

Abstract
As with all physiologic processes, chronic wounds are associated with unique intracellular and cellular/extracellular matrix (ECM) receptor types and signalling messages. These cellular receptors mediate responses of the epidermis to provisional wound matrix and change in form and number in cases of impaired wound healing. Integrins are the major cell‐surface receptors for cell adhesion and migration and epidermal keratinocytes express several integrins that bind ECM ligands in provisional wound ECM. Integrin receptors and more particularly integrin clusters and focal adhesion points appear to influence epidermal and dermal cell matrix interactions, cell motility, cell phenotype and ultimate healing trajectory. In chronic wounds, a variety of changes in receptors have been identified: decreased integrin α5β1 receptors affect the integration of fibronectin and subsequent keratinocyte migration; integrin αvβ6 stimulate transforming growth factor (TGF)‐β and may increase the susceptibility to ulceration and fibrosis; however, TGF‐β signal receptors have been found to be dysfunctional in many chronic wounds; additionally receptor interactions result in increased senescent cells including fibroblasts, myofibroblasts and even keratinocytes – this produces a degradative ECM and wound bed and corrosive chronic wound fluid. The activation or inhibition of integrin receptors by various agents may provide an excellent means of influencing wound healing. This process offers an earlier intervention into the wound healing cascade promoting intrinsic healing and elaboration of growth factors and ECM proteins, which may be more cost effective than the traditional attempts at extrinsic addition of these agents.
Keywords: Chronic wound, Focal adhesion, Integrin, Ligands, Signals
INTRODUCTION
Any interruption in the traditionally recognised phases of wound healing can result in a chronic wound. A chronic wound is one that has failed to heal in a timely fashion. The vast majority of chronic wounds fall into one of three categories: pressure sores, diabetic ulcers and venous ulcers (1). Although these wounds all have different aetiologies, chronic wound development is most often associated with a background pathophysiologic sequence of an ischemia–reperfusion injury often with biofilm/bacterial contamination resulting in prolonged inflammatory response 2, 3, 4. In many of these cases healing does not occur despite the best care 1, 4 Failure to re‐epithelialise can be a consequence of a number of factors, including prolonged inflammation, an imbalance of regulatory growth factors (GFs) and cytokines, defective extracellular matrix (ECM) that fails to support keratinocyte migration, modified fibroblast function and defective capillary function leading to inadequate tissue oxygenation (5). The ECM of these wounds has been referred to as ‘corrupt’5, 6 characterised by chronic wound fibroblasts unresponsive to GFs and other signals (7), containing high levels of matrix metalloproteinases (MMPs) and lacking of receptors such as integrin receptors for fibronectin (FN) binding and keratinocyte migration (8). New evidence is accumulating that chronic wound healing failure may be related to increased or reduced integrin receptors, production of senescent cells that are unresponsive to GFs and changes relating to cell signal or receptor dysfunction 1, 5, 7, 8, 9, 10, 11, 12, 13, 14, 15. Integrins appear to be the coordinating translator and effectors of signals that control cell proliferation, ECM maturation and cellular spreading. This is very relevant to the chronic wound state where specific changes in integrin type and receptors sensitivity may precipitate the changes in cellular phenotype that contribute to this chronicity.
Integrins are a large family of heterodimeric membrane glycoproteins involved in cell‐ECM and cell–cell interactions. Integrins consist of one α and one β subunit with an additional inserted domain (αI domain) in their α subunit 16, 17. Four out of the nine αI containing integrins, namely α1β1, α2β1, α10β1 and α11β1, are receptors for collagens 16, 17 but α1β1 and α2β1 integrins are the major cellular collagen receptors in fibroblasts 16, 17, 18. Integrin α1β1 regulates collagen synthesis and promotes cell growth, whereas integrin α2β1 is a functional cellular receptor for type I collagen fibrils, which mediates collagen gel contraction and regulates MMP‐1 expression 16, 17, 18, 19, 20. Integrins provide a mechanical connection between matrix components and the cytoskeleton and transduce an astonishing variety of signals either alone or in collaboration with GF receptors 16, 17, 18, 19, 20.
They were originally discovered as receptors that anchor cells to their surroundings by concomitantly binding to the cytoskeleton and ECM. Later, integrins were shown to activate cellular signalling pathways and, in that way, to contribute to the decisions about cell behaviour and fate 16, 21. Direct binding to GFs and regulation of endocytosis and trafficking of GFs make integrins even more multifunctional than previously appreciated 11, 16. α5β1 is considered the classical receptor for FN but other integrins (e.g. α3β1, α4β1 and αvβ1) also bind to FN with a lower affinity 8, 18. The combination of α and β subunits determines binding specificity for the ligands. The effect of multiple integrin binding that is, integrin clustering, increases the overall affinity for ligands (22). The cytoplasmic domain of integrins is anchored to the actin cytoskeleton through cytoskeletal linkage proteins such as vinculin and paxillin 17, 22. It is likely that collagen matrix remodelling is regulated through cell attachment to matrix molecules by integrin. Blocking of integrin subunits resulted in the detachment of fibroblasts from collagen (23).
CHRONIC WOUND INTEGRIN/RECEPTOR CHANGES
Decreased epithelial cell α5β1 integrin receptor, altered FN stimulation and keratinocyte migration
FN is a component of the initial blood clot and provisional matrix and is one of the key ECM proteins involved in wound healing (Figure 1) (8). It provides a matrix for deposition of other extracellular proteins and forms the framework over which epithelial cells, endothelial cells and fibroblasts migrate to accomplish wound closure. It thus constitutes a foundation for healing. Additionally it attracts monocytes, fibroblasts and endothelial cells to the site of the wound and opsonises debris 8, 18. Once the foundation and pathway for cellular migration has been established by FN, keratinocytes migrate over this foundation.
Figure 1.
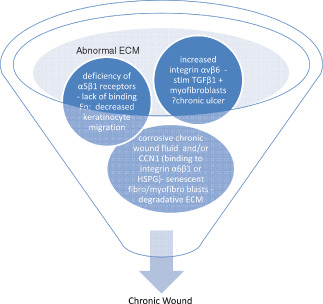
Integrin involvement in chronic wound pathophysiology: Various physiologic mechanisms (ischemia – reperfusion with reactive oxygen species, infection particularly biofilm, etc) may bring about a chronic wound status. The extracellular matrix (ECM) of these wounds has been referred to as ‘corrupt’ characterised by chronic wound fibroblasts unresponsive to growth factors (GFs) and other signals, containing high levels of matrix metalloproteinases (MMPs) and lacking of receptors such as integrin receptors for fibronectin (FN) binding and keratinocyte migration. The fragmentation of FN due to the corrosive wound fluid results in stimulation of mRNA FN but the deficiency of α5β1integrin receptors in chronic wound keratinocytes results in decreased production of complete FN. Additionally increased integrin αvβ6 in chronic wound epithelial cells appear to affect wound healing, by increasing transforming growth factor‐β (albeit unresponsive receptors) which stimulate myofibroblast formation particularly of the senescent phenotype. Additionally CCN1 and other chronic wound fluid components bind to specific chronic wound integrins resulting in senescent fibroblasts and myofibroblasts producing corrosive proteases and further aggravating the degradative ECM status. It is also likely that abnormal ECM composition fails to activate adhesion receptors on the fibroblasts preventing binding to ECM, a prerequisite for maximal GF stimulation.
Chronic wound fluid (CWF) is corrosive 3, 24 and appears to degrade FN resulting in its fragmentation. As a result, FN mRNA is stimulated to replace the degraded FN. High levels of FN mRNA have been showed in chronic leg ulcers (8). However, this does not translate to increased levels of complete FN and in fact; certain therapeutic interventions have suggested the application of FN to improve healing in chronic wounds (25). Why does the FN mRNA not translate to increased FN? It appears that the receptor responsible for converting FN mRNA to complete FN is deficient or altered in chronic wounds.
FN regulation of keratinocyte migration involves an interaction between integrin α5β1 receptors expressed on the surface of keratinocytes and FN localised in the basement membrane zone (26). A deficiency of α5β1 receptors would thus interfere with migration of epidermal keratinocytes to close the wound. It has been showed that although the level of FN mRNA and protein is heavily induced in chronic wounds, this integrin receptor, which was heavily induced in acute wounds, was not induced in the epidermis of chronic wounds (similar to normal non injured skin) (8). However, FN is known to bind to the other subspecies of integrins such as α3, α4 and αv with a lower affinity 8, 21. It is possible that other integrins with a lesser affinity for FN may be increased to compensate for the lack of α5 expression in chronic wounds. The failure to express appropriate levels of integrin α5 subunit on epidermal keratinocytes in chronic wounds may contribute to the healing defect in these wounds (8). Thus, prevention of FN degradation (modulation of chronic wound exudate) or stimulation of integrin α5 subunit in chronic wounds may be more important than inducing FN synthesis, as FN mRNA is abundantly expressed in chronic wounds.
Increased epithelial cell αvβ6‐integrin, TGF‐β and chronic fibrosis
Apart from the α5β1 subunit associated with FN, integrin αvβ6 also has implications in chronic wounds (27). This integrin is an epithelial cell‐specific receptor that is not normally expressed by resting epithelium but its expression is induced during wound healing (27). αvβ6‐integrin was shown to be strongly up regulated in the epidermis in human chronic wounds. Transgenic mouse lines were developed with constitutively expressed human 6‐integrin in the epithelium and during the breeding programme 16·1–27·0% of these mice developed spontaneous, progressing fibrotic chronic ulcers with severe fibrosis and numerous activated macrophages and fibroblasts expressing transforming growth factor (TGF)‐β (27). The findings suggest that increased v6‐integrin activation in keratinocytes is not important in normal wound repair but may play an active part in abnormal chronic wound healing possibly through a mechanism involving increased activation of TGF‐β with induction of fibroblast differentiation into myofibroblasts. This integrin activation may play a role when wound healing is compromised for example in conditions involving chronic inflammation or immunosupression.
Increased CCN1, stimulation of integrin α6β1, increased reactive oxygen species (ROS), premature senescent cellular (fibroblast, myofibroblast, endothelial, keratinocyte and macrophage) transformations, milieu of ECM degradation
Traditionally senescence in culture is associated with shortening of the telomeres during repeated cell divisions eventually leading to cell cycle arrest, known as replicative senescence (28). In the chronic wound, cellular senescence is not associated with telomere length but rather this phenotypic cell change is initiated by oxidative stress, activated oncogene supressor proteins and cyclin‐dependent kinase inhibitors (14). In the chronic wound environment, ROS attack DNA, causing an accumulation of lipofuscin (which is undegradable by the cell) and DNA damage‐induced cell cycle arrest (29). Fibroblast senescence in chronic wounds appears to be more related to chronic inflammation than telomere length (14). When exposed to CWF, normal fibroblasts in culture appear to switch to a senescent mode, showing changes in morphology consistent with senescent fibroblasts (14). CWF rapidly degrades exogenous GFs, decreases the production of cyclin D1, phosphorylated retinoblastoma protein RB and increases p21 (30). Since this form of senescence seems to be telomere independent, treatments aimed at increasing telomere length such as telomerase would be ineffective.
The transformation of fibroblast to myofibroblast plays an important part in the promotion of wound contraction and healing by producing ECM constituents to form granulation tissue. It appears that the system is kept in balance by myofibroblasts being driven into senescence at later stages of wound healing, thereby converting these ECM‐producing cells into ECM‐degrading cells, limiting fibrosis of the wound (28). In skin wound healing, myofibroblast senescence is triggered by the dynamically expressed matricellular protein CCN1 (also known as CYR61) through integrin signalling (28). CCN1 is an angiogenic matricellular protein of the CCN family (CCN is the acronym of the first three members: Cyr61, CTGF and Nov). CCN1 is normally expressed at a low level in most tissues but becomes highly expressed because of bacterial or viral infections 28, 31 or in tissue repair 28, 32, 33. Mechanistically, CCN 1 induces fibroblast senescence through its direct binding to integrin α6β1 and cell‐surface heparan sulphate (HS) proteoglycans (28) this triggers formation and accumulation of ROS, DNA damage and p53 activation resulting in degradation of the ECM. Additionally, through integrin signalling, myofibroblast senescence brings about increased expression of inflammatory cytokines/chemokines [e.g. Interleukin (IL)1, IL6, IL8, Monocyte chemotactic protein (MCP)2, MCP4, macrophage inflammatory protein (MIP)‐1a, MIP‐3a] and ECM‐degrading enzymes (e.g. MMPs), with down regulated expression of certain ECM components (e.g. collagen) (28). Thus, senescent myofibroblasts accumulate as part of the normal process of tissue repair, and function to limit the extent of fibrogenesis associated with wound healing 28, 34. However, premature cellular senescence is stimulated by events often present in the evolution of a chronic wound (infection, particularly biofilm, corrosive CWF, etc). This results in CCN1 binding to integrin α6β, ROS stimulation, fibroblast and then myofibroblast senescence, increased inflammation and degradation of the ECM.
In a clinical setting, these chronic wound bed changes have direct consequences on healing. The duration of venous leg ulcers (VLU) prior to first visit was shown to have a direct correlation with ultimate healing (5). It appears the longer a wound remains in the inflammatory phase the more cellular defects arise and accumulate rendering it less responsive to treatment. Premature senescence with ongoing ECM degradation has been suggested as a contributory factor in chronic wound occurrence 5, 14, 28. Accumulation of greater than 15% senescent fibroblasts has been described as a threshold beyond which wounds become hard to heal (5). Evidence for this is provided by the positive relationship between higher levels of fibroblasts with a senescent phenotype identified in VLU and a poor prognosis 5, 35.
While much attention has focused on the fibroblast senescence in the chronic wound, other cells have also been shown to be affected by cellular senescent transformation. Many of the same agents that cause abnormal changes in the fibroblast also affect the keratinocyte (14). Additionally, increases in proteolytic enzymes and inflammation may lead to a gradual loss of endothelial cell function mimicking replicative senescence (14). Therefore, induction of fibroblast senescence may have a direct effect on the induction of senescence in endothelial cells. The macrophage too, has been shown to produce significantly less vascular endothelial growth factor (VEGF) when isolated from aged skin with increased senescence compared to young controls (14).
Apart from cellular senescence induction, CCN1 can upregulate plasminogen activator inhibitor‐1 (PAI‐1), possibly through the activation of p53 14, 28, 36. Plasminogen activators activate circulating plasminogen to produce plasmin. Plasmin participates in the breakdown of other glycoproteins in the ECM and the activation of MMPs (28). Chronic wounds are associated with elevated levels of plasmin, collagenases and other degradative enzymes. Over‐expression of PAI‐1 is sufficient to drive fibroblasts into senescence in vitro 14, 28. It would appear that a dual process occurs in the ECM of a chronic wound. Fibroblasts secrete factors that stimulate matrix degradation yet they also produce matrix‐promoting factors (TGF‐β1 and PAI‐1). The PAI 1 and CCN1 may then stimulate senescent myofibroblast phenotype but resultant ratios of PAI‐1 and TGF‐β1 to plasmin decrease and dominant degradative ECM results (37). Thus, the association between fibroblast senescence and ECM degradation appears to be supported by changing ratios that shift the balance towards degradation of the ECM as senescence is reached (37). The synthetic function of fibroblasts may decrease with senescence, although Herrick et al.(38) have shown that chronic wound fibroblasts show no decrease in matrix secretion – thus the observed degradation of ECM appears to be related to increases in matrix remodelling enzymes.
Unresponsive fibroblast GF receptors, non binding to ECM, persistent ECM degradation
An additional reason for the dominance of ECM degradation is its unresponsiveness to the stimulatory action of TGF‐β. Researchers have showed that VLU fibroblasts have decreased TGF‐β Type II receptor expression (7)– this was accompanied by failure of ulcer fibroblasts to phosphorylate Smad 2, Smad 3 and p42/44 mitogen‐activating protein kinase, and was associated with a slower proliferative rate in response to TGF‐β (7). This further highlights the possibility that non healing in chronic wounds may well be related to a decrease in receptor expression and a failure in specific signal transduction.
Much of the destructive ECM milieu that is associated with cellular senescence relates to the increased presence of MMPs. In acute wounds, there is a balance between protease activity and ECM deposition encouraged by tissue inhibitors of metalloproteases (TIMPs). The ratio of MMP‐9/TIMP‐1 in chronic wounds is an important healing measurement parameter 14, 39. Chronic wound fibroblasts have a diminished capacity to react to GFs. This decreased response to bFGF, EGF and PDGF does not appear to be related to a decrease in receptor quantity, but rather a dysfunction in intracellular signalling (38). The logical approach to changing the degradative ECM wound milieu is wound bed preparation including debridement to stimulate vascularisation, and decrease bacterial burden 3, 4, 5. Importantly, removal of the senescent cells and their products may contribute greatly to this change. Only then can exogenously applied GFs and biologics be expected to stimulate the surrounding healthy tissue and promote wound healing.
Proteolysis by MMPs exposes RGD sites on ECM proteins (FN, fibrinogen, osteopontin, tenascin and vitronectin) facilitating cell‐surface integrin recognition and attachment of the cell to the ECM protein (40). This then initiates GF signalling (40). Chronic wounds are associated with increased inflammation, copious corrosive wound exudate, increased proteolysis and degradation of the ECM resulting in an abnormal wound matrix that induces less GF action because of diminished integrin binding 3, 4, 5, 9, 10, 11, 18, 19, 20, 21, 22, 23, 41, 42 Aside from increased proteinases (particularly MMP9) levels, chronic wound edge keratinocytes also display GF and ECM receptor dysfunction, which fails to convert them into a migratory phenotype (1). VEGF wound fluid levels were also significantly higher in non healing VLU than in normal wounds despite excessive degradation of VEGF by plasmin 1, 43. However, the VEGF inhibitor, soluble VEGFR‐1, was increased fourfold in wound fluids from chronic VLU compared with wound fluids from acute excisional wounds on the lower leg (1). Thus, it is likely that abnormal ECM composition fails to activate adhesion receptors on the fibroblasts preventing binding to ECM, a prerequisite for maximal GF stimulation 1, 9, 12.
INTEGRIN CLUSTERING
Complexes between receptors and their ECM ligands do not appear to be a one to one reaction (44). Instead, clusters of multiple ligands and signalling receptors interact with each other and the resulting cellular responses occur following integration of multiple incoming signals (44). The best‐characterised matrix receptors are the integrins, but syndecans, receptor tyrosine kinases and phosphotyrosine phosphatases as well as HS receptors and hyaluronan receptors have recently been shown to be equally important players (44).
The expression and action of integrins are predominantly localised to the focal adhesion complex as originally showed in fibroblasts. Focal adhesion complexes are the structural connection between the ECM and the cytoskeleton. Here, transmembrane signalling is mediated by integrins that cluster with dozens of cytoskeletal proteins and signalling molecules (20). An example of such ECM component clustering is seen with native collagen integrin receptor α2β1. Major functions of this receptor include the induction of MMP1, MMP13 and MT1‐MMP, as well as activation of proMMP2 44, 45, 46, 47. More importantly, the formation of a direct complex between α2β1 and pro‐MMP1 has been shown in keratinocytes plated on native type I collagen (48). It is postulated that tissue injury exposes epithelial cells to fibrillar collagen, which triggers integrin ligation. This collagen and integrin interaction induces transcriptional upregulation of MMP1 in the cell which then binds to the α2β1 receptor complex and is converted to its active form 44, 48. The activated MMP1 then cleaves type I collagen, which rapidly denatures reducing its integrin‐binding affinity. The focal adhesion complex then disassembles allowing the cell to move forward to its next ECM contact (44). Thus, cellular proliferation and movement takes place by sequential component clustering. The components of this focal adhesion largely determine the ECM remodelling and subsequent healing trajectory.
This integrin clustering in the chronic wound situation translates to a simultaneous transformation in integrin appearance and signal modulation described above with concurrent changes occurring predominantly in fibroblasts, keratinocytes and the ECM. The chronic wound milieu is self perpetuating with CWF and its corrosive nature (or biofilm) stimulating the formation of altered integrin clusters that transform the cellular cross talk and ‘dynamic reciprocity’ between cells and ECM.
REPROGRAMMING INTEGRINS AND/OR THEIR RECEPTORS
From the ECM changes described above, the extrinsic addition of GFs would not be expected to alter this wound milieu significantly. This has been borne out in the literature where results with PDGF and other GFs have been rather disappointing 49, 50. As a result, new therapeutic applications have been sought. Amelogenins (AMG), extracted from the developing porcine tooth, are local adjunctive proteins to periodontal surgery for advanced periodontitis (51). Topical AMG proteins promoted granulation tissue formation and accelerated the time of the closure of full‐thickness experimental skin wounds in rabbits (52). AMG have been identified as cell adhesion proteins. Endothelial cells do not produce significant amounts of VEGF and must therefore rely on VEGF supplied by neighbouring cells, mainly macrophages and fibroblasts, in early granulation tissue (52). Incubation of dermal fibroblasts with AMG significantly increased VEGF levels and MMP2 is expressed by endothelial cells during wound healing (52)– these both contribute to angiogenesis by cleavage of endothelial buds (MMP2) and promotion of VEGF (52). Van der Pauw et al.(53) proposed that the integrin subunit β‐1 is involved in the interaction between AMG and fibroblasts – GFs such as TGF‐β have high affinity for AMG (53). Additionally AMG was shown to increase endogenous production of TGF‐β by fibroblasts in a collagen matrix. AMG promote the adhesion of several cell lines and to a similar degree as FN (53). Although AMG lacks RGD sequences that bind to integrins, reports suggest that fibroblasts bind to AMG via integrins 53, 54, 55, 56. Thus, integrin character changes are assumed to have taken place reinvigorating GF sensitivity. In a study of treatment for hard‐to‐heal leg ulcers, wound area reduction in those treated with AMG (Xelma) was showed (54). However, this did not reach statistical significance when compared with the control (54). Although this is a step in the right direction, AMG may not adequately alter the chronic wound receptor status.
Other indirect attempts have been made to restimulate integrin receptors by adding ECM components that have been degraded in chronic wound beds. Thus, heparan sulphate glycosaminoglycan has been used as a fluid wound dressing (CACIPLIQ20), with the aim of replacing HS that has been destroyed, thus restoring tissue homeostasis and protecting the wound from further degradation (57). Integrins and HS proteoglycans act as master regulators of multiple ECM proteins and signalling pathways – they sense, integrate and respond to the physical and chemical environmental information either by directly connecting with the local adhesion sites or regulating global cellular processes through growth factor receptor signalling pathways, leading to the integration of both external and internal signals in space and time (57). The study showed an improvement in chronic wound healing and pain in recalcitrant wounds free of heavy purulent drainage or necrotic tissue (57).
It is apparent that limited therapeutic agents have been used and few related studies have been undertaken focusing on integrin receptor stimulation, signal alteration or cellular cross talk. It would appear that specific focus on integrin and cell receptor activation could be useful. In this regard, certain linker proteins have been identified that modulate integrin conformation. Agonist stimulation of integrin receptors, composed of transmembrane α and β subunits, leads cells to regulate integrin affinity (‘activation’), a process that controls cell adhesion and migration, and ECM assembly 58, 59. Talin and kindlin were recently described as key linker proteins responsible for integrin activation 58, 59. Activation results in increased affinity for ECM molecules and clustering into focal adhesions with rapid tyrosine phosphorylation of specific substrates and an increase in the concentration of lipid second messengers 58, 59. Activation is initiated through interactions at the integrin tails (60). Additionally in vivo experiments indicate that kindlins are also important in integrin activation (61). Talin is a major cytoskeletal protein that colocalises with and binds to integrins and to actin and actin‐binding proteins such as vinculin (59). The binding of talin to integrin β cytoplasmic domains appear to change the integrin conformation to an extended activated form (62). It appears that a bent conformation represents a low‐affinity state, whereas the extended conformation is associated with a high‐affinity state of integrins. Currently, three separate integrin conformational classes have been identified: inactive, active (or primed) and ligand bound. It is surmised that these states correspond to a bent conformation, an extended form with a closed headpiece and an extended form with an open headpiece, respectively (62). Thus, talin appears to play a general role in activating multiple classes of integrins.
Integrin expression and activity can be further modulated by other trans membrane proteins such as tetraspanins (and HS proteoglycans as noted above) (63). They appear to contribute to activation of TGF‐β by αv6‐integrins as noted earlier. Integrin αvβ6 is abundantly expressed by keratinocytes in chronic wound keratinocytes 27, 63. Binding of latent TGF‐β to this integrin induces a conformational change unmasking the mature TGF‐β moiety, which induces TGF‐β signalling in adjacent cells setting up myofibroblastic transformational changes and a probable chronic wound healing trajectory (63). Targeted receptor blocking may be a logical sequence in therapeutic options. Thus far, much of the focus on talin‐induced activation has been on platelet activation and clot retraction (64). Investigation of β integrin activation in relation to collagen matrix binding in bioengineered substitutes and chronic wound manipulation appear to be worth pursuing.
SUMMARY AND CONCLUSION
Wound chronicity is a timely process – the typical biochemical abnormalities described above occur over some time and the resulting wound milieu often render the wound non responsive to most treatments. Early focused and targeted approaches to these wound abnormalities would likely change the course of chronicity. This is especially relevant in patients at high risk of developing chronic wounds, including those with diabetic, venous or pressure ulcers who often do not respond adequately to several weeks of standard care. Because of the consequences of this ‘corrupt’ ECM, wound healing strategies that aim to correct dysfunctional signalling may be beneficial.
It has often been speculated that, given the relatively large number of receptors on the cell surface, a decrease in receptor number does not have important functional consequences. Results of studies indicate that this may not be the case and it has been suggested that tissue integrity and ulcer recurrence might be due to decreased expression of TGF‐β receptors and its consequences on signal transduction (7). As noted previously topically applied GFs, including TGF‐αs, have been tested extensively in chronic wounds with mixed results 49, 50, 65, 66. It has been speculated that topical application of these agents in the corrosive environment of the wound bed and CWF rapidly degrades these GFs 3, 24. However, an alternate or additional explanation for the relative lack of success of GFs in the treatment of chronic wounds may reside in the phenotypic abnormalities of wound cells and their relative unresponsiveness to stimulatory signals.
A key to understanding the pathogenesis of chronic wounds, and age‐related wound defects, is identifying cellular receptors that mediate responses of the epidermis to provisional wound matrix and determining how changes in these receptors contribute to impaired wound healing. Integrins are the major cell‐surface receptors for cell adhesion and migration 9, 10, 17, and epidermal keratinocytes express several integrins that bind ECM ligands in provisional wound ECM 26, 27, 48.
Much of the focus of this article is examining cellular extracellular communication and the changes in ‘conversation’ or signalling that occur in different pathological contexts, Integrin receptors and more particularly integrin clusters and focal adhesion points appear to influence cell matrix interaction, cell motility, cell phenotype and ultimate healing trajectory. Integrin type and status in terms of conformational expression may provide a good picture of wound bed healing status and may serve wound diagnostic purposes in the future. The integrin influence in cellular ECM interaction, fits in well with the concept of dynamic reciprocity that has been recently updated in relation to our improved understanding of molecular and cellular events (66). The integrin serves as the facilitator of this dynamic reciprocity, its extracellular component interacting with ECM molecules and its intracellular component interacting with signalling proteins that link to the cytoskeleton. Chronic wound characteristics likely alter the ability of cells to switch their integrin expression or type affecting their binding capacity and limiting their adaptation to changes within the ECM (66). Amongst a host of molecular or other biochemical and structural abnormalities, this results in the type of integrin and signalling changes detailed above.
The activation or inhibition of integrin receptors by various agents may provide an excellent means of influencing wound healing. This process offers an earlier intervention into the wound healing cascade promoting intrinsic healing and elaboration of GFs and ECM proteins, which may be more cost effective than the traditional attempts at extrinsic addition of these agents.
References
- 1. Agren MS, Werthen M. The extracellular matrix in wound healing: a closer look at therapeutics for chronic wounds. Int J Low Extrem Wounds 2007;6:82–97. [DOI] [PubMed] [Google Scholar]
- 2. Midwood KS, Williams LV, Schwarzbauer JE. Tissue repair and the dynamics of the extracellular matrix. Int J Biochem Cell Biol 2004;36:1031–7. [DOI] [PubMed] [Google Scholar]
- 3. Widgerow AD. Chronic wound exudate – thinking outside the box. Wound Repair Reg 2011;19:287–91. [DOI] [PubMed] [Google Scholar]
- 4. Widgerow AD. Deconstructing the ‘stalled wound’– Wounds. Accepted for publication October 2011. [PubMed]
- 5. Harding KG, Moore K, Phillips TJ. Wound chronicity and fibroblast senescence – implications for treatment. Int Wound J 2005;2:364–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Clark RA. Fibrin and wound healing. Ann NY Acad Sci 2001;936:355–67. [DOI] [PubMed] [Google Scholar]
- 7. Kim BC, Kim HT, Park SH, Cha JS, Yufit T, Kim SJ, Falanga V. Fibroblasts from chronic wounds show altered TGF‐beta‐signalling and decreased TGF‐beta Type II receptor expression. J Cell Physiol 2003;195:331–6. [DOI] [PubMed] [Google Scholar]
- 8. Ongenae KC, Phillips TJ, Park HY. Level of fibronectin mRNA is markedly increased in human chronic wounds. Dermatol Surg 2000;26: 447–51. [DOI] [PubMed] [Google Scholar]
- 9. Schwartz MA, Ginsberg MH. Networks and crosstalk: integrin signalling spreads. Nat Cell Biol 2002;4:E65–8. [DOI] [PubMed] [Google Scholar]
- 10. Lee JW, Juliano R. Mitogenic signal transduction by integrin‐and growth factor receptor‐mediated pathways. Mol Cells 2004;17:188–202. [PubMed] [Google Scholar]
- 11. Li W, Fan J, Chen M, Guan S, Sawcer D, Bokoch GM, Woodley DT. Mechanism of human dermal fibroblast migration driven by type I collagen and platelet‐derived growth factor‐BB. Mol Biol Cell 2004;15:294–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li W, Henry G, Fan J, Bandyopadhyay B, Pang K, Garner W, Chen M, Woodley DT. Signals that initiate, augment, and provide directionality for human keratinocyte motility. J Invest Dermatol 2004;123:622–33. [DOI] [PubMed] [Google Scholar]
- 13. Telgenhoff D, Shroot B. Cellular senescence mechanisms in chronic wound healing. Cell Death and Differentiation 2005;12:695–8. [DOI] [PubMed] [Google Scholar]
- 14. Ågren MS, Steenfos HH, Dabelsteen S, Hansen JB, Dabelsteen E. Proliferation and mitogenic response to PDGF‐BB of fibroblasts isolated from chronic venous leg ulcers is ulcer‐age dependent. J Invest Dermatol 1999;112:463–9. [DOI] [PubMed] [Google Scholar]
- 15. Ivaska J, Heino J. Cooperation between integrins and growth factor receptors in signaling and endocytosis. Annu Rev Cell Dev Biol 2011;27:291–320. [DOI] [PubMed] [Google Scholar]
- 16. Barczyk M, Carracedo S, Gullberg D. Integrins. Cell Tissue Res 2010;339:269–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Velling T, Risteli J, Wennerberg K, Mosher DF, Johansson S. Polymerization of type I and III collagens is dependent on fibronectin and enhanced by integrins alpha 11beta 1 and alpha 2beta 1. J Biol Chem 2002;277:37377–81. [DOI] [PubMed] [Google Scholar]
- 18. Xu J, Clark RAF. Extracellular matrix alters PDGF regulation of fibroblast integrins. J Cell Biol 1996;132:239–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu S, Shi‐wen X, Blumbach K, Eastwood M, Denton CP, Eckes B, Krieg T, Abraham DJ, Leask A. Expression of integrin β1 by fibroblasts is required for tissue repair in vivo. J Cell Sci 2010;123:3674–82. [DOI] [PubMed] [Google Scholar]
- 20. Hu S, Cui D, Yang X, Hu J, Wan W, Zeng J. The crucial role of collagen‐binding integrins in maintaining the mechanical properties of human scleral fibroblasts‐seeded collagen matrix. Molecular Vision 2011;17:1334–42. [PMC free article] [PubMed] [Google Scholar]
- 21. Gardner H, Broberg A, Pozzi A, Laato M, Heino J. Absence of integrin alpha1beta1 in the mouse causes loss of feedback regulation of collagen synthesis in normal and wounded dermis. J Cell Sci 1999;112:263–72. [DOI] [PubMed] [Google Scholar]
- 22. Rayment EA, Upton Z, Shoote GK. Increased matrix metalloproteinase‐9 (MMP‐9) activity observed in chronic wound fluid is related to the clinical severity of the ulcer. Br J Dermatol 2008;158:951–61. [DOI] [PubMed] [Google Scholar]
- 23. White DJ, Puranen S, Johnson MS, Heino J. The collagen receptor subfamily of the integrins. Int J Biochem Cell Biol 2004;36:1405–10. [DOI] [PubMed] [Google Scholar]
- 24. Schultz GS, Ladwig G, Wysocki A. Extracellular matrix: review of its roles in acute and chronic wounds, World wide wounds August 2005. URL www.worldwidewounds.com [accessed on November 2011].
- 25. Grinnell F, Ho C‐H, Wysocki A. Degradation of fibronectin and vitronectin in chronic wound fluid: analysis by cell blotting, immunoblotting, and cell adhesion assays. J Invest Dermatol 1992;98:410–6. [DOI] [PubMed] [Google Scholar]
- 26. Häkkinen l, Koivisto l, Gardner H, Saarialho‐Kere U, Carroll JM, Lakso M, Rauvala H, Laato M, Heino J, Larjava H. Increased expression of β6‐integrin in skin leads to spontaneous development of chronic wounds. Am J Pathol 2004;164:229–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jun J, Lau JF. Cellular senescence controls fibrosis in wound healing. AGING 2010;2:627–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bernard D, Gosselin K, Monte D, Vercamer C, Bouali F, Pourtier A, Vandenbunder B. Involvement of Rel/nuclear factor‐kappaB transcription factors in keratinocyte senescence. Cancer Res 2004;64:472–81. [DOI] [PubMed] [Google Scholar]
- 29. Van de Berg JS, Robson MC. Arresting cell cycles and the effect on wound healing.Surg. Clin North Am 2003;83:509–20. [DOI] [PubMed] [Google Scholar]
- 30. Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. Senescence‐associated secretory phenotypes reveal cell nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 2008;6:2853–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Erusalimsky JD, Kurz DJ. Cellular senescence in vivo: its relevance in ageing and cardiovascular disease. Exp Gerontol 2005;40:634–42. [DOI] [PubMed] [Google Scholar]
- 32. Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature 2008;453:314–21. [DOI] [PubMed] [Google Scholar]
- 33. Jun J‐I, Lau LF. The matricellular protein CCN1/ CYR61 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat Cell Biol 2010;12:676–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stanley A, Osler T. Senescence and the healing rates of venous ulcers. J Vasc Surg 2001;33:1206–11. [DOI] [PubMed] [Google Scholar]
- 35. Kortlever RM, Higgins PJ, Bernards R. Plasminogen activator inhibitor‐1 is a critical downstream target of p53 in the induction of replicative senescence. Nat Cell Biol 2006;8:877–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vande Berg JS, Rose MA, Haywood‐Reid PL, Rudolph R, Payne WG, Robson MC. Cultured pressure ulcer fibroblasts show replicative senescence with elevated production of plasmin, plasminogen activator inhibitor‐1, and transforming growth factor‐beta1. Wound Repair Regen 2005;13:76–83. [DOI] [PubMed] [Google Scholar]
- 37. Herrick SE, Sloan P, McGurk M, Freak L, McCollum CN, Ferguson MWJ. Sequential changes in histologic pattern and extracellular matrix deposition during the healing of chronic venous ulcers. Am J Pathol 1992;141:1085–95. [PMC free article] [PubMed] [Google Scholar]
- 38. Mirastschijski U, Haaksma CJ, Tomasek JJ, Ågren MS. Matrix metalloproteinase inhibitor GM 6001 attenuates keratinocyte migration, contraction and myofibroblast formation in skin wounds. Exp Cell Res 2004;299:465–75. [DOI] [PubMed] [Google Scholar]
- 39. Mirastschijski U, Schnabel R, Claes J, Schneider W, Ågren MS, Haaksma C, Tomasek JJ. Matrix metalloproteinase inhibition delays wound healing and blocks latent transforming growth factor‐β1 promoted myofibroblast formation and function. Wound Repair Regen 2010;18:223–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Drinkwater SL, Burnand KG, Ding R, Smith A. Increased but ineffectual angiogenic drive in nonhealing venous leg ulcers. J Vasc Surg 2003;38:1106–12. [DOI] [PubMed] [Google Scholar]
- 41. Hynes RO. Integrins: bidirectional, allosteric signalling machines. Cell 2002;110:673–87. [DOI] [PubMed] [Google Scholar]
- 42. Connors WL, Jokinen J, White DJ, Puranen JS, Kankaanpa P, Upla P, Tulla, M , Johnson MS, Heino J. Two synergistic activation mechanism of alpha 2 beta 1 integrin mediated collagen binding. Biol Chem 2007;282:14675–83. [DOI] [PubMed] [Google Scholar]
- 43. Vogel WF. Collagen‐receptor signalling in health and disease. Eur J Derm 2001;11:506–14. [PubMed] [Google Scholar]
- 44. Heino J. The collagen receptor integrins have distinct ligand recognition and signalling functions. Matrix Biol 2000;19:319–23. [DOI] [PubMed] [Google Scholar]
- 45. Eckes B, Zigrino P, Kessler D, Holtkotter O, Shephard P, Mauch C, Krieg T. Fibroblast‐matrix interactions in wound healing and fibrosis. Matrix Biol 2000;19:325–32. [DOI] [PubMed] [Google Scholar]
- 46. Zigrino P, Drescher C, Mauch C. Collagen‐induced proMMP‐2 activation by MT1‐MMP in human dermal fibroblasts and the possible role of alpha2beta1 integrins. Eur J Cell Biol 2001; 80:68–77. [DOI] [PubMed] [Google Scholar]
- 47. Dumin JA, Dickeson SK, Stricker TP, Bhattacharyya‐Pakrasi M, Roby JD, Santoro SA, Parks WC. Procollagenase‐1 (MMP‐1) Binds the alpha2 beta1integrin upon release from keratinocytes migrating on type I collagen. J Biol Chem 2001; 18:18. [DOI] [PubMed] [Google Scholar]
- 48. Veves A, Sheehan P, Pham HT. A randomized, controlled trial of Promogran (a collagen/oxidized regenerated cellulose dressing) vs standard treatment in the management of diabetic foot ulcers. Arch Surg 2002;137:822–7. [DOI] [PubMed] [Google Scholar]
- 49. Lobmann R, Zemlin C, Motzkau M, Reschke K, Lehnert H. Expression of matrix metalloproteinases and growth factors in diabetic foot wounds treated with a protease absorbent dressing. J Diabetes Complications 2006;20:329–35. [DOI] [PubMed] [Google Scholar]
- 50. Esposito M, Coulthard P, Thomsen P, Worthington HV. Enamel matrix derivative for periodontal tissue regeneration in treatment of intrabony defects: a Cochrane systematic review. J Dent Educ 2004;68:834–44. [PubMed] [Google Scholar]
- 51. Mirastschijski U, Konrad D, Lundberg E, Lyngstadaas SP, Jorgensen LN, Ågren MS. Effects of a topical enamel matrix derivative on skin wound healing. Wound Repair Regen 2004;12:100–8. [DOI] [PubMed] [Google Scholar]
- 52. Van der Pauw MT, Everts V, Beertsen W. Expression of integrins by human periodontal ligament and gingival fibroblasts and their involvement in fibroblast adhesion to enamel matrix‐derived proteins. J Periodontal Res 2002;37:317–23. [DOI] [PubMed] [Google Scholar]
- 53. Suzuki N, Ohyama M, Maeno M, Ito K, Otsuka K. Attachment of human periodontal ligament cells to enamel matrix‐derived protein is mediated via interaction between BSP‐like molecules and integrin alpha(v)beta3. J Periodontol 2001;72:1520–6. [DOI] [PubMed] [Google Scholar]
- 54. Vowden P, Romanelli M, Peter R, Boström A, Josefsson A, Stege H. The effect of amelogenins (Xelma) on hard‐to‐heal venous leg ulcers. Wound Repair Regen 2006;14:240–6. [DOI] [PubMed] [Google Scholar]
- 55. Grayson RE, Yamakoshi Y, Wood EJ, Ågren MS. The effect of the amelogenin fraction of enamel matrix proteins on fibroblast‐mediated collagen matrix reorganization. Biomaterials 2006;27:2926–33. [DOI] [PubMed] [Google Scholar]
- 56. Groah SL, Libin A, Spungen M, Nguyen K‐L, Woods E, Nabili M, Ramella‐Roman J, Barritault D. Regenerating matrix‐based therapy for chronic wound healing: a prospective within‐subject pilot study. Int Wound J 2010;1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ye F, Hu G, Taylor D, Ratnikov B, Bobkov AA, McLean MA, Sligar SG, Taylor KA, Ginsberg MH. Recreation of the terminal events in physiological integrin activation. J Cell Biol 2010;188:157–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Banno A, Ginsberg MH. Integrin activation. Biochem Soc Trans 2008;36:229–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nieswandt B, Moser M, Pleines I, Varga‐Szabo D, Monkley S, Critchley D, Fässler R. Loss of talin1 in platelets abrogates integrin activa‐tion, platelet aggregation, and thrombus formation in vitro and in vivo. J Exp Med 2007;204:3113–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Moser M, Legate KR, Zent R, Fässler R. The tail of integrins, talin, and kindlins. Science 2009;324:895–99. [DOI] [PubMed] [Google Scholar]
- 61. Askari JA, Tynan CJ, Webb SED, Martin‐Fernandez ML, Ballestrem C, Humphries MJ. Focal adhesions are sites of integrin extension. J Cell Biol 2010; 188:891–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Eckes B, Nischt R, Krieg T. Cell‐matrix interactions in dermal repair and scarring. Fibrogenesis and Tissue repair 2010;3:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Leisner TM, Parise LV. Talin's second act‐ivation: retraction. Blood 2011;117:1442–3. [DOI] [PubMed] [Google Scholar]
- 64. Robson MC, Phillips TJ, Falanga V, Odenheimer DJ, Parish LC, Jensen JL, Steed DL. Randomized trial of topically applied repifermin (recombinant human keratinocyte growth factor‐2) to accelerate wound healing in venous ulcers. Wound Repair Regen 2001;9:347–52. [DOI] [PubMed] [Google Scholar]
- 65. Smiell JM, Wieman TJ, Steed DL, Perry BH, Sampson AR, Schwab BH. Efficacy and safety of becapler‐min (recombinant human platelet‐derived growth factor‐BB) in patients with nonhealing, lower extremity diabetic ulcers: a combined analysis of four randomized studies. Wound Repair Regen 1999;7:335–46. [DOI] [PubMed] [Google Scholar]
- 66. Schultz GS, Davidson JM, Kirsner RS, Bornstein P, Herman IM. Dynamic reciprocity in the wound microenvironment. Wound Rep Reg 2011;19:134–48. [DOI] [PMC free article] [PubMed] [Google Scholar]