

Abstract
The prevalence of the chronic metabolic disorder, diabetes mellitus, is expected to increase in the coming years and worldwide pandemic levels are predicted. Inevitably, this will be accompanied by an increase in the prevalence of diabetic complications, including diabetic foot ulcers. At present, treatment options for diabetic foot ulcers are in many cases insufficient, and progression of the condition results in the requirement for limb amputation in a proportion of patients. To improve therapy, an increase in our understanding of the pathobiology of diabetic complications such as impaired wound healing is necessary. In this review, recent advances in molecular aspects of normal and impaired diabetic wound healing are discussed. Furthermore, investigations of the role of epigenetic processes in the pathogenesis of impaired diabetic wound healing are now emerging. Indeed, epigenetic changes have already been identified as key factors in diabetes and related complications and these are overviewed in this review.
Keywords: Diabetes, Diabetic ulcer, Epigenetics, Wound healing, Wounds
INTRODUCTION
Diabetes mellitus is one of the most common chronic metabolic disorders, with worldwide disease prevalence expected to rise further because of aging populations, an increase in obesity and physical inactivity, and the provision of improved health care and longevity for diabetic patients 1, 2. It has been estimated that there will be 285 million adults with diabetes in the year 2010, representing a global prevalence of 6·4%, which is expected to increase to 7·7% by the year 2030, to 439 million (2).
Diabetes is a group of heterogeneous metabolic disorders that arise as a result of underlying hyperglycaemia caused by defects in either insulin secretion and/or action 3, 4. This includes type 1 diabetes, most commonly diagnosed in childhood, and results from the destruction of pancreatic, insulin producing β‐cells. The most common form, type 2 diabetes, which occurs mostly in adults, is caused by the insulin resistance of peripheral tissues. All types of diabetes, if left untreated, can result in serious long‐term complications, including cardiovascular diseases, nephropathy, retinopathy, neuropathy and the formation of diabetic foot ulcers 5, 6, 7, 8.
Numerous molecular changes have been associated with diabetic complications. More recently, epigenetic events – changes in gene transcription or phenotype that are not the result of changes in the underlying DNA sequence – which are relatively well characterised in cancer, have been implicated in diabetes 9, 10. A number of recent studies have shown that epigenetic changes play a role in the pathogenesis of diabetic complications 11, 12, 13. For example, with respect to the formation of atherosclerotic plaques, some studies have found dysregulation of tri‐methylation of lysine residue 9 in histone H3 (H3K9me3) and dimethylation of lysine 4 on histone H3 (H3K4me2) in vascular smooth muscle cells (VSMC) in diabetic mice at genes encoding for various inflammatory compounds such as interleukin‐6 (IL‐6), macrophage colony stimulating factor‐1 (MCSF) (14). These results were also observed in human vascular smooth muscle cells (HVSMC) after incubation with high glucose levels. In addition, in diabetic nephropathy, it has been shown that elevated levels in the acetylation of H3K9 and H3K23, dimethylation of H3K4 and H3 phosphorylation at serine 10 were associated with increased severity of induced glomerulosclerosis in diabetic mice compared with non diabetic mice (15). Histone deacetylase 2 (HDAC‐2) was also found to promote extracellular matrix accumulation in renal disease (16). Together, these highlight the important role that epigenetic changes may play in the pathology of diabetic complications. The aim of this review is to discuss the genetic and epigenetic changes in the pathogenesis of diabetic wound healing and the formation of chronic diabetic foot ulcers.
NORMAL WOUND HEALING
Normal wound healing is essential for the replacement of lost tissue and the restoration of the tissue to a functional state. For example, healing of a cutaneous wound is essential for restoring the protective barrier provided by skin to external elements. Although superficial wounds may be healed by the process of regeneration, where injured or necrotic tissue is replaced by cells of the same type, larger wounds cannot be healed in this way and results in the replacement of functional tissue with connective tissue that continues to remodel and develop for many years after the insult.
Generally, wound healing involves three main phases: acute inflammation, proliferation and remodelling, resulting in the formation of a scar 17, 18. The first step involved in wound healing is the formation of a blood clot by thrombosis. The clot consists primarily of plasma fibrin as well as aggregated platelet cells 19, 20. This thrombotic clot is essential as it protects the wound from infection or exposure to exogenous substances, as well as preventing further blood loss and the loss of fluids essential for the healing process. The clot is eventually degraded by proteolysis and replaced by new epithelium. Platelet cells facilitate fibrin deposition and release platelet‐derived growth factor (PDGF), which along with the IL‐1 released by damaged keratinocytes (21), helps trigger the acute inflammatory response (22).
The acute inflammatory response is involved in the breakdown and removal of necrotic or injured tissue, as well as preventing infection of the wound (23). Vasodilation, which is triggered by the release of histamines, kinins and prostaglandins, is required for recruitment of cells from the bloodstream, such as neutrophils, to migrate into the injured tissue. Neutrophils are the first inflammatory cells to migrate to the site of injury and help to degrade necrotic tissue and infectious agents, followed by macrophages which phagocytose foreign and cellular debris, including neutrophils and the fibrin clot (24).
Macrophages release many chemokines and chemoattractants that assist in the formation of granulation tissue, which forms part of the proliferative phase of wound healing. This involves the replacement of the fibrin clot with a temporary, highly vascular tissue containing many different cell types and a provisional extracellular matrix (25). As part of the proliferative phase, new blood vessels are formed in the tissue by angiogenesis, a process essential for the delivery of oxygen and nutrients to the highly proliferative granulation tissue 26, 27. This process is triggered by the release of cytokines and growth factors produced by inflammatory cells, keratinocytes and fibroblasts including fibroblast growth factor (FGF) and vascular endothelial growth factor (VEGF). Reepithelialisation by keratinocytes also occurs in this phase, and involves the formation of a new epithelium to seal a wound and replace the fibrin clot. This process is triggered by endothelial growth factor (EGF) and transforming growth factor‐α (TGF‐α) produced by macrophages and keratinocytes. An essential component of granulation tissue are fibroblasts which differentiate into myofibroblasts (28). Myofibroblasts contribute to the strength and structural integrity of the wound. Furthermore, contraction of the myofibroblasts helps contract the wound (29). They are also one of the main secretors of extracellular matrix components such as collagen.
The final phase of wound healing is the remodelling phase. This involves the contraction of the wound, mediated by the myofibroblasts (30), and reorganisation of the extracellular matrix to provide more strength. By this stage, most of the new blood vessels would have undergone apoptosis, and the area develops into an avascular and acellular scar. This process may take many years to complete, depending on the type and size of the wound (31).
In contrast to an acute wound, a chronic wound does not follow the orderly process of wound healing, and may take considerably longer to heal, or may not heal at all. Chronic wounds differ from acute wounds at the molecular level; there is a difference in the expression of growth factors, cytokines and other proteins that help regulate and control the wound healing process. There are multiple types of chronic cutaneous wounds with many different causes, which usually take the form of ulcers, including pressure wounds, fungal wounds, ulcers resulting from venous diseases and diabetic foot ulcers. With adequate treatment, some ulcers may last only weeks; however, many ulcers are difficult to treat and may last months, in certain cases years.
CHANGES IN GENE EXPRESSION
Gene expression profiling has shown that even immediately after injury to normal skin, there is a change in gene expression. It has been shown that 3% of the 4000 genes studied were upregulated within 30 minutes of injury, and that these genes were mainly related to signalling and transcription (32). Furthermore, the findings from one particular study which investigated wound healing in human patients with basal cell carcinoma identified many gene expression changes compared with the pre‐injury state. Importantly it was shown that although there was an increase in the pro‐inflammatory genes early after a cutaneous punch biopsy wound was performed (2 days), there was also upregulation of some, mostly pro‐inflammatory genes for up to 8 days. This included genes encoding for caspases, collagen and hypoxia inducible factor (HIF). Upregulation of repair, angiogenic and remodelling genes such as genes encoding for matrix metalloprotease (MMP)‐9, granulin and type IV collagen were observed at later stages in the repair process (4–8 days), which is consistent with the delayed onset of these processes. It was also shown from the profiles of genes related to macrophage phenotypes that although there was a mixture of both M1 and M2 macrophages in the early phase of the repair process, in the later phases of repair there was a predominant expression of genes related to M2 macrophages. Unlike M1 macrophages, which release mainly pro‐inflammatory cytokines and are involved in killing microorganisms and tumour cells, M2 macrophages help to resolve the inflammatory response and contribute to angiogenesis and tissue remodelling (33). Other studies have also confirmed some of these findings using murine models of wound healing, including the increased gene expression of HIF and MMP‐9 34, 35. Furthermore, a separate study showed an upregulation of angiogenesis‐related genes in the late phase of repair, as well as upregulation of pro‐inflammatory genes in the early phase, and continued gene expression of inflammatory genes in later phases (34).
EPIGENETIC PROCESSES IN NORMAL WOUND HEALING
Furthermore, it has been shown that epigenetic changes play an important role in regulating normal wound healing. For example, it has been shown that knocking out dicer, an essential enzyme for producing microRNA, from endothelial cells, results in impaired angiogensis (Figure 1) (36). This result has also been replicated in vitro with human endothelial cells, where the knockout of dicer resulted in impaired ability of the cells to form blood vessels (37). This indicates that epigenetic changes are crucial for the regulation of angiogenesis, and by extension wound healing.
Figure 1.
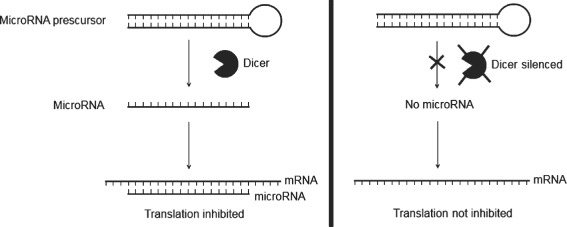
Dicer, a cytoplasmic RNaseIII enzyme essential for the formation of microRNAs, is involved in cleaving larger, hairpin‐shaped microRNA precursors into smaller, single‐stranded segments, which are typically 19–25 nucleotides in length. MicroRNAs inhibit translation of mRNA by binding to complementary sequences. It has been shown that the silencing of dicer results in impaired angiogenesis, possibly because of the dysregulation of anti‐angiogenesis genes such as TSP‐1.
Other epigenetic mechanisms, particularly histone and DNA methylation, have also been recently shown to play a role in the wound healing process (Figure 2). It has been shown that trimethylation of H3K27 (H3K27me3) was reduced in the wound edge in the epidermis in a mouse model (38). Trimethylation of H3K27 is known to repress gene expression. Furthermore, it was shown that the H3K27‐specific lysine demethylases Jmjd3 and Utx are upregulated during murine wound healing, while the components of the polycomb repressive complex 2 (PRC2) Eed, Ezh2 and Suz12, which methylate H3K27, were downregulated. It was shown that there is reduced Eed protein associated with the regulatory regions of the genes Egfr and Myc, which are two genes that are potential targets of polycomb group proteins and known to be upregulated during the repair process for which they are essential. This indicates that reduction in the methylation of H3K27 may be involved in the activation of genes required for the wound healing process.
Figure 2.
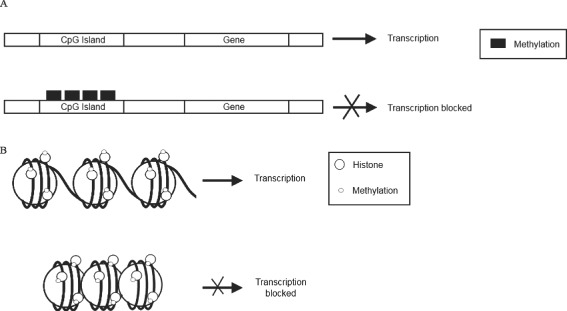
DNA and histone methylation status is important in the regulation of gene transcription during wound healing. The methylation of cytosine and guanine islands (CpG) residues, located in the gene promoter region, results in repressed gene transcription (A). Histone methylation can result in either gene expression or repression, depending on which histone residue has been methylated (B). For example, mono‐, di‐ or trimethylation of histone 3 on lysine residue 4 (H3K4) confers a transcriptionally active chromatin state. An example of this is H3K4me. In contrast, other histone methylation events, such as H3K9me and trimethylation of H3K27 (H3K27me3) are correlated with transcriptionally inactive chromatin.
Another study has investigated the role of DNA methylation in the transdifferentiation of hepatic stellate cells (HSC) to myofibroblasts (39). The study found that the addition of 5‐aza‐2′‐deoxycytidine (5‐azadC) to cultured HSC‐suppressed myofibroblast transdifferentiation in vitro. The epigenetic modifier, 5‐azadC is an inhibitor of DNA methytransferase‐1 (DNMT1), and therefore an inhibitor of DNA methylation. It was found that several days after the removal of the drug from the cultured cells, the cells began to show the initial morphological changes associated with myofibroblast transdifferentiation. Furthermore, it was shown that the transcription factors peroxisome proliferator‐activated receptor‐γ (PPARγ) and IκBα, which regulate the fibrogenic and pro‐inflammatory characteristics of myofibroblasts, respectively, were also downregulated, indicating that DNA methylation is also involved in the control of these genes. As transdifferentiation to produce myofibroblasts is a common feature in wound healing in various different types of tissue, it is possible that this epigenetic mechanism may also be involved in the differentiation of skin fibroblasts into myofibroblasts during cutaneous wound healing.
IMPAIRED WOUND HEALING IN DIABETES
Diabetic foot ulcers occur in up to 15% of patients with diabetes and occur as a result of impaired wound healing. The ulcers are chronic wounds that either do not heal or take many weeks or months to heal. In some cases, infection of the ulcer may occur, resulting in the possibility of limb amputation because of the risk of sepsis (40). As with other forms of diabetic complications, impaired wound healing is usually a result of macro‐ and micro‐angiopathy, as well as neuropathy (nerve damage that may occur in people with diabetes) 41, 42, 43. There is no single cause of impaired wound healing and defects or dysregulation are observed at both the cellular and molecular levels, and in all phases of the wound healing process.
The defects in diabetic wound healing include impairment of the inflammatory response, extra‐cellular matrix, macrophage function, angiogenesis, reepithelialisation, keratinocyte and fibroblast migration and proliferation and cytokine, chemokine and growth factor production 44, 45, 46. For example, macrophages have been shown to have impaired phagocytic function in diabetic patients (47). They play an integral role in the inflammatory phase of wound healing by clearing up necrotic tissue, without which repair is not properly initiated. Macrophages isolated from the wounds of diabetic mice have been shown to have a reduced ability of clearing necrotic tissue (46). This, along with other factors such as the dysregulation of cytokine and growth factor levels, may prolong the inflammatory phase, which in turn hinders the progression of subsequent phases of wound healing (48).
Keratinocytes at the wound site present impaired abilities to migrate, proliferate and differentiate. One possible reason for this is the stabilisation of β‐catenin, which blocks the EGF response and represses important cytoskeletal components in keratinocytes, thereby inhibiting migration (49). Furthermore, keratinocytes, as well as epithelial cells, fail to upregulate the expression of growth factors, including those which promote angiogenesis and immune cell chemoattractants, thus contributing to the impaired healing.
Endothelial progenitor cells (EPCs) are requited from the basement membrane as a response to injury and are homed to the site of injury to mediate neovasculogenisis (50). This is initiated by VEGF, which is released by cells such as fibroblasts, macrophages and epithelial cells at the site of injury. This triggers the activation of the eNOS in the bone marrow, to produce nitric oxide which is required to trigger the migration of EPCs into the bloodstream (51). The EPCs are then honed to the site of injury by the cytokine SDF‐1α. People with type 1 and type 2 diabetes can have decreased levels of EPCs, and the existing cells may have impaired function and diminished ability to migrate to the site of injury 52, 53, 54, 55, 56. Using a mouse model, it has been shown that this process is impaired in diabetes because of impairment in the phosphorylation of eNOS, and therefore the migration of the EPC into the bloodstream (57). The study also showed that decreased production of SDF‐1α from myofibroblasts and epithelial cells impaired recruitment of the EPCs to the site of injury.
DYSREGULATION OF GROWTH FACTORS AND CYTOKINES
Changes in the production of growth factors and cytokines have also been shown to have a negative effect on wound healing in diabetic patients (Table 1). These changes can affect the function of multiple cell types. For example, the lack of reepithelialisation may be linked to the decreased expression of insulin‐like growth factor‐1 (IGF‐1), which is decreased in both human diabetic skin and in diabetic mice (58). IGF‐1 has multiple roles, including the promotion of reepithelialisation, keratinocyte and fibroblast proliferation and the induction of endothelial cell chemotaxis 59, 60. Studies in mice have shown that IGF‐1 accelerates wound healing in diabetic animal models 61, 62. Numerous studies have showed an increased level of proteases in chronic diabetic wounds, including matrix metalloproteases such as MMP‐2, MMP‐8 and MMP‐9, along with reduced levels of MMP inhibitors (63). The increased level of proteases appears to be stimulated by a prolonged inflammatory response and by factors such as tumour necrosis factor (TNF)‐α and IL‐1. The increase in protease levels prolongs the healing process because of the destruction of proteins and growth factors that are required for normal healing (64).
Table 1.
Changes in expression of cytokines and growth factors in diabetic wound healing
| Cytokine and growth factors | Normal role in wound healing | Expression In diabetic wound healing | Reference |
|---|---|---|---|
| IGF‐1 | Promotion of reepithelialisation | Decreased | 58, 59, 60 |
| Keratinocyte and fibroblast proliferation | |||
| Endothelial cell activation | |||
| TGF‐β1 | Chemoattractant (keratinocytes, fibroblasts, inflamamtory cells) | Decreased | 62, 65, 66, 67, 68 |
| ECM deposition | |||
| Promotes angiogenesis | |||
| PDGF | Fibroblast activation | Decreased | (69) |
| Promotes angiogenesis | |||
| ECM deposition | |||
| MMP synthesis | |||
| EGF | ECM deposition | Decreased | (70) |
| Keratinocyte migration and proliferation | |||
| IL‐8 | Keratinocyte proliferation | Decreased | (71) |
| Mactrophage chemotaxis | |||
| Neutrophil chemotaxis | |||
| Angiopoietin‐2 | Disrupts blood vessel formation | Increased | (72) |
EGF, endothelial growth factor; IGF‐1, insulin‐like growth factor‐1; IL‐8, interleukin‐8; MMP, matrix metalloprotease; PDGF, platelet‐derived growth factor; TGF‐β1, transforming growth factor‐β1.
Many other cytokines and growth factors that are involved in stimulating epithelial cells are also downregulated in the foot ulcers of diabetic patients, including PDGF, IL‐8, IL‐10 and TGF‐β1. In fact, TGF‐β1 is a growth factor involved in cutaneous wound healing which affects different cell types involved in the various phases of wound healing 66, 67. The actions of TGF‐β1 include chemoattraction of multiple cell types including inflammatory cells, keratinocytes and fibroblasts, stimulation of angiogenesis and promoting the formation of extracellular matrix (ECM). TGF‐β1 has been shown to be decreased in the skin and foot ulcers of diabetic patients and in diabetic rats 68, 73. Topical application of TGF‐β1 has been shown to accelerate wound healing in diabetic rats (65).
In addition to TGF‐β1, various cytokines involved in the promotion of angiogenesis are downregulated in diabetic foot ulcers, including PDGF, IGF‐1, EGF and IL‐8 (71). Angiogenesis is a process essential for the delivery of nutrients and oxygen to the wound. In one particular study, the expression of various cytokines, growth factors and transcription factors which are involved in angiogenesis in diabetic mice compared with non diabetic mice has been compared (35). Differential regulation was observed for multiple genes, including the genes encoding for angiopoietin 2, HIF‐1α and osteopontin. Expression of angiopoietin 2, which has been shown to disrupt blood vessel formation and is known to be elevated in diabetic wounds, was shown to be increased early post‐injury in diabetic mice compared with non diabetic mice (72). This correlates with the delayed vascularisation observed in the diabetic wound. While HIF‐1α was expressed early in both diabetic and non diabetic mice, gene expression levels eventually dropped by day 11 post injury in non diabetic mice but not in diabetic mice. Furthermore, the increased expression of esteopontin was shown to be positively correlated with the formation of new blood vessels in both diabetic and non diabetic mice. The delayed expression of esteopontin in diabetic mice correlated with the delayed angiogenesis observed.
Experiments with diabetic rabbits have shown that the baseline levels of gene expression of cytokines IL‐8 and IL‐6 as well as their receptors in the skin were increased compared with normal rabbits, which is consistent with previous reports of chronic low‐grade inflammation caused by diabetes 63, 71, 74. However, unlike in non diabetic rabbits, it was found that there was no significant increase in gene expression of IL‐8 and IL‐6 and their receptors post‐injury. Given the possibility that non immune cells may be the source of the increased baseline cytokine production, it was suggested that the lack of increased gene expression may be a result of impaired or insufficient immune cell infiltration at the wound site, or because of immune cell dysfunction.
EPIGENETIC CHANGES IN DIABETIC WOUND HEALING
In addition to changes in gene expression, epigenetic changes also play a role in impaired diabetic wound healing. As mentioned earlier, microRNAs have been shown to be essential for angiogenesis and, by extension, wound healing. The role microRNAs might play in impaired diabetic wound healing has been investigated in numerous murine models. For example, one study found that the enzyme dicer was decreased by more than 40% in diabetic mice (type 2 diabetes, db/db) compared with non diabetic mice (db/+) (75). As a result, it was found that the microRNA miR‐27b expression was decreased by greater than 66% in diabetic mice. Conversely, an anti‐angiogenetic molecule, thrombospondin‐1 (TSP‐1), was found to be significantly unregulated in diabetic mice compared with non diabetic mice. However, transfection of a miR‐27b mimic reduced the levels of TSP‐1 in diabetic mice and promoted angiogenesis. Angiogenesis was also promoted by the silencing of TSP‐1. These results were also observed when miR‐27b was silenced in normal endothelial cells.
A similar effect with a different microRNA has also been observed in mice with type 1 diabetes. MicroRNA let7‐f was shown to be downregulated in type 1 diabetic mice compared with control mice, and in vitro transfection of diabetic EPC was shown to improve angiogenesis (76). This improvement was shown to be the result of activation of AMP‐activated protein kinase (AMPK), which improves endothelial function and angiogenesis in diabetes, and the induction of mitochondrial superoxide dismutase, which assists in the reducing oxidative stress.
Another way in which epigenetic changes may influence wound healing is by impaired keratinocyte proliferation. Some chronic foot ulcers are also ischaemic and are linked with higher mortality and amputation rates (77). Using a mouse model it has been shown that microRNA‐210 may play a role in wound impairment in ischaemic chronic ulcers (78). MicroRNA‐210 is transcriptionally regulated by HIF‐1α. The study showed that in the ischaemic model, HIF‐1α induces expression of microRNA‐210. One of the targets of miR‐210 is the gene encoding for transcription factor E2F3 (79). E2F3 is an important component of wound healing and has been shown to promote keratinocyte proliferation (80). In this study it was shown that HIF‐1α stabilisation resulted in expression of miR‐210, which then silenced the expression of E2F3. Thus it appears that epigenetic changes in keratinocytes as a result of miR‐210 may result in reduced proliferation and therefore impaired reepithelialisation of the wound.
CONCLUSION
In conclusion, diabetic foot ulcers are, and are expected to continue to be, a serious life‐changing complication of diabetes. While treatment options are available, they are suboptimal for a proportion of patients who may eventually require an amputation. Presently, there is an intense research effort aimed at developing novel therapeutic strategies for diabetic ulcers. This is tightly linked with investigations into the changes in wound healing processes as a consequence of diabetes. One research area which has only recently emerged is the investigation of the role of epigenetic changes in normal wound healing and impaired diabetic wound healing. The area of epigenetics, which has already proven to be of great importance in other diseases such as cancer, has the potential to offer not only an increased understanding of the underlying pathobiology of diabetic foot ulcers but also to provide new directions for therapeutic intervention. It is anticipated that research in this direction will intensify, culminating in the development and evaluation of new epigenetic therapies.
ACKNOWLEDGEMENTS
The support of the Australian Institute of Nuclear Science and Engineering is acknowledged. T.C.K was the recipient of AINSE awards. Epigenomic Medicine Lab is supported by the National Health and Medical Research Council of Australia (566559). H.R. is supported by an Australian post‐graduate and BakerIDI bright spark awards.
REFERENCES
- 1. Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care 2004;27:1047–53. [DOI] [PubMed] [Google Scholar]
- 2. Shaw JE, Sicree RA, Zimmet PZ. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res Clin Pract 2010;87:4–14. [DOI] [PubMed] [Google Scholar]
- 3. Choi K, Kim YB. Molecular mechanism of insulin resistance in obesity and type 2 diabetes. Korean J Intern Med 2010;25:119–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jones KL. The “other” forms of diabetes in children. Adolesc Med State Art Rev 2010;21:120–8, x. [PubMed] [Google Scholar]
- 5. Creager MA, Luscher TF, Cosentino F, Beckman JA. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: Part I. Circulation 2003;108:1527–32. [DOI] [PubMed] [Google Scholar]
- 6. Kollias AN, Ulbig MW. Diabetic retinopathy: early diagnosis and effective treatment. Dtsch Arztebl Int 2010;107:75–83. quiz 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schmieder RE, Martin S, Lang GE, Bramlage P, Bohm M. Angiotensin blockade to reduce microvascular damage in diabetes mellitus. Dtsch Arztebl Int 2009;106:556–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sego S. Pathophysiology of diabetic nephropathy. Nephrol Nurs J 2007;34:631–3. [PubMed] [Google Scholar]
- 9. Santos‐Reboucas CB, Pimentel MM. Implication of abnormal epigenetic patterns for human diseases. Eur J Hum Genet 2007;15:10–7. [DOI] [PubMed] [Google Scholar]
- 10. Cooney CA. Epigenetics – DNA‐based mirror of our environment? Dis Markers 2007;23:121–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ling C, Groop L. Epigenetics: a molecular link between environmental factors and type 2 diabetes. Diabetes 2009;58:2718–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Soldatos G, Cooper ME. Diabetic nephropathy: important pathophysiologic mechanisms. Diabetes Res Clin Pract 2008;82 Suppl 1:S75–9. [DOI] [PubMed] [Google Scholar]
- 13. Gray SG, De Meyts P. Role of histone and transcription factor acetylation in diabetes pathogenesis. Diabetes Metab Res Rev 2005;21:416–33. [DOI] [PubMed] [Google Scholar]
- 14. Villeneuve LM, Reddy MA, Lanting LL, Wang M, Meng L, Natarajan R. Epigenetic histone H3 lysine 9 methylation in metabolic memory and inflammatory phenotype of vascular smooth muscle cells in diabetes. Proc Natl Acad Sci U S A 2008;105:9047–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sayyed SG, Gaikwad AB, Lichtnekert J, Kulkarni O, Eulberg D, Klussmann S, Tikoo K, Anders HJ. Progressive glomerulosclerosis in type 2 diabetes is associated with renal histone H3K9 and H3K23 acetylation, H3K4 dimethylation and phosphorylation at serine 10. Nephrol Dial Transplant 2010;25:1811–7. [DOI] [PubMed] [Google Scholar]
- 16. Noh H, Oh EY, Seo JY, Yu MR, Kim YO, Ha H, Lee HB. Histone deacetylase‐2 is a key regulator of diabetes‐ and transforming growth factor‐beta1‐induced renal injury. Am J Physiol Renal Physiol 2009;297:F729–39. [DOI] [PubMed] [Google Scholar]
- 17. Watkins SA, Zippin JH. When wound healing goes awry: a review of normal and abnormal wound healing, scar pathophysiology, and therapeutics. J Drugs Dermatol 2008;7:997–1005. [PubMed] [Google Scholar]
- 18. Shaw TJ, Martin P. Wound repair at a glance. J Cell Sci 2009;122:Pt 18:3209–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Laurens N, Koolwijk P, de Maat MP. Fibrin structure and wound healing. J Thromb Haemost 2006;4:932–9. [DOI] [PubMed] [Google Scholar]
- 20. Nurden AT, Nurden P, Sanchez M, Andia I, Anitua E. Platelets and wound healing. Front Biosci 2008;13:3532–48. [DOI] [PubMed] [Google Scholar]
- 21. Barrientos S, Stojadinovic O, Golinko MS, Brem H, Tomic‐Canic M. Growth factors and cytokines in wound healing. Wound Repair Regen 2008;16:585–601. [DOI] [PubMed] [Google Scholar]
- 22. Bahou WF, Gnatenko DV. Platelet transcriptome: the application of microarray analysis to platelets. Semin Thromb Hemost 2004;30:473–84. [DOI] [PubMed] [Google Scholar]
- 23. Eming SA, Krieg T, Davidson JM. Inflammation in wound repair: molecular and cellular mechanisms. J Invest Dermatol 2007;127:514–25. [DOI] [PubMed] [Google Scholar]
- 24. Kim MH, Liu W, Borjesson DL, Curry FR, Miller LS, Cheung AL, Liu FT, Isseroff RR, Simon SI. Dynamics of neutrophil infiltration during cutaneous wound healing and infection using fluorescence imaging. J Invest Dermatol 2008;128: 1812–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Adamson R. Role of macrophages in normal wound healing: an overview. J Wound Care 2009;18:349–51. [DOI] [PubMed] [Google Scholar]
- 26. Adams RH, Alitalo K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol 2007;8:464–78. [DOI] [PubMed] [Google Scholar]
- 27. Tonnesen MG, Feng X, Clark RA. Angiogenesis in wound healing. J Investig Dermatol Symp Proc 2000;5:40–6. [DOI] [PubMed] [Google Scholar]
- 28. Hinz B. Formation and function of the myofibroblast during tissue repair. J Invest Dermatol 2007;127:526–37. [DOI] [PubMed] [Google Scholar]
- 29. Darby IA, Hewitson TD. Fibroblast differentiation in wound healing and fibrosis. Int Rev Cytol 2007;257:143–79. [DOI] [PubMed] [Google Scholar]
- 30. Desmouliere A, Chaponnier C, Gabbiani G. Tissue repair, contraction, and the myofibroblast. Wound Repair Regen 2005;13:7–12. [DOI] [PubMed] [Google Scholar]
- 31. Deonarine K, Panelli MC, Stashower ME, Jin P, Smith K, Slade HB, Norwood C, Wang E, Marincola FM, Stroncek DF. Gene expression profiling of cutaneous wound healing. J Transl Med 2007;5:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cole J, Tsou R, Wallace K, Gibran N, Isik F. Early gene expression profile of human skin to injury using high‐density cDNA microarrays. Wound Repair Regen 2001;9:360–70. [DOI] [PubMed] [Google Scholar]
- 33. Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci 2008;13:453–61. [DOI] [PubMed] [Google Scholar]
- 34. Roy S, Khanna S, Rink C, Biswas S, Sen CK. Characterization of the acute temporal changes in excisional murine cutaneous wound inflammation by screening of the wound‐edge transcriptome. Physiol Genomics 2008;34:162–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sharma A, Singh AK, Warren J, Thangapazham RL, Maheshwari RK. Differential regulation of angiogenic genes in diabetic wound healing. J Invest Dermatol 2006;126:2323–31. [DOI] [PubMed] [Google Scholar]
- 36. Suarez Y, Fernandez‐Hernando C, Yu J, Gerber SA, Harrison KD, Pober JS, Iruela‐Arispe ML, Merkenschlager M, Sessa WC. Dicer‐dependent endothelial microRNAs are necessary for postnatal angiogenesis. Proc Natl Acad Sci U S A 2008;105:14082–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kuehbacher A, Urbich C, Zeiher AM, Dimmeler S. Role of Dicer and Drosha for endothelial microRNA expression and angiogenesis. Circ Res 2007;101:59–68. [DOI] [PubMed] [Google Scholar]
- 38. Shaw T, Martin P. Epigenetic reprogramming during wound healing: loss of polycomb‐mediated silencing may enable upregulation of repair genes. EMBO Rep 2009;10:881–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mann J, Oakley F, Akiboye F, Elsharkawy A, Thorne AW, Mann DA. Regulation of myofibroblast transdifferentiation by DNA methylation and MeCP2: implications for wound healing and fibrogenesis. Cell Death Differ 2007;14:275–85. [DOI] [PubMed] [Google Scholar]
- 40. Bartus CL, Margolis DJ. Reducing the incidence of foot ulceration and amputation in diabetes. Curr Diab Rep 2004;4:413–8. [DOI] [PubMed] [Google Scholar]
- 41. La Fontaine J, Harkless LB, Davis CE, Allen MA, Shireman PK. Current concepts in diabetic microvascular dysfunction. J Am Podiatr Med Assoc 2006;96:245–52. [DOI] [PubMed] [Google Scholar]
- 42. Ngo BT, Hayes KD, DiMiao DJ, Srinivasan SK, Huerter CJ, Rendell MS. Manifestations of cutaneous diabetic microangiopathy. Am J Clin Dermatol 2005;6:225–37. [DOI] [PubMed] [Google Scholar]
- 43. Reiber GE, Vileikyte L, Boyko EJ, del Aguila M, Smith DG, Lavery LA, Boulton AJ. Causal pathways for incident lower‐extremity ulcers in patients with diabetes from two settings. Diabetes Care 1999;22:157–62. [DOI] [PubMed] [Google Scholar]
- 44. Lerman OZ, Galiano RD, Armour M, Levine JP, Gurtner GC. Cellular dysfunction in the diabetic fibroblast: impairment in migration, vascular endothelial growth factor production, and response to hypoxia. Am J Pathol 2003;162:303–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Galkowska H, Wojewodzka U, Olszewski WL. Chemokines, cytokines, and growth factors in keratinocytes and dermal endothelial cells in the margin of chronic diabetic foot ulcers. Wound Repair Regen 2006;14:558–65. [DOI] [PubMed] [Google Scholar]
- 46. Khanna S, Biswas S, Shang Y, Collard E, Azad A, Kauh C, Bhasker V, Gordillo GM, Sen CK, Roy S. Macrophage dysfunction impairs resolution of inflammation in the wounds of diabetic mice. PLoS One 2010;5:e9539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liu BF, Miyata S, Kojima H, Uriuhara A, Kusunoki H, Suzuki K, Kasuga M. Low phagocytic activity of resident peritoneal macrophages in diabetic mice: relevance to the formation of advanced glycation end products. Diabetes 1999;48:2074–82. [DOI] [PubMed] [Google Scholar]
- 48. Blakytny R, Jude EB. Altered molecular mechanisms of diabetic foot ulcers. Int J Low Extrem Wounds 2009;8:95–104. [DOI] [PubMed] [Google Scholar]
- 49. Stojadinovic O, Brem H, Vouthounis C, Lee B, Fallon J, Stallcup M, Merchant A, Galiano RD, Tomic‐Canic M. Molecular pathogenesis of chronic wounds: the role of beta‐catenin and c‐myc in the inhibition of epithelialization and wound healing. Am J Pathol 2005;167:59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gill M, Dias S, Hattori K, Rivera ML, Hicklin D, Witte L, Girardi L, Yurt R, Himel H, Rafii S. Vascular trauma induces rapid but transient mobilization of VEGFR2(+)AC133(+) endothelial precursor cells. Circ Res 2001;88:167–74. [DOI] [PubMed] [Google Scholar]
- 51. Abidia A, Laden G, Kuhan G, Johnson BF, Wilkinson AR, Renwick PM, Masson EA, McCollum PT. The role of hyperbaric oxygen therapy in ischaemic diabetic lower extremity ulcers: a double‐blind randomised‐controlled trial. Eur J Vasc Endovasc Surg 2003;25:513–8. [DOI] [PubMed] [Google Scholar]
- 52. Capla JM, Grogan RH, Callaghan MJ, Galiano RD, Tepper OM, Ceradini DJ, Gurtner GC. Diabetes impairs endothelial progenitor cell‐mediated blood vessel formation in response to hypoxia. Plast Reconstr Surg 2007;119:59–70. [DOI] [PubMed] [Google Scholar]
- 53. Tepper OM, Galiano RD, Capla JM, Kalka C, Gagne PJ, Jacobowitz GR, Levine JP, Gurtner GC. Human endothelial progenitor cells from type II diabetics exhibit impaired proliferation, adhesion, and incorporation into vascular structures. Circulation 2002;106:2781–6. [DOI] [PubMed] [Google Scholar]
- 54. Loomans CJ, de Koning EJ, Staal FJ, Rookmaaker MB, Verseyden C, de Boer HC, Verhaar MC, Braam B, Rabelink TJ, van Zonneveld AJ. Endothelial progenitor cell dysfunction: a novel concept in the pathogenesis of vascular complications of type 1 diabetes. Diabetes 2004;53:195–9. [DOI] [PubMed] [Google Scholar]
- 55. Ii M, Takenaka H, Asai J, Ibusuki K, Mizukami Y, Maruyama K, Yoon YS, Wecker A, Luedemann C, Eaton E, Silver M, Thorne T, Losordo DW. Endothelial progenitor thrombospondin‐1 mediates diabetes‐induced delay in reendothelialization following arterial injury. Circ Res 2006;98:697–704. [DOI] [PubMed] [Google Scholar]
- 56. Thum T, Fraccarollo D, Schultheiss M, Froese S, Galuppo P, Widder JD, Tsikas D, Ertl G, Bauersachs J. Endothelial nitric oxide synthase uncoupling impairs endothelial progenitor cell mobilization and function in diabetes. Diabetes 2007;56:666–74. [DOI] [PubMed] [Google Scholar]
- 57. Gallagher KA, Liu ZJ, Xiao M, Chen H, Goldstein LJ, Buerk DG, Nedeau A, Thom SR, Velazquez OC. Diabetic impairments in NO‐mediated endothelial progenitor cell mobilization and homing are reversed by hyperoxia and SDF‐1 alpha. J Clin Invest 2007;117:1249–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Blakytny R, Jude EB, Martin Gibson J, Boulton AJ, Ferguson MW. Lack of insulin‐like growth factor 1 (IGF1) in the basal keratinocyte layer of diabetic skin and diabetic foot ulcers. J Pathol 2000;190:589–94. [DOI] [PubMed] [Google Scholar]
- 59. Bhora FY, Dunkin BJ, Batzri S, Aly HM, Bass BL, Sidawy AN, Harmon JW. Effect of growth factors on cell proliferation and epithelialization in human skin. J Surg Res 1995;59:236–44. [DOI] [PubMed] [Google Scholar]
- 60. Grant M, Jerdan J, Merimee TJ. Insulin‐like growth factor‐I modulates endothelial cell chemotaxis. J Clin Endocrinol Metab 1987;65:370–1. [DOI] [PubMed] [Google Scholar]
- 61. Tsuboi R, Shi CM, Sato C, Cox GN, Ogawa H. Co‐administration of insulin‐like growth factor (IGF)‐I and IGF‐binding protein‐1 stimulates wound healing in animal models. J Invest Dermatol 1995;104:199–203. [DOI] [PubMed] [Google Scholar]
- 62. Bitar MS. Insulin‐like growth factor‐1 reverses diabetes‐induced wound healing impairment in rats. Horm Metab Res 1997;29:383–6. [DOI] [PubMed] [Google Scholar]
- 63. Stehouwer CD, Gall MA, Twisk JW, Knudsen E, Emeis JJ, Parving HH. Increased urinary albumin excretion, endothelial dysfunction, and chronic low‐grade inflammation in type 2 diabetes: progressive, interrelated, and independently associated with risk of death. Diabetes 2002;51:1157–65. [DOI] [PubMed] [Google Scholar]
- 64. Yager DR, Zhang LY, Liang HX, Diegelmann RF, Cohen IK. Wound fluids from human pressure ulcers contain elevated matrix metalloproteinase levels and activity compared to surgical wound fluids. J Invest Dermatol 1996;107:743–8. [DOI] [PubMed] [Google Scholar]
- 65. Broadley KN, Aquino AM, Hicks B, Ditesheim JA, McGee GS, Demetriou AA, Woodward SC, Davidson JM. Growth factors bFGF and TGB beta accelerate the rate of wound repair in normal and in diabetic rats. Int J Tissue React 1988;10:345–53. [PubMed] [Google Scholar]
- 66. Klass BR, Grobbelaar AO, Rolfe KJ. Transforming growth factor beta1 signalling, wound healing and repair: a multifunctional cytokine with clinical implications for wound repair, a delicate balance. Postgrad Med J 2009;85:9–14. [DOI] [PubMed] [Google Scholar]
- 67. Wang XJ, Han G, Owens P, Siddiqui Y, Li AG. Role of TGF beta‐mediated inflammation in cutaneous wound healing. J Investig Dermatol Symp Proc 2006;11:112–7. [DOI] [PubMed] [Google Scholar]
- 68. Wysocki J, Wierusz‐Wysocka B, Wykretowicz A, Wysocki H. The influence of thymus extracts on the chemotaxis of polymorphonuclear neutrophils (PMN) from patients with insulin‐dependent diabetes mellitus (IDD). Thymus 1992;20:63–7. [PubMed] [Google Scholar]
- 69. Grotendorst GR, Martin GR, Pencev D, Sodek J, Harvey AK. Stimulation of granulation tissue formation by platelet‐derived growth factor in normal and diabetic rats. J Clin Invest 1985;76:2323–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Laato M, Kahari VM, Niinikoski J, Vuorio E. Epidermal growth factor increases collagen production in granulation tissue by stimulation of fibroblast proliferation and not by activation of procollagen genes. Biochem J 1987;247:385–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Pradhan L, Cai X, Wu S, Andersen ND, Martin M, Malek J, Guthrie P, Veves A, Logerfo FW. Gene expression of pro‐inflammatory cytokines and neuropeptides in diabetic wound healing. J Surg Res 2009. doi: 10.1016/j.jss.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski C, McClain J, Aldrich TH, Papadopoulos N, Daly TJ, Davis S, Sato TN, Yancopoulos GD. Angiopoietin‐2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 1997;277:55–60. [DOI] [PubMed] [Google Scholar]
- 73. Bitar MS, Labbad ZN. Transforming growth factor‐beta and insulin‐like growth factor‐I in relation to diabetes‐induced impairment of wound healing. J Surg Res 1996;61:113–9. [DOI] [PubMed] [Google Scholar]
- 74. Duncan BB, Schmidt MI. The epidemiology of low‐grade chronic systemic inflammation and type 2 diabetes. Diabetes Technol Ther 2006;8:7–17. [DOI] [PubMed] [Google Scholar]
- 75. Wang J‐M, Tao J, Chen AF. Abstract 1958: microRNA Mir‐27b rescues impaired angiogenic function of endothelial progenitor cells and accelerates wound healing in type 2 diabetes. Circulation 2008;118:S_412. [Google Scholar]
- 76. Wang X‐R, Zhang Y, Chen AF. Abstract 3383: microRNA Let‐7f augments angiogenic properties of endothelial progenitor cells through AMPK activation and MnSOD induction in type 1 diabetes. Circulation 2008;118:S_415–6. [Google Scholar]
- 77. Moulik PK, Mtonga R, Gill GV. Amputation and mortality in new‐onset diabetic foot ulcers stratified by etiology. Diabetes Care 2003;26: 491–4. [DOI] [PubMed] [Google Scholar]
- 78. Biswas S, Roy S, Banerjee J, Hussain SR, Khanna S, Meenakshisundaram G, Kuppusamy P, Friedman A, Sen CK. Hypoxia inducible microRNA 210 attenuates keratinocyte proliferation and impairs closure in a murine model of ischemic wounds. Proc Natl Acad Sci U S A 2010;107:6976–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Giannakakis A, Sandaltzopoulos R, Greshock J, Liang S, Huang J, Hasegawa K, Li C, O’Brien‐Jenkins A, Katsaros D, Weber BL, Simon C, Coukos G, Zhang L. miR‐210 links hypoxia with cell cycle regulation and is deleted in human epithelial ovarian cancer. Cancer Biol Ther 2008;7:255–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Chang WY, Bryce DM, D’Souza SJ, Dagnino L. The DP‐1 transcription factor is required for keratinocyte growth and epidermal stratification. J Biol Chem 2004;279:51343–53. [DOI] [PubMed] [Google Scholar]