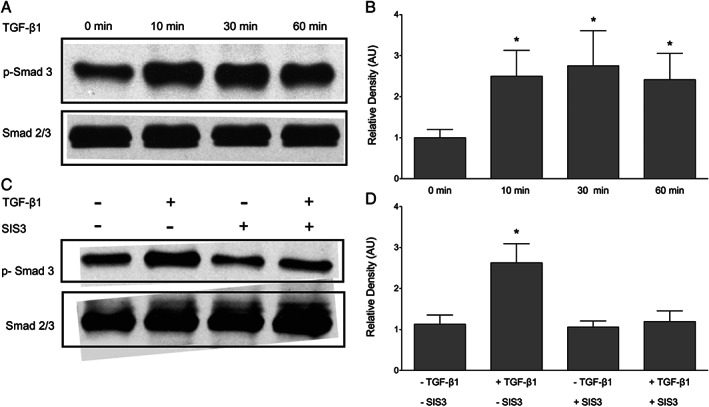
Transforming growth factor β1 (TGF‐β1) activates SMAD signalling in keratinocytes. Keratinocytes were incubated with 10 ng/ml TGF‐β1 for 0–60 minutes, and phosphorylated‐Smad 3 was measured. (A) TGF‐β1 induced the phosphorylation of Smad 3. Maximum phosphorylation was seen after 30 minutes. (B) The relative density of the bands was normalised to total Smad. (C) Pre‐treatment with 10‐μM Smad 3 inhibitor (SIS3) for 60 minutes blocked TGF‐β1‐induced Smad 3 phosphorylation. (D) The relative density of the bands was normalised to total Smad. One representative western blot out of three independently performed experiments is shown. Graphs show mean ± SD from three independent experiments, n = 6. *P < 0·05 versus untreated control (0 minutes).
TGF‐β1‐induced CCN2 production in human keratinocytes is in part mediated by Smad 3
To determine whether TGF‐β1‐induced CCN2 expression in human keratinocytes was mediated through Smad signalling, cells were incubated with 10 μM SIS3 for 60 minutes prior to the addition of 10 ng/ml TGF‐β1 for 30 minutes. Inhibition of Smad 3 only somewhat reduced TGF‐β1‐induced CCN2 mRNA expression, and this reduction was not statistically significant (Figure 6A). In accordance with the mRNA data, TGF‐β1‐induced CCN2 protein production is slightly impaired by SIS3 (Figure 6B and C). Taken together, the above findings indicate that TGF‐β1‐induced CCN2 production in human keratinocytes is only partially mediated by Smad 3, indicating that TGF‐β1 might act through alternative pathways.
Figure 6.
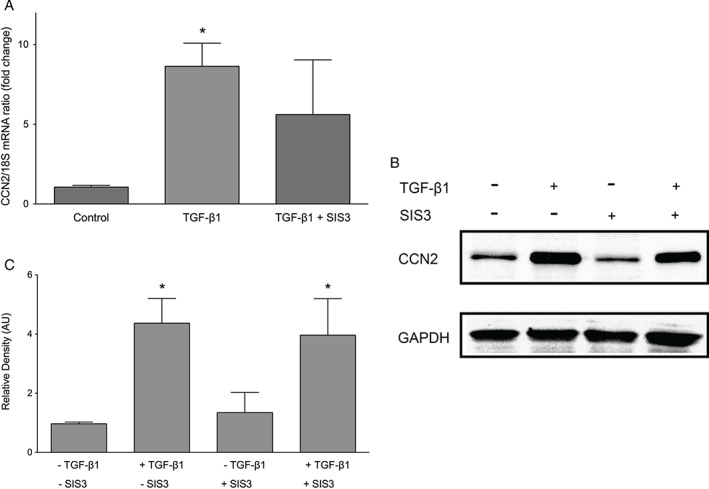
Effect of SIS3 on transforming growth factor β1 (TGF‐β1)‐induced connective tissue growth factor (CCN2) expression in keratinocytes. Keratinocytes were cultured with or without pre‐treatment with 10‐μM Smad 3 inhibitor (SIS3) prior to incubation with 10 ng/ml TGF‐β1 for 30 minutes. (A) CCN2 mRNA expression was analysed by quantitative real‐time PCR. Inhibition of Smad 3 only partially reduced TGF‐β1‐induced CCN2 mRNA expression. Graphs show mean ± SD from three independent experiments, n = 9. *P < 0·05 versus untreated controls. (B) Western blot results mirrored the mRNA data. One representative western blot out of three independently performed experiments is shown. (C) The relative density of the bands was normalised to GAPDH. Graphs show mean ± SD from three independent experiments, n = 6. *P < 0·05 versus untreated control (0 minute).
CCN2 induction by TGF‐β1 in human keratinocytes requires ERK activity
Given that pathways other than Smad are known to mediate TGF‐β1 signalling, we investigated the contribution of ERK to TGF‐β1‐induced CCN2. TGF‐β1 activates MAPKs, including ERK, p38 and JNK, in a cell type‐dependent manner, and ERK has been shown to be required for TGF‐β1‐induced CCN2 expression in corneal epithelial cells 17. To confirm that TGF‐β1 activates the ERK pathway in keratinocytes, immunoblotting was performed to detect phosphorylated ERK. Incubation with 10 ng/ml TGF‐β1 induced phosphorylation of ERK measurable after 10 minutes (Figure 7A). At 60 minutes, phosphorylation of ERK in response to TGF‐β1 was still evident (Figure 7B).
Figure 7.
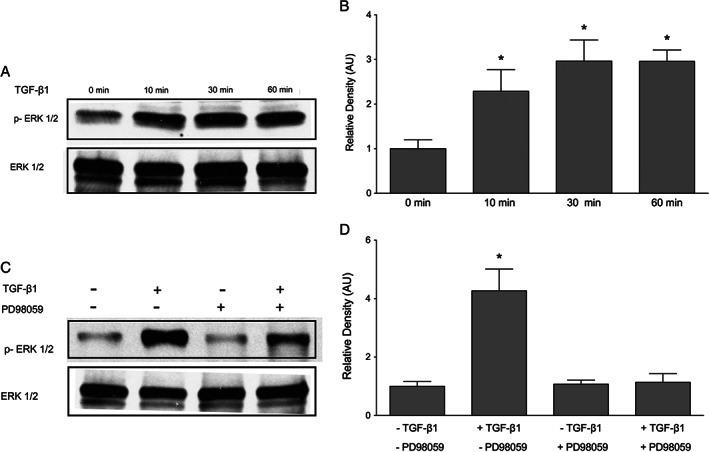
Transforming growth factor β1 (TGF‐β1)‐induced ERK phosphorylation in keratinocytes. Keratinocytes were incubated with 10 ng/ml TGF‐β1 for 0–60 minutes, and phosphorylated ERK was measured. (A) TGF‐β1 induced the phosphorylation of ERK as early as 10 minutes of incubation. One representative western blot out of three independently performed experiments is shown. (B) The relative density of the bands was normalised to total ERK. (C) Pre‐treatment with MEK inhibitor PD98059 for 60 minutes blocked TGF‐β1‐induced ERK phosphorylation after 30 minutes' incubation with 10 ng/ml TGF‐β1. (D) The relative density of the bands was normalised to total ERK. Graphs show mean ± SD from three independent experiments, n = 6.
PD98059 is a specific pharmacological inhibitor targeting MEK‐ERK signalling. PD98059 prevents the phosphorylation of MEK, thereby inhibiting ERK 41. In keratinocytes, 60 minutes of pre‐treatment with 20‐μM PD98059 efficiently prevented TGF‐β1‐induced ERK activation at 30 minutes (Figure 7C). Therefore, this time point was chosen for all subsequent experiments involving ERK signalling in the induction of CCN2 expression. To determine whether the ERK signalling pathway plays a role in TGF‐β1‐induced CCN2 expression, keratinocytes were pre‐treated with 20‐μM PD98059 for 60 minutes prior to incubation with 10 ng/ml TGF‐β1 for 30 minutes. Blockage of MEK phosphorylation potently reduced the ability of TGF‐β1 to induce CCN2 mRNA and protein expression (Figure 8A–C). Collectively, these data suggest that activation of ERK signalling is necessary for TGF‐β1‐induced CCN2 expression.
Figure 8.
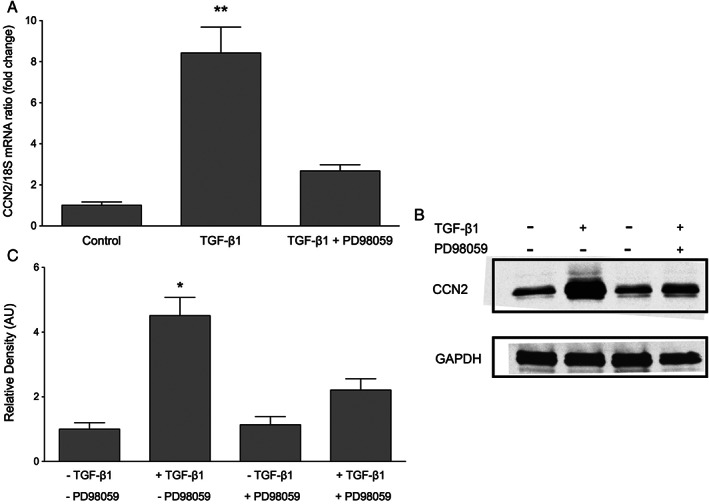
Effect of PD98059 on transforming growth factor β1 (TGF‐β1)‐induced connective tissue growth factor (CCN2) expression in keratinocytes. Keratinocytes were cultured with or without pre‐treatment with MEK 1 inhibitor PD98059 prior to incubation with 10 ng/ml TGF‐β1 for 30 minutes. (A) CCN2 mRNA expression was analysed by quantitative real‐time PCR. Inhibition of MEK/ERK blocked TGF‐β1‐induced CCN2 mRNA expression. Data represent the mean ± SD for three separate experiments. Total n = 9, *P < 0·05 versus untreated controls. (B) Western blotting confirmed the mRNA data. One representative western blot out of three independently performed experiments is shown. (C) The relative density of the bands was normalised to GAPDH. Graphs show mean ± SD from three independent experiments, n = 6. *P < 0·05 versus untreated control.
Inhibiting ERK does not prevent Smad phosphorylation or nuclear translocation
As both Smad and ERK signalling pathways were found to be involved in TGF‐β1‐induced CCN2 expression in keratinocytes, we investigated the potential cross‐talk between the Smad and ERK signalling pathways. Recent studies have suggested that ERK modifies TGF‐β1‐induced Smad signalling by directly phosphorylating Smad 3 42. To investigate whether ERK is involved in Smad phosphorylation, TGF‐β1‐induced Smad 3 phosphorylation was studied after inhibition of ERK signalling. Keratinocytes were pre‐treated with 20‐μM PD98059 for 60 minutes prior to incubation with 10 ng/ml TGF‐β1 for 30 minutes. TGF‐β1‐induced phosphorylation of Smad 3 was not blocked by PD98059 (Figure 9A and B), suggesting that ERK does not mediate TGF‐β1‐induced Smad 3 phosphorylation in keratinocytes.
Figure 9.
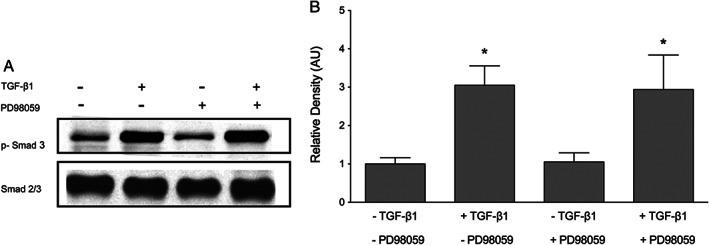
Inhibition of ERK does not prevent transforming growth factor β1 (TGF‐β1)‐induced Smad phosphorylation. Keratinocytes were cultured with or without pre‐treatment with MEK 1 inhibitor PD98059 prior to incubation with 10 ng/ml TGF‐β1 for 30 minutes. (A) Blocking ERK did not prevent TGF‐β1‐induced Smad phosphorylation. (B) The relative density of the bands was normalised to total Smad. One representative western blot out of three independently performed experiments is shown. Graphs show mean ± SD from three independent experiments, n = 6. *P < 0·05 versus untreated control.
Although inhibition of ERK did not reduce TGF‐β1‐induced Smad 3 phosphorylation, ERK might still regulate Smad signalling by affecting the nuclear translocation of phosphorylated Smad 3 complex. To examine the effect of ERK inhibition on the nuclear translocation of phosphorylated Smad 3, immunofluorescent stainings were conducted to visualise the location of phosphorylated Smad 3 following incubation with TGF‐β1. Nuclei were stained with DAPI (blue) to correlate with the nuclear localisation of phosphorylated Smad 3 (red). The fluorescent signal for phosphorylated Smad 3 was undetectable in control keratinocytes cultured in medium (Figure 10A and D). After 30 minutes' incubation with 10 ng/ml TGF‐β1, an intense fluorescent signal for phosphorylated Smad 3 was detected in the cell nuclei of keratinocytes (Figure 9B and E). Inhibition of ERK activation by PD98059 has no effect on the nuclear localisation of phosphorylated Smad 3 (Figure 10C and F). In addition to the immunofluorescence studies, nuclear translocation of phosphorylated Smad 3 was measured using western blot. Lamin A/C, a nuclear marker, was used as a control for sample loading and transfer. Keratinocytes were cultured with 10 ng/ml TGF‐β1 for 30 minutes with or without PD98059 pre‐treatment. Blocking ERK activation with PD98059 did not prevent the nuclear accumulation of phosphorylated Smad (Figure 11A and B). Collectively, these results suggest that inhibiting ERK does not prevent Smad phosphorylation or nuclear translocation.
Figure 10.
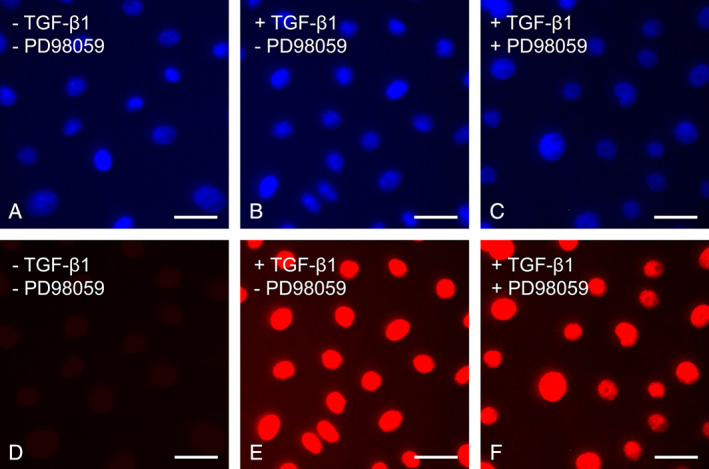
Inhibition of ERK does not prevent Smad nuclear translocation. Nuclear translocation of Smad was detected by immunofluorescence. Phosphorylated Smad 3 was labelled with red fluorescence and cell nuclei with blue fluorescence. (A, C) After 24 hours' culture in a serum‐free control culture media, keratinocytes expressed low levels of phosphorylated Smad 3. (B, E) After the addition of 10 ng/ml transforming growth factor β1 (TGF‐β1), phosphorylated Smad 3 was detected in the cell nuclei of keratinocytes. (C, F) Inhibition of ERK did not prevent nuclear translocation of phosphorylated Smad 3. The experiments were repeated three times with similar results. Scale bar equals 20 µm in A–F.
Figure 11.
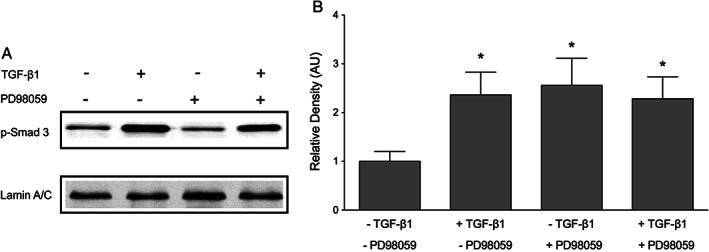
The effect of PD98059 on nuclear phosphorylated Smad 3. Nuclear translocation of phosphorylated Smad 3 was analysed using western blot. Keratinocytes were cultured with or without pre‐treatment with 10‐μM Smad 3 inhibitor (SIS3) prior to incubation with 10 ng/ml transforming growth factor β1 (TGF‐β1) for 30 minutes. Nuclear protein was extracted as described in the Methods section, and lamin A/C was used as a control for sample loading and transfer. (A) TGF‐β1 induced the phosphorylation of Smad 3. Inhibition of ERK did not prevent nuclear translocation of phosphorylated Smad 3. (B) The relative density of the bands was normalised to laminin A/C. One representative western blot out of three independently performed experiments is shown. Graphs show mean ± SD from three independent experiments, n = 6. *P < 0·05 versus untreated control.
Discussion
The skin's response to injury is rapid wound closure facilitated by the proliferation and migration of keratinocytes. The directed migration of keratinocytes is particularly important as reepithelialisation yields a cellular interface capable of maintaining homeostasis in a changing environment. While numerous signalling molecules contribute to reepithelialisation, CCN2 has emerged as a regulator of cell motility in addition to its well‐established roles in proliferation, differentiation, angiogenesis and maintenance of the extracellular matrix 17, 43, 44, 45, 46, 47, 48. The goal of the present study was to examine CCN2 expression in epidermal wound healing and potential involvement of the SMAD and ERK signalling pathways in response to TGF‐β1. Using an in vivo porcine model, our study demonstrates that CCN2 is transiently expressed by the in‐growing epidermis, strongly implicating a role for CCN2 in epidermal regeneration. We also demonstrate that in keratinocytes, CCN2 mRNA and protein are induced by TGF‐β1 signalling through dynamic interactions between the SMAD and ERK signalling pathways.
With regard to wound healing, CCN2 is best known as a profibrotic factor that stimulates the production of matricellular proteins for dermal regeneration. It is currently being explored as a potential target for pro‐ and anti‐fibrotic therapy, with the aims of achieving closure of non‐healing wounds and reducing hypertrophic scarring, respectively. Thus, our findings are clinically significant as they confirm a contribution of CCN2 to epidermal wound healing and provide a foundation for two of the major signalling pathways involved. Attempts to alter the wound‐healing process by modulating expression of CCN2 in the dermis must also consider possible unfavourable effects on the epidermis.
It has traditionally been thought that CCN2 is a downstream mediator of TGF‐β1 in mesenchymal cells but not epithelial cells 9, 16, 39. For instance, in an early study by Fraizer et al., TGF‐β1 injections into the dermis of NIH Swiss mice did not result in any detectable CCN2 staining in the epidermis or endothelial cells of large vessels and capillaries 12. However, the authors expressed some doubt regarding the validity of their results, speculating that their injection model might not effectively mirror the wound‐healing milieu. Another influential study by Grotendorst et al. showed that TGF‐β1 induced a 25‐ to 30‐fold increase in CCN2‐tethered luciferase activity in transfected NIH/3T3 fibroblasts and foetal bovine smooth muscle cells but not the two epithelial cell lines tested, namely HepG2 hepatocytes and human breast epithelial cells 14. The notion that CCN2 expression is confined to mesenchymal cells was challenged when a number of studies documented CCN2 expression in epithelial cells, although these initial results were too inconsistent to immediately reverse the traditional perception. Conflicting data regarding Grotendorst's HepG2 findings were reported, with additional studies showing no CCN2 mRNA expression 49, no appreciable CCN2 transcript staining 50 or marked CCN2 expression in hepatocytes 51. More recently, the ability of hepatocytes to synthesise CCN2 was confirmed by detailed cell culture studies that showcased hepatic CCN2 expression in a TGF‐β1‐free environment. These results were strengthened when the expression of CCN2 was found to be up‐regulated by TGF‐β1 as expected 25, 52. Similarly, another key finding that has been heavily explored is that CCN2 is not expressed by human breast epithelial cells 14. In a follow‐up study, Frazier and Grotendorst assayed human invasive mammary ductal carcinomas with in situ hybridisation for CCN2. Again, transcripts were found in the fibroblasts of the stroma but not in tumour epithelial cells or leukocytes 13. Studies by others have produced conflicting data, suggesting that CCN2 mRNA and protein expression occurs in both normal and malignant breast epithelial cells 53. Collectively, these more recent studies appear to suggest that CCN2 is expressed and plays a role in the cellular activities of epithelial cells, and the present study contributes to this observation in the context of the keratinocyte.
CCN2 holds great therapeutic potential not just for aberrant wound healing but also for the development of novel diagnostics and treatments for cancer. Studies have associated CCN2 with breast cancer 54, prostate cancer 55, glioma 56, lung cancer 57 and, most notably, pancreatic cancer 58 and metastatic melanoma 20. Clinical specimens of select pancreatic carcinomas display up to 50‐fold over‐expression of CCN2 secondary to aberrant TGF‐β signalling 59, 60. This over‐expression manifests as a desmoplastic reaction that facilitates unregulated proliferation of the stromal microenvironment. The end result is a walled‐off tumour with a reservoir of matricellular growth factors to sustain growth and development 61. Targeting the stromal microenvironment by modulating expression of CCN2 may thus offer a personalised approach for treatment and diagnosis. Similarly, clinical specimens of metastatic melanoma have also been associated with over‐expression of CCN2 secondary to aberrant TGF‐β signalling. One recent study found that melanoma cell lines with upregulated CCN2 displayed more invasive and migratory behaviour compared to melanoma cell lines expressing normal levels of CCN2 19. Non‐malignant melanocytes displayed similar changes in behaviour when transfected with recombinant CCN2, indicating that the presence or absence of CCN2 may orchestrate cellular activity in otherwise normal cells. Another study similarly found that CCN2 was up‐regulated in the majority of tested melanoma cell lines, which were amenable to treatment with anti‐CCN2 antibody FG‐3019 in vivo 20.
The present study not only highlights the basic mechanism of TGF‐β‐induced CCN2 expression in keratinocytes but also underscores the potential implications for CCN2‐centred therapies in wound healing and cancer. While CCN2 has great therapeutic potential, its diversity of functions and variable expression between cell types indicates that multiple molecular pathways are likely involved in regulating CCN2 activity. Additional studies focusing on the role of alternative molecular pathways in keratinocyte as well as other cell types will be critical in developing therapeutic agents targeting CCN2.
References
- 1. Chuong CM, Nickoloff BJ, Elias PM, Goldsmith LA, Macher E, Maderson PA, Sundberg JP, Tagami H, Plonka PM, Thestrup‐Pederson K, Bernard BA, Schröder JM, Dotto P, Chang CM, Williams ML, Feingold KR, King LE, Kligman AM, Rees JL, Christophers E. What is the ‘true’ function of skin? Exp Dermatol 2002;11:159–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fuchs E, Byrne C. The epidermis: rising to the surface. Curr Opin Genet Dev 1994;4:725–36. [DOI] [PubMed] [Google Scholar]
- 3. Kielty CM, Shuttleworth CA. Microfibrillar elements of the dermal matrix. Microsc Res Tech 1997;38:413–27. [DOI] [PubMed] [Google Scholar]
- 4. Kiwanuka E, Junker J, Eriksson E. Harnessing growth factors to influence wound healing. Clin Plast Surg 2012;39:239–48. [DOI] [PubMed] [Google Scholar]
- 5. Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature 2008;453:314–21. [DOI] [PubMed] [Google Scholar]
- 6. Sen CK, Gordillo GM, Roy S, Kirsner R, Lambert L, Hunt TK, Gottrup F, Gurtner GC, Longaker MT. Human skin wounds: a major and snowballing threat to public health and the economy. Wound Repair Regen 2009;17:763–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Perbal B. CCN proteins: multifunctional signalling regulators. Lancet 2004;363:62–4. [DOI] [PubMed] [Google Scholar]
- 8. Igarashi A, Okochi H, Bradham DM, Grotendorst GR. Regulation of connective tissue growth factor gene expression in human skin fibroblasts and during wound repair. Mol Biol Cell 1993;4:637–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Grotendorst GR. Connective tissue growth factor: a mediator of TGF‐beta action on fibroblasts. Cytokine Growth Factor Rev 1997;8:171–9. [DOI] [PubMed] [Google Scholar]
- 10. Rachfal AW, Brigstock DR. Structural and functional properties of CCN proteins. Vitam Horm 2005;70:69–103. [DOI] [PubMed] [Google Scholar]
- 11. Brigstock DR. The CCN family: a new stimulus package. J Endocrinol 2003;178:169–75. [DOI] [PubMed] [Google Scholar]
- 12. Frazier K, Williams S, Kothapalli D, Klapper H, Grotendorst GR. Stimulation of fibroblast cell growth, matrix production, and granulation tissue formation by connective tissue growth factor. J Invest Dermatol 1996;107:404–11. [DOI] [PubMed] [Google Scholar]
- 13. Frazier KS, Grotendorst GR. Expression of connective tissue growth factor mRNA in the fibrous stroma of mammary tumors. Int J Biochem Cell Biol 1997;29:153–61. [DOI] [PubMed] [Google Scholar]
- 14. Grotendorst GR, Okochi H, Hayashi N. A novel transforming growth factor beta response element controls the expression of the connective tissue growth factor gene. Cell Growth Differ 1996;7:469–80. [PubMed] [Google Scholar]
- 15. Kiwanuka E, Hackl F, Caterson EJ, Nowinski D, Junker JP, Gerdin B, Eriksson E. CCN2 is transiently expressed by keratinocytes during re‐epithelialization and regulates keratinocyte migration in vitro by the ras‐MEK‐ERK signaling pathway. J Surg Res 2013;185:e109–19. [DOI] [PubMed] [Google Scholar]
- 16. Leask A, Holmes A, Black CM, Abraham DJ. Connective tissue growth factor gene regulation. Requirements for its induction by transforming growth factor‐beta 2 in fibroblasts. J Biol Chem 2003;278:13008–15. [DOI] [PubMed] [Google Scholar]
- 17. Secker GA, Shortt AJ, Sampson E, Schwarz QP, Schultz GS, Daniels JT. TGFbeta stimulated re‐epithelialisation is regulated by CTGF and Ras/MEK/ERK signalling. Exp Cell Res 2008;314:131–42. [DOI] [PubMed] [Google Scholar]
- 18. Sha W, Leask A. CCN2 expression and localization in melanoma cells. JCell Commun Signal 2011;5:219–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Braig S, Wallner S, Junglas B, Fuchshofer R, Bosserhoff AK. CTGF is overexpressed in malignant melanoma and promotes cell invasion and migration. Br J Cancer 2011;105:231–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Finger EC, Cheng CF, Williams TR, Rankin EB, Bedogni B, Tachiki L, Spong S, Giaccia AJ, Powell MB. CTGF is a therapeutic target for metastatic melanoma. Oncogene 2014;33(9):1093–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Leask A, Sa S, Holmes A, Shiwen X, Black CM, Abraham DJ. The control of ccn2 (ctgf) gene expression in normal and scleroderma fibroblasts. Mol Pathol 2001;54:180–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Leask A, Abraham DJ. TGF‐beta signaling and the fibrotic response. FASEB J 2004;18:816–27. [DOI] [PubMed] [Google Scholar]
- 23. Shi Y, Massague J. Mechanisms of TGF‐beta signaling from cell membrane to the nucleus. Cell 2003;113:685–700. [DOI] [PubMed] [Google Scholar]
- 24. Li J, Zhao Z, Liu J, Huang N, Long D, Wang J, Li X, Liu Y. MEK/ERK and p38 MAPK regulate chondrogenesis of rat bone marrow mesenchymal stem cells through delicate interaction with TGF‐beta1/Smads pathway. Cell Prolif 2010;43:333–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gressner OA, Lahme B, Demirci I, Gressner AM, Weiskirchen R. Differential effects of TGF‐beta on connective tissue growth factor (CTGF/CCN2) expression in hepatic stellate cells and hepatocytes. J Hepatol 2007;47:699–710. [DOI] [PubMed] [Google Scholar]
- 26. Laug R, Fehrholz M, Schütze N, Kramer BW, Krump‐Konvalinkova V, Speer CP, Kunzmann S. IFN‐gamma and TNF‐alpha synergize to inhibit CTGF expression in human lung endothelial cells. PLoS One 2012;7:e45430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Derynck R, Zhang YE. Smad‐dependent and Smad‐independent pathways in TGF‐beta family signalling. Nature 2003;425:577–84. [DOI] [PubMed] [Google Scholar]
- 28. Mori Y, Ishida W, Bhattacharyya S, Li Y, Platanias LC, Varga J. Selective inhibition of activin receptor‐like kinase 5 signaling blocks profibrotic transforming growth factor beta responses in skin fibroblasts. Arthritis Rheum 2004;50:4008–21. [DOI] [PubMed] [Google Scholar]
- 29. Kretzschmar M, Doody J, Timokhina I, Massague J. A mechanism of repression of TGFbeta/Smad signaling by oncogenic Ras. Genes Dev 1999;13:804–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lutz M, Knaus P. Integration of the TGF‐beta pathway into the cellular signalling network. Cell Signal 2002;14:977–88. [DOI] [PubMed] [Google Scholar]
- 31. Hu X, Wang H, Liu J, Fang X, Tao K, Wang Y, Li N, Shi J, Wang Y, Ji P, Cai W, Bai X, Zhu X, Han J, Hu D. The role of ERK and JNK signaling in connective tissue growth factor induced extracellular matrix protein production and scar formation. Arch Dermatol Res 2013;305:433–45. [DOI] [PubMed] [Google Scholar]
- 32. Kawaki H, Kubota S, Suzuki A, Suzuki M, Kohsaka K, Hoshi K, Fujii T, Lazar N, Ohgawara T, Maeda T, Perbal B, Takano‐Yamamoto T, Takigawa M. Differential roles of CCN family proteins during osteoblast differentiation: Involvement of Smad and MAPK signaling pathways. Bone 2011;49:975–89. [DOI] [PubMed] [Google Scholar]
- 33. Ponticos M, Holmes AM, Shi‐wen X, Leoni P, Khan K, Rajkumar VS, Hoyles RK, Bou‐Gharios G, Black CM, Denton CP, Abraham DJ, Leask A, Lindahl GE. Pivotal role of connective tissue growth factor in lung fibrosis: MAPK‐dependent transcriptional activation of type I collagen. Arthritis Rheum 2009;60:2142–55. [DOI] [PubMed] [Google Scholar]
- 34. Kiwanuka E, Hackl F, Philip J, Caterson EJ, Junker JP, Eriksson E. Comparison of healing parameters in porcine full‐thickness wounds transplanted with skin micrografts, split‐thickness skin grafts, and cultured keratinocytes. J Am Coll Surg 2011;213:728–35. [DOI] [PubMed] [Google Scholar]
- 35. Hackl F, Bergmann J, Granter SR, Koyama T, Kiwanuka E, Zuhaili B, Pomahac B, Caterson EJ, Junker JP, Eriksson E. Epidermal regeneration by micrograft transplantation with immediate 100‐fold expansion. Plast Reconstr Surg 2012;129:443e–52. [DOI] [PubMed] [Google Scholar]
- 36. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) Method. Methods 2001;25:402–8. [DOI] [PubMed] [Google Scholar]
- 37. Tsai NP, Lin YL, Tsui YC, Wei LN. Dual action of epidermal growth factor: extracellular signal‐stimulated nuclear‐cytoplasmic export and coordinated translation of selected messenger RNA. J Cell Biol 2010;188:325–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zuehlke J, Ebenau A, Krueger B, Goppelt‐Struebe M. Vectorial secretion of CTGF as a cell‐type specific response to LPA and TGF‐beta in human tubular epithelial cells. Cell Commun Signal 2012;10:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Holmes A, Abraham DJ, Sa S, Shiwen X, Black CM, Leask A. CTGF and SMADs, maintenance of scleroderma phenotype is independent of SMAD signaling. J Biol Chem 2001;276:10594–601. [DOI] [PubMed] [Google Scholar]
- 40. Jinnin M, Ihn H, Tamaki K. Characterization of SIS3, a novel specific inhibitor of Smad3, and its effect on transforming growth factor‐beta1‐induced extracellular matrix expression. Mol Pharmacol 2006;69:597–607. [DOI] [PubMed] [Google Scholar]
- 41. Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, Copeland RA, Magolda RL, Scherle PA, Trzaskos JM. Identification of a novel inhibitor of mitogen‐activated protein kinase kinase. J Biol Chem 1998;273:18623–32. [DOI] [PubMed] [Google Scholar]
- 42. Hough C, Radu M, Dore JJ. Tgf‐beta induced Erk phosphorylation of smad linker region regulates smad signaling. PLoS One 2012;7:e42513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kireeva ML, Latinkić BV, Kolesnikova TV, Chen CC, Yang GP, Abler AS, Lau LF. Cyr61 and Fisp12 are both ECM‐associated signaling molecules: activities, metabolism, and localization during development. Exp Cell Res 1997;233:63–77. [DOI] [PubMed] [Google Scholar]
- 44. Surveyor GA, Brigstock DR. Immunohistochemical localization of connective tissue growth factor (CTGF) in the mouse embryo between days 7.5 and 14.5 of gestation. Growth Factors 1999;17:115–24. [DOI] [PubMed] [Google Scholar]
- 45. Ito Y, Aten J, Bende RJ, Oemar BS, Rabelink TJ, Weening JJ, Goldschmeding R. Expression of connective tissue growth factor in human renal fibrosis. Kidney Int 1998;53:853–61. [DOI] [PubMed] [Google Scholar]
- 46. Blalock TD, Duncan MR, Varela JC, Goldstein MH, Tuli SS, Grotendorst GR, Schultz GS. Connective tissue growth factor expression and action in human corneal fibroblast cultures and rat corneas after photorefractive keratectomy. Invest Ophthalmol Vis Sci 2003;44:1879–87. [DOI] [PubMed] [Google Scholar]
- 47. Kantarci A, Black SA, Xydas CE, Murawel P, Uchida Y, Yucekal‐Tuncer B, Atilla G, Emingil G, Uzel MI, Lee A, Firatli E, Sheff M, Hasturk H, Van Dyke TE, Trackman PC. Epithelial and connective tissue cell CTGF/CCN2 expression in gingival fibrosis. J Pathol 2006;210:59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rittie L, Perbal B, Castellot JJ Jr, Orringer JS, Voorhees JJ, Fisher GJ. Spatial‐temporal modulation of CCN proteins during wound healing in human skin in vivo. JCell Commun Signal 2011;5:69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hayashi N, Kakimuma T, Soma Y, Grotendorst GR, Tamaki K, Harada M, Igarashi A. Connective tissue growth factor is directly related to liver fibrosis. Hepatogastroenterology 2002;49:133–5. [PubMed] [Google Scholar]
- 50. Morikawa H, Tamori A, Nishiguchi S, Enomoto M, Habu D, Kawada N, Shiomi S. Expression of connective tissue growth factor in the human liver with idiopathic portal hypertension. Mol Med 2007;13:240–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kobayashi H, Hayashi N, Hayashi K, Yamataka A, Lane GJ, Miyano T. Connective tissue growth factor and progressive fibrosis in biliary atresia. Pediatr Surg Int 2005;21:12–16. [DOI] [PubMed] [Google Scholar]
- 52. Weng HL, Ciuclan L, Liu Y, Hamzavi J, Godoy P, Gaitantzi H, Kanzler S, Heuchel R, Ueberham U, Gebhardt R, Breitkopf K, Dooley S. Profibrogenic transforming growth factor‐beta/activin receptor‐like kinase 5 signaling via connective tissue growth factor expression in hepatocytes. Hepatology 2007;46:1257–70. [DOI] [PubMed] [Google Scholar]
- 53. Jiang WG, Watkins G, Fodstad O, Douglas‐Jones A, Mokbel K, Mansel RE. Differential expression of the CCN family members Cyr61, CTGF and Nov in human breast cancer. Endocr Relat Cancer 2004;11:781–91. [DOI] [PubMed] [Google Scholar]
- 54. Diaz de Leon A, Cronkhite JT, Yilmaz C, Brewington C, Wang R, Xing C, Hsia CC, Garcia CK. Subclinical lung disease, macrocytosis, and premature graying in kindreds with telomerase (TERT) mutations. Chest 2011;140:753–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yang F, Tuxhorn JA, Ressler SJ, McAlhany SJ, Dang TD, Rowley DR. Stromal expression of connective tissue growth factor promotes angiogenesis and prostate cancer tumorigenesis. Cancer Res 2005;65:8887–95. [DOI] [PubMed] [Google Scholar]
- 56. Goodwin CR, Lal B, Zhou X, Ho S, Xia S, Taeger A, Murray J, Laterra J. Cyr61 mediates hepatocyte growth factor‐dependent tumor cell growth, migration, and Akt activation. Cancer Res 2010;70:2932–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chen PP, Li WJ, Wang Y, Zhao S, Li DY, Feng LY, Shi XL, Koeffler HP, Tong XJ, Xie D. Expression of Cyr61, CTGF, and WISP‐1 correlates with clinical features of lung cancer. PLoS One 2007;2:e534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dornhöfer N, Spong S, Bennewith K, Salim A, Klaus S, Kambham N, Wong C, Kaper F, Sutphin P, Nacamuli R, Höckel M, Le Q, Longaker M, Yang G, Koong A, Giaccia A. Connective tissue growth factor‐specific monoclonal antibody therapy inhibits pancreatic tumor growth and metastasis. Cancer Res 2006;66:5816–27. [DOI] [PubMed] [Google Scholar]
- 59. Hartel M, Di Mola FF, Gardini A, Zimmermann A, Di Sebastiano P, Guweidhi A, Innocenti P, Giese T, Giese N, Büchler MW, Friess H. Desmoplastic reaction influences pancreatic cancer growth behavior. World J Surg 2004;28:818–25. [DOI] [PubMed] [Google Scholar]
- 60. Behrens ME, Grandgenett PM, Bailey JM, Singh PK, Yi CH, Yu F, Hollingsworth MA. Expression and differential regulation of connective tissue growth factor in pancreatic cancer cells. Oncogene 1999;18:1073–80. [DOI] [PubMed] [Google Scholar]
- 61. Korc M. Pancreatic cancer‐associated stroma production. Am J Surg 2007;194(4 Suppl):S84–6. [DOI] [PMC free article] [PubMed] [Google Scholar]