

Abstract
The molecular mechanism associated with mammalian meiosis has yet to be fully explored, and one of the main reasons for this lack of exploration is that some meiosis-essential genes are still unknown. The profiling of gene expression during spermatogenesis has been performed in previous studies, yet few studies have aimed to find new functional genes. Since there is a huge gap between the number of genes that are able to be quantified and the number of genes that can be characterized by phenotype screening in one assay, an efficient method to rank quantified genes according to phenotypic relevance is of great importance. We proposed to rank genes by the probability of their function in mammalian meiosis based on global protein abundance using machine learning. Here, nine types of germ cells focusing on continual substages of meiosis prophase I were isolated, and the corresponding proteomes were quantified by high-resolution MS. By combining meiotic labels annotated from the mouse genomics informatics mouse knockout database and the spermatogenesis proteomics dataset, a supervised machine learning package, FuncProFinder (https://github.com/sjq111/FuncProFinder), was developed to rank meiosis-essential candidates. Of the candidates whose functions were unannotated, four of 10 genes with the top prediction scores, Zcwpw1, Tesmin, 1700102P08Rik, and Kctd19, were validated as meiosis-essential genes by knockout mouse models. Therefore, mammalian meiosis-essential genes could be efficiently predicted based on the protein abundance dataset, which provides a paradigm for other functional gene mining from a related abundance dataset.
Keywords: meiosis, spermatogenesis, proteome, machine learning
Abbreviations: DAPI, 4',6-diamidino-2-phenylindole; DEPs, differentially expressed proteins; DSB, double-strand break; KEGG, Kyoto Encyclopedia of Genes and Genomes; LFQ, label-free quantification; MGI, mouse genomics informatics; NBM, naive Bayesian model; PDC, pyruvate dehydrogenase complex; RA, retinoic acid; RBF, radial basis function; RS, round spermatid; SC, synaptonemal complex; sgRNAs, single guide RNAs; SVM, support vector machine
Graphical Abstract
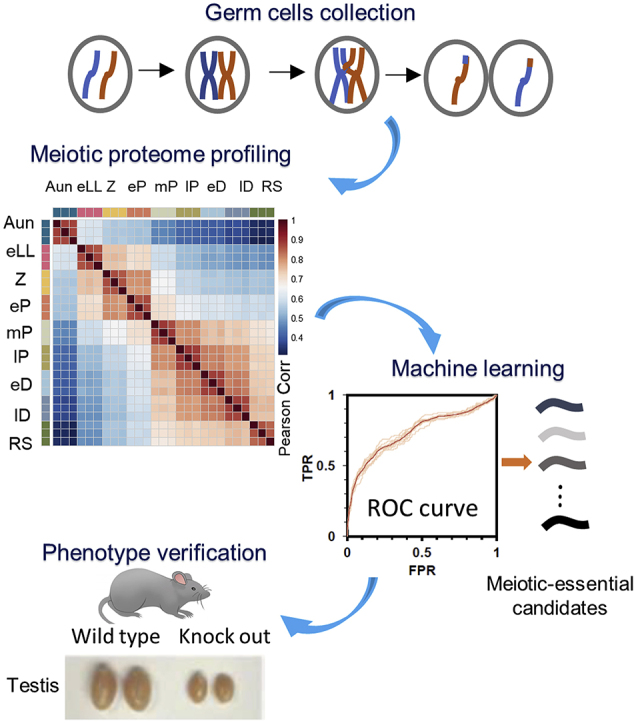
Highlights
-
•
Quantitative proteomics of germ cells at nine substages of spermatogenesis.
-
•
The protein abundance responses imply midpachytene as a checkpoint.
-
•
Machine learning is used to predict meiosis-essential genes.
-
•
The meiotic roles of the five candidates are verified by knockout mice.
In Brief
A comprehensive proteomic characterization of the germ cells at different meiotic stages of spermatogenesis was investigated, the dynamical abundances of a total of 8002 proteins formed four proteomic clusters across these meiotic substages, offering novel insights into meiotic protein expression. A model of supervised machine learning based on the mouse genomics informatics and the spermatogenesis proteomic dataset was established to predict meiosis-essential genes, while the prediction was experimentally verified by knockout mice.
Meiosis is a cell division process specific to germ cells, in which DNA replicates once and divides twice to generate four gametes. It is accepted that mammalian meiosis is a complex process that includes several molecular events, including homologous recombination, synapsis, and so on, whereas the genes involved in these events are not well characterized in mammals (1, 2). In contrast, the genes participating in yeast meiosis are well studied, and thus, homology comparisons between yeast and mammals is a common strategy to investigate meiotic genes, such as Spo11, Dmc1, Psmc3ip, and Rnf212 (3, 4, 5, 6, 7). Since the regulatory mechanism of mammalian meiosis is more complicated than that of yeast, the genes specifically related to mammalian meiosis cannot be found through such a comparison strategy. The knockout of genes with testis- or oocyte-specific expression patterns is another approach to identify meiosis-related genes in mammals. This approach can provide a clear answer once a gene knockout leads to a phenotypic change; however, this strategy fails in a lethal effect or a noninfluence on meiosis. For example, the group by Miyata et al. (8) knocked out 54 testis-specific genes and found none of them with a meiotic phenotype. An efficient approach to search for mammalian meiosis-essential genes is thus desperately required.
Since the status of gene expression is a fundamental characteristic tightly associated with physiological functions, the dynamic information of gene expression throughout spermatogenesis would be extremely useful for exploring meiosis-essential genes. Up until now, transcriptomes in thousands of germ cells covering various developmental stages of spermatogenesis were implemented using a single-cell sequencing technique (9, 10, 11, 12, 13, 14, 15, 16), resulting in the transcriptome landscape throughout spermatogenesis. There is a solid evidence that demonstrates a poor correlation between mRNA and protein abundance in many tissues, including testes (17, 18). Although 90% of protein-coded genes were identified, the spermatogenesis-related transcriptomes could not well explain the meiotic functions that are performed by proteins. A global and dynamic profiling of proteins in response to spermatogenesis is of great meaning to unravel the meiotic molecules with certain functions. The investigation toward spermatogenesis using proteomics is still unsatisfactory, even though a report exists regarding the quantitative proteomics in relation to pachytene spermatocytes (7, 18). Hence, a comprehensive proteomics study of the different cell types of male meiosis is proposed.
For the sake of exploring meiosis-essential genes through proteomics, seven consecutive types of meiotic cells plus premeiotic spermatogonia and postmeiotic round spermatids (RSs) were first isolated using flow cytometry, and the proteome in each cell type was quantitatively identified by high-resolution MS in the label-free mode. The meiosis-dependent proteins were defined based upon the proteomic characteristics at different substages of spermatogenesis. A supervised ensemble machine learning package, FuncProFinder (https://github.com/sjq111/FuncProFinder), was developed to predict the meiosis-essential genes on account of meiosis-dependent proteins, and the knockout experiment was conducted to verify the five candidate meiosis-essential genes, Pdha2, Zcwpw1, Tesmin, Kctd19, and 1700102P08Rik. The results integrated from proteomics, machine learning, and knockout mice indeed pave a pathway to discover meiosis-essential genes and to uncover their functions in meiosis.
Experimental Procedures
Experimental Design and Statistical Rationale
A total of nine types of germ cells were separated from different numbers of mice, and the cells at the same substage were pooled before protein extraction to obtain enough materials for the deep proteomic assay. The digested peptides from each type of germ cells were separated into five fractions, and each fraction was delivered to MS with triplicate injection. The proteome quantification was estimated by MaxQuant (19) using the MaxLFQ algorithm (20), and only proteins identified with at least two unique peptides were used for quantification. Differentially expressed proteins (DEPs) among the nine substages were defined by two-way ANOVA in Perseus (21) filtered with an adjusted q value <0.001 and protein abundance fold change ≥2. For each meiotic candidate gene, over 30 born F1 mice were surveyed for the genotyping (supplemental Table S5), and 3 to 10 infertile mice for each meiotic essential gene were surveyed for the phenotype investigation.
Mice for Experiments
WT C57BL/6Slac mice were purchased from SLAC China. All animal experiments were conducted in accordance with the guidelines of the Animal Care and Use Committee at Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Science.
Spermatogenesis Synchronization
Spermatogenesis was synchronized as previous reported with slight modifications (9, 22, 23). Briefly, C57BL/6Slac mice were fed on WIN 18446 at 100 μg/g body weight from P2 to P8, which could block spermatogonia differentiation. Spermatogonia differentiation was reinitiated in these mice at P9 through the intraperitoneal injection of retinoic acid (RA) at 35 μg/g body weight. The testes of P37 to P46 mice were collected for synchronization efficiency evaluation through histological staining and cell sorting.
Isolation of Mouse Spermatocytes
Spermatocytes were isolated by fluorescence-activated cell sorting as previously described with minor modifications (24). Briefly, the testes of an individual spermatogenesis-synchronized mouse were collected in Gibco balanced salt solution. The tunica albuginea was removed, and the testes were digested with collagenase type I (120 U/ml) at 32 °C with gentle agitation for 15 min, then further digested with 0.15% trypsin and DNase I (10 μg/ml) at 32 °C for 30 min, and the digestion was terminated with 10% fetal bovine serum. The suspension was then filtered through a cellular filter, and the flow through was centrifuged at 500g for 5 min at 4 °C to collect the cell pellet. The cells were resuspended in Dulbecco's modified Eagle's medium and stained with Hoechst 33342 and propidium iodide for sorting by a FACSAria II cell sorter (BD Biosciences).
Isolation of Mouse THY1+ c-KIT− Spermatogonia
Undifferentiated spermatogonia (THY1+ c-KIT− spermatogonia) were isolated as previously stated (25), and the workflow is summarized in supplemental Fig. S1C. Briefly, testes of P7 mice were digested with collagenase type I and trypsin to obtain separate cells, and the cell mixture was suspended in PBS, layered on 2 ml of 30% Percoll and centrifuged at 600g for 8 min. The cell pellets were resuspended and incubated in PBS containing anti-c-KIT magnetic microbeads for 20 min and were then loaded on a magnetic device to collect the flow through, which are c-KIT− cells. These c-KIT− cells were then incubated with anti-THY1 magnetic microbeads for 20 min, and the bound THY1+ c-KIT− cells were enriched using an MS column (130-042-201; Miltenyi Biotech) and a MiniMACS separator (130-042-102; Miltenyi Biotech). Their purity was estimated by anti-PLZF (SC-22839; Santa Cruz Biotechnology), (Plzf, a spermatogonia-specific transcription factor in the testis) and 4',6-diamidino-2-phenylindole (DAPI) staining.
Histological Analysis
Histological analysis was performed following a common protocol. Briefly, testes were fixed in Bouin's solution, embedded in paraffin, and sectioned. Then the sections were dewaxed with xylene and rehydrated in a series of ethanol concentrations. The treated sections were stained with H&E and sealed with nail polish. Spermatogenesis stages in seminiferous tubule crosssections were recognized as previously described (26).
Meiotic Chromosome Spreading and Immunofluorescence
The meiotic chromosome spreading assay was performed following a previous protocol with some modifications (27). Briefly, cells were sequentially resuspended in hypotonic extraction buffer and 100 mM sucrose, and then, the suspension was pipetted onto a glass slide and dried slowly in a humid chamber. For immunofluorescence staining, the slides were washed with PBS, blocked with 3% bovine serum albumin, and incubated with different antibodies, including anti-SCP1 (ab15090; Abcam), anti-SCP3 (sc-74569; Santa Cruz), anti-SCP3 (ab15093; Abcam), anti-γH2AX (05-636; Millipore), anti-γH2AX (#9718; CST), and anti-MLH1 (550838; BD Pharmingen). Finally, the slides were incubated with Alexa Fluor 488- or 594-conjugated secondary antibodies (711545152 and 711585152; Jackson ImmunoResearch Laboratories) and were treated with DAPI for defining nuclei. The spread cells were monitored undera FV3000 confocal laser scanning microscope (Olympus).
Protein Extraction and Digestion
The pooled cells were homogenized in the lysis buffer containing 7 M urea, 2 M thiourea, 0.2% SDS in 100 mM Tris-HCl, and 1× cocktail-free protease inhibitor (Promega) at a pH of 7.4. The proteins in lysates were reduced with 5 mM DTT and alkylated with 55 mM 2-iodoacetamide, and then, they were further purified by cold acetone precipitation. The precipitated proteins were resolved in 7 M urea lysis buffer, and the protein concentrations were estimated using a Bradford protein assay (Bio-Rad). Proteins were digested using a filter-assisted sample preparation strategy (28). For each sample, 200 μg of protein was loaded into a 10 kD spin filter (Millipore) and washed in order with the urea lysis buffer and 1 M triethylamine bicarbonate, followed by treatment with trypsin (Promega) at a ratio of 1:50 at 37 °C for 16 h. Tryptic peptides were collected by centrifugation and quantified using the Quantitative Colorimetric Peptide Assay (Thermo Scientific).
Peptide Fractionation on Reversed-Phase HPLC
For each sample, approximately 100 μg of peptides was dissolved in elution buffer A containing 20 mM ammonium bicarbonate and 5% acetonitrile at a pH of 9.8. The dissolved peptides were loaded on a Phenomenex C18 column (5 μm particle, 110 Å pore, and 250 × 4.6 mm) that was mounted on a Shimadzu liquid chromatography system and pre-equilibrated with elution buffer B containing 20 mM ammonium bicarbonate and 90% acetonitrile at a pH of 9.8. The peptides were eluted through a stepped gradient program as follows: 0 to 3 min, 5% B; 3 to 7 min, 9% B; 7 to 11 min, 13% B; 11 to 15 min, 19% B; 15 to 19 min, 80% B; 19 to 21 min, 5% B; 21 to 21.5 min, 5 to 80% B; 21.5 to 22.5 min, 80% B; 22.5 to 23 min, 80% B to 5% B, and 23 to 29 min, 5% B at a flow of 1 ml/min. Twenty-four fractions from 3 to 26 min were collected, and these fractions were further combined into five fractions according to the absorption peaks at 214 nm during chromatography as follows: fractions 1 to 5 as F1, 6 to 10 as F2, 11 to 14 as F3, 15 to 18 as F4, and 19 to 24 as F5.
Peptide Detection by LC–MS/MS
The identification of peptides was conducted on a quadrupole Orbitrap mass spectrometer (Q Exactive HF; Thermo Fisher Scientific) coupled to an ultra HPLC system (Dionex Ultimate 3000; Dionex) via a nanospray Flex ion source. Approximately 1 μg of peptides was loaded on a C18 trap column (75 μm I.D. × 1.5 cm; in-house packed using Welch C18 3 μm silica beads) and was directly loaded into a C18 analysis column (75 μm I.D. × 20 cm; in-house packed using Welch C18 3 μm silica beads). The peptides used for MS were eluted at 300 nl/min with the following two elution buffers: buffer A, 0.1% formic acid and 2% acetonitrile and buffer B, 0.1% formic acid and 98% acetonitrile, following a gradient program of 0 to 5 min, 5% B; 5 to 7 min, 5 to 7% B; 7 to 67 min, 7 to 28% B; 67 to 80 min, 28 to 43% B; 80 to 82 min, 43 to 98% B; 82 to 84 min, 98% B; 84 to 85 min, 5% B; and 85 to 90 min, 5% B. The mass spectrometer was operated in the top-30 data-dependent mode collecting MS spectra in the Orbitrap mass analyzer (120,000 resolution in the 350–1500 m/z range) with an automatic gain control target of 3E6 and a maximum ion injection time of 50 ms. The ions with higher intensities were isolated with an isolation width of 1.6 m/z and fragmented through higher-energy collisional dissociation with a normalized collision energy of 28%. The MS/MS spectra were collected at 15,000 resolution with an automatic gain control target of 1E5 and a maximum ion injection time of 45 ms. Precursor dynamic exclusion was enabled with a duration of 60 s.
Peptide Search and Protein Quantification by MaxQuant
Tandem mass spectra were searched against the Swiss-Prot mouse databases (downloaded on November 19, 2018; 17,000 entries) using MaxQuant (version 1.5.3.30) (19) with a 1% false discovery rate at the peptide and protein levels. The search parameters for a peptide were set as trypsin digestion only, maximum of two missed cleavages, minimum length of six amino acids, cysteine carbamidomethylation as a fixed modification, and N-terminal acetylation and methionine oxidations as variable modifications. The precursor mass tolerance was set to 20 ppm, and the fragment mass tolerance was 0.05 Da. The “Match Between Run” option was used. Label-free quantification (LFQ) was estimated with the MaxLFQ algorithm (20) using a minimum ratio count of 1. The protein was identified with at least two unique peptides (protein false discovery rate <0.01). The specifically relative LFQ for a protein was defined by the ratio of the protein LFQ at a certain substage being divided by the protein maximum LFQ among all nine substages.
Proteomic Informatics Analysis
Bioinformatics analysis of the identified and quantified proteins was performed with Perseus software (version 1.6.1.3) (21), R statistical software (http://www.R-project.org/), and Excel. DEPs among the nine substages were defined by two-way ANOVA in Perseus filtered with an adjusted q value <0.001. To look for the DEP groups with protein abundance changes during spermatogenesis that share similar patterns, the DEPs were first evaluated by the NbClust package (29) in R (version 3.5.1) to find the optimum K value, and the relative LFQs of DEPs were clustered using K-means analysis. Gene Ontology and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses were performed using David (version 6.8) (30) and Enrichr (31), in which an enriched function was accepted upon an adjusted p value less than 10e−5, and only the Gene Ontology Biological Process terms with <250 genes were included.
Comparison of the Abundance Changes of Transcriptomes and Proteomes in Spermatogenesis
The transcriptomic data (GSE107644) in response to the substages of spermatogenesis measured by the single-cell sequencing technique was downloaded from Gene Expression Omnibus of National Center for Biotechnology Information (9). The CPM for a gene was calculated from the raw counts in the dataset, and the expression status for a gene at a certain substage was represented by the median CPM of the gene. For convenient comparison, the gene symbols in RNA-Seq data were converted to the Swiss-Prot accession numbers using the gene ID conversion tool in DAVID. The Pearson correlation for all the substages of spermatogenesis was implemented for the abundances between the transcriptomes and proteomes with log2-transformed CPM and LFQ. The concordance of a gene's abundance change pattern between the transcriptional and translational levels during spermatogenesis was estimated by Pearson correlation, which was based on the scaled CPMs and LFQs in each corresponding substage.
Machine Learning for the Prediction of Meiosis-Essential Proteins
The quantified protein abundances in nine types of germ cells served as inputs to the classifiers. The meiosis-essential proteins in this study, defined as the overlapping proteins of these proteomic data and the proteins whose knockout leads to the male meiotic arrest phenotype annotated in the mouse genomics informatics (MGI) database, were labeled as positive, and the meiosis nonessential proteins were labeled as negative. Three algorithms, the regularized radial basis function (RBF) (32), naive Bayesian model (NBM), and support vector machine (SVM) (33), were employed to gain the predictions for meiosis-essential protein candidates. The details of regularized RBF, NBM, and SVM are presented in the Supplemental Methods section. In this study, the prediction learning process was conducted, which was ensembled into an in-house Matlab package, termed as FuncProFinder. Generally, the prediction is conducted in two steps:
Step 1
The nine substage protein abundances of the labeled positive and negative proteins were randomly divided into two sets, training (80%) and testing (20%). The training set was used by the machine learning algorithm to construct a subclassifier, and the subclassifier was then used for prediction of the testing set. This process was repeated 1000 times, and the final predicted score of each protein, called the meiotic confidence score, is the mean value of the individual scores.
Step 2
A Monte Carlo crossvalidation was applied (34) to evaluate the performance of each algorithm according to the precision-recall curve and receiver operating characteristic curve. The algorithm with the best prediction efficiency for the dataset could be selected for the unknown gene prediction, following the same workflow as in step 1, where the subclassifier produced by the training set was used for the prediction of both the testing and unknown datasets.
Generation of Gene Knockout Mice With the CRISPR/Cas9 System
To generate knockout mice corresponding to the genes of interest in this study, single guide RNAs (sgRNAs) were designed based on their genome structures (supplemental Table S5). The T7-Cas9 PCR product was gel purified and used as the template for in vitro transcription using the mMESSAGE mMACHINE T7 ULTRA transcription kit (AM1345; Thermo Fisher Scientific). The T7-sgRNA PCR product was gel purified and used as the template for in vitro transcription with a MEGAshortscript T7 Transcription Kit (AM1354; Thermo Fisher Scientific). Both the Cas9 mRNA and the sgRNAs were purified using the MEGAclear Transcription Clean-Up Kit (AM1908; Thermo Fisher Scientific) and eluted in RNase-free water (10977015; Thermo Fisher Scientific). The Cas9 mRNA and sgRNAs were injected into one-cell embryos as previously described (35, 36, 37). The injected embryos were cultured in vitro to develop to two-cell embryos and transplanted into oviducts to generate knockout pups. After pups were born, genotyping was performed by direct sequencing following PCR to validate the knockout consequences. The genotyping primer sequences that were used are listed in supplemental Table S5.
Results
Isolation of Mouse Spermatogenic Cells
To quantify changes in protein abundance and closely monitor the molecular events in response to mouse meiosis, we isolated spermatogenic cells before, during, and after meiosis from C57BL/6 mouse testes, including premeiotic type A undifferentiated spermatogonia, consecutive types of meiotic cells, and postmeiotic RSs.
The isolation workflow of spermatogenic cells is illustrated in Figure 1A. Type A undifferentiated THY1+ c-KIT− spermatogonia (Aun) were isolated from the testes of postnatal day 7 (P7) mice using magnetic-activated cell sorting according to an established method (25) (supplemental Fig. S1, A and C). Immunofluorescence staining of PLZF, a well-known Aun marker, revealed that the percentage of PLZF+ cells increased from 10% to 70% after purification (Fig. 1; supplemental Fig. S1, B and D), implying that Aun cells were greatly enriched. The haploid RSs were purified by DNA content–based cell sorting from the testes of P28 mice (supplemental Fig. S1, E and G). DAPI staining images indicated that the purity of isolated RS almost reached 100% (Fig. 1; supplemental Fig. S1, F and H).
Fig. 1.

Isolation of the meiotic prophase I spermatocytes.A, experimental design and workflow. B, strategy to obtain the spermatogenesis-synchronized mice. C–G, cross sections of H&E-stained testes obtained from spermatogenesis-synchronized mice on postnatal day 37 (P37) to P46. Roman numerals in each tubule designate the seminiferous tubule stage judging by the criteria described previously (26). The scale bar represents 50 μm. H–L, FACS plots of Hoechst 33342 stained testes cells obtained from the spermatogenesis-synchronized mice, their corresponding seminiferous tubule stages were labeled in C–G. M–T, chromatin spreading of the FACS-sorted spermatocytes that were costained with DAPI (blue), anti-γH2AX (green), and anti-SYCP3 (red). The scale bar represents 5 μm. FACS, fluorescence-activated cell sorting; MACS, magnetic-activated cell sorting; RS, round spermatid.
In the seminiferous tubules of mouse testes, consecutive types of meiotic cells are mixed and difficult to separate. To simplify the types of spermatocytes in testes, we applied a spermatogenesis synchronization method described previously (9, 22, 23): mouse spermatogonia differentiation was inhibited by WIN 18446 for 7 days and was reactivated synchronously by RA injection on P9 (Fig. 1B). Four weeks after RA treatment, the testes of P37 to P46 mice exhibited only one or two types of meiotic spermatocytes at a given time point (Fig. 1, C–G), greatly facilitating DNA content–based cell sorting for purification (Fig. 1, H–L). To assess the purity of the isolated meiotic cells, we performed immunofluorescence staining with antibodies against the synaptonemal complex (SC) marker SYCP3 and the DNA damage marker γH2AX (Fig. 1, M–T) and recognized spermatocyte cell types with the criteria described previously (27). Based on the quantitative evaluation of fluorescence, most of the isolated meiotic cells were of high purity (Fig. 2). Considering that the protein amounts of isolated early leptotene and leptotene were less than 120 μg, we mixed these two adjacent cell types together as an earlyL/L (eLL) group for the following proteomic analysis. Thus, a total of seven types of meiotic cells, early leptotene and leptotene (eLL), zygotene (Z), early pachytene (eP), middle pachytene (mP), late pachytene (lP), early diplotene (eD), and late diplotene (lD) were prepared for further proteomic study.
Fig. 2.

The purity of the separated spermatogenic cells of different stages around meiosis. RS, round spermatid.
Quantitative Proteomic Atlas of Mouse Meiosis During Spermatogenesis
In the nine types of spermatogenic cells isolated previously, a total of 8002 proteins were identified (unique peptides ≥2), with 6000 to 7000 proteins in each cell type (supplemental Table S1). Within the identified proteins, 7742 were only detected in the seven substages of meiosis, and 5108 proteins were globally identified over all nine cell types. To obtain high-quality quantification data, each sample was analyzed in triplicate by LC–MS/MS. The Pearson correlation coefficients for all the technical triplicates in the same sample reached 0.85 (supplemental Fig. S2), indicating that proteomic quantification was highly replicated. In addition, the comparison of protein expression correlation among the nine different cell types revealed that the protein abundance changed dramatically between eP and mP, which strongly implied that spermatocytes undergo a cell state transition after passing of the mP checkpoint.
Next, to evaluate the consistency of our protein quantification with previous knowledge, we tracked the protein abundance changes of 15 well-known cell type–specific biomarkers throughout spermatogenesis (e.g., LIN28A, STRA8, SPO11, TNP2, etc). The protein abundances of these biomarkers appeared to be typically phase dependent (Fig. 3B), which was basically in agreement with previous studies (38, 39, 40, 41). In addition, proteins in several meiosis-related processes, such as meiotic DNA double-strand break (DSB) formation, chiasma assembly, and SC, are generally recognized as meiotic phase dependent, and the proteomic evidence in this study further implied their functions (Fig. 3C). For example, the SC mainly formed from Z to lP and decreased after lP (1), and most SC components in proteomics data were consistent with previous results. However, the protein abundance of SYCE1, a key SC component, remained stable from lP to RS. A similar Syce1 transcriptional expression pattern was observed from a single-cell RNA transcriptome dataset (9), implying that SYCE1 might perform additional functions except synapsis after meiosis prophase I.
Fig. 3.

Proteomic informatics of the mouse germ cells.A, Pearson correlation coefficients of the log-transformed protein LFQ intensities among the cells at nine substages. The color key represents the value of Pearson correlation coefficients. B, relative abundances of the typical biomarkers in the nine substages of germ cells. Error bars represent SEM in triplicates. C, heat maps of dynamic abundances of the proteins involved in the five representative meiotic pathways. D, K-means clusters of the relative protein abundances elicited from the differentially expressed proteins. The color key represents the relative protein abundances. E, Gene Ontology analysis of the enriched biological processes of the four differentially expressed protein clusters. ER, endoplasmic reticulum; FDR, false discovery rate; GOBP, Gene Ontology Biological Process; LFQ, label-free quantification; piRNA, Piwi-interacting RNA; RS, round spermatid.
To further explore the phase-dependent dynamic processes related to meiosis, the abundances of all the 8002 identified proteins in nine cell types were input into a statistical software, Perseus (21), for DEP analysis. A DEP was defined as a protein with a significant change in its abundance between any two substages with a q value less than 0.001. A total of 6020 proteins were determined to be DEPs, and these DEPs were divided into four groups by K-means analysis: proteins in C1 showing high expression in Aun, C2 in eLL, C3 in Z-eP, and C4 in mP-RS (Fig. 3D; supplemental Table S2). Gene Ontology analysis toward DEPs in each cluster led to the uncovering of the biological processes enriched in different phases around meiosis. As illustrated in Figure 3E, cell–cell adhesion and actin cytoskeleton organization–related proteins were enriched in Aundiff cells. Nucleic acid–related processes such as rRNA processing and DNA replication were enriched in earlyL/L cells. Meiotic cell cycle–related proteins were enriched in Z-early P cells, and Piwi-interacting RNA metabolism and sperm function–related proteins were enriched in midP-RS cells. Taking all the aforementioned information, both the qualitative and quantitative proteomic information not only highly agreed with prior knowledge but also offered new clues to understand meiotic molecules, approaching the additional functions of meiotic proteins, functionally categorizing the meiotic DEPs and uncovering previously uncharacterized molecular signatures and dynamic processes from the protein abundance changes during meiosis.
Comparison of the Meiosis-Related Transcriptomes and Proteomes in Response to Spermatogenesis
The transcriptomic responses to spermatogenesis development have been reported. Recently, Chen et al. (9) utilized single-cell technology to monitor the transcriptomic changes during spermatogenesis and revealed several uncharacterized dynamic processes and molecular signatures in the substages of male germ cell development. As described previously, the dynamic changes of proteomes were systematically evaluated in this study (supplemental Table S3), and hence, a question was naturally raised whether the transcriptomic and translational responses to spermatogenesis kept the same rates or not. The transcriptomic data were collected from the published sources and subjected to normalization and correlation analysis, which was described in the Experimental Procedures section.
The Pearson correlation analysis of the transcriptomes and proteomes in all the spermatogenesis substages shown in Figure 4A unraveled two sets of gene expressions having a poor correlation with lower correlation coefficients of approximately 0.30 to 0.45. To evaluate the consistency of the dynamic changes in transcription and translation for individual genes during spermatogenesis, Pearson correlation was conducted between corresponding substages based on the scaled abundances. As illustrated in Figure 4B, the interquantile ranges for the coefficients of such Pearson correlation in all the coquantified genes were −0.97 to 0.06, 0.06 to 0.48, 0.48 to 0.75, and 0.75 to 1. The corresponding dynamic abundance changes for the coquantified genes in transcripts and proteins are presented in Figure 4C. It exhibits that the genes with correlation coefficients of 0.75 to 0.99 shared similar abundance trends in transcripts and proteins during spermatogenesis, whereas those with coefficients of −0.97 to 0.48 displayed divergent modes, and the genes with the correlation coefficients between 0.48 and 0.75 appeared to have transition patterns of transcript and protein abundances. Therefore, there were approximately 25% of genes that could remain with similar abundance trends for the transcriptional and translational products in response to spermatogenesis. The Gene Ontology analysis of the genes in each quantile group suggested that the genes with coefficients over 0.75 were enriched in the functions related to meiosis division and spermatogenesis, whereas those with coefficients less than 0.06 were involved in mRNA processing and proteasome splicing (Fig. 4D). Taking all the correlation information together, the abundance responses of the transcriptome and proteome in spermatogenesis were generally not at the same pace. The observation of the spermatogenesis-dependent transcriptome and proteome endorses the hypothesis that transcriptional and translational regulations toward spermatogenesis are complementary and irreplaceable.
Fig. 4.

Dynamical transcriptomes and proteomes abundances correlation analysis of the germ cells during spermatogenesis.A, Pearson correlations of the expression abundances among all quantified transcriptomes and proteomes of the spermatogenic germ cells. B, distribution of the genes versus the genes' Pearson correlation coefficients between mRNA and protein abundances. The interquantile ranges of the coefficients were −0.98 to 0.06, 0.06 to 0.48, 0.48 to 0.75, and 0.75 to 0.99. C, heat maps of the abundance changes of the transcripts (left) and the proteins (right) during spermatogenesis. The genes were quantiled according to their Pearson correlation coefficients. D, enriched biological processes of the genes sets with the corresponding Pearson correlation coefficients. FDR, false discovery rate; GOBP, Gene Ontology Biological Process; LFQ, label-free quantification; RS, round spermatid.
Prediction of Meiosis-Essential Candidates Using Supervised Machine Learning Analysis Based on Proteomic Data
Although 8002 proteins were identified in spermatogenesis and further divided into four groups with relevant biological processes, the proteins essential to meiosis were still unclear. Recently, supervised machine-learning approaches were applied to systemically predict functional genes (42). Here, we established a supervised ensemble machine learning Matlab package called FuncProFinder to predict meiosis-essential candidates based on the meiotic proteomic data (Experimental Procedures and Supplemental Methods sections). According to phenotype annotation in the MGI database, a protein is meiosis essential if knockout of the protein leads to meiosis arrest, whereas a nonessential protein is defined by that the protein knockout mice do not have the lethal or meiosis-arrest phenotype. From the 8002 identified proteins in this study, a total of 159 proteins were essential and 2151 were nonessential (Fig. 5A; supplemental Table S4).
Fig. 5.

Prediction of the meiotic-essential candidates using machine learning. A, workflow to build machine-learning algorithms based on the proteome data.B, comparison of the prediction results, precision-recall curves (upper panels), and receiver operating characteristic curves (lower panels) generated from the three subclassifiers, regularized RBF, NBM, and SVM. The Monte Carlo crossvalidation (MCCV). C, distribution of meiosis-essential, meiosis-nonessential, lethal, and unknown proteins in the 8002 quantified proteins (left panel) and the top 500 meiosis candidates derived from the RBF prediction (right panel). D, heat map of dynamic abundances of the top 500 meiosis candidates derived from the RBF prediction. E, comparison of the distribution of the DEP clusters treated with/without RBF filtration. AUC, area under the curve; DEP, differentially expressed protein; LFQ, label-free quantification; MGI, mouse genomics informatics; NBM, naive Bayesian model; RBF, radial basis function; RS, round spermatid; SVM, support vector machine.
With the abundances of these proteins as training sets, three methods of the FuncProFinder, RBF (32), NBM, and SVM (33), were used to construct classifiers to predict whether a given protein was meiosis essential. Based on the FuncProFinder package, the prediction precision tested by Monte Carlo cross validation (34) reached 47.70% (RBF), 30.97% (NBM), and 20.71% (SVM) with the recall setting at 0.2. The area under a curve for the receiver operating characteristic curve of the predictions was 0.7364 (RBF), 0.7150 (NBM), and 0.6711 (SVM), respectively (Fig. 5B). As the prediction performance of RBF with regard to both the precision and area under the curve was superior to the other two algorithms in this dataset, FuncProFinder-RBF was accepted to predict the meiosis-essential possibility of a given protein. The abundances of all identified proteins in the nine substages, labeled with positive (meiosis essential), negative (meiosis nonessential), and unknown (lethal genes and genes without MGI mammalian phenotype annotation) were set as inputs of the FuncProFinder-RBF algorithm for the calculation of the meiotic confidence scores. The higher the meiotic confidence score, the more likely a protein was meiosis essential (supplemental Table S4). A total of 500 proteins with the top scores were filtered, and their functional information in MGI is shown in Figure 5C. The ratio of meiosis-essential proteins against nonessential proteins was 0.07 (159:2151) for all the 8002 proteins, whereas the ratio changed to 0.97 (35:36) for the top 500 selected candidates, indicating that meiosis-essential proteins could be greatly enriched. The top 500 candidates exhibited dynamic protein abundance changes during spermatogenesis (Fig. 5D), containing more DEPs (94.00%) compared with the total 8002 proteins (75.23%). In addition, in the top 500 candidates, the ratio of genes showing higher expression in the three meiotic clusters (C2–C4) was also higher than the ratio in the global proteome (Fig. 5E), consistent with the hypothesis that a meiosis-essential candidate could be a DEP with higher expression abundance in subphases of meiosis. Based on these results, the RBF algorithm is a potential method to select meiosis-dependent candidates from a large pool of identified proteins.
Pyruvate Dehydrogenase Alpha 2 (PDHA2) Is Essential for Meiosis
To further characterize the predicted candidate meiotic genes, Enrichr (31) was implemented to identify enriched pathways of these top 500 candidates based on a KEGG 2019 mouse database. The top six functional categories after enrichment analysis were determined (Fig. 6A), and there was one protein, PDHA2, enriched in both the pyruvate metabolism pathway and tricarboxylic acid cycle pathway (supplemental Table S4). Moreover, it had been reported that pyruvate metabolism was required in the isolated pachytene spermatocytes cultured in vitro (43). However, whether pyruvate-related proteins were essential in meiosis has not been verified in vivo.
Fig. 6.

Phenotypic validation of the PDHA2 in meiosis.A, Kyoto Encyclopedia of Genes and Genomes pathways enriched in the top 500 candidate proteins. B, dynamic abundances of the six proteins of the pyruvate dehydrogenase complex. The error bars represent SEM from triplicate samples. C, comparison of the weights of the Pdha2+/− and Pdha2−/− mice testes (n = 10, unpaired two-tailed t test, p < 0.0001). D, cross sections of the H&E-stained seminiferous tubules from the 8-week-old Pdha2+/− (upper panel) and Pdha2−/− mice (lower panel); the insets denote the specific seminiferous tubules under a higher magnification. The arrow points to a pachytene-like cell. E, cross sections of the H&E-stained epididymis from the 8-week-old Pdha2+/− (upper panel) and Pdha2−/− (lower panel) mice. The scale bar represents 50 μm. PDHA2, pyruvate dehydrogenase alpha 2; RS, round spermatid; TCA, tricarboxylic acid.
PDHA2 is a catalytic subunit of the pyruvate dehydrogenase complex (PDC), associated with four other proteins, PDHB, DLAT, DLD, and PDHX (44). The lack of any component in the complex could lead to activity loss. In a previous study, the transcription status of the Pdha2 gene was shown to be dynamically changed during spermatogenesis, increasing from the pachytene and gradually decreasing in spermatids (45). With our proteomic data, the expression changes of all proteins in the PDC during spermatogenesis are further illustrated (Fig. 6B). The protein abundances of the two catalytic subunits of PDC, PDHA1 and PDHA2, changed in opposite directions: X-chromosome-linked protein PDHA1 decreased during meiosis because of meiotic sex chromosome inactivation, whereas PDHA2 increased from eP to lD. For the other four components of PDC, the changes in their abundances were similar to PDHA2, implying that the PDC had structural integrity of catalytic functions during meiotic development.
To further verify the physiological roles of PDHA2 during meiosis, a Pdha2 knockout mouse model was generated by CRISPR/Cas9-mediated genome engineering. A 13 base pair deletion was induced into the Pdha2 exon, which led to a reading-frame shift of Pdha2 and early termination (supplemental Fig. S3A); the knockout result was examined by genotyping (supplemental Fig. S3B). The weights of testes of 8-week-old adult Pdha2−/− mice were significantly smaller than those of Pdha2+/− mice (Fig. 6C), and the testes histology of these mice was further examined by H&E staining of cross sections (Fig. 6, D and E). In Pdha2+/− mice, the testes were comparable to WT mice, in which all types of germ cells were observed, and mature sperm had fully filled their epididymides. In contrast to their heterozygous littermates, Pdha2−/− mice entirely lacked postmeiotic cells, and pachytene-like spermatocytes were accumulated in their testes. Furthermore, no spermatozoa were observed in their epididymides. Thus, with the help of FuncProFinder-RBF prediction, this study provides evidence that PDHA2 is a meiotic-regulation factor, the knockout of which is likely to stop the meiosis process at the pachytene stage. As PDC catalyzes pyruvate to acetyl-CoA and determines the energy level in a cell, it is a reasonable deduction that PDHA2, as a key component of PDC, could regulate ATP generation in spermatocytes and affect the meiotic process.
Phenotype Verification of the Top 10 Male Meiosis-Essential Candidates Without Functional Annotation
Of the top 500 candidates, a total of 176 proteins have never been knocked out for a phenotype examination using the mouse model according to the MGI database; furthermore, 41 proteins appeared to lack KEGG pathway annotation (Fig. 7A). Whether they are essential for meiosis needs to be further verified by experiments. The top 10 candidates based on the meiotic confidence score were selected and knocked out in mice using CRISPR/Cas9-mediated genome engineering (supplemental Fig. S4, A–J). Abundance changes of the 10 proteins are shown in Figure 7B. After knockout treatment, we obtained surviving homozygous deficient pups from eight of the 10 genes, and no born mouse was homozygous deficient in Gapvd1 and 1700037H04Rik; therefore, we assumed that the homozygous deficiency in the two genes would be lethal before birth. The reproductive anatomy of these nonlethal mice was carefully examined by the weight of the testis and histological analysis. In the Txnl1−/−, AA467197−/−, Lrrc40−/−, and Naxe−/− mice, no significant changes were observed in the weights or histology of the testes (supplemental Fig. S5, A–H). However, knockout of the other four genes affected the meiosis process of the homozygous deficient mice. Generally, the weights of the testes of the Zcwpw1−/−, Tesmin−/−, Kctd19−/−, and 1700102P08Rik−/− mice were significantly lighter than their heterozygous littermates (Fig. 7, C–F). Specifically, H&E-stained images of the testes derived from Zcwpw1−/−, Tesmin−/−, and 1700102P08Rik−/− mice appeared to have a pachytene-arrested phenotype, pachytene spermatocytes with condensed nuclei, a lack of postmeiotic cells, and tubules that were highly vacuolized (Fig. 7, G–I). The Kctd19−/− mice exhibited a typical metaphase I–arrested phenotype, containing spermatocytes from leptotene to metaphase I, but with no postmeiotic cells (Fig. 7J).
Fig. 7.

Phenotypic validation of the meiotic-essential proteins predicted by the FuncProFinder-RBF.A, distribution of the phenotype annotations and the KEGG pathway annotations in the top 500 meiosis candidates. B, dynamic abundances of the top 10 RBF-ranked meiosis candidates. The error bars represent SEM in triplicates. The bulbs on the figure (right) indicate the validated phenotypes of KO mice, orange as meiosis-essential, blue as meiosis nonessential, and gray as lethal. C–F, comparison of the weights of the testes derived from the 8-week-old Zcwpw1+/− and Zcwpw1−/− mice (C), Tesmin+/− and Tesmin−/− mice (D), 1700102P08Rik+/− and 1700102P08Rik−/− mice (E), and Kctd19+/− and Kctd19−/− mice (F). ∗∗∗∗ represents p < 0.0001 in unpaired two-tailed t test. G–J, cross sections of the H&E-stained seminiferous tubules from the heterozygous and homozygous knockout mice of the aforementioned four genes; the insets denote the specific seminiferous tubules under a higher magnification. The filled arrows point to the pachytene-like cells. The hollow arrows point to the metaphase I-like cells. The scale bar represents 50 μm. KEGG, Kyoto Encyclopedia of Genes and Genomes; RBF, radial basis function; RS, round spermatid.
The molecular mechanism of pachytene arrest is hypothesized to result from failure of DNA repair or incomplete synapsis (46, 47). As the knock out of Zcwpw1, Tesmin, or 1700102P08Rik led to pachytene arrest, the molecular mechanisms underlying the pachytene arrest phenotype in these three knockout mice lines need to be verified. To address this question, the spermatocytes in Zcwpw1−/−, Zcwpw1+/−, Tesmin−/−, Tesmin+/−, 1700102P08Rik−/−, or 1700102P08Rik+/− mice were chromosome spread and immunostained with antibodies against DNA repair and synapsis events, including SYCP3 and SYCP1 as components of the SC, γH2AX as an indicator of DNA damage, and MLH1 as a marker of crossover formation. The immunostaining of the four different antibodies against the spermatocytes from TESMIN and 1700102P08RIK in both heterozygous and homozygous knockout mice exhibited no difference (Fig. 8, F–N), implying that neither DNA repair nor synapsis was affected by gene knockout. However, the immunostaining of these antibodies against the Zcwpw1−/− spermatocytes was quite different from Zcwpw1+/− (Fig. 8, A–E). The staining signal distribution of γH2AX in the leptotene spermatocytes of the Zcwpw1−/− mice was comparable with that of Zcwpw1+/−, suggesting that the formation of DSBs was not affected by the absence of ZCWPW1 (Fig. 8A). In the pachytene spermatocytes, the γH2AX staining was only seen in the sex body region, whereas it was still spread out in the autosome regions in Zcwpw1−/−, indicating that the DSB repair was not finished on the autosome without ZCWPW1 (Fig. 8B). Coimmunostaining of SYCP3 and SYCP1 indicated that they were highly merged in the chromosomes in Zcwpw1+/− mice, whereas the locations of the two SC components were not fully overlapped in Zcwpw1−/− mice, suggesting that the SCs were not fully formed because of the lack of ZCWPW1 (Fig. 8C). Furthermore, the MLH1 loci were perceived in Zcwpw1+/− pachytene spermatocytes and counted within a normal range, whereas the Zcwpw1−/− pachytene spermatocytes appeared to have nearly no MLH1 loci, implicating that crossovers were not formed without ZCWPW1 (Fig. 8, D and E). Based on these results, ZCWPW1 functions directly in both DNA repair and synapsis, but the functions of 1700102P08RIK and TESMIN are not clearly clarified even though the two proteins participate in the regulation of meiosis.
Fig. 8.

Imaging of homologous recombination and synapsis in the heterozygous and homozygous gene knockout mice.Zcwpw1 (A–D), Tesmin (F–I), and 1700102P08Rik (K–N). Confocal images of the cells coimmunostained with anti-SYCP3 (red) and anti-γH2AX (green) for the display of DSB formation in leptotene (A, F, K) and sex body formation in pachytene (B, G, L), anti-SYCP3 (red) and anti-SYCP1 (green) for the examination of synapsis in pachytene (C, H, M) and anti-SYCP3 (red) and anti-MLH1 (green) for the display of crossover formation in pachytene (D, I, N). The scale bar represents 50 μm. E, J, O, comparison of the MLH1 foci number in spermatocytes derived from Zcwpw1+/+ and Zcwpw1−/− (E), Tesmin−/− and Tesmin+/− (J), and 1700102P08Rik+/− and 1700102P08Rik−/− (O) mice. ∗∗∗∗ represents p < 0.0001 in unpaired two-tailed t test. DSB, double-strand break; n.s., not significant.
To summarize the knockout experiments for predicting the meiosis-essential proteins, FuncProFinder-RBF offered a set of satisfactory candidates. Of the 10 candidates, at least 40% were verified as meiosis essential. As their functions are not annotated yet, their involvement in meiosis would be an interesting direction for functional exploration. For example, the pachytene arrest in Zcwpw1−/− mice was found to result from failed DSB repair and incomplete synapsis.
Discussion
In this study, one of the fundamental goals was to acquire global and quantitative proteomic profiles during different stages of mouse meiosis. How to obtain such information is a long-standing question. Proteomic investigations of the entire mouse testis and one type of meiotic cells, pachytene spermatocytes, have been accomplished in several laboratories (7, 18, 48). However, without isolation of different types of meiotic cells, these studies could not generate a precise picture of the different stages of mouse meiosis.
Here, first, we designed a project that enabled comprehensive proteomic profiling around meiosis. To reach our goal, seven consecutive types of meiotic cells plus premeiotic spermatogonia and postmeiotic RSs were isolated, and the proteins in each cell type were identified and quantified by high-resolution MS. A total of 8002 proteins were identified, including 6020 DEPs, which is the largest dataset of proteomics related to meiosis, offering global information of protein quantities in different stages of the mammalian meiosis process.
Second, this comprehensive proteomics study is likely to provide new views for the understanding of meiosis. For example, it is generally accepted that spermatocytes are categorized in several substages judged by the status of chromosome morphology (24, 26). Based on this criterion, eP and mP are categorized to the similar group of pachytene cells; nevertheless, the protein abundance correlation revealed that these two types of spermatocytes with similar appearance were totally different (Fig. 3, A and D). As another example, components of the SC were assumed to mainly exist from Z to lP and decreased after lP. However, SYCE1, a key SC component, was consistently detected after lP to RS with a relatively high abundance (Fig. 3C), implying that SYCE1 might perform additional functions (except synapsis) after meiosis prophase I. Therefore, the refined profile of the quantitative proteome provided a new assessment of molecular events related to meiosis.
Third, informatics analysis of proteomic data related to spermatogenesis is likely to provide a basis for the functional exploration of mouse meiotic genes. Genes involved in critical meiotic events, such as homologous recombination and synapsis, are not well identified (1, 2). Miyata et al. (8) selected 54 testis-specific genes and constructed the corresponding gene knockout mice, but unfortunately, they did not find any genes essential to meiosis. In this study, an ensemble strategy of machine learning was used to predict meiosis-essential proteins based on the different abundance patterns between meiosis-essential or nonessential proteins. With this strategy, the enrichment of meiosis-essential proteins was raised from 6.54% to 49.30% after prediction on the test set (Fig. 5). Moreover, at least four of the top 10 candidates without a KEGG pathway annotation were confirmed as meiosis-essential proteins by gene knockout mice (Fig. 7, C–J). Hence, functional exploration based upon proteomics seems to significantly improve the prediction efficiency.
In addition, with the machine learning prediction and gene knockout validation, the meiosis development of three genes, Zcwpw1, Tesmin, and 1700102P08Rik, was found to be arrested at pachytene in homologous gene knockout mice. Immunofluorescence images in this study revealed that Zcwpw1 functioned in DNA repair and synapsis. Very recently, Zcwpw1 was identified as a histone H3K4me3 reader required for the repair of PR domain zinc finger protein 9-dependent DNA DSBs and synapsis by three independent research groups (49, 50, 51, 52), which was consistent with our observations. Tesmin and 1700102P08Rik have also recently been identified as essential genes in spermatogenesis (53, 54), yet their mechanisms of involvement in meiosis-essential biological events still require further exploration. In addition, a list of meiosis-essential candidates without KEGG annotation was presented here (supplemental Table S4). Based on this list, more meiosis-essential genes could be verified, and new biological events could be uncovered concerning the molecular basis of mouse meiosis.
Data Availability
Supplemental figures and tables are available. The MS proteomics raw data and MaxQuant search result files have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD017284. The FuncProFinder Matlab package was uploaded on https://github.com/sjq111/FuncProFinder.
Conflict of interest
The authors declare no competing interests.
Acknowledgments
We thank Dr Timothy Karr for helpful comments and the two reviewers for constructive suggestions and comments. We thank the histology, flow cytometry, and animal services at Shanghai Institute of Biochemistry and Cell Biology.
Funding and additional information
C. D. C. was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB19000000) and the National Natural Science Foundation of China (91753128 and 81772472). S. L. was supported by the National Key R&D Program of China (2017YFC0908400 and 2017YFC0906703), the National Natural Science Foundation of China (31700728), and the Shenzhen Engineering Laboratory for Proteomics (DRC-SZ[2016]749). H. Y. was supported by the R&D Program of China (2018YFC2000100 and 2017YFC1001302), the CAS Strategic Priority Research Program (XDB32060000), the National Natural Science Foundation of China (31871502 and 31522037), the Shanghai Municipal Science and Technology Major Project (2018SHZDZX05), and the Shanghai City Committee of Science and Technology Project (18411953700 and 18JC1410100).
Author contribtions
C. D. C. and K. F. conceived the project; K. F. performed the synchronization of mouse spermatogenesis, isolated different types of germ cells, validated the phenotype of knockout mice, and contributed to the article; W. G. performed the experiments to produce the proteomic data and contributed to proteomic data analysis; Q. L. performed the proteomic data analysis and their correlation with mRNA analysis and wrote the article; J. S. developed the machine learning package FuncProFinder; Y. W., C. Z., R. W., and W. Y. constructed all the knockout mouse models; L. Y. and Y. Z. helped with the visualization of proteomic data; H. Y. supervised the knockout mouse production; S. L. supervised the proteomic data production, data analysis, and refined the article; and C. D. C. supervised the project. All authors contributed to the article.
Footnotes
This article contains supporting information.
Contributor Information
Hui Yang, Email: huiyang@ion.ac.cn.
Siqi Liu, Email: siqiliu@genomics.cn.
Charlie Degui Chen, Email: cdchen@sibcb.ac.cn.
Supporting Information
References
- 1.Cahoon C.K., Hawley R.S. Regulating the construction and demolition of the synaptonemal complex. Nat. Struct. Mol. Biol. 2016;23:369–377. doi: 10.1038/nsmb.3208. [DOI] [PubMed] [Google Scholar]
- 2.Keeney S., Lange J., Mohibullah N. Self-organization of meiotic recombination initiation: General principles and molecular pathways. Annu. Rev. Genet. 2014;48:187–214. doi: 10.1146/annurev-genet-120213-092304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Romanienko P.J., Camerini-Otero R.D. The mouse Spo11 gene is required for meiotic chromosome synapsis. Mol. Cell. 2000;6:975–987. doi: 10.1016/s1097-2765(00)00097-6. [DOI] [PubMed] [Google Scholar]
- 4.Baudat F., Manova K., Yuen J.P., Jasin M., Keeney S. Chromosome synapsis defects and sexually dimorphic meiotic progression in mice lacking Spo11. Mol. Cell. 2000;6:989–998. doi: 10.1016/s1097-2765(00)00098-8. [DOI] [PubMed] [Google Scholar]
- 5.Bannister L.A., Pezza R.J., Donaldson J.R., de Rooij D.G., Schimenti K.J., Camerini-Otero R.D., Schimenti J.C. A dominant, recombination-defective allele of Dmc1 causing male-specific sterility. PLoS Biol. 2007;5:e105. doi: 10.1371/journal.pbio.0050105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Petukhova G.V., Romanienko P.J., Camerini-Otero R.D. The Hop2 protein has a direct role in promoting interhomolog interactions during mouse meiosis. Dev. Cell. 2003;5:927–936. doi: 10.1016/s1534-5807(03)00369-1. [DOI] [PubMed] [Google Scholar]
- 7.Wang L., Guo Y., Liu W., Zhao W., Song G., Zhou T., Huang H., Guo X., Sun F. Proteomic analysis of pachytene spermatocytes of sterile hybrid male mice. Biol. Reprod. 2016;95:52. doi: 10.1095/biolreprod.116.138644. [DOI] [PubMed] [Google Scholar]
- 8.Miyata H., Castaneda J.M., Fujihara Y., Yu Z., Archambeault D.R., Isotani A., Kiyozumi D., Kriseman M.L., Mashiko D., Matsumura T., Matzuk R.M., Mori M., Noda T., Oji A., Okabe M. Genome engineering uncovers 54 evolutionarily conserved and testis-enriched genes that are not required for male fertility in mice. Proc. Natl. Acad. Sci. U. S. A. 2016;113:7704–7710. doi: 10.1073/pnas.1608458113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen Y., Zheng Y., Gao Y., Lin Z., Yang S., Wang T., Wang Q., Xie N., Hua R., Liu M., Sha J., Griswold M.D., Li J., Tang F., Tong M.H. Single-cell RNA-seq uncovers dynamic processes and critical regulators in mouse spermatogenesis. Cell Res. 2018;28:879–896. doi: 10.1038/s41422-018-0074-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Green C.D., Ma Q., Manske G.L., Shami A.N., Zheng X., Marini S., Moritz L., Sultan C., Gurczynski S.J., Moore B.B., Tallquist M.D., Li J.Z., Hammoud S.S. A comprehensive roadmap of murine spermatogenesis defined by single-cell RNA-seq. Dev. Cell. 2018;46:651–667.e610. doi: 10.1016/j.devcel.2018.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lukassen S., Bosch E., Ekici A.B., Winterpacht A. Characterization of germ cell differentiation in the male mouse through single-cell RNA sequencing. Sci. Rep. 2018;8:6521. doi: 10.1038/s41598-018-24725-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jung M., Wells D., Rusch J., Ahmad S., Marchini J., Myers S.R., Conrad D.F. Unified single-cell analysis of testis gene regulation and pathology in five mouse strains. Elife. 2019;8 doi: 10.7554/eLife.43966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ernst C., Eling N., Martinez-Jimenez C.P., Marioni J.C., Odom D.T. Staged developmental mapping and X chromosome transcriptional dynamics during mouse spermatogenesis. Nat. Commun. 2019;10:1251. doi: 10.1038/s41467-019-09182-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo J., Grow E.J., Mlcochova H., Maher G.J., Lindskog C., Nie X., Guo Y., Takei Y., Yun J., Cai L., Kim R., Carrell D.T., Goriely A., Hotaling J.M., Cairns B.R. The adult human testis transcriptional cell atlas. Cell Res. 2018;28:1141–1157. doi: 10.1038/s41422-018-0099-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hermann B.P., Cheng K., Singh A., Roa-De La Cruz L., Mutoji K.N., Chen I.C., Gildersleeve H., Lehle J.D., Mayo M., Westernstroer B., Law N.C., Oatley M.J., Velte E.K., Niedenberger B.A., Fritze D. The mammalian spermatogenesis single-cell transcriptome, from spermatogonial stem cells to spermatids. Cell Rep. 2018;25:1650–1667.e1658. doi: 10.1016/j.celrep.2018.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xia B., Yan Y., Baron M., Wagner F., Barkley D., Chiodin M., Kim S.Y., Keefe D.L., Alukal J.P., Boeke J.D., Yanai I. Widespread transcriptional scanning in the testis modulates gene evolution rates. Cell. 2020;180:248–262.e221. doi: 10.1016/j.cell.2019.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vicens A., Borziak K., Karr T.L., Roldan E.R.S., Dorus S. Comparative sperm proteomics in mouse species with divergent mating systems. Mol. Biol. Evol. 2017;34:1403–1416. doi: 10.1093/molbev/msx084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gan H., Cai T., Lin X., Wu Y., Wang X., Yang F., Han C. Integrative proteomic and transcriptomic analyses reveal multiple post-transcriptional regulatory mechanisms of mouse spermatogenesis. Mol. Cell. Proteomics. 2013;12:1144–1157. doi: 10.1074/mcp.M112.020123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cox J., Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 20.Cox J., Hein M.Y., Luber C.A., Paron I., Nagaraj N., Mann M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics. 2014;13:2513–2526. doi: 10.1074/mcp.M113.031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M.Y., Geiger T., Mann M., Cox J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods. 2016;13:731–740. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- 22.Hogarth C.A., Evanoff R., Mitchell D., Kent T., Small C., Amory J.K., Griswold M.D. Turning a spermatogenic wave into a tsunami: Synchronizing murine spermatogenesis using WIN 18,446. Biol. Reprod. 2013;88:40. doi: 10.1095/biolreprod.112.105346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Romer K.A., de Rooij D.G., Kojima M.L., Page D.C. Isolating mitotic and meiotic germ cells from male mice by developmental synchronization, staging, and sorting. Dev. Biol. 2018;443:19–34. doi: 10.1016/j.ydbio.2018.08.009. [DOI] [PubMed] [Google Scholar]
- 24.Gaysinskaya V., Soh I.Y., van der Heijden G.W., Bortvin A. Optimized flow cytometry isolation of murine spermatocytes. Cytometry A. 2014;85:556–565. doi: 10.1002/cyto.a.22463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kubota H., Avarbock M.R., Brinster R.L. Growth factors essential for self-renewal and expansion of mouse spermatogonial stem cells. Proc. Natl. Acad. Sci. U. S. A. 2004;101:16489–16494. doi: 10.1073/pnas.0407063101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ahmed E.A., de Rooij D.G. Staging of mouse seminiferous tubule cross-sections. Methods Mol. Biol. 2009;558:263–277. doi: 10.1007/978-1-60761-103-5_16. [DOI] [PubMed] [Google Scholar]
- 27.Peters A.H., Plug A.W., van Vugt M.J., de Boer P. A drying-down technique for the spreading of mammalian meiocytes from the male and female germline. Chromosome Res. 1997;5:66–68. doi: 10.1023/a:1018445520117. [DOI] [PubMed] [Google Scholar]
- 28.Wisniewski J.R., Zougman A., Nagaraj N., Mann M. Universal sample preparation method for proteome analysis. Nat. Methods. 2009;6:359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 29.Charrad M., Ghazzali N., Boiteau V., Niknafs A. NbClust: An R package for determining the relevant number of clusters in a data set. J. Stat. Softw. 2014;61 doi: 10.18637/jss.v061.i06. [DOI] [Google Scholar]
- 30.Huang D.W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 31.Kuleshov M.V., Jones M.R., Rouillard A.D., Fernandez N.F., Duan Q.N., Wang Z.C., Koplev S., Jenkins S.L., Jagodnik K.M., Lachmann A., McDermott M.G., Monteiro C.D., Gundersen G.W., Ma'ayan, Enrichr A. A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44:W90–W97. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Poggio T., Girosi F. Networks for approximation and learning. Proc. IEEE. 1990;78:1481–1497. [Google Scholar]
- 33.Cortes C., Vapnik V. Support-vector Networks. Machine Learn. 1995;20:273–297. [Google Scholar]
- 34.Picard R.R., Cook R.D. Cross-validation of regression models. J. Am. Stat. Assoc. 1984;79:575–583. [Google Scholar]
- 35.Wang H., Yang H., Shivalila C.S., Dawlaty M.M., Cheng A.W., Zhang F., Jaenisch R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-Mediated genome engineering. Cell. 2013;153:910–918. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang H., Wang H., Shivalila C.S., Cheng A.W., Shi L., Jaenisch R. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell. 2013;154:1370–1379. doi: 10.1016/j.cell.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang H., Wang H., Jaenisch R. Generating genetically modified mice using CRISPR/Cas-mediated genome engineering. Nat. Protoc. 2014;9:1956–1968. doi: 10.1038/nprot.2014.134. [DOI] [PubMed] [Google Scholar]
- 38.Bellani M.A., Boateng K.A., McLeod D., Camerini-Otero R.D. The expression profile of the major mouse SPO11 isoforms indicates that SPO11beta introduces double strand breaks and suggests that SPO11alpha has an additional role in prophase in both spermatocytes and oocytes. Mol. Cell Biol. 2010;30:4391–4403. doi: 10.1128/MCB.00002-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chakraborty P., Buaas F.W., Sharma M., Snyder E., de Rooij D.G., Braun R.E. LIN28A marks the spermatogonial progenitor population and regulates its cyclic expansion. Stem Cells. 2014;32:860–873. doi: 10.1002/stem.1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kleene K.C., Flynn J.F. Characterization of a cDNA clone encoding a basic protein, TP2, involved in chromatin condensation during spermiogenesis in the mouse. J. Biol. Chem. 1987;262:17272–17277. [PubMed] [Google Scholar]
- 41.Zhou Q., Nie R., Li Y., Friel P., Mitchell D., Hess R.A., Small C., Griswold M.D. Expression of stimulated by retinoic acid gene 8 (Stra8) in spermatogenic cells induced by retinoic acid: An in vivo study in vitamin A-sufficient postnatal murine testes. Biol. Reprod. 2008;79:35–42. doi: 10.1095/biolreprod.107.066795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krishnan A., Zhang R., Yao V., Theesfeld C.L., Wong A.K., Tadych A., Volfovsky N., Packer A., Lash A., Troyanskaya O.G. Genome-wide prediction and functional characterization of the genetic basis of autism spectrum disorder. Nat. Neurosci. 2016;19:1454–1462. doi: 10.1038/nn.4353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grootegoed J.A., Jansen R., Van der Molen H.J. The role of glucose, pyruvate and lactate in ATP production by rat spermatocytes and spermatids. Biochim. Biophys. Acta. 1984;767:248–256. doi: 10.1016/0005-2728(84)90194-4. [DOI] [PubMed] [Google Scholar]
- 44.Patel M.S., Nemeria N.S., Furey W., Jordan F. The pyruvate dehydrogenase complexes: Structure-based function and regulation. J. Biol. Chem. 2014;289:16615–16623. doi: 10.1074/jbc.R114.563148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Takakubo F., Dahl H.H. The expression pattern of the pyruvate dehydrogenase E1 alpha subunit genes during spermatogenesis in adult mouse. Exp. Cell Res. 1992;199:39–49. doi: 10.1016/0014-4827(92)90459-l. [DOI] [PubMed] [Google Scholar]
- 46.Roeder G.S., Bailis J.M. The pachytene checkpoint. Trends Genet. 2000;16:395–403. doi: 10.1016/s0168-9525(00)02080-1. [DOI] [PubMed] [Google Scholar]
- 47.Ashley T., Gaeth A.P., Creemers L.B., Hack A.M., de Rooij D.G. Correlation of meiotic events in testis sections and microspreads of mouse spermatocytes relative to the mid-pachytene checkpoint. Chromosoma. 2004;113:126–136. doi: 10.1007/s00412-004-0293-5. [DOI] [PubMed] [Google Scholar]
- 48.Shao B., Guo Y., Wang L., Zhou Q., Gao T., Zheng B., Zheng H., Zhou T., Zhou Z., Guo X., Huang X., Sha J. Unraveling the proteomic profile of mice testis during the initiation of meiosis. J. Proteomics. 2015;120:35–43. doi: 10.1016/j.jprot.2015.02.015. [DOI] [PubMed] [Google Scholar]
- 49.Li M., Huang T., Li M.J., Zhang C.X., Yu X.C., Yin Y.Y., Liu C., Wang X., Feng H.W., Zhang T., Liu M.F., Han C.S., Lu G., Li W., Ma J.L. The histone modification reader ZCWPW1 is required for meiosis prophase I in male but not in female mice. Sci. Adv. 2019;5 doi: 10.1126/sciadv.aax1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huang T., Yuan S., Li M., Yu X., Zhang J., Yin Y., Liu C., Gao L., Li W., Liu J., Chen Z.-J., Liu H. The histone modification reader ZCWPW1 links histone methylation to repair of PRDM9-induced meiotic double stand breaks. bioRxiv. 2019 doi: 10.1101/836023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mahgoub M., Paiano J., Bruno M., Wu W., Pathuri S., Zhang X., Ralls S., Cheng X., Nussenzweig A., Macfarlan T. Dual histone methyl reader ZCWPW1 Facilitates repair of meiotic double strand breaks. bioRxiv. 2019 doi: 10.1101/821603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wells D., Bitoun E., Moralli D., Zhang G., Hinch A.G., Donnelly P., Green C., Myers S.R. ZCWPW1 is recruited to recombination hotspots by PRDM9, and is essential for meiotic double strand break repair. bioRxiv. 2019 doi: 10.1101/821678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oji A., Isotani A., Fujihara Y., Castaneda J.M., Oura S., Ikawa M. Tesmin. Metallothionein-like 5, is required for spermatogenesis in micedagger. Biol. Reprod. 2020;102:975–983. doi: 10.1093/biolre/ioaa002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu X.-L., Yun D.-M., Gao S., Liang A.J., Duan Z.-Z., Wang H.-S., Wang G.-S., Sun F. The testis-specific gene 1700102P08Rik is essential for male fertility.pdf. Mol. Reprod. Dev. 2020;87:231–240. doi: 10.1002/mrd.23314. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Supplemental figures and tables are available. The MS proteomics raw data and MaxQuant search result files have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD017284. The FuncProFinder Matlab package was uploaded on https://github.com/sjq111/FuncProFinder.