

ABSTRACT
Dressing materials involve conventional gauzes and modern materials such as hydrogels and foam‐based biomaterials. Although the choice of dressing material depends on the type of wound, a dressing material is expected to be non‐cytotoxic. Additionally, moist dressing is considered appropriate to accelerate epithelialisation, while dry dressing may cause tissue damage during removal. An ideal dressing material is expected to provide a moist environment and degrade and release the drug for faster wound healing. Thus, we have designed a hydrogel‐based biodegradable dressing material to provide the moist environment with no cytotoxic effect in vitro. The design of the hydrogel involved alginate–collagen reinforced with whisker cellulose derived from cotton. The hydrogel was prepared via amide linkage in the presence of 1‐ethyl‐(dimethylaminopropyl) carbodiimide (EDC) and N‐hydroxysulfosuccinimide (NHS), followed by divalent cationic cross‐linking of alginate and hydrogen bonding with cellulose. The high water retention capability of the hydrogel enables a moist environment to be maintained in the wounded area. The constituents of the hydrogel provided a microenvironment that was suitable for cell proliferation in the vicinity of the hydrogel but inhibited cell attachment on it. The MTT assay results indicated a higher fibroblast proliferation and viability in the presence of the hydrogel.
Keywords: Alginate, Cellulose, Collagen, Hydrogel, Skin tissue engineering
Introduction
Wound healing is a complex physiological process, involving sequential overlapping events including haemostasis followed by an inflammatory response, cell proliferation including angiogenesis and tissue remodelling 1. The process of tissue repair involves direct cell‐to‐cell and cell–matrix interaction and can be accelerated by suitable internal and external cues that depend on the properties of extracellular matrix 2. The efficacy of a therapeutic approach depends on wound and tissue properties. For instance, for skin tissue, wounds can be incisional, excisional, acute and chronic 3.
The skin is the largest organ of the body and protects the body from external environmental threats, such as injury and bacterial infection. The prevention of microbial infection and trans‐epidermal water loss is required to accelerate wound healing 4, 5. This helps in the restoration of the skin barrier, which plays a very crucial role in the treatment of injuries 6. Moreover, wound healing is a dynamic process, and the performance of a dressing material is influenced by the progression of the healing phase. In this context, a warm and moist environment promotes faster wound healing 4, 7, 8. Moist dressing not only increases the rate of epithelialisation but also prevents the disruptive damage to the wound. In this context, dry dressing may cause tissue damage during its removal 9. Thus, an ideal wound dressing material is characterised by high porosity and high surface‐to‐volume ratio required for oxygen permeability and absorption of exudates 10, 11, 12. In this regard, hydrogel‐based dressing materials are considered potentially suitable and constitute the focus of the study described here. Hydrogels are made of hydrophilic polymers that may absorb a large quantity of water, up to thousands of times their dry weight 13. Hydrogels are considered ‘physical’ or ‘reversible’ gels because of molecular entanglements of polymeric networks and the presence of secondary forces, such as hydrophobic forces and ionic and hydrogen bonding 14, 15. Depending on the application, hydrogel properties can be tailored. For instance, hydrogels can be made chemically stable or degradable with a controlled degradation rate.
A number of natural and synthetic materials have been studied as hydrogel‐based wound dressings. With regards to this, natural biopolymers, collagen, alginate, chitosan, cellulose acetate and hyaluronic acid 16, 17, 18, 19, 20, 21 were explored in terms of biocompatible properties and their potential role as a wound dressing material. In this context, alginate‐based dressing material has a pharmacological function, which is because of calcium ions used as a cross‐linking agent. Studies have shown that the presence of calcium alginate can promote the proliferation of mouse fibroblast 22. Also, alginate is biodegradable and can be washed away with saline solution during the replacement of dressings 23. Similarly, collagen is a natural protein and primarily used as a biomaterial because of its unique properties, such as non‐antigenicity, biodegradability and biocompatibility 21. Collagen‐based dressings are beneficial in the treatment of severe burns and wounds, such as pressure sores and diabetic foot ulcers 24, 25, 26. However, untreated natural collagen cannot be directly used because of its high thrombogenicity and inferior mechanical properties, and it also promotes the deposition of calcium 27. While collagen provides a protein matrix for the tissue growth, chitosan provides a non‐protein matrix that activates macrophages for tumouricidal activity. Chitosan stimulates cell proliferation and helps in natural blood clotting 28. Thus, different materials in dressings play a different role and collectively help in wound healing.
Recent studies focused on developing wound dressing materials that are biocompatible and can accelerate wound repair by providing an ideal environment for epithelialisation 29, 30, 31. In this regard, the present study is focused on exploring alginate‐, collagen‐ and cellulose‐based hydrogel dressing material that provides a moist environment to the wounded area without any cytotoxic effect. A two‐step cross‐linking process was used to synthesise the hydrogel. Collagen was conjugated with alginate by cross‐linking with N‐(3‐dimethylaminopropyl)‐N′‐ethylcarbodiimide (EDC) and N‐hydroxysuccinimide (NHS). This approach minimises the direct interaction of cells with the collagen, which is one of the main causes for calcification and thrombogenicity. Furthermore, cellulose was used to enhance the mechanical strength of the hydrogel.
Experimental
Materials
Collagen and bovine achilles tendon powder (Cat. No. 9007345) was procured from Alfa Aesar (USA). Alginate (alginic acid sodium salt from Brown Algae, Cat. No. A0682) and N‐hydroxy‐succinimide (NHS, Cat. No. 130672) were obtained from Sigma‐Aldrich (USA); 1‐ethyl‐3‐(3‐dimethylaminopropyl) carbodiimide (EDC, Cat. No. 22980) was purchased from Thermo Scientific (USA). Whisker cellulose was synthesised using acid hydrolysis of cellulose from cotton.
Synthesis of whisker cellulose from cotton
Whisker cellulose was prepared by acid hydrolysis of cellulose from cotton. First, cotton was washed by 2 wt% NaOH solution for 1 hour using a magnetic stirrer at room temperature. Next, the cotton was soaked in deionised water at 80 °C for 1 hour and dried overnight in air. After drying, the cellulose powder was hydrolysed, while vigorous magnetic stirring at 40 °C was carried out for 1 hour using concentrated hydrochloric acid at 20 ml/g cellulose‐to‐hydrochloric acid ratio. Subsequently, the mixed solution was centrifuged at 10 000 rpm for 5 minutes and washed with deionised water until a pH of 7 was obtained. Finally, the hydrolysed cellulose was stored at −80 °C for overnight followed by lyophilisation for 24 hours using a freeze dryer. The lyophilised powder was ground into fine whisker powder using agate mortar and pestle. This fine powder was characterised by X‐ray diffraction (Bruker's D8 Discover; Bruker, Germany) and FT‐IR (FT/IR‐4600LE; Jasco, Japan). Furthermore, scanning electron microscopy (SEM; S‐4800; Hitachi, Japan) was used to characterise the morphology of cellulose.
Synthesis of hybrid pre‐hydrogel reinforced with cellulose
First, pre‐hydrogel was synthesised using NHS and EDC as cross‐linkers, followed by final cross‐linking with CaCl2. To synthesise pre‐hydrogel, first, an aqueous solution of collagen was prepared by mixing 50 mg pepsin and 500 mg collagen powder in 15 ml high‐purity deionised water (DI), and the solution was kept on a magnetic stirrer (500 rpm) at 37 °C. The pH was adjusted to approximately 1·0 using concentrated HCl to activate pepsin. After 2 hours, when collagen was completely digested, pH was further adjusted to 7·4 using ammonium hydroxide (NH4OH) to deactivate the pepsin. The concentration of prepared collagen solution was considered 3·33 wt%. Next, 3·33 wt% alginate (500 mg) was mixed in powder form to 3·33 wt% aqueous solution of collagen at 37 °C using a magnetic stirrer. Subsequently, 6·66 wt% cellulose (1000 mg) was mixed in the powder form to the solution of alginate and collagen at 37 °C using a magnetic stirrer. This led to a 10‐ml solution with a final ratio of alginate, collagen and cellulose of 1:1:2. To prepare the pre‐hydrogel, 15 mg NHS and 25 mg EDC was added to the 10‐ml solution of alginate, collagen and cellulose (1:1:2) and stirred at 37 °C for 2 hours. The prepared pre‐hydrogel reinforced with cellulose was filled in a sterilised syringe and stored at 6 °C in the refrigerator until use.
The pre‐hydrogel was cast in a 24‐well plate, followed by cross‐linking with 0·05 M and 1 M CaCl2 for 10 minutes to prepare a hydrogel with the strength appropriate for handling. After cross‐linking, the hydrogel samples were immersed in 1× PBS solution for 15 minutes to remove excess CaCl2. These samples were freeze dried for further characterisation to study the phase assemblage and pore architecture.
Phase assemblage and pore architecture
To study the phase assemblage and microstructure of the hydrogel, samples were analysed using XRD, FT‐IR and SEM in secondary electron (SE) mode. For XRD and FT‐IR, the freeze‐dried hydrogel samples were ground to powder using agate mortar and pestle. XRD of hydrogel powder as well as starting powders was carried out using CuKα as a monochromatic X‐ray source of wavelength 1·54056 Å at 40 kV voltage and 40 mA current. The samples were scanned in locked‐coupled mode from (2θ) 10° to 90° at a scanning rate of 2°/minutes and increment (step size) of 0·03°. To conduct FT‐IR studies, thin pellets of samples were prepared by mixing KBr and the sample in the ratio of 200:1, followed by pressing the mixture into a 3‐mm diameter mould. To acquire data, thin sample pellets were scanned from 4000 to 400 cm−1. For SEM studies, the freeze‐dried samples were first gold‐coated to minimise charging during imaging. Samples were imaged in the SE mode at an accelerating voltage of 20 kV for microstructural characterisation of the hydrogel. Energy dispersive spectroscopy (EDS) analysis was carried out for the elemental analysis of hydrogel.
Effect of cross‐linking time on the dissolution behaviour of hydrogel
To study the stability of hydrogel in aqueous media, the samples prepared by cross‐linking for 10 and 15 minutes using 0·05 M CaCl2 were cut to dimensions of 8 × 8 × 2 mm and immersed in 2 ml 1× PBS in a 24‐well plate. Before immersion, samples were sterilised for 30 minutes using UV light. The well plate was sealed with parafilm and kept at 37 °C for 2, 4 and 6 days to study dissolution. To obtain statistically relevant data, the experiments were carried out in triplicates and repeated at least thrice. After incubation for 2, 4 and 6 days, 200‐µl solution from each well was transferred to a 96‐well plate, and absorption was measured at 630 nm using ELISA plate reader; 1× PBS was used as a control.
A standard curve was plotted between the amount of cellulose dissolved in 1× PBS (5, 10, 25, 20, 25 and 30 mg/ml) and corresponding optical density (OD) at 630 nm. A trend line on the plotted curve was used to obtain an equation to estimate the amount of dissolved cellulose from the hydrogel during dissolution.
Study of sorption kinetics of hydrogel in PBS
To study the sorption kinetics, pre‐hydrogels were cast in an eight‐well plate and cross‐linked with 0·05 M and 1 M CaCl2 for 10 minutes. After this, hydrogel samples were washed with 1× PBS for 15 minutes to remove excess CaCl2. Next, samples were cut into a rectangular shape with dimensions of 8 × 8 × 4 mm thickness, followed by freeze drying. Next, the freeze‐dried samples were weighed and immersed in 1× PBS. The samples immersed in 1× PBS were kept at 37 °C and constant humidity. The weight of swollen samples was measured every 30 minutes. Before weight measurement, excess water on the sample surface was removed using filter paper. This process was repeated, and final weight was taken at equilibrium swelling. The weight change during the sorption process was used to calculate the swelling ratio, equilibrium swelling ratio, swelling rate and percentage equilibrium water content and data were plotted as a function of time using Equations (1–4). To obtain statistically relevant data, all measurements were carried out in triplicate, and the study was repeated at least thrice.
| (1) |
| (2) |
| (3) |
| (4) |
where m t, m d, m equ and Δt are the weight of the swollen hydrogel at time ‘t’, weight of the dried hydrogel and weight of the swollen hydrogel at equilibrium state and time interval between two measurements, respectively.
PBS desorption kinetics for swollen hydrogel
In continuation of Section 2·6, after attaining equilibrium swelling, samples were removed from 1× PBS. After removing the excess water from the sample surface using a filter paper, the swollen weight (w ∞) of samples was measured. After this, samples were dried at 37 °C with constant humidity for 30 minutes, and the weight was measured. This process of weight measurement after every 30 minutes was repeated until constant weight (weight of completely dried sample) was obtained. The obtained PBS desorption data was plotted with respect to time, and Equation (5) was used to estimate the PBS desorption ratio.
| (5) |
where mt, m 0 and m ∞ are the weight of hydrogel at time ‘t’, initial time ‘0’, and time when the sample was completely dried, respectively.
Cytocompatibility assessment
Hydrogel was cast in an eight‐well plate using the method described in Section 2·5. The cast hydrogel sheet was cut into small pieces of dimensions 8 × 8 × 2 mm for cell culture studies. These samples were UV sterilised for 30 minutes and washed with 1× PBS. The NIH3T3 fibroblast cells (ATCC, USA) with a cell density of approximately 20,000 cells/ml were added to each well in an MEM (Minimum Essential Media) medium and incubated at 37 °C in a humidified atmosphere (95% RH) and 5% CO2. During incubation, old culture media were replaced with fresh culture media every second day.
Cell–cell and cell–material interaction
To study cell–cell and cell–material interaction, after 4 days of incubation, the samples were stained for expression of actin, vinculin and nucleus. After staining, samples were removed from the well plate, and cells adhered to the well plate surface were observed from the bottom of the well plate using a fluorescence microscope. Well plate surface was used as a control. The details of immunofluorescence microscopy protocol used in this study have been reported elsewhere 32. Briefly, before fluorescence microscopy, cells were fixed with 4% formaldehyde for 20 minutes at room temperature, followed by washing with 1× PBS. Next, cells were treated with 0·1% triton×100 for 6 minutes to permeabilise the cell membrane. Furthermore, after washing with 1× PBS, samples were treated with 5% FBS for 30 minutes to minimise the non‐specific binding of antibodies. After this, cells were treated for 60 minutes with a staining agent for study of expression of actin. After washing with 1× PBS, samples were stained for 60 minutes for expression of actin stress fibres. Finally, after washing with 1× PBS, samples were incubated with DAPI to stain the cell nucleus. After staining, samples were washed at least thrice with 1× PBS. The stained samples were stored in the dark at 6 °C in 1× PBS until imaging by a fluorescence microscope.
MTT assay/cell proliferation assay
MTT assay protocol used in this study has been previously reported 33. Briefly, after 1, 2 and 4 days of incubation, the culture media were replaced with 10% MTT solution, followed by incubation for 4 hours in a CO2 incubator. The MTT solution was prepared by dissolving MTT salt (3(4, 5‐dimethylthiazol‐2‐yl)‐2, 5‐diphenyl tetrazolium bromide)) reagent in 1× PBS 33. This leads to the reduction of MTT salt into insoluble formazan crystals by the mitochondrial succinate dehydrogenase enzyme of viable cells. After 4 hours of incubation, cells were removed from the sample surface using slow up–down pipetting, and the cell containing the MTT reagent was transferred to a centrifuge tube, followed by centrifugation for 5 minutes at 10 000 rpm. After centrifugation, the supernatant was discarded, and a violet‐colour pellet of formazan crystals was dissolved in 400 µl dimethyl sulfoxide solution (DMSO; D8418; Sigma‐Aldrich, France). About 150 µl of this violet‐colour solution was transferred to a 96‐well plate, and optical density (OD) was measured at 570 nm wavelength using an ELISA plate reader (ELx800; BioTek, USA). The MTT assay result of hydrogel was compared with the control (well plate surface) samples. The MTT data were represented as mean ± standard error of mean and plotted as a function of incubation of time using the Origin software (version 8·5; Origin Lab Corporation, USA).
Statistical analysis
The MTT data were analysed using commercial software (SPSS 19·0; IBM, USA). For statistical analysis, a multivariate post‐hoc test was applied, and significant difference between the sample mean and control at P < 0·05 was measured using Dunnett t (represented by symbol *) tests. Also, a Dunnett C (represented by symbol **) test was used to compare the samples' means on different incubation time.
Results
Hydrogel synthesis
To synthesise the hydrogel, first, whisker cellulose was prepared by acid hydrolysis of cellulose from cotton. Synthesis of hydrogel involved a two‐step process, pre‐hydrogel prepared by cross‐linking using NHS and EDC followed by final cross‐linking with CaCl2 for 10 minutes prior to application (i.e., cell culture), as shown in Figure 1. Two different concentrations of CaCl2 (0·05 and 1 M) were used to obtain hydrogels of different pore architecture.
Figure 1.
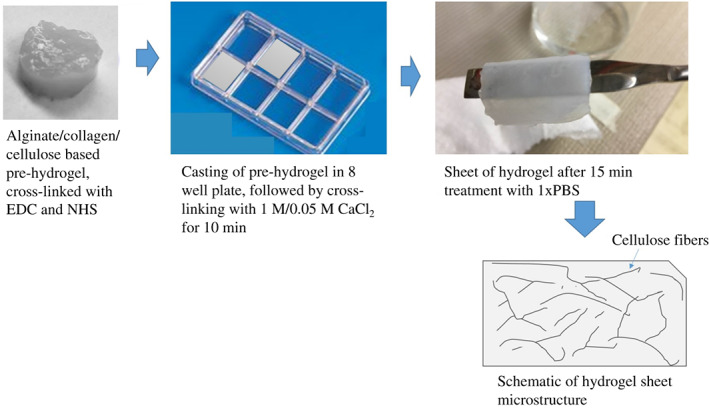
Illustration of preparation method of cellulose‐/collagen‐/alginate‐based hydrogel sample preparation.
Microstructure and phase assemblage
The secondary electron microscopy (SEM) of freeze‐dried cellulose powder, hydrogel cross‐linked with 0·05 M CaCl2 and hydrogel cross‐linked with 1 M CaCl2 was carried out to study the morphology of cellulose powder and the pore architecture and phase distribution (through atomic number contrast) in cross‐linked hydrogel (Figure 2). SEM revealed a whisker morphology of cellulose powder with no evidence of agglomeration (Figures 2 A and B). The low and high magnification micrographs of hydrogel cross‐linked by 0·05 and 1 M CaCl2 confirmed a highly porous microstructure with bimodal distribution of pores (Figures 2 C–F). Moreover, pores were larger in size in the case of 1 M CaCl2 than 0·05 M CaCl2.
Figure 2.

Representative scanning electron micrographs of (A, B) whisker cellulose powder, (C, D) freeze‐dried hydrogel cross‐linked for 10 minutes with 0·05 M CaCl2 and (E, F) freeze‐dried hydrogel cross‐linked for 10 minutes with 1 M CaCl2.
XRD analysis of hydrogel‐1 (cross‐linked with 1 M CaCl2) confirmed the existence of diffraction peaks corresponding to alginate, collagen and cellulose, and a few peaks were associated with sodium chloride (Figure 3). In contrast, no diffraction peaks corresponding to sodium chloride was observed in the case of hydrogel‐0·05 (cross‐linked with 0·05 M CaCl2) (Figure 3). In case of hydrogel‐0·05, only diffraction peaks of alginate, collagen and cellulose were observed. A comparison of hydrogel‐1 and hydrogel‐0·05 revealed the greater presence of collagen and alginate in hydrogel‐0·05 based on the intensity of XRD diffraction peaks. The EDS analysis confirmed the presence of approximately 58% and 17% calcium in hydrogel‐1 and hydrogel‐0·05, respectively.
Figure 3.
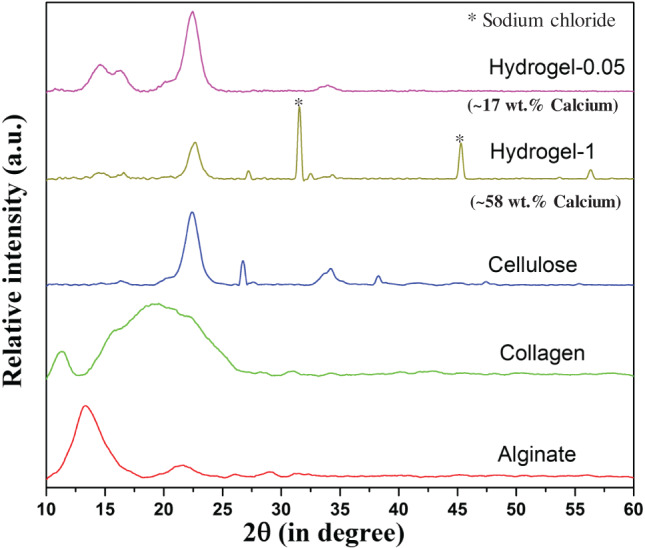
XRD of alginate, collagen, cellulose powder and freeze‐dried hydrogel cross‐linked for 10 minutes with 0·05 M CaCl2 (hydrogel‐0·05) and 1 M CaCl2 (hydrogel‐1). Hydrogel‐1 confirmed the presence of sodium chloride and an excessive amount of calcium (based on EDS results).
FT‐IR studies were carried out only with hydrogel‐0·05 sample to confirm the presence of alginate and collagen in hydrogel (Figure 4). The presence of alginate in the hydrogel was confirmed from the existence of the absorption peaks at 3450, 1618, 1440 and 1050 cm−1 corresponding to —OH group, —COO—, —COO— and C—O, respectively. The presence of absorption peaks at 1658 and 1551 cm−1 were associated with amide‐I and amide‐II, respectively. This confirmed the presence of collagen in the hydrogel. No conformational change in the structure of alginate and collagen was observed.
Figure 4.
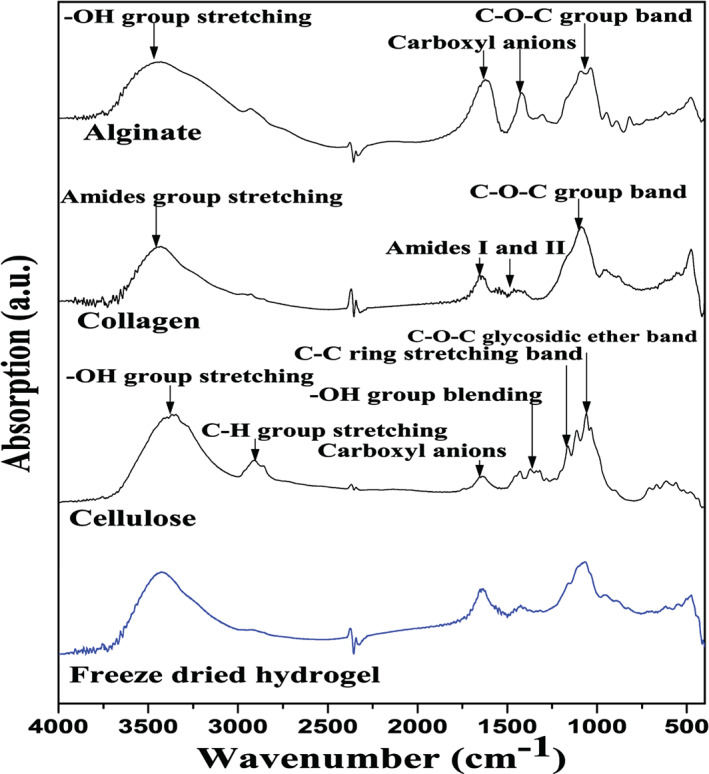
FT‐IR of freeze‐dried hydrogel (cross‐linked for 10 minutes with 0·05 M CaCl2), cellulose, collagen, and alginate, gelatin and freeze‐dried hydrogel. KBr was used as a reference.
Sorption and desorption kinetics of hydrogel
Swelling ratio (Figure 5), swelling rate (Figure 6) and desorption ratio (Figure 7) of cross‐linked hydrogel was studied to understand sorption and desorption kinetics in 1× PBS. A strong effect of CaCl2 (final cross‐linker) concentration on the swelling ratio was noted, with a higher swelling ratio in the case of hydrogel cross‐linked with 1 M CaCl2 than 0·05 m CaCl2. In both the cases, an increasing trend in swelling ratio was observed with time.
Figure 5.
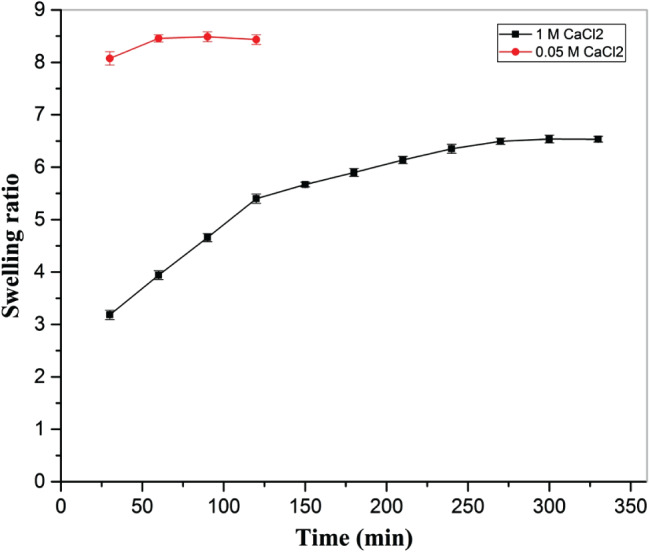
Effect of cross‐linker amount (0·05 M and 1 M CaCl2) on swelling ratio of hydrogel, immersed in 1× PBS at 37 °C at constant humidity maintained in a hot air oven.
Figure 6.
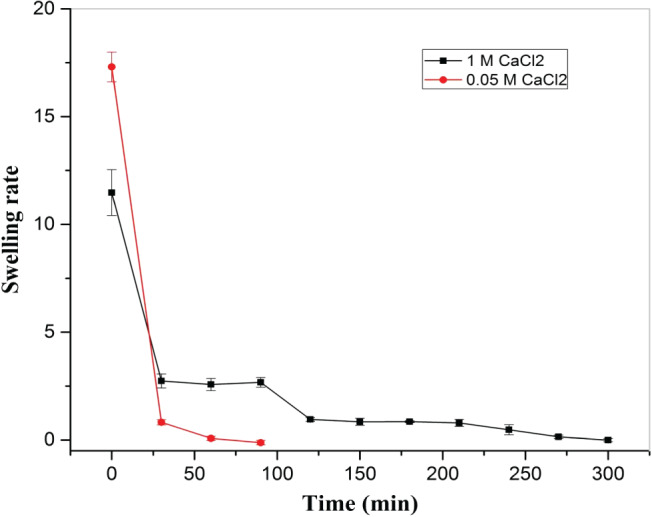
Effect of cross‐linker amount (0·05 M and 1 M CaCl2) on hydrogel swelling rate with time in 1× PBS at 37 °C at constant humidity maintained in a hot air oven.
Figure 7.
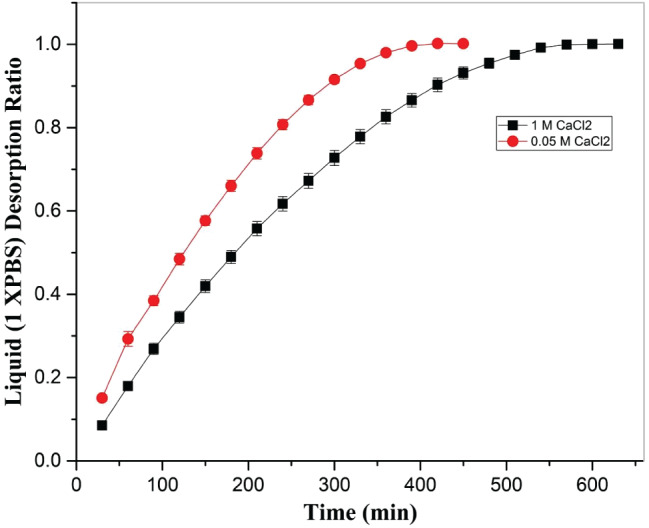
Effect of cross‐linker amount (0·05 M and 1 M CaCl2) on desorption rate of 1× PBS from hydrogel with time at 37 °C at constant humidity maintained in a hot air oven.
Furthermore, as compared to 1 M CaCl2, a higher swelling rate was observed in the case of 0·05 M CaCl2 during initial stage of swelling, with a decreasing trend in both cases. However, after 25 minutes of immersion of hydrogel in 1× PBS, a lower swelling rate was noted in the case of 0·05 M CaCl2 than 1 M CaCl2, and equilibrium swelling was reached significantly earlier in hydrogel cross‐linked with 0·05 M CaCl2 than hydrogel cross‐linked with 1 M CaCl2. In the case of 0·05 M CaCl2, the values of equilibrium swelling ratio and percentage equilibrium water (1× PBS) content were 8·46 ± 0·08 and 89·43 ± 0·01, respectively, while for 1 M CaCl2, the values of equilibrium swelling ratio and percentage equilibrium water (1× PBS) content were 6·52 ± 0·06 and 86·70 ± 0·01, respectively.
After the sorption study, samples were dried at 37 °C at constant humidity, and progressive change in weight was recorded to study the desorption kinetics. In both the cases (0·05 M and 1 M CaCl2), a similar trend in the desorption ratio of 1× PBS was observed with a lower value of liquid desorption ratio at a given time (Figure 7). However, during the later stage of drying, a stable desorption ratio was observed.
Dissolution study of hydrogel in 1× PBS
To study the dissolution behaviour of hydrogel, first, a standard curve was obtained (Figure 8) by dissolving different amount of cellulose (5, 10, 15, 20, 25 and 30 mg) in 1 ml 1× PBS. The corresponding optical density was measured at 630 nm using an ELISA plate reader. For optical density measurements, 200 μL solution was transferred to a 96‐well plate. Optical density was plotted as a function of dissolved amount of cellulose in 1× PBS and a trend line obtained. This equation (absorption = 0·05377 × dissolved amount of cellulose) was used to calculate the dissolved amount of cellulose in 1× PBS at 37 °C at a given time at a given optical density.
Figure 8.
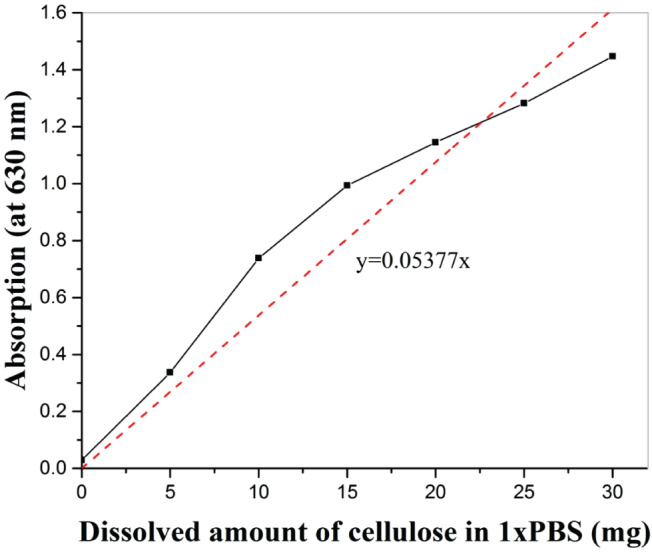
Standard plot between dissolved cellulose in 1× PBS and absorption at 630 nm depicting relation between absorption and dissolved amount of cellulose.
Next, pre‐hydrogel samples cross‐linked with 0·05 M CaCl2 for two different time periods (10 and 15 minutes) were kept in a 24‐well plate, with one sample in each well. Samples were incubated in 1 ml 1× PBS at 37 °C for 2, 4 and 6 days. After incubation, 200 μL solution from each well was transferred to a 96‐well plate without disturbing the integrity of the hydrogel, followed by measuring the absorption of the solution at 630 nm (Figure 9). The equation obtained from the standard curve was used to calculate the dissolved amount of cellulose in 1× PBS at a given time. Considering that the cellulose was homogenously distributed in the hydrogel, the dissolved amount of cellulose was used to understand the dissolution behaviour of hydrogel in 1× PBS. There was no evidence of burst release (corresponding to the dissolution of hydrogel components in the solution) during the entire duration of the dissolution study. There was no effect of cross‐linking time on the dissolution profile. However, in both cases (10 and 15 minutes), there was an increasing tendency in the dissolution of hydrogel with time, followed by a decrease in the dissolution rate after 4 days of incubation.
Figure 9.
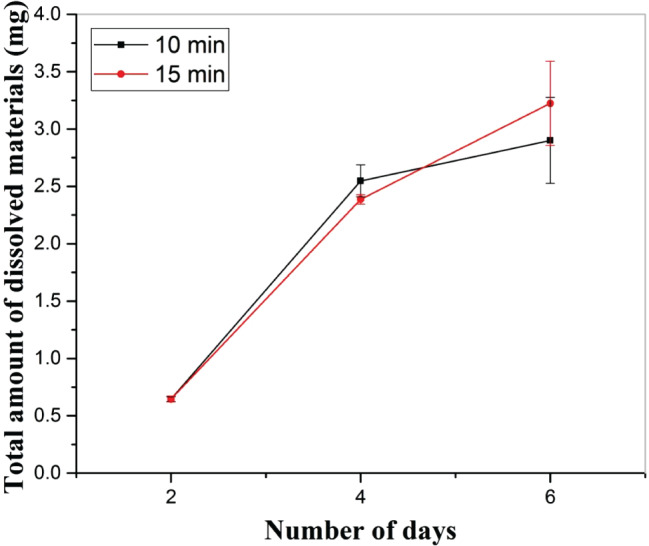
Effect of immersion time and cross‐linking time on the dissolution behaviour of hydrogel, cross‐linked with 0·05 M CaCl2.
Cell adhesion and proliferation
We studied cell adhesion and proliferation of fibroblasts in the presence of hydrogel cross‐linked with 0·05 M CaCl2 for 10 minutes. The objective was to explore the ability of hydrogel in not supporting cell adhesion without having any cytotoxic effect. Cells were found on the hydrogel surface after 4 days of incubation, as confirmed from the staining of nucleus and actin filaments (Figure 10). However, a high density of cells was noted in the well plate area around the hydrogel. As shown in Figure 10, the black area in Figures 10 B, C, E and F is hydrogel and shows no evidence of staining of nucleus and actin.
Figure 10.

Fluorescence micrographs showing proliferation of cells on the well plate surface in the presence of the hydrogel cross‐linked with 0·05 M CaCl2. (A, D) An artistic representation of presence of hydrogel sample and cells in the well plate after 4 days of incubation. Cells were seeded on both sample and well plate surface, but no cell adhesion was noted on the sample surface. (B) and (C) are the cells on well plate surface, stained for the nucleus and actin, respectively. (D), (E) and (F) are high magnification micrographs of (A), (B) and (C), respectively.
Furthermore, an MTT assay was used to study the viability/proliferation of fibroblasts in the presence of hydrogel (cross‐linked with 0·05 M CaCl2 for 10 minutes) and hydrogel‐0·2 gelatin (cross‐linked with 0·05 M CaCl2 for 10 minutes and then coated with 0·2 wt % gelatin), and results were compared with control (well plate surface). This study was carried out for 1, 2 and 4 days (Figure 11). In all the cases, an increase in cell proliferation with time was noted. After the first day, a relatively higher cell density was noted on control than hydrogel‐1 and hydrogel‐2. However, after the second and fourth day of incubation, cell density became higher on the hydrogel surface as compared to control. Interestingly, no significant difference in the cell viability on the hydrogel‐1 and hydrogel‐2 was observed after 1, 2 and 4 days of incubation. In contrast, a Dunnett t‐test revealed a significant difference in cell proliferation when hydrogel‐1 and hydrogel‐2 were compared with the control after 2 and 4 days of incubation. Moreover, a comparison of cell proliferation on a given sample on different days (1, 2 and 4 days) using a Dunnett C‐test indicated significant difference in proliferation on different days (1,1,2,2,4,4), marked with ** in Figure 11.
Figure 11.
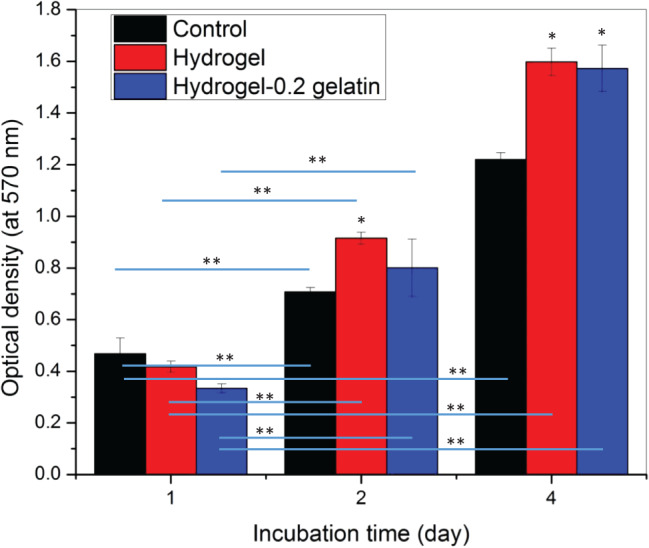
MTT assay results showing cell proliferation after 1, 2 and 4 days of incubation on well plate surface in the presence of hydrogel cross‐linked with 0·05 M CaCl2. The optical density corresponding to cell number is reported as mean ± standard error of mean at n = 3. The * represents significant difference in the viable cell density on samples compared with the control (well plate surface without hydrogel) on the same day using a Dunnett t‐test at P < 0·05. The ** shows significant difference in viable cell density at P < 0·05 when comparison is made between same sample on different days using a Dunnett C‐test.
Discussion
Collagen is an attractive natural polymer found as a structural protein in mammalian tissues 34. As it is a main component of ECM 35, there is a great potential to use collagen as a scaffolding biomaterial. Additionally, collagen can be blended with other biomaterials, such as acrylonitrile butadiene styrene (ABS) fibres to increase its mechanical properties without affecting its biological properties 36, 37, 38, 39. Therefore, in the present study, cellulose extracted from the cotton was used as supporting material for the collagen. Also, alginate was used as a carrier material for collagen and cellulose to make it flowable during extrusion. In the past, alginate‐based biomaterials were studied mainly as a drug delivery system and as a support material because of its outstanding biocompatibility 40. In the context of skin tissue application, alginate is widely used as a dressing material to treat acute and chronic wounds 7. Alginate is a linear copolymer of β‐(1‐4)‐linked d‐mannuronic acid and β‐(1‐4)‐linked l‐guluronic acid units. Alginate is an anionic polysaccharide, present in the cell wall of brown algae. It is hydrophilic in nature and exhibits chelating ability in the presence of divalent cations such as Ca2+. Therefore, alginate can effectively be used to synthesise hydrogel because of its gelation properties. The hydrogel made of collagen and gelatin was observed to be weaker in strength and fractured easily during folding or wrapping after freeze drying. Thus, to make this hydrogel more elastically compliant and stronger, cellulose whisker was added in the blend of collagen and alginate during hydrogel preparation. Cellulose is abundantly available, has low density, is biodegradable and exhibits good mechanical properties 41, 42. Therefore, we prepared a hydrogel of alginate and collagen, reinforced with cellulose, by using a two‐step cross‐linking method. EDC and NHS were used in the first‐step cross‐linking, followed by cross‐linking with CaCl2 in the second step. During the cross‐linking process, EDC was bonded to the carboxyl group of alginate, followed by NHS bonding with alginate by replacing EDC. The NHS‐bonded alginate product bonded together through the collagen to make the hydrogel, referred as pre‐hydrogel (Figure 12). Furthermore, in the second step, this pre‐hydrogel was cross‐linked with CaCl2 to obtain the final hydrogel (Figure S1, Supporting information). The two‐stage cross‐linking process provides the flexibility of fabricating samples of different size and shape as well as incorporation of cells and growth factors.
Figure 12.
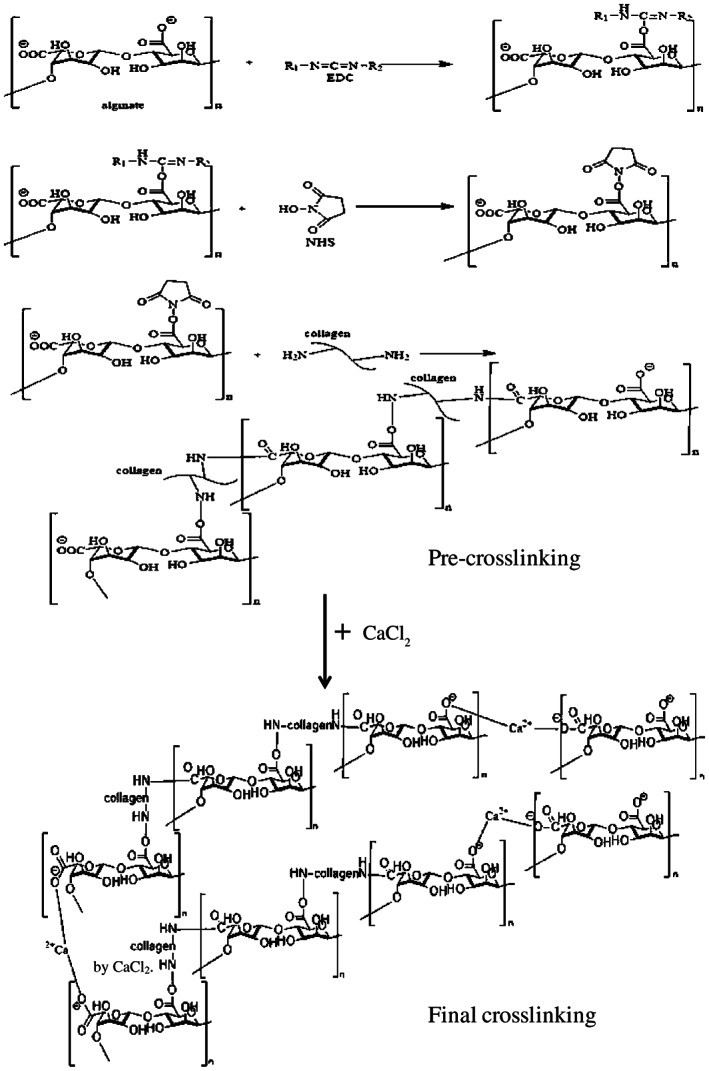
Mechanism of cross‐linking of alginate and collagen through EDC and EHS, followed by CaCl2.
This study encompasses many advantageous properties of the synthesised hydrogel in the context of non‐adherent wound dressing with high absorbent properties. These important properties have been discussed in the following paragraphs on the basis of results obtained from the various characterisation tools.
Microstructure, chemical and physical properties
It has been reported that the long‐term stability of alginic acid (alginate) cross‐linked with divalent metal ions is poor because of the unintended tendency of leaching of ions (used in cross‐linking) from the hydrogel to the solution 43. In addition to this, an excessive amount of these metallic ion‐based cross‐linkers, such as CaCl2, are toxic to cells 44. Therefore, a 0·05 M CaCl2 solution was used in the present study. However, a higher concentration of CaCl2 (1 M) was used from the viewpoint of comparison. To accomplish gelation in the first step, EDC and NHS were used. The addition of EDC and NHS is expected to improve the hydrogel's life because of the stabilisation of amide linkage, thereby preventing the hydrolysis. In addition to this, these chemicals (EDC and NHS) are beneficial over toxic and expensive cross‐linking agents, such as glutaraldehyde and genipin 45, 46.
As shown in Figure 2, an SEM study revealed a highly porous microstructure in the hydrogel cross‐linked with 0·05 M CaCl2 for 10 minutes (labelled as sample A), with the average pore size varying from 50 µm to 250 µm. However, a higher porosity with pore size in the range of 150–400 µm was observed in samples cross‐linked with 1 M CaCl2 for 10 minutes (referred as sample B). It is obvious that a larger pore size (void space) yields inferior mechanical properties in the tensile mode 47. Thus, in the context of dressing material where wrapping and bending is frequent, the sample A is expected to perform better than the sample B. Results of extensive XRD and FT‐IR confirmed the presence of cellulose, alginate and collagen, with no evidence of conformational changes in the structure. However, distinct diffraction peaks of sodium chloride were noticed in the case of sample B. The presence of sodium chloride is associated with the reaction product of calcium chloride and sodium alginate. A 1900‐times higher concentration of calcium chloride in sample B than sample A is an obvious reason for the presence of sodium chloride in sample B. It is important to note that the presence of a higher content of sodium chloride in the sample is expected to further impair the dissolution and pore size in the aqueous media. Thus, to avoid change in the properties of hydrogel during the study because of dissolution of phases like sodium chloride, sample A was selected over sample B for the dissolution and cell culture studies.
Sorption and desorption kinetics
The study of sorption kinetics revealed that, with a high swelling rate to start with, a decrease in swelling rate was observed for both samples A and B. In the case of sample A, equilibrium was attained at least 3 hours earlier than sample B. However, desorption kinetics showed a similar profile. It took at least 8 hours to attain equilibrium in sample A, while sample B took 2 hours more to attain equilibrium. Although there is no information on the effect of sorption and desorption kinetics of dressing material on wound healing, we believe that a higher swelling rate may allow the removal of exudate, and the negative pressure because of sorption may further act as a barrier to inhibit bacterial intervention from the outside environment. The large‐size pores in the hydrogel may provide a pathway for pathogens in air to establish a contact with the wound. In addition to this, slower desorption of absorbed exudate provides a moist environment, which is required for healing of wound.
Dissolution behaviour and in vitro cytocompatibility of the hydrogel
Dissolution behaviour of sample A cross‐linked for 10 and 15 minutes was investigated in 1× PBS. There was no significant difference in the dissolution behaviour of samples cross‐linked for 10 and 15 minutes in the study carried out for 6 days, and data were collected every second day. During dissolution, a constant dissolution rate was observed in sample cross‐linked for 15 minutes. However, in case of a sample cross‐linked for 10 minutes, a slower rate of dissolution was observed after 4 days. An intact structure was noted after the dissolution in 1× PBS. It is important to mention that the dissolution was carried out by the complete immersion of completely wet hydrogel in 1× PBS for 6 days. This condition is harsher than the normal wound. Therefore, we can expect that the developed hydrogel can provide a moist environment for a longer period of time without becoming structurally deformed and disintegrated in the fluid environment. As the developed hydrogel is very stable in the moist condition, we can expect enhanced cell proliferation in the presence of hydrogel without inflammation.
The overall objective of this study was to develop a cytocompatible hydrogel with a large water retention property, which does not encourage cell adhesion and proliferation on it. However, it should promote the cell adhesion and proliferation in its vicinity without causing cytotoxicity. In this context, in our study, no evidence of cells was found on the hydrogel surface even after 4 days of incubation (Figure 10). However, a high density of cells was noted in the well plate area around the hydrogel. In Figure 10, the black area in upper left corner denotes the sample position during the cell culture study (Figure S2).
In addition to this, cell proliferation and cell viability assessment based on MTT assay results showed a similar cell density (directly proportional to the optical density) of fibroblasts in the presence of hydrogel (cross‐linked with 0·05 M CaCl2 for 10 minutes) and hydrogel‐0·2 gelatin (cross‐linked with 0·05 M CaCl2 for 10 minutes and then coated with 0·2 wt% gelatin) (Figure 11). However, the cell density on hydrogel and hydrogel‐0·2 gelatin was higher than the control sample. Here, the well plate surface was used as control. The gelatin‐coated hydrogel was used to study the effect of bare hydrogel on cytotoxicity. In all the cases, an increase in cell density with time was found. The MTT assay and cell adhesion results confirmed that the hydrogel itself did not allow the cell adhesion on its surface but promoted cell proliferation in its vicinity. This is an important outcome and is relevant to wound dressing application, where the dressing material should not allow cell adhesion on its surface but should accelerate wound healing by promoting cell proliferation in the wound area 48. As compared to the well plate surface, a higher proliferation of cells in the presence of hydrogel can be correlated to bovine achilles tendon collagen, present as a major component in the hydrogel. The collagen from tendon is known for its iso‐ionic pH (5·1) and is beneficial when haemostat is required, such as wound area 49.
Conclusions
The proposed collagen–alginate–cellulose hydrogel was characterised by a high water retention ability, with an equilibrium water content of approximately 89%. Collagen and alginate promoted cell proliferation, while EDC/NHS in the presence of CaCl2 allowed effective cross‐linking with no detectable toxicity. The presence of cellulose whisker positively contributed to the structural stability of the hydrogel and prevented splintering because of folding or wrapping after freeze drying.
The developed hydrogel did not encourage cell adhesion. This characteristic ensures the easy removal of dressing material without damaging the wound. Additionally, inherent cytocompatible properties of alginate and collagen enable faster recovery of the damaged tissue because of high cell proliferation in the wound area. The developed hybrid hydrogel can be a cost‐effective solution for dressing of wound, where the removal of exudate and thereafter a moist environment are required for the wound healing, although a rigorous in vivo study and clinical trial are required.
Supporting information
Figure S1. Schematic showing the cross‐linking of alginate and collagen with EDC, NHS and CaCl2.
Figure S2. Schematic illustration of the placement of samples in the 24‐well plate for the in vitro cytocompatibility assessment.
Acknowledgements
Authors acknowledge the funding for the research provided by the Department of Metallurgical, Materials and Biomedical Engineering, University of Texas at El Paso, Texas, USA.
References
- 1. Enoch S, Leaper DJ. Basic science of wound healing. Surgery (Oxford) 2008;26:31–7. [Google Scholar]
- 2. Witte MB, Barbul A. General principles of wound healing. Surg Clin North America 1997;77:509–28. [DOI] [PubMed] [Google Scholar]
- 3. Fu X, Li H. Mesenchymal stem cells and skin wound repair and regeneration: possibilities and questions. Cell Tissue Res 2008;335:317. [DOI] [PubMed] [Google Scholar]
- 4. Field CK, Kerstein MD. Overview of wound healing in a moist environment. Am J Surg 1994;167:S2–6. [DOI] [PubMed] [Google Scholar]
- 5. Depan D, Misra RDK. Hybrid nanoscale architecture of wound dressing with super hydrophilic, antimicrobial, and ultralow fouling attributes. J Biomed Nanotechnol 2015;11:306–18. [DOI] [PubMed] [Google Scholar]
- 6. Liu S‐J, Kau Y‐C, Chou C‐Y, Chen J‐K, Wu R‐C, Yeh W‐L. Electrospun PLGA/collagen nanofibrous membrane as early‐stage wound dressing. J Membr Sci 2010;355:53–9. [Google Scholar]
- 7. Boateng JS, Matthews KH, Stevens HNE, Eccleston GM. Wound healing dressings and drug delivery systems: a review. J Pharm Sci 2008;97:2892–923. [DOI] [PubMed] [Google Scholar]
- 8. Unnithan AR, Nejad AG, Sasikala ARK, Thomas RG, Jeong YY, Murugesan P, Nasseri S, Wu D, Park CH, Kim CS. Electrospun zwitterionic nanofibers with in situ decelerated epithelialization property for non‐adherent and easy removable wound dressing application. Chem Eng J 2016;287:640–8. [Google Scholar]
- 9. Jones V, Grey JE, Harding KG. Wound dressings. Br Med J 2006;7544:777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. GhavamiNejad A, Rajan Unnithan A, Ramachandra Kurup Sasikala A, Samarikhalaj M, Thomas RG, Jeong YY, Nasseri S, Murugesan P, Wu D, Hee Park C. Mussel‐inspired electrospun nanofibers functionalized with size‐controlled silver nanoparticles for wound dressing application. ACS Appl Mater Interfaces 2015;7:12176–83. [DOI] [PubMed] [Google Scholar]
- 11. Agarwal S, Wendorff JH, Greiner A. Use of electrospinning technique for biomedical applications. Polymer 2008;49:5603–21. [Google Scholar]
- 12. GhavamiNejad A, Sasikala ARK, Unnithan AR, Thomas RG, Jeong YY, Vatankhah Varnoosfaderani M, Stadler FJ, Park CH, Kim CS. Mussel inspired electrospun smart magnetic nanofibers for hyperthermic chemotherapy. Adv Funct Mater 2015;25:2867–75. [Google Scholar]
- 13. Hoffman AS. Hydrogels for biomedical applications. Adv Drug Deliv Rev 2012;64:18–23. [DOI] [PubMed] [Google Scholar]
- 14. Campoccia D, Doherty P, Radice M, Brun P, Abatangelo G, Williams DF. Semisynthetic resorbable materials from hyaluronan esterification. Biomaterials 1998;19:2101–27. [DOI] [PubMed] [Google Scholar]
- 15. Prestwich GD, Marecak DM, Marecek JF, Vercruysse KP, Ziebell MR. Controlled chemical modification of hyaluronic acid: synthesis, applications, and biodegradation of hydrazide derivatives. J Control Release 1998;53:93–103. [DOI] [PubMed] [Google Scholar]
- 16. Kumar PTS, Raj NM, Praveen G, Chennazhi KP, Nair SV, Jayakumar R. In vitro and in vivo evaluation of microporous chitosan hydrogel/nanofibrin composite bandage for skin tissue regeneration. Tissue Eng Part A 2012;19:380–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zarandi MA, Zahedi P, Rezaeian I, Salehpour A, Gholami M, Motealleh B. Drug release, cell adhesion and wound healing evaluations of electrospun carboxymethyl chitosan/polyethylene oxide nanofibres containing phenytoin sodium and vitamin C. IET Nanobiotechnol 2015;9:191–200. [DOI] [PubMed] [Google Scholar]
- 18. Srinivasan S, Jayasree R, Chennazhi KP, Nair SV, Jayakumar R. Biocompatible alginate/nano bioactive glass ceramic composite scaffolds for periodontal tissue regeneration. Carbohydr Polym 2012;87:274–83. [DOI] [PubMed] [Google Scholar]
- 19. Unnithan AR, Gnanasekaran G, Sathishkumar Y, Lee YS, Kim CS. Electrospun antibacterial polyurethane–cellulose acetate–zein composite mats for wound dressing. Carbohydr Polym 2014;102:884–92. [DOI] [PubMed] [Google Scholar]
- 20. Anisha BS, Biswas R, Chennazhi KP, Jayakumar R. Chitosan–hyaluronic acid/nano silver composite sponges for drug resistant bacteria infected diabetic wounds. Int J Biol Macromol 2013;62:310–20. [DOI] [PubMed] [Google Scholar]
- 21. Sripriya R, Kumar MS, Sehgal PK. Improved collagen bilayer dressing for the controlled release of drugs. J Biomed Mater Res B Appl Biomater 2004;70:389–96. [DOI] [PubMed] [Google Scholar]
- 22. Schmidt RJ, Turner TD. Calcium alginate dressings. Pharm J 1986;236:578. [Google Scholar]
- 23. Gilchrist T, Martin AM. Wound treatment with Sorbsan—an alginate fibre dressing. Biomaterials 1983;4:317–20. [DOI] [PubMed] [Google Scholar]
- 24. Rho KS, Jeong L, Lee G, Seo B‐M, Park YJ, Hong S‐D, Roh S, Cho JJ, Park WH, Min B‐M. Electrospinning of collagen nanofibers: effects on the behavior of normal human keratinocytes and early‐stage wound healing. Biomaterials 2006;27:1452–61. [DOI] [PubMed] [Google Scholar]
- 25. Chvapil M. Considerations on manufacturing principles of a synthetic burn dressing: a review. J Biomed Mater Res 1982;16:245–63. [DOI] [PubMed] [Google Scholar]
- 26. Donaghue VM, Chrzan JS, Rosenblum BI, Giurini JM, Habershaw GM, Veves A. Evaluation of a collagen‐alginate wound dressing in the management of diabetic foot ulcers. Adv Skin Wound Care 1998;11:114–9. [PubMed] [Google Scholar]
- 27. Nam K, Kimura T, Kishida A. Physical and biological properties of collagen‐phospholipid polymer hybrid gels. Biomaterials 2007;28:3153–62. [DOI] [PubMed] [Google Scholar]
- 28. Jayakumar R, Prabaharan M, Kumar PTS, Nair SV, Tamura H. Biomaterials based on chitin and chitosan in wound dressing applications. Biotechnol Adv 2011;29:322–37. [DOI] [PubMed] [Google Scholar]
- 29. Shibata H, Shioya N, Kuroyanagi Y. Development of new wound dressing composed of spongy collagen sheet containing dibutyryl cyclic AMP. J Biomater Sci Polym Ed 1997;8:601–21. [DOI] [PubMed] [Google Scholar]
- 30. Ulubayram K, Cakar AN, Korkusuz P, Ertan C, Hasirci N. EGF containing gelatin‐based wound dressings. Biomaterials 2001;22:1345–56. [DOI] [PubMed] [Google Scholar]
- 31. Draye J‐P, Delaey B, Van de Voorde A, Van Den Bulcke A, De Reu B, Schacht E. In vitro and in vivo biocompatibility of dextran dialdehyde cross‐linked gelatin hydrogel films. Biomaterials 1998;19:1677–87. [DOI] [PubMed] [Google Scholar]
- 32. Kumar A, Nune KC, Misra RDK. Biological functionality and mechanistic contribution of extracellular matrix ornamented three dimensional Ti6Al4 V mesh scaffolds. J Biomed Mater Res Part A 2016;104:2751–63. [DOI] [PubMed] [Google Scholar]
- 33. Kumar A, Mallik AK, Acikbas NC, Yaygıngol M, Kara F, Mandal H, Basu D, Biswas K, Basu B. Cytocompatibility property evaluation of gas pressure sintered SiAlON–SiC composites with L929 fibroblast cells and Saos‐2 osteoblast‐like cells. Mater Sci Eng C 2012;32:464–9. [Google Scholar]
- 34. Lee CH, Singla A, Lee Y. Biomedical applications of collagen. Int J Pharm 2001;221:1–22. [DOI] [PubMed] [Google Scholar]
- 35. Bruce Alberts, Alexander Johnson, Julian Lewis, Martin Raff, Keith Roberts, Peter Walter; Molecular Biology of the Cell, 5th Edition; Garland Science, Taylor & Francis Group Place: New York, USA, 2008; pages 1464. [Google Scholar]
- 36. Tan W, Krishnaraj R, Desai TA. Evaluation of nanostructured composite collagen‐chitosan matrices for tissue engineering. Tissue Eng 2001;7:203–10. [DOI] [PubMed] [Google Scholar]
- 37. Chen G, Ushida T, Tateishi T. Development of biodegradable porous scaffolds for tissue engineering. Mater Sci Eng C 2001;17:63–9. [Google Scholar]
- 38. Huang L, Nagapudi KP, Apkarian R, Chaikof EL. Engineered collagen–PEO nanofibers and fabrics. J Biomater Sci Polym Ed 2001;12:979–93. [DOI] [PubMed] [Google Scholar]
- 39. Mozdzen LC, Rodgers R, Banks JM, Bailey RC, Harley BAC. Increasing the strength and bioactivity of collagen scaffolds using customizable arrays of 3D‐printed polymer fibers. Acta Biomater 2016;33:25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sun J, Tan H. Alginate‐based biomaterials for regenerative medicine applications. Materials 2013;6:1285–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kumar A, Negi YS, Choudhary V, Bhardwaj NK. Characterization of cellulose nanocrystals produced by acid‐hydrolysis from sugarcane bagasse as agro‐waste. J Mater Phys Chem 2014;2:1–8. [Google Scholar]
- 42. Habibi Y, Lucia LA, Rojas OJ. Cellulose nanocrystals: chemistry, self‐assembly, and applications. Chem Rev 2010;110:3479–500. [DOI] [PubMed] [Google Scholar]
- 43. Lee KY, Mooney DJ. Alginate: properties and biomedical applications. Prog Polym Sci 2012;37:106–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Amin AK, Huntley JS, Bush PG, Simpson A, Hall AC. Chondrocyte death in mechanically injured articular cartilage—the influence of extracellular calcium. J Orthop Res 2009;27:778–84. [DOI] [PubMed] [Google Scholar]
- 45. Wen C, Lu L, Li X. Mechanically robust gelatin–alginate IPN hydrogels by a combination of enzymatic and ionic crosslinking approaches. Macromol Mater Eng 2014;299:504–13. [Google Scholar]
- 46. Rosellini E, Cristallini C, Barbani N, Vozzi G, Giusti P. Preparation and characterization of alginate/gelatin blend films for cardiac tissue engineering. J Biomed Mater Res A 2009;91:447–53. [DOI] [PubMed] [Google Scholar]
- 47. Yu H, Matthew HW, Wooley PH, Yang SY. Effect of porosity and pore size on microstructures and mechanical properties of poly ϵ caprolactone hydroxyapatite composites. J Biomed Mater Res B Appl Biomater 2008;86:541–7. [DOI] [PubMed] [Google Scholar]
- 48. Dhivya S, Padma VV, Santhini E. Wound dressings – a review. BioMedicine 2015;5: 24–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Radhika M, Babu M, Sehgal PK. Cellular proliferation on desamidated collagen matrices. Comp Biochem Physiol C Pharmacol Toxicol Endocrinol 1999;124:131–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Schematic showing the cross‐linking of alginate and collagen with EDC, NHS and CaCl2.
Figure S2. Schematic illustration of the placement of samples in the 24‐well plate for the in vitro cytocompatibility assessment.