

SUMMARY
Indisulam and related sulfonamides recruit the splicing factor RBM39 to the CRL4-DCAF15 E3 ubiquitin ligase, resulting in RBM39 ubiquitination and degradation. Here, we used a combination of domain mapping and random mutagenesis to identify domains or residues that are necessary for indisulam-dependent RBM39 ubiquitination. DCAF15 mutations at Q232 or D475 prevent RBM39 recruitment by indisulam. RBM39 is recruited to DCAF15 by its RRM2 (RNA recognition motif 2) and is ubiquitinated on its N terminus. RBM23, which is an RBM39 paralog, is also recruited to the CRL4-DCAF15 ligase through its RRM2 domain and undergoes sulfonamide-dependent degradation. Indisulam alters the expression of more than 3,000 genes and causes widespread intron retention and exon skipping. All of these changes can be attributed to RBM39, and none are the consequence of RBM23 degradation. Our findings demonstrate that indisulam selectively degrades RBM23 and RBM39, the latter of which is critically important for splicing and gene expression.
In Brief
Ting et. al. demonstrate that indisulam and related sulfonamides recruit either RBM39 or RBM23 to the CRL4-DCAF15 ubiquitin ligase, leading to polyubiquitination and proteasomal degradation. Gene expression changes and splicing abnormalities resulting from indisulam treatment are the consequence of RBM39 degradation and not the result of RBM23 degradation.
Graphical Abstract
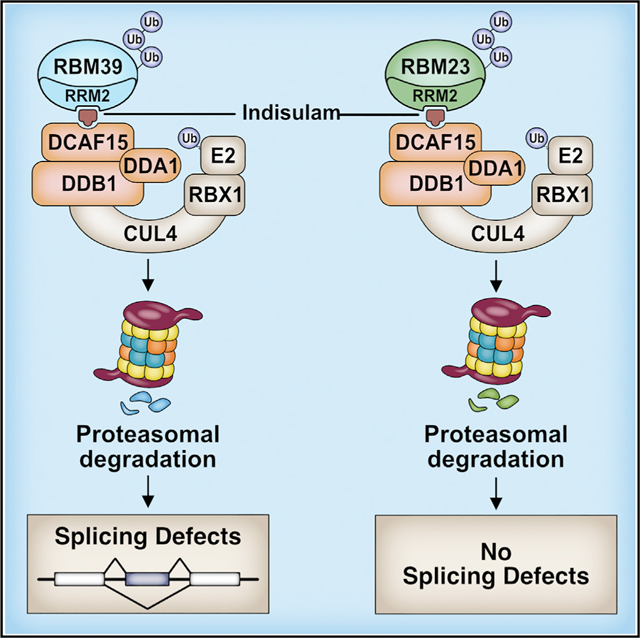
INTRODUCTION
The development of new small molecule therapies has been limited by the challenge of targeting proteins that lack known binding pockets, such as receptors or enzymes. Without a rationale for designing small molecule binders, these proteins are sometimes referred to as “undruggable” (Huang and Dixit, 2016; Salami and Crews, 2017). Recent discoveries of compounds that mediate protein degradation have launched a new field of pharmacotherapeutics for targeting previously undruggable proteins. These compounds are often referred to as “molecular glues” because they induce proximity between a target protein and an E3 ubiquitin ligase receptor. A notable example is the immunomodulatory drugs (IMiDs), which include thalidomide, lenalidomide, and pomalidomide. IMiDs bind the E3 ubiquitin ligase receptor cereblon (CRBN) and lead to the recruitment of numerous neo-substrates.
IMiDs recruit a wide range of neo-substrates with broad cellular functions. IMiDs are efficacious in the treatment of multiple myeloma by catalyzing the degradation of two transcription factors that contain zinc finger domains, Ikaros (IKZF1) and Aiolos (IKZF3) (Krönke et al., 2014; Lu et al., 2014). Specific IMiD derivatives can degrade unique proteins. For example, lenalidomide but not thalidomide induces degradation of casein kinase 1 alpha (CK1α), leading to clinical efficacy in the treatment of del(5q) myelodysplastic syndrome (Krönke et al., 2015). In addition to its effects on immune cell development and function, IMiDs also mediate degradation of another zinc-finger-domain-containing transcription factor, Sal-like protein 4 (SALL4), which leads to teratogenicity (Donovan et al., 2018; Matyskiela et al., 2018).
X-ray crystallography studies of CRBN in complex with an IMiD and different neo-substrates suggest that IMiDs can recruit a common motif referred to as a “degron” (Chamberlain et al., 2014; Fischer et al., 2014; Petzold et al., 2016). The degron for IMiDs within IKZF1, IKZF3, SALL4, and ZFP91 contains a conserved Cys2-His2 zinc finger motif (Petzold et al., 2016; Sievers et al., 2018). Similar structural features in proteins without zinc finger domains can also serve as IMiD degrons. For example, the IMiD derivative CC-885 recruits the translation termination factor GSPT1, which has little homology to other known IMiD substrates except for a beta hairpin containing a key glycine residue (Matyskiela et al., 2016). The ability of IMiD derivatives to target a wide range of proteins for degradation and their established clinical success provides a rationale for the development of molecular glues as therapy.
Recently, a second class of molecule glues was discovered by our lab and others (Han et al., 2017; Uehara et al., 2017). These compounds are all aryl sulfonamides and include indisulam, tasisulam, E7820, and chloroquinoxaline sulfonamide (CQS; hereafter simply referred to as sulfonamides). These sulfonamides recruit the splicing factor RBM39 to the E3 ligase substrate receptor DCAF15. Recruitment of RBM39 to DCAF15 leads to RBM39 ubiquitination and degradation, which results in altered RNA splicing and death in a number of cancer cell lines. Hematopoietic or lymphoid malignancies are more sensitive to indisulam cytotoxity, highlighting the susceptibility of these cancers to splicing changes (Han et al., 2017). Likewise, a recent CRISPR/Cas9 screen demonstrated that RBM39 orchestrates a splicing program that is critical for the survival of acute myeloid leukemia cells (Wang et al., 2019).
RBM39 is the only known indisulam substrate. It is unknown whether sulfonamides like IMiDs degrade a wide variety of substrates leading to differential cellular consequences. Many other RNA-binding proteins share homology with RBM39 and, thus, may also contain a degron that targets them for sulfonamide-dependent degradation. Here, we identify the degron within RBM39 as well as the residues in DCAF15 that are required for sulfonamide-dependent complex formation. Furthermore, we define the biochemical requirements for RBM39 recruitment and ubiquitination and introduce RBM23 as another previously unreported neo-substrate of the CRL4-DCAF15 E3 ligase.
RESULTS
In Vitro Reconstitution of RBM39 Ubiquitination
DCAF15 is a component of the CRL4-DCAF15 E3 ubiquitin ligase, which is composed of a Cullin-4 scaffold, the RING finger protein Rbx1, the substrate receptor DCAF15, the adaptor protein DDB1, and a regulatory protein, DDA1. CRL4 complexes catalyze the transfer of ubiquitin from an E2 enzyme to a substrate bound to DCAF15. Accordingly, RBM39 recruitment to CRL4-DCAF15 leads to RBM39 polyubiquitination and degradation by the proteasome. Consistent with this model, genetic disruption of DCAF15, DDB1, or DDA1 or expression of a dominant-negative CUL4 impair RBM39 degradation in vivo, providing evidence that CRL4-DCAF15 is necessary for RBM39 ubiquitination (Han et al., 2017). Here, we sought to determine if CRL4-DCAF15 is sufficient for RBM39 ubiquitination by establishing an in vitro biochemical system.
To reconstitute RBM39 ubiquitination in vitro, we utilized an SF9 insect cell system to express and purify recombinant RBM39 and the CRL4-DCAF15 E3 ligase (Figure 1A). These purified E3 ligase components included a complex consisting of the Cul4a scaffold and a truncated version of the RING protein Rbx1 Δ1–14 and a second complex consisting of full-length DCAF15, DDB1, and DDA1 (Figure 1B). To determine the competence of our recombinant system to ubiquitinate RBM39, we added our recombinant proteins to a ubiquitination reaction that included an ATP regeneration system, ubiquitin, E1 enzyme, and one of 11 different E2 enzymes (Figure 1C). Western blotting with RBM39 antibodies revealed a higher molecular weight species in the presence of any one of the following E2 enzymes: UbcH5a, UbcH5b, or UbcH5c (Figure 1C). To confirm that this species was indeed polyubiquitination, we repeated the reaction with the removal of one of the following components: indisulam, ubiquitin, E1 enzyme, E2 enzyme, the Cul4a-Rbx1 complex, or the DCAF15-DDB1-DDA1 complex (Figure 1D). Elimination of any one of these components either blocked or substantially reduced the formation of the larger molecular weight RBM39 species. A low level of RBM39 ubiquitination was detected in the absence of added E1. We suspect that the low activity observed in the absence of the E1 enzyme is the result of a contaminating E1 that co-purified with one of our recombinant proteins. Based on these observations, we concluded that RBM39 is polyubiquitinated in vitro by CRL4A-DCAF15.
Figure 1. In Vitro Reconstitution of RBM39 Ubiquitination by the CRL4-DCAF15 E3 Ligase.

(A) Schematic diagram illustrating a model of indisulam-dependent ubiquitination of RBM39 by the CRL4-DCAF15 E3 Ubiquitin Ligase.
(B) Stain-free gel of recombinant proteins expressed and purified from SF9 insect cells.
(C) RBM39 can be ubiquitinated in vitro by E2 enzymes UbcH5a, UbcH5b, and UbcH5c. Purified RBM39 was combined with CRL4-DCAF15 E3 ligase complex and screened in a ubiquitination assay against a panel of E2 enzymes (Enzo). Samples were analyzed by western blot with anti-RBM39.
(D) Dropout of individual components of the RBM39 ubiquitination mixture was performed to determine the minimal components required for RBM39 ubiquitination, which was analyzed by western blot with anti-RBM39.
(E) RBM39 ubiquitination in vitro occurs in a dose-dependent manner with the three sulfonamides indisulam, chloroquinoxaline sulfonamide (CQS), and tasisulam.
We then compared the ability of different indisulam-related sulfonamides to mediate RBM39 ubiquitination. Dose-dependent ubiquitination of RBM39 occurred with any one of three sulfonamides: indisulam, tasisulam, and CQS (Figure 1E). Taken together, our results demonstrate that RBM39 ubiquitination can be reconstituted in vitro and that the CRL4A-DCAF15 E3 ligase is sufficient to mediate this ubiquitination reaction in the presence of a sulfonamide, E1 and E2 enzymes, and energy.
N-Terminal Lysines Are Ubiquitinated in RBM39
Next, we mapped the residues that are ubiquitinated on RBM39. The majority of lysines in RBM39 are located in clusters within the first 135 amino acids at the N terminus (Figure 2A). Therefore, we generated RBM39 constructs in which groups of potential lysine ubiquitination sites were mutated to arginine residues. We then transiently transfected these lysine-to-arginine mutants into 293T cells and evaluated levels with and without indisulam treatment (Figure 2B). Each mutant was tagged with a hemagglutinin (HA) epitope tag, which lacks lysines and, therefore, would not confound interpretation by adding potential ubiquitination sites to the protein. Lysine-to-arginine mutations within the first 60 amino acids of RBM39 did not affect RBM39 degradation (Figure 2B). By contrast, mutating the lysines between amino acids 60 and 120 blocked the ability of RBM39 to undergo indisulam-dependent degradation in vivo (Figure 2B). This loss of RBM39 degradation was not due to a defect in indisulam-dependent interaction with DCAF15 because all RBM39 lysine-to-arginine mutants we tested were still able to complex with DCAF15 when treated with indisulam (Figure 2C). To test whether N-terminal lysines are substrates for ubiquitination in vitro, we purified an RBM39 ubiquitination mutant in which all the lysine residues within the first 120 amino acids were mutated to arginine residues (RBM39 K(1–120)R) (Figure S1A). In vitro ubiquitination of the RBM39 K(1–120)R mutant was substantially reduced in comparison to wild-type RBM39 (Figure 2D). Both in vitro and in vivo observations indicate that the primary ubiquitination site of RBM39 is located within the N-terminal region of 120 amino acids.
Figure 2. Identification of the RBM39 Ubiquitination Site.

(A) Schematic diagram illustrating all potential ubiquitination site lysine residues in RBM39.
(B) Mutating lysine residues between amino acids 60–120 of RBM39 blocks indisulam-dependent degradation in vivo. Lysine residues within the indicated RBM39 amino acid regions were mutated to arginine residues. Each mutant was tagged with a lysine-free HA epitope and transfected into 293Ts. Cells were treated with 10 μM indisulam for 6 h, and whole-cell lysates were analyzed by western blotting with anti-HA to assess indisulam-dependent degradation.
(C) RBM39 lysine-to-arginine mutants from Figure 2B still retain the ability to interact with DCAF15. 293T cells were co-transfected with RBM39-HA and DCAF15–3xFLAG and treated with 1 μM MLN-4924 for 2 h, followed by 10 μM indisulam for 4 h. FLAG immunoprecipitation experiments were performed to examine indisulam-dependent interaction between RBM39 and DCAF15.
(D) Mutating lysines within the first 120 amino acids of RBM39 prevents RBM39 ubiquitination in vitro. 3XFLAG tagged wild-type RBM39 or the RBM39 K(1–120)R mutant were purified from 293T cells by FLAG immunoprecipitation and subjected to an in vitro ubiquitination assay with the indicated concentrations of indisulam. RBM39 ubiquitination was assessed by western blot with anti-RBM39.
See also Figure S1.
The RBM39 RRM2 Domain Is Required for Indisulam-Dependent Interaction with DCAF15
To identify the minimal binding site of RBM39 required for indisulam-dependent interaction with DCAF15, we generated a panel of RBM39 truncation constructs that lacked either N-terminal or C-terminal sequences (Figures 3A and 3B). We co-expressed these mutants with FLAG-tagged DCAF15 in 293T cells and then treated the cells with either DMSO or indisulam. We investigated the ability of each truncated RBM39 protein to co-purify with DCAF15 in comparison to wild-type RBM39. An indisulam-dependent interaction between DCAF15 and RBM39 was not affected by either deletion of the first 249 amino acids of RBM39 (Figure 3A) or by deletion of the last 200 amino acids of RBM39 (Figure 3B). Any truncation of RBM39 that included an amino acid between 250 and 330 resulted in a decreased interaction with DCAF15. Taken together, we concluded that amino acids 250–330 of RBM39, which corresponds to the RNA recognition motif 2 (RRM2) domain of RBM39, are necessary to interact with DCAF15. To test whether this domain is sufficient, we compared the interaction of the RBM39 RRM2 to full-length RBM39 (Figure 3C). The RBM39 RRM2 domain is sufficient for recruitment to DCAF15 in the presence of indisulam.
Figure 3. Identification of RBM39-Binding Site Required for Indisulam-Dependent Interaction with DCAF15.

(A) The RBM39-binding site for indisulam-dependent interaction with DCAF15 begins at amino acid 250. HA-tagged truncation mutants of RBM39 were co-expressed with DCAF15–3xFLAG in 293T cells. Cells were treated with 1 μM MLN-4924 for 2 h, followed by 10 μM indisulam for 4 h. FLAG immunoprecipitation was performed to identify truncation mutants that still interact with DCAF15.
(B) The RBM39-binding site for indisulam-dependent interaction with DCAF15 ends at amino acid 330. FLAG immunoprecipitation experiments were performed with RBM39 truncation mutants, as described in Figure 3A.
(C) DCAF15 interacts with full-length (FL) wild-type RBM39 and the isolated RBM39 RRM2 domain but not with RBM39 G268V. DCAF15–3xFLAG was co-expressed with RBM39-HA in 293Ts. Cells were treated with 1 μM MLN-4924 for 2 h, followed by 10 μM indisulam for 4 h. FLAG immunoprecipitation and western blots were performed to examine RBM39-indisulam-DCAF15 interactions.
(D) Wild-type RBM39 RRM2 but not RBM39 G268V RRM2 binds to DCAF15-DDA1-DDB1 in vitro. Time-resolved FRET (TR-FRET) assays were performed by titrating indisulam with RBM39(RRM2)-3xFLAG (amino acids 250–330) purified from E. coli and His-DCAF15-DDA1-DDB1 complex purified from SF9 insect cells. The experiment was performed in triplicate, and data are plotted as the mean ± SEM. Some error bars are not visible at the scale of the figure.
See also Figure S2.
To quantify the binding affinity of this interaction, we used an Escherichia coli system to purify the RBM39 RRM2 domain (Figure S2A). We used time-resolved Förster resonance energy transfer (TR-FRET) to measure the impact of indisulam on the binding of RBM39 RRM2 to recombinant DCAF15-DDB1-DDA1. Titration of indisulam to a fixed equimolar ratio of RBM39 RRM2 and DCAF15-DDB1-DDA1 led to a dose-dependent increase in TR-FRET signal (half maximal effective concentration (EC50), 2.27 μM) (Figure 3D). We have previously demonstrated that G268V mutations within the RBM39 RRM2 block recruitment to DCAF15 (Han et al., 2017). Accordingly, we found that recombinant RRM2 with a G268V mutation does not bind to DCAF15 in the presence of indisulam concentrations as high as 50 μM (Figure 3D). These observations collectively demonstrate that the RBM39 RRM2 is the minimal binding domain required for indisulam-dependent recruitment to CRL4-DCAF15.
DCAF15 Residues Required for RBM39 Ubiquitination Were Identified by Random Mutagenesis
We also sought to define the amino acids in DCAF15 that are important for recruitment of RBM39 by sulfonamides. Unlike RBM39, the expression of DCAF15 truncation mutants led to protein instability, precluding us from using this strategy to map the sulfonamide binding domain. Therefore, we performed an error-prone PCR mutagenesis screen as an alternative approach to identify residues important for RBM39 recruitment.
Cells in which we previously used CRISPR/Cas9 to silence DCAF15 (DCAF15 knockout [KO]) are resistant to indisulam, and complementation with functional DCAF15 restores indisulam sensitivity (Han et al., 2017). Hence, we reasoned that we could use indisulam to select for loss-of-function DCAF15 mutants among DCAF15 KO cells complemented with a library of random DCAF15 mutants. Importantly, indisulam sensitivity is also influenced by the level of DCAF15 expression, which introduces a possible confounder in our system. Mutants with reduced expression might be enriched regardless of their ability to bind indisulam. First, we generated a library of DCAF15 mutants by using error-prone PCR (Figure 4A). To minimize the impact of expression differences, we then cloned DCAF15-P2A-GFP into a lentiviral vector, so that the levels of GFP were expected to correlate with the amount of DCAF15 expression. Lentivirus generated from this library was transduced into HCT-116 DCAF15 KO cells at conditions that were expected to lead to one integrant per cell. To normalize for DCAF15 expression levels, we sorted infected cells for a narrow range of GFP expression. GFP-positive cells were then treated with either DMSO or indisulam. To evaluate which mutations were enriched in the indisulam condition, we used deep massively parallel sequencing to analyze the sequence of DCAF15 PCR amplicons derived from either DMSO or indisulam-treated cells. We tabulated the number of sequencing reads with a mutation in both conditions and ranked mutations by the odds ratio (OR), reflecting their association with the indisulam treatment group. The 50 highest ranking missense mutations were chosen for further evaluation (Table 1).
Figure 4. Identification of DCAF15 Residues Required for RBM39 Recruitment and Degradation.

(A) Schematic diagram illustrating the error-prone PCR mutagenesis approach to identifying DCAF15 residues that protect against indisulam-mediated cytotoxicity.
(B) DCAF15 Q232R and D475N/H/V mutants cannot interact with RBM39 but retain their ability to interact with DDB1 and DDA1. Cells co-transfected with DCAF15–3xFLAG and RBM39-HA were treated with 1 μM MLN-4924 for 2 h, followed by 10 μM indisulam for 4 h. FLAG immunoprecipitation experiments were performed to examine indisulam-dependent interaction between RBM39 and DCAF15.
(C) Summary table of DCAF15 mutations thatprevent RBM39 degradation. From the immunoprecipitation experiments shown in Figure S4, DCAF15 mutations can be categorized into three groups based on their ability to interact with DDB1, DDA1, and/or RBM39. Arrows indicate decreased interaction of DCAF15 with one of these proteins by co-immunoprecipitation.
(D) Introducing the D475N mutation in DCAF15 prevents indisulam-dependent interaction with RBM39 in vitro. FRET assays were performed by titrating indisulam to purified RBM39–3xFLAG (amino acid [aa] 250–330) and His-DCAF15-DDA1-DDB1 complex purified from SF9 insect cells. The experiment was performed in triplicate, and data are plotted as the mean ± SEM. Some error bars are not visible at the scale of the figure.
(E) Introducing the D475N mutation in DCAF15 results in impaired RBM39 ubiquitination in vitro. Purified DCAF15-DDA1-DDB1 complex and full-length RBM39 were combined in an in vitro ubiquitination assay. RBM39 ubiquitination was analyzed by western blot. Stain-free gel demonstrates the purity of the DCAF15-DDB1-DDA1 complexes expressed in SF9 insect cells.
Table 1.
DCAF15 Mutations Discovered in Error-Prone PCR Mutagenesis Screen
| Nucleotide Mutation | Protein Mutation | Result | WTa Reads (DMSO) | WT Reads (Indisulam) | Mutant Reads (DMSO) | Mutant Reads (Indisulam) | Odds Ratio |
|---|---|---|---|---|---|---|---|
| c.1282C > T | p.R428W | missense | 441,001 | 303,481 | 852 | 109,260 | 186.299 |
| c.1202G > A | p.G401E | missense | 447,510 | 307,468 | 1,046 | 111,109 | 154.542 |
| c.389A > G | p.Q130R | missense | 443,681 | 336,151 | 1,350 | 111,988 | 109.428 |
| c.653T > C | p.L218P | missense | 372,754 | 272,630 | 1,381 | 107,628 | 106.475 |
| c.1423G > C | p.D475H | missense | 448,480 | 371,080 | 382 | 31,596 | 99.94 |
| c.1111C > G | p.R371G | missense | 380,273 | 322,008 | 340 | 28,166 | 97.807 |
| c.704T > C | p.F235S | missense | 412,092 | 317,957 | 1,525 | 102,219 | 86.803 |
| c.1430T > G | p.V477G | missense | 447,276 | 397,352 | 99 | 7,341 | 83.45 |
| c.628C > T | p.Q210X | nonsense | 384,107 | 378,035 | 140 | 10,081 | 73.164 |
| c.1201G > A | p.G401R | missense | 446,851 | 398,700 | 405 | 19,333 | 53.471 |
| c.1397A > G | p.H466R | missense | 452,211 | 388,919 | 434 | 18,971 | 50.791 |
| c.875C > T | p.P292L | missense | 339,503 | 290,758 | 388 | 14,962 | 44.987 |
| c.980T > C | p.I327T | missense | 372,097 | 316,134 | 508 | 18,338 | 42.445 |
| c.1436T > C | p.L479P | missense | 453,357 | 369,692 | 1,232 | 40,398 | 40.247 |
| c.1520A > G | p.Q507R | missense | 472,719 | 394,034 | 1,301 | 33,378 | 30.779 |
| c.1411T > C | p.F471L | missense | 447,821 | 390,826 | 562 | 13,217 | 26.95 |
| c.1330G > A | p.E444K | missense | 423,505 | 386,196 | 260 | 5,922 | 24.979 |
| c.161T > C | p.F54S | missense | 373,112 | 364,151 | 362 | 8,593 | 24.323 |
| c.545T > A | p.V182D | missense | 378,739 | 374,058 | 190 | 4,546 | 24.227 |
| c.731T > C | p.L244P | missense | 421,869 | 395,621 | 1,018 | 22,407 | 23.474 |
| c.1792A > G | p.I598V | missense | 286,644 | 266,085 | 582 | 12,439 | 23.027 |
| c.67G > A | p.A23T | missense | 305,888 | 291,633 | 653 | 12,852 | 20.646 |
| c.702C > T | p.A234A | synonymous | 420,278 | 420,773 | 329 | 6,463 | 19.623 |
| c.1424A > T | p.D475V | missense | 446,109 | 398,927 | 184 | 3,195 | 19.421 |
| c.269T > A | p.V90D | missense | 481,319 | 473,794 | 252 | 4,748 | 19.143 |
| c.1550T > C | p.L517P | missense | 476,579 | 402,386 | 2,116 | 32,015 | 17.926 |
| c.560T > C | p.V187A | missense | 372,191 | 361,449 | 569 | 9,800 | 17.738 |
| c.119T > G | p.L40R | missense | 292,518 | 286,667 | 394 | 6,839 | 17.715 |
| c.653T > G | p.L218R | missense | 372,754 | 272,630 | 60 | 760 | 17.319 |
| c.386T > C | p.F129S | missense | 435,229 | 425,855 | 844 | 13,669 | 16.555 |
| c.317T > C | p.I106T | missense | 449,404 | 420,480 | 1,880 | 28,063 | 15.959 |
| c.1320C > T | p.Y440Y | synonymous | 420,107 | 385,434 | 220 | 3,085 | 15.288 |
| c.587G > A | p.C196Y | missense | 373,388 | 368,361 | 243 | 3,539 | 14.766 |
| c.1014C > T | p.V338V | synonymous | 356,956 | 320,587 | 184 | 2,367 | 14.329 |
| c.1753T > C | p.S585P | missense | 389,195 | 350,692 | 2,535 | 30,404 | 13.308 |
| c.1344A > T | p.L448L | synonymous | 414,689 | 381,083 | 112 | 1,334 | 12.956 |
| c.272T > C | p.L91P | missense | 479,719 | 455,497 | 1,268 | 14,774 | 12.267 |
| c.696G > A | p.Q232Q | synonymous | 412,171 | 415,728 | 374 | 4,557 | 12.084 |
| c.955A > G | p.K319E | missense | 389,836 | 342,974 | 289 | 3,056 | 12.018 |
| c.299A > G | p.D100G | missense | 467,809 | 457,473 | 472 | 5,544 | 12.007 |
| c.277T > C | p.Y93H | missense | 473,526 | 450,008 | 1,445 | 14,737 | 10.731 |
| c.1248C > G | p.P416P | synonymous | 448,094 | 423,002 | 73 | 704 | 10.216 |
| c.594T > C | p.D198D | synonymous | 370,909 | 365,526 | 574 | 5,656 | 9.998 |
| c.1645A > G | p.S549G | missense | 404,504 | 365,095 | 1,437 | 12,866 | 9.92 |
| c.1162A > G | p.N388D | missense | 427,846 | 388,954 | 722 | 6,330 | 9.644 |
| c.1410T > C | p.S470S | synonymous | 446,092 | 395,970 | 646 | 5,524 | 9.633 |
| c.385T > G | p.F129V | missense | 431,972 | 435,108 | 105 | 993 | 9.389 |
| c.203T > G | p.I68S | missense | 446,891 | 444,926 | 96 | 890 | 9.312 |
| c.1175T > C | p.L392P | missense | 445,628 | 400,585 | 1,421 | 11,614 | 9.092 |
| c.695A > G | p.Q232R | missense | 409,264 | 399,176 | 934 | 8,196 | 8.997 |
| c.202A > G | p.I68V | missense | 446,102 | 441,142 | 390 | 3,305 | 8.57 |
| c.1665C > T | p.C555C | synonymous | 410,435 | 382,169 | 229 | 1,825 | 8.559 |
| c.176C > T | p.P59L | missense | 419,232 | 413,591 | 266 | 2,238 | 8.528 |
| c.11G > T | p.S4I | missense | 264,754 | 269,833 | 47 | 407 | 8.497 |
| c.1423G > A | p.D475N | missense | 448,480 | 371,080 | 312 | 2,150 | 8.328 |
| c.334T > C | p.W112R | missense | 461,571 | 458,173 | 382 | 3,155 | 8.32 |
| c.1070C > A | p.A357E | missense | 353,901 | 324,481 | 78 | 583 | 8.152 |
| c.1358T > A | p.V453D | missense | 416,321 | 379,517 | 179 | 1,330 | 8.151 |
| c.1704C > T | p.S568S | synonymous | 374,733 | 362,876 | 362 | 2,808 | 8.01 |
| c.1614G > A | p.E538E | synonymous | 427,210 | 392,194 | 198 | 1,456 | 8.01 |
| c.1370T > A | p.V457D | missense | 427,845 | 389,424 | 318 | 2,289 | 7.908 |
| c.1258A > C | p.T420P | missense | 431,301 | 405,772 | 133 | 934 | 7.464 |
DCAF15 mutations were compared between DMSO and indisulam treatment conditions as outlined in Figure 4A. Mutations were ranked by odds ratio, and the 50 highest ranking mutations were chosen for validation studies.
WT, wild-type.
To investigate these DCAF15 mutations, we transfected each DCAF15 mutant into cells to compare their expression levels and their ability to degrade RBM39 (Figure S3A). DCAF15 mutants that were unable to mediate indisulam-dependent RBM39 degradation were further examined for their ability to form a complex with either DDB1 or DDA1 and to recruit RBM39 in the presence of indisulam (Figure S4A). DCAF15 mutations that decreased interaction with DDA1, DDB1, and RBM39 were identified within amino acids 90–250 (Figure 4C; Figure S4A). By contrast, DCAF15 mutations that only impaired interaction with DDA1 were clustered within amino acids 390–460, suggesting that this region might directly interact with DDA1 (Figure 4C; Figure S4A).
We sought to identify those mutations that did not influence DCAF15 binding to DDB1 or DDA1 but specifically blocked recruitment of RBM39 to DCAF15 by indisulam. From our analysis, the following four DCAF15 mutations representing two amino acids met this criteria: Q232R, D475N, D475H, and D475V (Figures 4B and 4C; Figure S4). These structure-function studies of DCAF15 suggest that D475 and Q232 are important residues for binding either RBM39 or sulfonamides. We sought to test this hypothesis in vitro. We were able to express and purify the D475N (but not the Q232R) mutation from SF9 insect cells (Figures 4D and 4E). In comparison to wild-type DCAF15, we found that DCAF15 D475N did not interact with the RBM39 RRM2 in the presence of indisulam at concentrations as high as 50 μM (Figure 4D). Concordant with the binding assay, the ability of the DCAF15 D475N mutant to mediate RBM39 ubiquitination in vitro was substantially diminished compared to wild-type DCAF15 (Figure 4E).
RBM23 Is a Paralog of RBM39 That Undergoes Indisulam-Dependent Degradation
Substrate receptors, including DCAFs, often recruit substrates with similar sequences or structural features, known as degrons (Guharoy et al., 2016; Sievers et al., 2018). RBM23 is a known paralog of RBM39 (Dowhan et al., 2005). The RBM23 RRM2 domain has an 88% identity to the RBM39 RRM2, which is sufficient for recruitment to DCAF15 (Figure 5A). Therefore, we hypothesized that RBM23 can also be recruited to CRL4-DCAF15 by sulfonamides, ubiquitinated, and degraded in vivo. To test this, we endogenously tagged RBM23 with a C-terminal 3xFLAG sequence in order to monitor RBM23 protein levels (Figure S5A). Treatment with three different sulfonamides led to a dose-dependent decrease in RBM23–3xFLAG protein (Figure 5B). In addition to the predominant FLAG-antibody-reactive band, we observed a lower molecular weight band that migrated at 27 kD and decreased with indisulam treatment (Figure 5B). This proteoform may be a cleavage product or alternatively spliced product of RBM23 that still retains its ability to undergo indisulam-dependent degradation. To determine whether degradation of RBM23, like RBM39, is also dependent on DCAF15, we ablated DCAF15 from the endogenously tagged RBM23–3xFLAG line by using CRISPR/Cas9 editing (Figure 5C). RBM23 degradation was significantly impaired in these cells, demonstrating that degradation of RBM23, like RBM39, is DCAF15 dependent (Figure 5C). We then tested whether RBM23 degradation was independent of RBM39 degradation. Using CRISPR/Cas9, we engineered an epitope tag into the C terminus of RBM23 in a previously identified HCT-116 clone that expresses RBM39 with a G268V mutation (Han et al., 2017) (Figures S5B–S5D). Consistent with our previous findings, RBM39 G268V is not degraded by indisulam (Figure 5D). By contrast, RBM23 degradation still occurred in an indisulam-dependent manner, demonstrating that RBM23 degradation is independent of RBM39 levels (Figure 5D).
Figure 5. RBM23 Is an Indisulam-Dependent Neo-substrate That Is Recruited to DCAF15.

(A) Schematic alignment of full-length RBM39 and RBM23 proteins. The percentage identity between the serine-arginine (SR) domains and RNA recognition motifs (RRMs) of the proteins is indicated.
(B) Endogenous RBM23 degrades in a dose-dependent manner with indisulam, CQS, and tasisulam treatment. CRISPR/Cas9 was used to tag the C terminus ofRBM23 with 3xFLAG in the HCT-116 cell line. Cells were treated with the indicated compound doses for 24 h and analyzed by western blot using FLAG-horseradish peroxidase (HRP) antibody.
(C) DCAF15 is required for RBM23 degradation with indisulam treatment. A CRISPR guide against DCAF15 was expressed in an endogenously tagged HCT-116 RBM23–3xFLAG cell line. Cells were treated with the indicated doses of indisulam for 24 h. RBM23 levels were analyzed by western blot using FLAG-HRP antibody.
(D) RBM23 degradation is not dependent on RBM39 recruitment to the CRL4-DCAF15. RBM23 was tagged in cell lines with wild-type RBM39 or RBM39 G268V. Cells were treated with the indicated doses of indisulam for 24 h, and RBM23 protein levels were analyzed by western blot using FLAG-HRP antibody.
(E) Alignment of the RRM2 domains of RBM39 and RBM23. Differences in the amino acid sequences of RBM39 and RBM23 are indicated by asterisks.
(F) Amino acids 210–330 of RBM23 encompassing the RRM2 interacts with DCAF15-DDB1-DDA1 in vitro. TR-FRET assays were performed by titrating indisulam to purified RBM23–3xFLAG (amino acids 210–330) and His-DCAF15-DDA1-DDB1 complex. The experiment was performed in triplicate, and data are plotted as the mean ± SEM. Some error bars are not visible at the scale of the figure.
See also Figure S6.
Based on these findings, we predicted that the region of RBM23 that interacts with DCAF15 also maps to a region containing the RRM2 domain of RBM23 (Figure 5E). To test this hypothesis, we generated a panel of HA-tagged RBM23 truncation mutants and analyzed their ability to co-immunoprecipitate with DCAF15 (Figure S6A). We found that the RBM23-binding site mapped to amino acids 210–330, which encompass the RBM23 RRM2 domain (Figures S6A and S6B). We then expressed and purified the RBM23 minimal binding site and tested its ability to interact with the DCAF15-DDB1-DDA1 complex by TR-FRET (Figure S2A). Titration of indisulam to a fixed equimolar amount of the RBM23 (amino acids 210–330) domain and DCAF15-DDB1-DDA1 resulted in an EC50 of 2.58 μM (Figure 5F). Introduction of either the RBM39 G268V mutation or the equivalent G265V mutation in RBM23 prevented indisulam-dependent interaction with DCAF15-DDB1-DDA1 by TR-FRET (Figure 5F). Together, these results demonstrate that, similar to RBM39, RBM23 is a neo-substrate that can be recruited to DCAF15 and undergo sulfonamide-dependent degradation.
Indisulam Alters Gene Expression and Alternative Splicing through RBM39 but Not RBM23
RBM39 and RBM23 have previously been reported to regulate transcriptional activation and alternative splicing, but their distinct roles in these processes have not been clearly defined (Dowhan et al., 2005). We previously demonstrated that indisulam causes splicing defects by degrading RBM39 (Han et al., 2017). We reasoned that indisulam would also cause changes in gene expression and possibly splicing as a result of degradation of RBM23 and perhaps yet unidentified substrates.
We developed an isogenic system to assay gene expression and splicing changes that are the consequence of RBM39, RBM23, and putative substrates (Figure 6A). The first cell line (cell line #1) is an HCT-116 cell line that expresses wild-type RBM39 and a 3XFLAG-tagged RBM23 (Figure S5A). We tested indisulam treatment in this cell line and observed that RBM39 was degraded, as expected (Figure S7A). The second cell line is an HCT-116 cell line with RBM39 G268V in which we used CRISPR/Cas9 to engineer an auxin-inducible degron (AID) and 3XFLAG epitope into the C terminus of both alleles of RBM23 (Figures S5A–S5D). The latter cell line also expresses the plant E3 ubiquitin ligase TIR1, so that in the presence of indoleacetic acid (IAA), RBM23-AID-3XFLAG is recruited to TIR1 and degraded (Figure S7B). We used massively parallel RNA sequencing to compare gene expression changes for each cell line following 12-h treatment with DMSO, 10 μM indisulam, or 100 μM IAA (Figure 6B). Using this system, we can identify changes in gene expression or splicing that are specific to either RBM39, RBM23, or as yet an unknown indisulam neo-substrate.
Figure 6. Indisulam-Mediated Gene Expression Is Dependent on RBM39.

(A) Schematic diagram of cell lines used to examine RNA expression and splicing. Endogenously tagged RBM23 cell lines in the background of wild-type RBM39 or RBM39 G268V were treated with DMSO, indisulam (10 μM), or IAA (100 μM) for 12 h. Two clones per cell line were used for gene expression analysis.
(B) MA plots of indisulam-specific, RBM39-specific, and RBM23-specific changes in differential gene expression. Upregulated or downregulated genes with each condition are indicated in red (p < 0.05, log2 fold change > 1). See also Figure S7.
(C) Table of splicing events that are dependent on indisulam, RBM39, or RBM23. Treatment with indisulam, as outlined in Figure 6A, leads to exon skipping and intron retention events that are dependent on RBM39 degradation.
Indisulam treatment altered the expression of 3,080 genes (Figure 6B). By contrast, we found no significant gene expression changes in RBM39 G268V cells (Figure 6B). This observation suggests that RBM23 and any yet unidentified substrate have no impact on gene expression 12 h following 10 μM of indisulam. To confirm that RBM23 had no impact on gene expression, we used IAA to selectively degrade RBM23 to even lower levels than with indisulam-mediated degradation (Figure S7B). Auxin treatment alone in a cell line without AID-tagged proteins did not lead to any gene expression changes (Figure S7C). Auxin-induced RBM23 degradation also did not result in any gene expression changes, suggesting that RBM23 has minimal to no impact on gene expression in HCT-116 cells (Figure 6B). Taken together, we concluded that all of the gene expression changes that we identified following indisulam treatment are due to RBM39 degradation.
Similar to our gene expression analysis, indisulam treatment resulted in multiple splicing events that were all dependent on RBM39. Indisulam treatment for 12 h resulted in 1,640 exon-skipping events, 132 intron retention events, and 22 alternative splice site events (Figure 6C). However, no RBM39-independent splicing changes occurred following indisulam treatment in cells expressing the RBM39 G268V allele. In addition, no RBM23-dependent splicing events were detected following auxin treatment in the RBM23-AID cell line (Figure 6C). Taken together, these results suggest that the observed effect of indisulam on splicing is mediated exclusively through RBM39 and not through degradation of RBM23 or any other substrate.
DISCUSSION
Using a combination of biochemical and cellular assays, our results demonstrate that the CRL4-DCAF15 E3 ligase is sufficient for sulfonamide-dependent neo-substrate ubiquitination. Structure-function studies of the complex revealed the RRM2 domain of RBM39 is both necessary and sufficient for compound-dependent recruitment to CRL4-DCAF15. These observations are consistent with our prior studies in which we identified mutations in the RRM2 that render RBM39 resistant to sulfonamide-emediated degradation. A random mutagenesis screen revealed DCAF15 mutants important for DDB1 or DDA1 binding and RBM39 recruitment. Mutations within amino acids 90–250 of DCAF15 weakened the interaction of DCAF15 with both DDB1 and DDA1, suggesting that this region is important for adaptor-receptor binding. By contrast, residues located in amino acids 390–460 of DCAF15 appeared to solely disrupt an interaction between DCAF15 and DDA1.
DDA1 is a component of several CRL4 E3 ligases and is required for efficient RBM39 degradation, but its precise function is unknown (Jin et al., 2006; Olma et al., 2009). Previous crystallography studies revealed that the N-terminal region of DDA1 binds to the β-propeller A domain of DDB1 (Shabek et al., 2018). However, this crystal structure did not include the C-terminal domain of DDA1, which may be capable of interacting with other proteins in the E3 ubiquitin ligase. Our results suggest that a region of DCAF15 between amino acids 390–460 may directly interact with DDA1.
Finally, DCAF15 D475H/N/V and Q232R were identified as mutants that form a complex with DDB1 and DDA1 but fail to recruit RBM39 in the presence of sulfonamides. Our hypothesis is that these residues are important for either making contacts between DCAF15 and indisulam or DCAF15 and RBM39. Future structural studies of the DDB1-DDA1-DCAF15-sulfonamideRBM39 complex may elucidate the role of these two residues.
Discovery of the aryl sulfonamide class of molecular glues has raised the hypothesis that sulfonamide derivatives might target RNA-binding proteins (RBPs) containing RRM domains, much like the IMiDs target transcription factors with zinc finger domains (Petzold et al., 2016; Sievers et al., 2018). RBM39, the first discovered indisulam-dependent substrate, belongs to a large family of more than 450 RBPs with diverse roles in human disease (Dvinge et al., 2016; Lee and Abdel-Wahab, 2016; Wang et al., 2019). RBPs have been shown to be involved in several aspects of post-transcriptional regulation and are implicated in multiple diseases, including cancer and neurodegenerative disease. Small molecules that degrade specific RBPs would have multiple potential clinical applications, and thus, there is enthusiasm for using sulfonamide derivatives to target RBP neo-substrates other than RBM39.
We identified RBM23 as an additional sulfonamide neo-substrate. The RBM23 RRM2 domain has high sequence identity to the RBM39 RRM2 degron sequence. Similar to RBM39, RBM23 is recruited to the CRL4-DCAF15 ubiquitin ligase and undergoes indisulam-dependent degradation. Mutations in RBM39 that block degradation also rescue cells from indisulam, demonstrating that RBM39 degradation is the cause of cancer cell death. We expected that cells with RBM39 mutations might still exhibit gene expression changes that are the result of degradation of RBM23 or other yet undefined substrates. Strikingly, we found that gene expression was essentially unchanged in cells that cannot degrade RBM39. At this depth of coverage, it is possible that our analysis did not detect rare transcripts influenced by other substrates. Notwithstanding, we concluded that RBM23 is not essential for the proliferation of HCT-116 cells and has minimal impact on gene expression or splicing. Furthermore, we found no gene expression or splicing changes that might be explained by an alternative neo-substrate.
The absence of any gene expression changes in RBM39 G268V cells suggests that indisulam may be remarkably selective for RBM39 and RBM23 degradation. This is especially surprising considering that a structurally conserved RRM2 domain of both proteins is sufficient for recruitment to DCAF15. Structural studies of the complex are necessary to understand the mechanism by which the RBM39 and RBM23 RRM2 bind to DCAF15 in the presence of the aryl sulfonamide. Structural studies might be able to predict how derivatives can be used to target alternative RRM2 domains to DCAF15.
STAR⋆METHODS
Detailed methods are provided in the online version of this paper and include the following:
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Deepak Nijhawan (Deepak.Nijhawan@utsouthwestern.edu). All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Reagents and Cell Culture
HCT-116 (male, ATCC) and Lenti-X 293T cells (sex unreported, Clontech) were maintained at 37°C and 5% CO2 in DMEM media supplemented with 10% FBS and 2 mM L-glutamine. SF9 insect cells (sex unreported, gift from H. Yu at UT Southwestern) were maintained at 27°C, 135 rpm, in SF900 II serum-free media (Life Technologies).
Endogenous tagging of RBM23
For endogenous auxin-inducible degron (AID) tagging, cells were first engineered to express the auxin-specific substrate receptor TIR1. For lentiviral expression of TIR1, 293T cells were seeded in a 6-well plate at 200,000 cells per well, and allowed to attach overnight. The next day, transfection complexes were prepared in 200 μL Opti-MEM and included 6 μL TransIT-Lenti (Mirus), 1 μg pLVX_TIR-V5-IRES-hygro, 100 ng pMD2.g (Addgene #12259), and 1 μg psPAX2 (Addgene #12260). Complexes were allowed to incubate at room temperature for 15 min, and added dropwise to the 293T cells. Lentivirus was collected and filtered 48 hr and 72 hr after transfection. Polybrene was added to the lentivirus at a final concentration of 8 μg/mL, and the lentivirus was used to infect the previously generated HCT116 RBM39-G269V cell line (Han et al., 2017). Three days after infection, the cells underwent selection with 500 μg/mL hygromycin for 7 days. To generate the AID-tagged lines, the genomic region surrounding the RBM23 stop codon was targeted using three individual guide RNAs, which were each cloned into the px330 vector (Addgene #42230). Repair templates were cloned into the pGEM-T-Easy vector (Promega) and included an AID-3xFLAG tag, an IRES-Neo cassette flanked by LoxP sites, and 1 kB homology arms encoding regions upstream and downstream of the stop codon. Repair templates included the last intron directly before the stop codon. HCT116 RBM39-G268V TIR1-expressing cell line were transfected with 1 μg repair template and 500 ng of mastermix containing equal amounts of the three RBM23 guides. Cells underwent selection with 1 mg/ml G418, and individual clones were isolated. Integration of the tags was validated by western blot using FLAG-HRP antibody (1:3000, Sigma-Aldrich). Clones with homozygous integration of the AID-FLAG tag were identified through PCR analysis using the following primers: 5′ CCTCAGTGAGCATACAATTTTCC 3′ and 5′CCAGGTGTGAGTTTTCATTTTTC 3′. Homozygously-tagged clones were then subjected to dose responses with indisulam and IAA, and evaluated by western blot for TIR1-V5 expression and RBM23 degradation using V5-HRP antibody (Sigma-Aldrich) and FLAG-HRP antibody (Sigma-Aldrich), respectively. Clones were then further validated by Sanger sequencing.
To generate the RBM23–3xFLAG endogenously-tagged lines, the genomic region surrounding the RBM23 stop codon was targeted using three individual guide RNAs, which were each cloned into the px330 vector. Templates were cloned into the pGEM-T-Easy vector and included a 3xFLAG tag, an IRES-Neo cassette flanked by LoxP sites, and 1 kB homology arms encoding regions upstream and downstream of the stop codon. Repair templates did not include the last intron directly before the stop codon. The HCT116 parental cell line was transfected with 1 μg repair template and 500 ng of mastermix containing equal amounts of the three RBM23 guides. Cells underwent selection with 1 mg/ml G418, and individual clones were isolated. Integration of the tags was validated by western blot using FLAG-HRP antibody. Clones expressing the tag were identified through PCR analysis using the following primers: 5′-CCTCAGTGAGCATACAATTTTCC-3′ and 5′-CCAGGTGTGAGTTTTCATTTTTC-3′. Clones were then further validated by Sanger sequencing.
For expression of DCAF15 CRISPR guides, the following DCAF15 guide was cloned into LentiCRISPRv2 (Addgene #52961): TGCTGACGTAGATGTCACGG. Lentivirus was generated by transfecting 293T cells with a mixture containing 200 μL Opti-MEM, LentiCRISPRv2 sgDCAF15 plasmid and lentiviral packaging plasmids. Lentivirus was then used to infect the HCT-116 RBM23–3xFLAG cell line. Cells were selected with 2 μg/ml puromycin for 7 days.
METHOD DETAILS
Protein Expression and Purification
For protein expression, the human genes encoding full-length RBM39 (GenBank: NM_184234.2), full-length DCAF15 (GenBank: NM_138353), full-length DDB1 (GenBank: NM_001923), and Rbx1 lacking the first 14 amino acids (Rbx1Δ1–14) were cloned into pFastBac vectors for the production of N-terminal His-tagged or untagged proteins. For Cul4a expression, attempts to generate baculovirus of full-length Cul4 were unsuccessful until synonymous mutations were introduced into the nucleotide sequence encoding the first 44 amino acids. Cul4a (GenBank: NM_001008895) amino acids 2–44 were mutated to the following sequence: GCAGA CGAAGCACCACGTAAAGGTAGTTTTTCAGCCTTAGTTGGTCGTACTAATGGTTTAACGAAACCAGCAGCATTAGCAGCAGCACCGG CAAAACCAGGTGGTGCAGGCGGCTCCAAGAAGCTGGTG. Mutated Cul4a was then His-tagged at the N terminus and cloned into pFastBac for SF9 insect cell expression. Baculoviruses were generated according to vendor’s instructions, and used to infect SF9 cells for expression. Cells were harvested 48 hr after baculovirus infection.
For purification of His-tagged RBM39, cells were resuspended in buffer containing 40 mM Tris, 1M NaCl, 0.1% NP40, 5 mM DTT, 1x SigmaFAST Protease Inhibitors, pH 7.5. The cell suspension was sonicated for 12 cycles at 10 s per cycle, with 30 s of cooling between cycles at 4°C. Lysate was centrifuged at 35000 rpm for 30 min, and incubated with Ni-NTA agarose for 1 hr with rotation at 4°C. Beads were washed three times in wash buffer (40 mM Tris, 1M NaCl, 20 mM imidazole, 5 mM DTT, pH 7.5). Protein was then eluted with elution buffer (40 mM Tris, 1M NaCl, 300 mM imidazole, 5 mM DTT, pH 7.5) and concentrated to 500 μL. Concentrated protein was injected onto a Superdex200 10/300 column (GE Healthcare). RBM39 protein eluted at 14 mL and was stored at −80°C in 40 mM Tris, 1M NaCl, 1mM TCEP, pH 7.5.
For purification of the DCAF15-DDB1-DDA1 complex, His-tagged DCAF15 was co-expressed with untagged DDB1 and DDA1 in SF9 cells. Cells were resuspended in buffer containing 20 mM Tris, 200 mM NaCl, 0.5% NP40, 1mM TCEP, 1x SigmaFAST protease inhibitors, pH 7.5. Cell suspension was sonicated for 12 cycles at 10 s per cycle, with 30 s of cooling between cycles at 4°C. Lysate was clarified by centrifugation at 35000 rpm for 30 min, and incubated with Ni-NTA agarose for 1 hr at 4°C. Beads were washed three times in wash buffer (20 mM Tris, 200 mM NaCl, 20 mM imidazole, 1mM TCEP, pH 7.5) and eluted with elution buffer (20 mM Tris, 200 mM NaCl, 300 mM imidazole, 1 mM TCEP, pH 7.5). Protein was concentrated and injected onto a Superdex200 Increase 10/300 column (GE Healthcare). The DCAF15-DDB1-DDA1 complex eluted at 13 mL. This peak was collected and further purified by anion exchange chromatography using a HiTrapQ column (GE Healthcare). The protein complex was immediately flash-frozen and stored at 80°C.
For purification of the Cul4a-Rbx1 complex, synonymously mutated His-Cul4a was coexpressed with His-Rbx1Δ1–14 in SF9 cells. Cells were resuspended in buffer containing 50 mM Tris, 200 mM NaCl, 0.1% Triton X-100, 1mM TCEP, and 1x SigmaFAST Protease Inhibitors, pH 8.0. Cell suspension was sonicated for 12 cycles at 10 s per cycle, with 30 s of cooling between cycles at 4°C. Lysate was centrifuged at 35000 rpm for 30 min, and incubated with Ni-NTA agarose for 1 hr with rotation at 4°C. Beads were washed with wash buffer (50 mM Tris, 200 mM NaCl, 10 mM imidazole, 1mM TCEP, pH 8.0) and eluted with elution buffer (50 mM Tris, 200 mM NaCl, 300 mM imidazole, 1mM TCEP, pH 8.0). Eluate was injected onto a Superdex200 Increase 10/300 column (GE Healthcare), and the protein complex eluted at 13.8 mL. Protein was concentrated and stored at −80°C in 50 mM Tris, 200 mM NaCl, 1mM TCEP, pH 8.0.
For FRET assays, RBM39 amino acids 250–330 or RBM23 amino acids 210–330 were tagged with an N-terminal 6xHis tag and C-terminal 3xFLAG tag. Constructs were transformed into Escherichia coli BL21(DE3), and cultures were grown at 37°C to an OD600 of 0.6–0.8. IPTG was added to a final concentration of 0.4 mM, and the cultures were incubated overnight at 16°C. Bacteria was pelleted and lysed in a buffer containing 40 mM Tris (pH 7.5), 225 mM NaCl, 1mM TCEP, and 1x SigmaFAST protease inhibitors. Cell suspension was sonicated, centrifuged at 35000 rpm for 30 min, filtered, and incubated with Ni-NTA agarose (QIAGEN) for 2h with rotation at 4°C. Beads were washed three times with wash buffer (40 mM Tris, 225 mM NaCl, 1 mM TCEP, 20 mM imidazole, pH 7.5) and eluted with elution buffer (40 mM Tris, 225 mM NaCl, 1 mM TCEP, 300 mM imidazole, pH 7.5). His tags were removed by overnight cleavage at 4°C with TEV protease. Cleaved proteins were incubated with nickel-NTA resin to remove cleaved His tags, and further purified by size-exclusion chromatography using a Superdex200 Increase 10/300 column (GE Healthcare). The protein eluted at 18–19 mL, and was stored at −80°C in 40 mM Tris, 225 mM NaCl, 1 mM TCEP, pH 7.5.
Ubiquitination Assays
Recombinant human Cul4a-Rbx1(Δ1–14), DCAF15-DDB1-DDA1, and RBM39 (full-length) were combined with E1 enzyme (UBA1, Boston Biochem), E2 enzyme (UbcH5a and UbcH5b, Boston Biochem), and ubiquitin (Boston Biochem). For E2 enzyme optimizations (Figure 1C), individual enzymes from the E2 panel (Enzo) were added to the reaction at concentrations recommended by the vendor. Final concentrations of proteins in the ubiquitination reaction were the following: 0.2 μM UBA1, 0.5 μM UbcH5a, 0.5 μM UbcH5b, 0.8 μM Cul4a-Rbx1, 1 μM DCAF15-DDB1-DDA1, 0.1 μM RBM39, and 100 μM ubiquitin. Proteins were incubated at 30°C for 1.5hr with agitation in a reaction buffer containing 50 mM HEPES (pH 7.5), 5 mM MgCl2, 5 mM ATP, 75 mM sodium citrate, 0.1% Tween20, 1x energy regeneration solution (Boston Biochem) and 0.05% BSA. Reaction mixtures were quenched with loading dye and DTT, followed by SDS-PAGE and immunoblot analysis using RBM39 antibody (Atlas, 1:3000).
Immunoprecipitation Assays
Anti-FLAG antibody (Clone M2, Sigma-Aldrich) was coupled to Dynabeads M-270 epoxy (Life Technologies) at the concentration of 10 μg of antibody per mg of beads. RBM39 (GenBank: NM_184234.2) and RBM23 (GenBank: NM_018107.4) sequences were cloned into the pcDNA3.1+ vector and tagged with a C-terminal HA tag. 293T cells were seeded in 6-well plates at a density of 0.3 million cells per well, and allowed to attach overnight. For each well, transfection complexes were prepared by combining 200 μL Opti-MEM, 3 μL TransIT LT1 (Mirus), and 1 μg DNA (200 ng pcDNA3-RBM23-HA or pcDNA3-RBM39-HA, 400 ng pcDNA3-DCAF15–3xFLAG, 350 ng pcDNA3–3xFLAG, and 50 ng pmaxGFP). Forty-eight hours after transfection, cells were treated with 1 μM MLN-4924 for 2 hr followed by 10 μM indisulam for 4 hr. Cell pellets were collected, washed with PBS, and resuspended in immunoprecipitation (IP) buffer containing 50 mM NaCl, 50 mM NaPO4, 50 mM sodium citrate, 20 mM HEPES pH 7.4, 0.1% Tween, and 1 3 SigmaFAST protease inhibitor (Sigma-Aldrich). Lysates were centrifuged at 12000 rpm for 10 min at 4°C. Clarified lysates were incubated with 1.5 mg of anti-FLAG magnetic beads for 1 min at 4°C with rotation. Beads were washed three times with IP buffer, followed by elution with 1 mg/ml 3xFLAG peptide (Sigma-Aldrich). Inputs and eluates were analyzed by SDS-PAGE and western blot analysis using FLAG-HRP (1:3000, Clone M2, Sigma-Aldrich) and HA-HRP (1:3000, Clone 6E2, Cell Signaling Technology) antibodies.
TR-FRET Assay
TR-FRET reactions were performed in triplicate in a 384-well plate at a final volume of 10 μL/well. Final assay concentrations were the following: 5 ng/μL anti-FLAG M2-XL665 (CisBio), 1 nM anti-6xHis LANCE Eu-W1024 (Perkin Elmer), 0.1 μM RBM39250–330-3xFLAG or RBM23210–330-3xFLAG protein, and 0.1 μM His-DCAF15/DDB1/DDA1 complex. Reactions were carried out in 25 mM HEPES pH 7.4, 100 mM NaCl, 0.1 mg/ml BSA, 0.005% Tween, and 0.5 mM TCEP. Samples were treated with DMSO or indisulam, incubated 5 min at room temperature, and read using a Cytation 5 instrument (Biotek). The Eu-labeled donor was excited at 360 nm, and its emission was monitored at 620 nm. Emission of the XL665-labeled acceptor was monitored at 665 nm. The TR-FRET ratio was calculated as the signal at 665 nm divided by the signal at 620 nm, and plotted as the mean ± SEM. The EC50 was calculated from a four parameter dose-response curve generated with the Prism 8 software package.
Error-Prone PCR Mutagenesis Screen
DCAF15 (GenBank: NM_138353) was amplified from a plasmid using Taq polymerase (NEB), which lacks proofreading activity. DCAF15 was amplified by PCR in a 50 μl reaction with 10 ng of plasmid template, 200 μM dNTP and 20 μM of primers using the following cycling conditions: 95°C for 30 s, 59°C for 1 minute, and 72°C for 30 s for 25 cycles followed by a 72°C 3 min extension cycle. These conditions were predicted to yield one mutation per DCAF15 ORF. PCR primers were the following: 5′TCTACTAGAGG ATCTATTTCCGGTGNNNNNNGCCGCCatggcgcccagc3′ and 5′ AGGGGGGGGGGAGGGAGAGGGGCGGTTACTTGTACAG 3′. Of note, the forward primer contained a NNNNNN which could be used as a barcode sequence, if necessary. The resulting PCR product was cloned using Gibson assembly to yield approximately 55,000 colonies. A plasmid prep representing the DCAF15 mutant library was used to generate lentivirus. 7.5 million cells were infected with the resulting lentivirus at a multiplicity of infection (MOI) of 0.20 to yield 1.5 million infected cells. 200,000 cells were sorted by FACS for a narrow, average intensity of GFP fluorescence. After expansion, 350,000 cells were plated on a 10 cm dish and treated with either DMSO or 10 μM indisulam with media and compound changes every three days. Nine million cells were collected after 6 days of incubation for the DMSO condition. Eleven million cells were collected from the indisulam condition 16 days after starting treatment. DCAF15 derived from the plasmid was amplified from genomic DNA using Herculase polymerase (Agilent) in 96 50-μl reactions, each with 100 ng of genomic DNA for 27 cycles (DMSO condition) and 30 cycles (Indisulam condition) using the following primers: 5′ CGTTTAGTGAACCGTCAGATCGCCTGGAGA 3′ and 5′ CAGCAGGCTGAAGTTAGTAGCTCCGCTTC 3′. The PCR product was purified using the Clontech nucleospin kit, and the amplicon was used for next generation sequencing.
The quality of sequencing reads was evaluated using the NGS QC Toolkit (v2.3.3) (Patel and Jain, 2012) and high-quality reads were extracted. Burrows-Wheeler Aligner (BWA, v0.7.15a) (Li et al., 2009) was employed to map the reads onto the DCAF15 protein coding sequence (GenBank: NM_138353.3), and SAMtools (v1.4) (Li et al., 2009) was employed to sort the alignments by position. The reads per position and base were counted and the base change frequencies were calculated using SAMtools mpileup and a custom Perl script. The significances of the base change differences between samples were calculated by Fisher’s exact test in R statistical package.
Compound-induced degradation assays
For monitoring indisulam-dependent degradation of mutants, RBM39 and RBM23 sequences were cloned into the pcDNA3.1+ vector and tagged with a C-terminal HA tag. 293Ts were seeded in a 6-well plate at 0.5 million cells per well one day prior to transfection. For each well, TransIT:DNA complexes were prepared by combining 200 μL Opti-MEM, 3 μL TransIT-LT1 (Mirus), and 1 μg of DNA (200 ng pcDNA3-RBM23-HA or pcDNA3-RBM39-HA, 400 ng pcDNA3-DCAF15–3xFLAG, 350 ng pcDNA3–3xFLAG, and 50 ng pmaxGFP). Complexes were allowed to incubate at room temperature for 15 min, and then added dropwise to 293T cells. Forty-eight hours after transfection, cells were treated with the indicated doses of indisulam or indoleacetic acid (IAA). Cells were lysed in 1% SDS, with benzonase (Sigma) diluted 1:10,000 in Buffer A (50 mM HEPES 7.4, 10 mM KCl, and 2 mM MgCl2). Samples were analyzed by western blot using HA-HRP antibody (1:3000, 6E2, Cell Signaling Technology) and FLAG-HRP antibody (1:3000, Clone M2, Sigma-Aldrich).
RNA Sequencing and Bioinformatic Analysis of Splicing Events
RNA sequencing analysis was performed in HCT116 cell lines in which RBM23 was endogenously tagged with 3xFLAG or AID-3xFLAG at the C terminus. In brief, cells were treated with DMSO, 10 μM indisulam, or 100 μM IAA for 12 hours. RNA was extracted using the Quick-RNA MiniPrep kit (Zymo) and treated with TURBO DNase (Ambion) to remove genomic DNA. Following mRNA enrichment, cDNA libraries were generated using the TruSeq Stranded mRNA Sample Prep Kit (Illumina). Strand-specific single-end RNA sequencing was performed using Illumina NextSeq. The quality of the sequencing reads was evaluated using NGS QC Toolkit (v2.3.3) (Patel and Jain, 2012) and high-quality reads were extracted. The data files of the reference genome, hg19, were downloaded from Illumina iGenomes (https://support.illumina.com/sequencing/sequencing_software/igenome.html). The quality of the RNA-sequencing libraries was estimated by mapping the reads onto human transcript and ribosomal RNA sequences (Ensembl release 89) using Bowtie (v2.3.2) (Langmead and Salzberg, 2012). STAR (Dobin et al., 2013) was employed to align the reads onto the reference genome and HTSeq Python package (Anders et al., 2015) was employed to count reads per gene. DESeq R Bioconductor package (Anders and Huber, 2010; Gentleman et al., 2004) was used to normalize read counts and identify differentially expressed (DE) genes. KEGG (Kanehisa et al., 2017) pathway data was downloaded using KEGG API (https://www.kegg.jp/kegg/rest/keggapi.html) and gene ontology (GO) data was downloaded from NCBI FTP (ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/gene2go.gz). The enrichment of DE genes to pathways and GOs were calculated by Fisher’s exact test in R statistical package. Differential alternative splicing events were identified using SpliceFisher (https://github.com/jiwoongbio/SpliceFisher).
QUANTIFICATION AND STATISTICAL ANALYSIS
DCAF15 mutagenesis sequencing data was analyzed using the NGS QC Toolkit (v2.3.3), Burrows-Wheler Aligner (BWA, v0.7.15a), and SAMtools. Significance was calculated by Fisher’s exact test. For RNA sequencing analyses, data was analyzed using NGS QC Toolkit (v2.3.3), Bowtie (v2.23.2), HTSeq python package, and DESeq R Bioconductor package. Significance was calculated by Fisher’s exact test. For FRET assays, experiments were performed in triplicate and plotted as mean ± SEM. The EC50 was calculated from a four parameter dose response curve generated with the Prism 8 software package.
DATA AND CODE AVAILABILITY
Original data for the DCAF15 mutagenesis screen and RNA sequencing from this study have been deposited at the National Center for Biotechnology Information Sequence Read Archive (Accession number: SRP222366).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-RBM39 | ProteinTech | Cat# HPA001591, RRID:AB_1079749 |
| Anti-FLAG (M2), HRP-linked | Sigma-Aldrich | Cat# A8592, RRID:AB_439702 |
| Anti-HA (6E2), HRP-linked | Cell Signaling Technology | Cat# 2999, RRID:AB_1264166 |
| Anti-V5, HRP-linked | Sigma-Aldrich | Cat# V2260, RRID:AB_261857 |
| Anti-β-Actin (C4), HRP-linked | Santa Cruz | Cat# sc-47778 HRP, RRID:AB_2714189 |
| Anti-DDB1 (D4C8) | Cell Signaling Technology | Cat# 6998, RRID:AB_10829458 |
| Anti-DDA1 | ProteinTech | Cat# 14995-1-AP, RRID:AB_10859095 |
| Anti-FLAG (M2), XL665-linked | CisBio | Cat# 61FG2XLA, RRID:AB_2811260 |
| Anti-FLAG (M2) | Sigma-Aldrich | Cat# F3165, RRID:AB_259529 |
| Anti-His, Europium-labeled | Perkin Elmer | Cat# AD0205, RRID:AB_2811261 |
| Bacterial and Virus Strains | ||
| DH5α Competent E. coli | Generated in-lab | N/A |
| Stbl3 Competent E. coli | Generated in-lab | N/A |
| DH10Bac Competent E. coli | Generated in-lab | N/A |
| BL21Competent E. coli | Generated in-lab | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Indisulam | MedKoo Biosciences | Cat# 201540, CAS:165668-41-7 |
| Chloroquinoxaline sulfonamide (CQS) | NCI Drug Collection | CAS:97919-22-7 |
| Tasisulam | AdooQ | Cat# A13070, CAS:519055-62-0 |
| 3-Indoleacetic acid (IAA) | Sigma-Aldrich | Cat# I2886, CAS:87-51-4 |
| MLN-4924 | Active Biochem | Cat# A-1139, CAS:905579-51-3 |
| G-418 disulfide salt | Sigma-Aldrich | Cat# A1720 |
| 3XFLAG peptide | Sigma-Aldrich | Cat# F4799 |
| Ubiquitin Activating Enzyme (E1) | Boston Biochem | Cat# E-305 |
| UbcH5a/UBE2D1 | Boston Biochem | Cat# E2-616 |
| UbcH5b/UBE2D2 | Boston Biochem | Cat# E2-622 |
| Ubiquitin | Boston Biochem | Cat# U-100H |
| Energy Regeneration Solution | Boston Biochem | Cat# B-10 |
| Ubiquitinylation kit (E2 panel) | Enzo | Cat# BML-UW9920-0001 |
| Benzonase | Sigma | Cat# E1014 |
| Herculase II Fusion DNA polymerase | Agilent Technologies | Cat# 600679 |
| Taq DNA polymerase | New England BioLabs | Cat# M0273S |
| TURBO DNase | Ambion | Cat# AM2238 |
| Critical Commercial Assays | ||
| Quick-RNA Miniprep Kit | Zymo Research | Cat# R1054 |
| Deposited Data | ||
| DCAF15 mutagenesis screen data | This paper | Data available under NCBI SRA accession SRP222366 |
| RNA sequencing data | This paper | Data available under NCBI SRA accession SRP222366 |
| Experimental Models: Cell Lines | ||
| HCT-116 cell line | ATCC | Cat# CCL-247, RRID:CVCL_0291 |
| Lenti-X 293T cell line | Clontech | Cat# 632180 |
| SF9 insect cell line | Gift from H. Yu lab (UTSW) | N/A |
| Recombinant DNA | ||
| px330 | Addgene | Cat# 42230, RRID:Addgene_42230 |
| pcDNA3.1+ | Gift from McKnight lab | N/A |
| pGEM-T-Easy | Promega | Cat# A3600 |
| pFastBac1 | Gift from Brown and Goldstein lab |
N/A |
| pMD2.G | Addgene | Cat# 12259, RRID:Addgene_12259 |
| psPAX2 | Addgene | Cat# 12260, RRID:Addgene_12260 |
| lentiCRISPR v2 | Addgene | Cat# 52961, RRID:Addgene_52961 |
| Software and Algorithms | ||
| NGS QC Toolkit (v2.3.3) | Patel and Jain, 2012 | http://www.nipgr.ac.in/ngsqctoolkit.html |
| Burrows-Wheeler Aligner (BWA, v0.7.15a) | Li and Durbin, 2009 | http://bio-bwa.sourceforge.net |
| SAMtools (v1.4) | Li et al., 2009 | http://www.htslib.org |
| Fisher’s Exact Test | R | https://www.r-project.org |
| hg19 reference genome | Illumina iGenomes; UCSC | https://support.illumina.com/sequencing/sequencing_software/igenome.html |
| Human transcript and ribosomal RNA sequences | Ensembl release 89 | https://www.ensembl.org/biomart/martview/34f859a4d8b6147d0a4bda896fadd954 |
| STAR (Spliced Transcripts Alignment to a Reference) (v2.5.2b) |
Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| Bowtie (v2.3.2) | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Illumina iGenomes | Illumina | https://support.illumina.com/sequencing/sequencing_software/igenome.html |
| HTSeq Python package | Anders et al., 2015 | https://htseq.readthedocs.io/en/release_0.11.1/ |
| DESeq2 R Bioconductor package |
Anders and Huber, 2010; Gentleman et al., 2004 |
https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| KEGG pathway | Kanehisa et al., 2017 | https://www.kegg.jp/kegg/rest/keggapi.html |
| Gene ontology | NCBI FTP | ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/gene2go.gz |
| Prism 8 | Graphpad | https://www.graphpad.com/scientific-software/prism/ |
| SpliceFisher | Han et al., 2017 | https://github.com/jiwoongbio/SpliceFisher |
| Other | ||
| SF900 II SFM (Serum-free media) | Life Technologies | Cat# 10902104 |
| SIGMAFAST Protease Inhibitor Tablets | Sigma-Aldrich | Cat# S8820 |
| Ni-NTA Agarose | QIAGEN | Cat# 30230 |
| Dynabeads M-270 Epoxy | Life Technologies | Cat# 14302D |
| TransIT®-LT1 Transfection Reagent | Mirus Bio | Cat# MIR 2300 |
| TransIT®-Lenti Transfection Reagent | Mirus Bio | Cat# MIR 6600 |
| Opti-MEM® Reduced Serum Medium | Invitrogen | Cat# 31985–070 |
| Superdex 200 Increase 10/300 GL Column | GE Healthcare | Cat# 28990944 |
| HiTrap Q HP anion exchange chromatography column | GE Healthcare | Cat# 29051325 |
Highlights.
RBM39 is recruited to DCAF15 by indisulam through its RRM2 domain
DCAF15 mutations in Q232 and D475 prevent indisulam-dependent RBM39 recruitment
RBM23 is an indisulam-dependent neo-substrate for CRL4-DCAF15
Indisulam-related mRNA expression and RNA splicing changes are RBM39 dependent
ACKNOWLEDGMENTS
The authors thank Nicholas Gaskill for his optimizations for the in vitro ubiquitination reaction, including buffer composition, protein reagent preparation, design of constructs for insect cell expression, and protocol development. We also thank Thu Nguyen for helpful discussions regarding RBM23 and David McFadden for reviewing the manuscript. T.C.T. is supported by a McKnight graduate student fellowship. This work was supported by grants issued to D.N. from the Welch Foundation (I-1879) and the National Cancer Institute (R37CA226771 and RO1CA217333).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.09.079.
REFERENCES
- Anders S, and Huber W. (2010). Differential expression analysis for sequence count data. Genome Biol. 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W. (2015). HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain PP, Lopez-Girona A, Miller K, Carmel G, Pagarigan B, Chie-Leon B, Rychak E, Corral LG, Ren YJ, Wang M, et al. (2014). Structure of the human Cereblon-DDB1-lenalidomide complex reveals basis for responsiveness to thalidomide analogs. Nat. Struct. Mol. Biol 21, 803–809. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan KA, An J, Nowak RP, Yuan JC, Fink EC, Berry BC, Ebert BL, and Fischer ES (2018). Thalidomide promotes degradation of SALL4, a transcription factor implicated in Duane Radial Ray syndrome. eLife 7, e38430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowhan DH, Hong EP, Auboeuf D, Dennis AP, Wilson MM, Berget SM, and O’Malley BW (2005). Steroid hormone receptor coactivation and alternative RNA splicing by U2AF65-related proteins CAPERalpha and CAPERbeta. Mol. Cell 17, 429–439. [DOI] [PubMed] [Google Scholar]
- Dvinge H, Kim E, Abdel-Wahab O, and Bradley RK (2016). RNA splicing factors as oncoproteins and tumour suppressors. Nat. Rev. Cancer 16, 413–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer ES, Böhm K, Lydeard JR, Yang H, Stadler MB, Cavadini S, Nagel J, Serluca F, Acker V, Lingaraju GM, et al. (2014). Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 512, 49–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. (2004). Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5, R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guharoy M, Bhowmick P, Sallam M, and Tompa P. (2016). Tripartite degrons confer diversity and specificity on regulated protein degradation in the ubiquitin-proteasome system. Nat. Commun 7, 10239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han T, Goralski M, Gaskill N, Capota E, Kim J, Ting TC, Xie Y, Williams NS, and Nijhawan D. (2017). Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science 356, eaal3755. [DOI] [PubMed] [Google Scholar]
- Huang X, and Dixit VM (2016). Drugging the undruggables: exploring the ubiquitin system for drug development. Cell Res. 26, 484–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Arias EE, Chen J, Harper JW, and Walter JC (2006). A family of diverse Cul4-Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol. Cell 23, 709–721. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Furumichi M, Tanabe M, Sato Y, and Morishima K. (2017). KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45 (D1), D353–D361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krönke J, Udeshi ND, Narla A, Grauman P, Hurst SN, McConkey M, Svinkina T, Heckl D, Comer E, Li X, et al. (2014). Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 343, 301–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krönke J, Fink EC, Hollenbach PW, MacBeth KJ, Hurst SN, Udeshi ND, Chamberlain PP, Mani DR, Man HW, Gandhi AK, et al. (2015). Lenalidomide induces ubiquitination and degradation of CK1a in del(5q) MDS. Nature 523, 183–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SC, and Abdel-Wahab O. (2016). Therapeutic targeting of splicing in cancer. Nat. Med 22, 976–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu G, Middleton RE, Sun H, Naniong M, Ott CJ, Mitsiades CS, Wong KK, Bradner JE, and Kaelin WG Jr. (2014). The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 343, 305–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matyskiela ME, Lu G, Ito T, Pagarigan B, Lu CC, Miller K, Fang W, Wang NY, Nguyen D, Houston J, et al. (2016). A novel cereblon modulator recruits GSPT1 to the CRL4(CRBN) ubiquitin ligase. Nature 535, 252–257. [DOI] [PubMed] [Google Scholar]
- Matyskiela ME, Couto S, Zheng X, Lu G, Hui J, Stamp K, Drew C, Ren Y, Wang M, Carpenter A, et al. (2018). SALL4 mediates teratogenicity as a thalidomide-dependent cereblon substrate. Nat. Chem. Biol 14, 981–987. [DOI] [PubMed] [Google Scholar]
- Olma MH, Roy M, Le Bihan T, Sumara I, Maerki S, Larsen B, Quadroni M, Peter M, Tyers M, and Pintard L. (2009). An interaction network of the mammalian COP9 signalosome identifies Dda1 as a core subunit of multiple Cul4-based E3 ligases. J. Cell Sci 122, 1035–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel RK, and Jain M. (2012). NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS One 7, e30619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petzold G, Fischer ES, and Thomä NH (2016). Structural basis of lenalidomide-induced CK1α degradation by the CRL4(CRBN) ubiquitin ligase. Nature 532, 127–130. [DOI] [PubMed] [Google Scholar]
- Salami J, and Crews CM (2017). Waste disposal—an attractive strategy for cancer therapy. Science 355, 1163–1167. [DOI] [PubMed] [Google Scholar]
- Shabek N, Ruble J, Waston CJ, Garbutt KC, Hinds TR, Li T, and Zheng N. (2018). Structural insights into DDA1 function as a core component of the CRL4-DDB1 ubiquitin ligase. Cell Discov. 4, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers QL, Petzold G, Bunker RD, Renneville A, S1abicki M, Liddicoat BJ, Abdulrahman W, Mikkelsen T, Ebert BL, and Thomä NH (2018). Defining the human C2H2 zinc finger degrome targeted by thalidomide analogs through CRBN. Science 362, eaat0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehara T, Minoshima Y, Sagane K, Sugi NH, Mitsuhashi KO, Yamamoto N, Kamiyama H, Takahashi K, Kotake Y, Uesugi M, et al. (2017). Selective degradation of splicing factor CAPERα by anticancer sulfonamides. Nat. Chem. Biol 13, 675–680. [DOI] [PubMed] [Google Scholar]
- Wang E, Lu SX, Pastore A, Chen X, Imig J, Chun-Wei Lee S., Hockemeyer K, Ghebrechristos YE, Yoshimi A, Inoue D, et al. (2019). Targeting an RNA-Binding Protein Network in Acute Myeloid Leukemia. Cancer Cell 35, 369–384.e367. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original data for the DCAF15 mutagenesis screen and RNA sequencing from this study have been deposited at the National Center for Biotechnology Information Sequence Read Archive (Accession number: SRP222366).