

Abstract
Chitosan (CS) aqueous salt blended with polyvinyl alcohol (PVA) nanofibre mats was prepared by electrospinning. CS was dissolved with hydroxybenzotriazole (HOBt), thiamine pyrophosphate (TPP) and ethylenediaminetetraacetic acid (EDTA) in distilled water without the use of toxic or hazardous solvents. The CS aqueous salts were blended with PVA at different weight ratios, and the effect of the solution ratios was investigated. The morphologies and mechanical and swelling properties of the generated fibres were analysed. Indirect cytotoxicity studies indicated that the CS/PVA nanofibre mats were non‐toxic to normal human fibroblast cells. The CS‐HOBt/PVA and CS‐EDTA/PVA nanofibre mats demonstrated satisfactory antibacterial activity against both gram‐positive and gram‐negative bacteria, and an in vivo wound healing test showed that the CS‐EDTA/PVA nanofibre mats performed better than gauze in decreasing acute wound size during the first week after tissue damage. In conclusion, the biodegradable, biocompatible and antibacterial CS‐EDTA/PVA nanofibre mats have potential for use as wound dressing materials.
Keywords: Chitosan; Electrospinning; Nanofibre; Polyvinyl alcohol; Wound healing
Introduction
Chitosan (CS) is a natural polysaccharide with biodegradable, biocompatible properties and non toxic effects; therefore, it has been proposed as a safer material for use in biomedical applications such as tissue engineering, delivery of pharmaceutical agent and wound dressing 1, 2. CS has been widely investigated as wound dressing material because it can function as proliferation promoters, antibacterial agents and macrophage activator. CS gradually depolymerises to N‐acetyl‐d‐glucosamine, which initiates fibroblast proliferation, aids in the ordered deposition of collagen and stimulates increased synthesis of natural hyaluronic acid at the wound site. CS is a haemostatic agent, which helps to promote natural blood clotting and blocks nerve endings to reduce pain. Recently, CS‐based materials have been prepared as fibres, hydrogels, membranes, sponges and scaffolds for wound dressing applications 3, 4.
Electrospun nanofibres are appropriate for use as a wound dressing material because of their useful properties, which include oxygen permeability, high porosity, variable pore size distribution and a high surface‐to‐volume ratio that can promote haemostasis and absorb wound exudates. Furthermore, the morphology of electrospun nanofibres is similar to that of the natural extracellular matrix (ECM) in the skin that promotes cell adhesion, migration and proliferation 3, 5. The fabrication of CS nanofibres via electrospinning techniques has recently been actively investigated for the development of wound dressings. CS nanofibres have been created from the electrospinning of pure CS, CS derivatives and CS blended with other polymers. However, some organic solvents or toxic acids, such as trifluoroacetic acid (TFA) (6), chloroform (7) and acetic acid 8, 9, 10 may be leftover from the CS nanofibre fabrication process, and these trace residues may have undesirable effects on wound healing. The types of solvents used are often classified as toxic and hazardous and have potential long‐term impacts on the environment and the health of the user. To eliminate toxicity from residual solvent exposure, water‐soluble CS has been used to prepare nanofibres using quaternary CS (11) and carboxyethyl chitosan/polyvinyal alcohol (CS/PVA) (12) for wound dressing applications.
We recently prepared CS as aqueous salts to allow the formation of nanofibres without the use of organic solvents or toxic acids. Nanofibres were generated using CS‐hydroxybenzotriazole (HOBt)/PVA (13) and CS‐ethylenediaminetetraacetic acid (EDTA)/ PVA (14) by blending the CS salts with PVA. Another salt was generated by dissolving CS with thiamine pyrophosphate (TPP) in distilled water. Owing to the phosphate groups of TPP, the molecule can form a salt with the amine groups of CS, improving the polymer's aqueous solubility. The amine groups of TPP, and especially that of thiazolium can be deprotonated, and are always positive, even at physiological pH. Chitosan thiamine pyrophosphate (CS‐TPP) was successfully prepared as a novel carrier for siRNA delivery (15).
In this study, CS‐HOBt, CS‐TPP and CS‐EDTA blended in PVA solution were prepared as nanofibre mats via the electrospinning technique with different CS/PVA weight ratios. The morphology, structure, mechanical properties and swellability of the nanofibre mats were characterised, and the effect of the viscosity, conductivity and surface tension of the electrospinning solution on the morphology of these nanofibre mats was evaluated. (3‐(4,5‐Dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide) MTT assays using normal human fibroblast cells were performed to study the cytotoxicity of the CS/PVA nanofibre mats. The antibacterial activities of the mats against gram‐positive and gram‐negative bacteria were investigated, and the in vivo healing activity of these nanofibre mats was evaluated using an animal model.
EXPERIMENTAL
Materials
CS (degree of deacethylation = 0·85, molecular weight = 110 kDa), hydroxybenzotriazole monohydrate (HOBt·H2O), TPP and EDTA were purchased from Sigma‐Aldrich Chemical Company, Saint Louis, MO. PVA (degree of polymerisation ≅ 1600, degree of hydrolysis ≅97·5–99·5 mol%) was purchased from Fluka, Buchs, Switzerland. Normal human foreskin fibroblast (NHF) cells were obtained from the American Type Culture Collection (ATCC, Rockville, MD). All other reagents and solvents were commercially available and were of analytical grade.
Preparation of spinning solutions
CS solutions (2% w/v) were prepared by dissolving CS with HOBt, TPP and EDTA at weight ratios of 1:1, 1:1 and 2:1, respectively. Briefly, HOBt (2 g), TPP (2 g) or EDTA (1 g) was dissolved with CS (2 g) in 100 ml of distilled water, and the solutions were continuously stirred with a magnetic stirrer at ambient temperature until the solutions became clear. The PVA solution (10% w/v) was prepared by dissolving PVA in distilled water at 80°C and then allowing the solution to stir for 4 h. The 2% CS‐HOBt, CS‐TPP and CS‐EDTA solutions were mixed with a 10% PVA solution at weight ratios of 10/90, 20/80, 30/70, 40/60, 50/50, 60/40, 70/30, 80/20 and 90/10. The viscosity, conductivity and surface tension of the solutions were measured using a Brookfield viscometer (Model DV–III ultra, Brookfield Engineering Laboratories Inc., Middleboro, MA), a EUTECH ECtestr11+ conductivity meter (Eutech Instruments Pte Ltd., Ayer Rajah Crescent, Singapore) and a Drop Shape Analyzer (FTA 100, First Ten Angstroms Inc, Portsmouth, VA), respectively.
Electrospinning process
The solutions were taken up in a 5 ml glass syringe equipped with a 20‐gauge, stainless steel needle (diameter = 0·9 mm) at the nozzle. The needle was connected to the emitting electrode of positive polarity of a Gamma High Voltage Research device. The electric potential was fixed at 15 kV and was electrospun at room temperature. The nanofibres were collected as‐spun on an aluminium sheet that was wrapped on a rotating collector. The solution feed was driven by a syringe pump with a controlled flow rate of approximately 0·25 ml/ h. The collection distance was fixed at approximately 20 cm. The process duration was fixed at 24 h for each CS/PVA weight ratio to provide mats with a 20–30 µm thickness.
Characterisations of nanofibre mats
The morphology of the nanofibre mats was observed under scanning electron microscopy (SEM; Camscan Mx2000, Obducat Camscan Ltd, Cambridge, UK). Randomly selected areas of the fibres were cut into squares and coated with a thin layer of gold. The average diameter of the nanofibre mats was analysed by randomly measuring the diameters of the nanofibres at 100 different points from SEM images using the image analysis software (JMicroVision V.1.2.7, University of Geneva, Geneva, Switzerland).
The tensile strength of the nanofibre mats was evaluated using a texture analyser (TA.XT plus, Stable Micro Systems, Godalming, UK) with a 5‐kg load cell equipped with a tensile grip holder. The samples were cut into a rectangular shape (5 mm × 25 mm2). The thicknesses of these samples ranged from 20 to 30 µm.
The degree of swelling of the nanofibre mats was investigated in phosphate buffer saline (PBS, pH 7·4) at room temperature for 1 h according to Equation (1):
| (1) |
where M is the weight of each sample after submersion in the buffer solution for 1 h, and M d is the initial weight of the sample in its dry state.
Indirect cytotoxicity of nanofibre mats
The cytotoxicity of the nanofibre mats was evaluated on the basis of a procedure adapted from the ISO10993‐5 standard test method (indirect contact) (16). The nanofibre mats were sterilised by ultraviolet radiation for 1 h. The mats were then immersed in a serum‐free medium [SFM; containing Dulbecco's modified Eagle's medium (DMEM), 1% v/v l‐glutamine, 1% v/v lactalbumin and 1% v/v antibiotic and antimycotic formulation] in an incubator for 24 h to produce extraction media of different concentrations (10, 7·5, 5, 2·5 and 1 mg/ml). NHF cells were plated in 100 µl of DMEM, supplemented with 10% fetal bovine serum (FBS), at a density of 8000 cells/well in 96‐well plates. When the cultures reached confluence (typically 48 h after plating), the cells were incubated with extraction media of varying concentrations for 24 h. After treatment, the tested extraction solutions were removed, and the cells were incubated with 100 µl of an MTT‐containing medium (1 mg/ml) for 4 h. Then, the MTT medium was removed, the cells were rinsed with PBS (pH 7·4) and the formazan crystals formed in living cells were dissolved in 100 µl dimethylsulfoxide per well. The relative cell viability (%) was calculated on the basis of the absorbance at 550 nm using a microplate reader (Universal Microplate Analyzer, Model AOPUS01 and AI53601, Packard BioScience, Meriden, CT). The viability of non‐treated control cells was defined as 100%.
Determination of antibacterial activity
The antibacterial activity of nanofibre mats was tested against Staphylococcus aureus ATCC 6538P and Escherichia coli ATCC 10536. For the minimum inhibitory concentration (MIC) test, S. aureus and E. coli were cultivated in tryptone soy broth (TSB) in a shaking incubator at 37°C and 0.14 g for 24 h. The bacterial suspension was diluted until the bacterial concentration reached 1 × 106 colony‐forming unit (CFU)/ml and was pipetted into a 24‐well plate at 1 ml/well. Different amounts of the nanofibre mats (1–10 mg) were placed into wells containing bacterial suspension and were incubated at 37°C for 24 h. The MIC was defined as the minimum concentration of mats for which no growth was observed after a 24‐h incubation. The optical density (OD) at 550 nm was measured using a microplate reader. For the determination of the minimum bactericidal concentration (MBC), the media mixtures from wells with no growth (100 µl) were spread onto agar plates. The MBC was defined as the minimum concentration of mats for which no colony growth was observed on agar plates after a 24‐h incubation at 37°C. The MIC and MBC determinations were carried out in triplicate. Penicillin (1 mg/ml) was used as a positive control.
Wound‐healing activity of nanofibre mats
Male Wistar rats (240–280 g) were used in this study. This study was approved by an Investigational Review Board (Animal Studies Ethics Committee, Faculty of Pharmacy, Silpakorn University, Approval No. 2‐2553). After anesthetisation, the neck area of the dorsal of each rat was shaved and wiped with 70% ethanol. Two wounds were created on the neck area of each rat using a skin biopsy punch (wound area of 0·8 cm2). The wound was treated by placing an equal size of nanofibre mat, gauze and commercial antibacterial gauze dressing (Sofra‐tulle®, Sanofi Aventis, Guildford, UK) (n = 6) over it without removing the test material throughout the study period. The area of each wound was measured every day by the planimetry method until the wound completely healed. The percentage of wound healing is defined in Equation (2).
| (2) |
where A i is the initial wound area and A is the wound area after a fixed time interval.
Statistical analysis
All data for the experiment were collected from triplicate samples and are expressed as the mean ± standard deviation (SD). Statistically significant differences in cell viability and wound area were analysed using the Student's t‐test. The significance level was set at P < 0·05.
Results and discussion
Morphology of nanofibres
The SEM images of CS‐HOBt/PVA, CS‐TPP/PVA and CS‐EDTA/PVA with different weight ratios are shown in Figure 1. When the CS/PVA weight ratio was increased to 30/70, the SEM micrographs demonstrated no beading. However, at CS/PVA 40/60, fibres with beads were observed. The maximum content of CS aqueous salt was 50/50 in the electrospinning mixture. When the CS content exceeded 50/50, nanofibres were unable to be formed; this may have been because of unsuitable solution parameters. Table 1 shows the solution parameters and average diameter of CS‐HOBt/PVA, CS‐TPP/PVA and CS‐EDTA/PVA fibres generated at different CS/PVA weight ratios. When the content of CS salt was increased from 10/90 to 50/50, the average diameter of the CS‐HOBt/PVA, CS‐TPP/PVA and CS‐EDTA/PVA nanofibres decreased from 228 ± 37 to 146 ± 33 nm, from 216 ± 34 to 100 ± 19 nm and from 221 ± 34 to 94 ± 20 nm. This decrease in the average fibre diameter was mainly ruled by solution viscosity and conductivity. The viscosity of the CS‐HOBt/PVA, CS‐TPP/PVA and CS‐EDTA/PVA solutions decreased with increasing CS salt content (Table 1). The changes in the viscosity of the CS‐TPP/PVA and CS‐EDTA/PVA solutions were greater than those of the CS‐HOBt/PVA solution, so the average diameters of the CS‐TPP/PVA and CS‐EDTA/PVA fibres were smaller than those of the CS‐HOBt/PVA nanofibres. The reduced viscosity of solutions affected bead formation when the CS salt content was higher than 30/70. Increases in solution conductivity also contributed to decreases in nanofibre diameter. This result was in good accordance with our previous work (13). The surface tension increased slightly when the CS salt content was increased from 10/90 to 50/50. These differences could have led to bead and droplet formation during the electrospinning process (8). Nanofibres with CS/PVA weight ratios of 30/70 that did not contain beads were selected for further investigation of antibacterial and wound healing activity.
Figure 1.
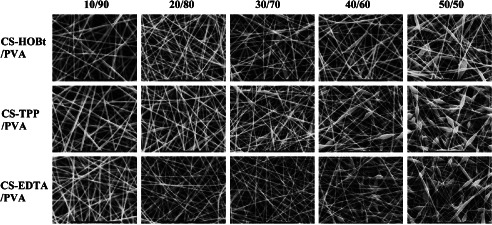
The SEM image of CS/PVA nanofibre mats with different salt and weight ratios.
Table 1.
Electrospun solution parameters of CS/PVA solution and average fibre diameter at each weight ratio
| Samples | Weight ratio | Viscosity (MPa s) | Conductivity (µS/cm) | Surface tension (mN/m) | Average diameter (nm) |
|---|---|---|---|---|---|
| CS‐HOBt/PVA | 10/90 | 531·57 ± 2·08 | 1036·7 ± 0·58 | 43·77 ± 1·04 | 228·06 ± 37·20 |
| 20/80 | 550·23 ± 3·92 | 1078·0 ± 2·65 | 43·63 ± 0·44 | 186·72 ± 32·93 | |
| 30/70 | 529·97 ± 1·99 | 1185·0 ± 3·61 | 44·57 ± 0·23 | 179·34 ± 39·36 | |
| 40/60 | 510·57 ± 2·96 | 1268·3 ± 2·52 | 45·45 ± 0·26 | 176·46 ± 28·86 | |
| 50/50 | 487·20 ± 1·30 | 1500·3 ± 1·53 | 46·48 ± 0·41 | 146·34 ± 33·23 | |
| CS‐TPP/PVA | 10/90 | 568·04 ± 0·38 | 1160·7 ± 9·71 | 46·62 ± 0·37 | 216·42 ± 34·45 |
| 20/80 | 459·54 ± 2·48 | 1268·7 ± 6·51 | 48·62 ± 0·24 | 188·66 ± 33·69 | |
| 30/70 | 346·98 ± 5·29 | 1573·7 ± 9·02 | 49·95 ± 0·58 | 141·51 ± 29·39 | |
| 40/60 | 260·45 ± 0·38 | 1771·3 ± 2·08 | 51·55 ± 0·22 | 126·77 ± 32·46 | |
| 50/50 | 123·54 ± 0·12 | 2110·0 ± 10·0 | 52·61 ± 0·23 | 99·91 ± 18·67 | |
| CS‐EDTA/PVA | 10/90 | 511·43 ± 3·46 | 1011·7 ± 2·52 | 46·46 ± 0·36 | 220·94 ± 34·19 |
| 20/80 | 387·17 ± 1·31 | 1014·3 ± 3·06 | 48·47 ± 0·31 | 184·63 ± 29·13 | |
| 30/70 | 254·74 ± 0·76 | 1022·3 ± 3·79 | 50·00 ± 0·38 | 160·96 ± 32·93 | |
| 40/60 | 223·67 ± 1·31 | 1032·0 ± 3·61 | 51·42 ± 0·83 | 125·87 ± 37·96 | |
| 50/50 | 181·27 ± 0·68 | 1036·7 ± 1·53 | 52·07 ± 1·77 | 94·09 ± 20·27 |
CS/PVA, chitosan aqueous salt blended with polyvinyl alcohol; EDTA, ethylenediaminetetraacetic acid; HOBt, hydroxybenzotriazole; TPP, thiamine pyrophosphate.
The data are expressed as mean ± standard deviation (n = 3).
Mechanical properties of nanofibre mats
When wound dressings interact with the wound, they must possess mechanical properties sufficiently similar to those of skin in order to function properly until the wound is healed. The maximum CS/PVA weight ratio that provided nanofibre mats from the electrospinning process was 50/50. However, the tensile strength was only measured for the nanofibre mats that could be peeled off of the aluminium foil collector. The 10/90‐30/70 CS/PVA nanofibre mats were easily peeled off. As the content of CS salt increased to 40/60, the mats became difficult to peel. Owing to the fibre structure of the 40/60 and 50/50 CS/PVA nanofibre mats, they contained many beads with very fragile fibres. Figure 2 shows the tensile strength of the CS/PVA nanofibre mats generated using different weight ratios. The tensile strength of all of the tested CS/PVA nanofibres mats ranged from 8·9 to 1·5 MPa, whereas the tensile strength of neat PVA nanofibre mats was 12·8 MPa. The results indicated that as the content of CS salt increased, the tensile strength decreased, the diameter of the nanofibres decreased and bead formation occurred in the mats. These findings demonstrated that these CS/PVA nanofibre mats possess tensile strength that is nearly equal to that of commercial microfibrous dressings, which have tensile strengths on the order of 10 MPa (17).
Figure 2.
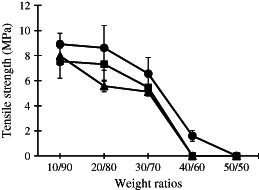
Tensile strength (MPa) of CS/PVA nanofibre mats with different weight ratios; ( ) CS‐HOBt/PVA, (
) CS‐HOBt/PVA, ( ) CS‐TPP/PVA and (
) CS‐TPP/PVA and ( ) CS‐EDTA/PVA. The data are expressed as mean ± standard deviation (n = 3).
) CS‐EDTA/PVA. The data are expressed as mean ± standard deviation (n = 3).
Swelling of nanofibre mats
The swelling of CS is the most important property that characterises its use for wound dressing applications. Figure 3 shows the degree of swelling of CS/PVA nanofibre mats with various weight ratios after immersion in a PBS (pH 7·4) at room temperature for 1 h. All the CS/PVA nanofibre mats demonstrated swelling over 100%, and this effect was slightly increased with increases in the CS content. The swelling of the 10/90 CS/PVA was approximately 100%, which increased to approximately 170% when the content of CS salt reached 50/50 in the blend. CS is a hydrophilic polymer, and water diffuses very rapidly through the material before CS degradation (18). The results indicated that increases in the degree of nanofibre mat swelling depended on the CS salt content. This observation is in agreement with Meng et al., who found that the swelling ratio of polylactic‐co‐glycolic acid (PLGA)/CS nanofibres was higher than that of neat PLGA nanofibres and increased with CS content (19).
Figure 3.
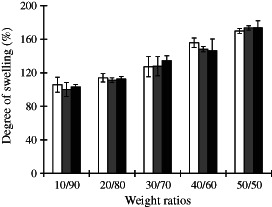
Degree of swelling (%) of CS/PVA nanofibre mats with different weight ratios; ( ) CS‐HOBt/PVA, (
) CS‐HOBt/PVA, ( ) CS‐TPP/PVA and (
) CS‐TPP/PVA and ( ) CS‐EDTA/PVA. The data are expressed as mean ± standard deviation (n = 3).
) CS‐EDTA/PVA. The data are expressed as mean ± standard deviation (n = 3).
Indirect cytotoxicity of nanofibre mats
The indirect cytotoxicity of various concentrations of medium extracts from CS/PVA nanofibre mats composed of 10/90, 30/70 and 50/50 weight ratios is shown in Figure 4. There was no significant decrease in cell viability when the NHF cells were incubated with various concentrations of the extraction media of CS‐HOBt/PVA, CS‐TPP/PVA and CS‐EDTA/PVA nanofibre mats when compared with the control (P < 0·05). The average cell viability decreased slightly when the concentration of the extract increased, but these changes were not statistically significant. From these data, the CS‐HOBt/PVA, CS‐TPP/PVA and CS‐EDTA/PVA nanofibre mats appear to be non‐toxic to NHF cells and have the potential to be developed as wound dressings.
Figure 4.
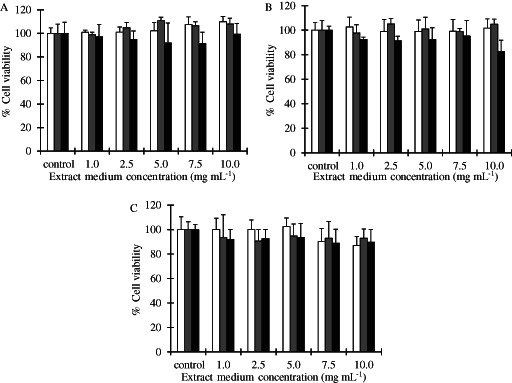
Cell viability in NHF cells at various concentrations of the extract of CS/PVA nanofibre mats (A) CS‐HOBt/PVA, (B) CS‐TPP/PVA and (C) CS‐EDTA/PVA with different weight ratios; ( ) 10/90, (
) 10/90, ( ) 30/70 and (
) 30/70 and ( ) 50/50. The data are expressed as mean ± standard deviation (n = 5).
) 50/50. The data are expressed as mean ± standard deviation (n = 5).
Determination of antibacterial activity
The antibacterial mechanism of CS is generally attributed to the amino group at C‐2 position of the glucosamine residue that is responsible for the cationic nature of CS in acidic conditions (20). When the CS/PVA nanofibre mats were immersed in a TSB solution containing a bacteria suspension, the CS was dissolved and showed a cationic charge in solution. Figure 5 shows the inhibition of bacterial growth by CS/PVA nanofibre mats, compared with neat PVA nanofibre mats, after a 24‐h incubation with S. aureus and E. coli. The CS/PVA nanofibre mats exhibited concentration‐dependent antibacterial activity against S. aureus and E. coli. The 30/70 CS‐EDTA/PVA and 30/70 CS‐HOBt/PVA nanofibre mats inhibited S. aureus growth at 5 and 7·5 mg/ml, respectively, and the 30/70 CS‐TPP/PVA almost inhibited S. aureus growth at 10 mg/ml (Figure 5A). The 30/70 CS‐EDTA/PVA and 30/70 CS‐HOBt/PVA nanofibre mats also inhibited E. coli growth at 5 and 10 mg/ml, respectively, but 30/70 CS‐TPP/PVA did not do so (Figure 5B). The neat PVA nanofibre mats did not show any effect on bacteria growth. The MIC and MBC of the 30/70 CS salt/PVA nanofibre mats are shown in Table 2. The results showed that the different antibacterial activities of the CS/PVA nanofibre mats were dependent on the type of CS salt used. The 30/70 CS‐EDTA/PVA nanofibre mats showed the highest antibacterial activity at 5 mg/ml, likely because of the intrinsic antibacterial activity of EDTA. Several recent publications have indicated that EDTA (alone and in combination with antibiotics) was an effective antimicrobial agent, with a spectrum covering both gram‐positive and gram‐negative bacteria. The antimicrobial activity of an EDTA–CS acetic acid combination has been tested, and the study showed that while EDTA possessed a weaker bacteriostatic effect than CS acetic acid, the combination of CS acetic acid and EDTA elicited synergistic activity against S. aureus (21). All the CS/PVA nanofibre mats tested at 1–10 mg in this study showed higher antibacterial activity against gram‐positive bacteria (S. aureus) than against gram‐negative bacteria (E. coli). The mode of action of antimicrobial agents depends on the nature of the target microorganisms, and differences between the response of gram‐positive and gram‐negative bacteria to various antibiotics are mainly related to their cell wall structure and the arrangement of their outer membrane 20, 22. Different theories have been proposed to explain CS's antimicrobial mode of action. In one mechanism 23, 24, positively charged CS molecules interfere with negatively charged residues on the bacterial surface. CS could interact with the membrane of the bacteria to alter cell permeability. Other studies (25) demonstrated that lower molecular weight CS may have intracellular targets. Dissociated CS molecules in solution could bind with DNA and inhibit the synthesis of mRNA and proteins. Xing et al. investigated the antimicrobial mode of oleoyl‐CS nanoparticles against E. coli and S. aureus and showed that the particles achieve their antibacterial activity by damaging the structures of the cell membrane and by putative binding to extracellular targets such as phosphate groups or to intracellular targets such as DNA and RNA 26, 27.
Figure 5.
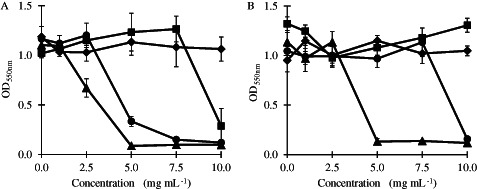
The growth of (a) S. aureus and (b) E. coli in the presence of ( ) 30/70 CS‐HOBt/PVA, (
) 30/70 CS‐HOBt/PVA, ( ) 30/70 CS‐TPP/PVA, (
) 30/70 CS‐TPP/PVA, (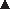 ) 30/70 CS‐EDTA/PVA and (
) 30/70 CS‐EDTA/PVA and ( ) PVA nanofibre mats, after 24‐h incubation. The data are expressed as mean ± standard deviation (n = 3).
) PVA nanofibre mats, after 24‐h incubation. The data are expressed as mean ± standard deviation (n = 3).
Table 2.
Minimum inhibition concentrations (MIC) and minimum bactericidal concentrations (MBC) of 30/70 CS‐HOBt/PVA, CS‐TPP/PVA and CS‐EDTA/PVA nanofibre mats against S. aureus and E. coli compared with PVA nanofibre mats
| Nanofibre mats | MIC (mg/ml) | MBC (mg/ml) | ||
|---|---|---|---|---|
| S. aureus | E. coli | S. aureus | E. coli | |
| PVA | >10·0 | >10·0 | >10·0 | >10·0 |
| 30/70 CS‐HOBt/PVA | 7·5 | 10·0 | 7·5 | 10·0 |
| 30/70 CS‐TPP/PVA | >10·0 | >10·0 | >10·0 | >10·0 |
| 30/70 CS‐EDTA/PVA | 5·0 | 5·0 | 5·0 | 5·0 |
CS, chitosan; EDTA, ethylenediaminetetraacetic acid; HOBt, hydroxybenzotriazole; PVA, polyvinyl alcohol; TPP, thiamine pyrophosphate.
Wound‐healing testing of nanofibre mats
In the wound‐healing test, two full‐thickness round wounds with surface areas of 0·8 cm2 were created on the ventral neck of each rat. Figure 6 shows the images of wound appearances and the percentage wound areas at 1, 4, 7 and 10 days after treatment with 30/70 CS‐HOBt/PVA, 30/70 CS‐TPP/PVA, 30/70 CS‐EDTA/PVA nanofibre mats, gauze (negative control) or commercial antibacterial gauze dressing (positive control). The wound areas decreased gradually and reached 2% of the original wound size after 10 days when treated with the five different wound dressings. Wound closure was achieved within 10 days of all treatments. However, at day 1 after the operation, the percentage wound area of wounds treated with CS‐HOBt/PVA, CS‐EDTA/PVA nanofibre mats and commercial antibacterial gauze dressing was smaller than those of the gauze treatment group (P < 0·05). This might be involved with the high surface area of the nanofibre mats provided high wound exudates absorption. The wounds treated with nanofibre mats were dry and smaller in wound size than the gauze treatment. At 4 days after the operation, wound healing with the CS‐EDTA/PVA nanofibre mat dressing was the greatest (P < 0·05), and in the first week, the 30/70 CS‐EDTA/PVA nanofibre mats exhibited excellent wound healing activity. This result was in accordance with the high antibacterial activity of the CS‐EDTA/PVA nanofibre mats. N‐acetyl‐d‐glucosamine from CS depolymerisation is reported to enhance wound healing by promoting fibroblast proliferation 21, 28. The percentage wound area of all the treatments until recovery was similar, which suggests that wound healing was progressed by some mechanism of the body that is independent of the effects of the treatment. This result is in agreement with a study by Chen et al. on electrospun collagen/CS nanofibrous membranes. The wound areas decreased gradually, and the membranes were found to be better than gauze and collagen sponge in promoting wound healing (16).
Figure 6.
Appearance of would healings and wound area (%) at 1, 4, 7 and 10 day after the treatment with (A,  ) gauze (negative control), (B,
) gauze (negative control), (B,  ) 30/70 CS‐HOBt/PVA nanofibre mats, (C,
) 30/70 CS‐HOBt/PVA nanofibre mats, (C,  ) 30/70 CS‐TPP/PVA nanofibre mats, (D,
) 30/70 CS‐TPP/PVA nanofibre mats, (D,  ) 30/70 CS‐EDTA/PVA nanofibre mats and (E,
) 30/70 CS‐EDTA/PVA nanofibre mats and (E,  ) commercial antibacterial gauze dressing (Sofra‐tulleregister) (positive control). Difference values * were statistically significant (P < 0·05) compared with gauze. The data are expressed as mean ± standard deviation (n = 6).
) commercial antibacterial gauze dressing (Sofra‐tulleregister) (positive control). Difference values * were statistically significant (P < 0·05) compared with gauze. The data are expressed as mean ± standard deviation (n = 6).
Conclusions
In this study, we successfully produced composite nanofibrous membranes composed of CS salt and PVA using water as a solvent, avoiding the use of toxic and hazardous solvents. These fibre mats were in the nanometre range and provided suitable tensile strength and swelling properties. The CS‐EDTA/PVA nanofibre mats showed the greatest antibacterial and wound‐healing activity during the first 4 days after wounds. The biodegradable, biocompatible and antibacterial CS‐EDTA/PVA nanofibre mats have immense potential for use as wound dressing materials.
Acknowledgements
The authors wish to thank the Commission of Higher Education (Thailand), the Thailand Research Funds through the Golden Jubilee Ph.D. Program (Grant No. PHD/0183/2550 and Project No. DBG5480004) for financial support.
References
- 1. Jayakumar R , Menona D , Manzoora K , Naira SV , Tamurab H. Biomedical applications of chitin and chitosan based nanomaterials ‐ a short review. Carbohydr Polym 2010. ; 82 : 227 – 32. [Google Scholar]
- 2. Dash M , Chiellini F , Ottenbrite RM , Chiellini E. Chitosan‐A versatile semi‐synthetic polymer in biomedical applications. Prog Polym Sci 2011. ; 36 : 981 – 1014. [Google Scholar]
- 3. Jayakumar R , Prabaharan M , Kumar PTS , Nair SV , Tamura H. Biomaterials based on chitin and chitosan in wound dressing applications. Biotechnol Adv 2011. ; 29 : 322 – 37. [DOI] [PubMed] [Google Scholar]
- 4. Paul W , Sharma CP. Chitosan and alginate wound dressings: a short review. Trends Biomater Artif Organs 2004. ; 18 : 18 – 23. [Google Scholar]
- 5. Zahedi P , Rezaeian I , Ranaei‐Siadat S‐O , Jafari S‐H , Supaphol P. A review on wound dressings with an emphasis on electrospun nanofibrous polymeric bandages. Polym Adv Technol 2010. ; 21 : 77 – 95. [Google Scholar]
- 6. Sangsanoh P , Supaphol P. Stability improvement of electrospun chitosan nanofibrous membranes in neutral or weak basic aqueous solutions. Biomacromolecules 2006. ; 7 : 2710 – 4. [DOI] [PubMed] [Google Scholar]
- 7. Neamnark A , Rujiravanit R , Supaphol P. Electrospinning of hexanoyl chitosan. Carbohydr polym 2006. ; 66 : 298 – 305. [Google Scholar]
- 8. Geng X , Kwon O‐H , Jang J. Electrospinning of chitosan dissolved in concentrated acetic acid solution. Biomaterials 2005. ; 26 : 5427 – 32. [DOI] [PubMed] [Google Scholar]
- 9. Bhattarai N , Edmondson D , Veiseh O , Matsen FA , Zhang M. Electrospun chitosan‐based nanofibers and their cellular compatibility. Biomaterials 2005. ; 26 : 6176 – 84. [DOI] [PubMed] [Google Scholar]
- 10. Jia Y‐T , Gong J , Gu X‐H , Kim H‐Y , Dong J , Shen X‐Y. Fabrication and characterization of poly (vinyl alcohol)/chitosan blend nanofibers produced by electrospinning method. Carbohydr Polym 2007. ; 67 : 403 – 9. [Google Scholar]
- 11. Ignatova M , Manolova N , Rashkov I. Novel antibacterial fibers of quaternized chitosan and poly(vinyl pyrrolidone) prepared by electrospinning. Eur Polym J 2007. ; 43 : 1112 – 22. [Google Scholar]
- 12. Zhou Y , Yang D , Chen X , Xu Q , Lu F , Nie J. Electrospun water‐soluble carboxyethyl chitosan/poly(vinyl alcohol) nanofibrous membrane as potential wound dressing for skin regeneration. Biomacromolecules 2008. ; 9 : 349 – 54. [DOI] [PubMed] [Google Scholar]
- 13. Charernsriwilaiwat N , Opanasopit P , Rojanarata T , Ngawhirunpat T , Supaphol P. Preparation and characterization of chitosan‐hydroxybenzotriazole/polyvinyl alcohol blend nanofibers by the electrospinning technique. Carbohydr Polym 2010. ; 81 : 675 – 80. [Google Scholar]
- 14. Charernsriwilaiwat N , Opanasopit P , Rojanarata T , Ngawhirunpat T. Fabrication and characterization of chitosan‐ethylenediaminetetra‐ acetic acid/polyvinyl alcohol blend electrospunnanofibers. Adv Mater Res 2011. ; 195–196 : 648 – 51. [Google Scholar]
- 15. Rojanarata T , Opanasopit P , Techaarpornkul S , Ngawhirunpat T , Ruktanonchai U. Chitosan‐thiamine pyrophosphate as a novel carrier for siRNA delivery. Pharma Res 2008. ; 25 : 2807 – 14. [DOI] [PubMed] [Google Scholar]
- 16. Chen J‐P , Chang G‐Y , Chen J‐K. Electrospun collagen/chitosan nanofibrous membrane as wound dressing. Colloids Surf A 2008. ; 313–314 : 183 – 8. [Google Scholar]
- 17. Leung V , Hartwell R , Yang H , Ghahary A , Ko F. Bioactive nanofibres for wound healing applications. Journal Fiber Bioeng Inform 2011. ; 4 : 1 – 14. [Google Scholar]
- 18. Baskar D , Kumar TSS. Effect of deacetylation time on the preparation, properties and swelling behavior of chitosan films. Carbohydr Polym 2009. ; 78 : 767 – 72. [Google Scholar]
- 19. Meng ZX , Zheng W , Li L , Zheng YF. Fabrication, characterization and in vitro drug release behavior of electrospun PLGA/chitosan nanofibrous scaffold. Mater Chem Phys 2011. ; 125 : 606 – 11. [Google Scholar]
- 20. Xie W , Xu P , Wang W , Liu Q. Preparation and antibacterial activity of a water‐soluble chitosan derivative. Carbohydr Polym 2002. ; 50 : 35 – 40. [Google Scholar]
- 21. El‐Sharif AA , Hussain MHM. Chitosan–EDTA new combination is a promising candidate for treatment of bacterial and fungal infections. Curr Microbiol 2011. ; 62 : 739 – 45. [DOI] [PubMed] [Google Scholar]
- 22. Li Y , Chen XG , Liu N , Liu CS , Liu CG , Meng XH , Yu LJ , Kenendy JF. Physicochemical characterization and antibacterial property of chitosan acetates. Carbohydr Polym 2007. ; 67 : 227 – 32. [Google Scholar]
- 23. Helander IM , Nurmiaho‐Lassila E‐L , Ahvenainen R , Rhoades J , Roller S. Chitosan disrupts the barrier properties of the outer membrane of Gram‐negative bacteria. Int J Food Microbiol 2001. ; 71 : 235 – 44. [DOI] [PubMed] [Google Scholar]
- 24. Sudarshan NR , Hoover DG , Knorr D. Antibacterial action of chitosan. Food Biotechnol 1992. ; 6 : 257 – 72. [Google Scholar]
- 25. Rabea EI , Badawy ME‐T , Stevens CV , Smagghe G , Steurbaut W. Chitosan as antimicrobial agent: Applications and mode of action. Biomacromolecules 2003. ; 4 : 1457 – 65. [DOI] [PubMed] [Google Scholar]
- 26. Xing K , Chen XG , Kong M , Liu CS , Cha DS , Park HJ. Effect of oleoyl‐chitosan nanoparticles as a novel antibacterial dispersion system on viability, membrane permeability and cell morphology of Escherichia coli and Staphylococcus aureus. Carbohydr Polym 2009. ; 76 : 17 – 22. [Google Scholar]
- 27. Xing K , Chen XG , Liu CS , Cha DS , Park HJ. Oleoyl‐chitosan nanoparticles inhibits Escherichia coli and Staphylococcus aureus by damaging the cell membrane and putative binding to extracellular or intracellular targets. Int J Food Microbiol 2009. ; 132 : 127 – 33. [DOI] [PubMed] [Google Scholar]
- 28. Muzzarelli RAA. Chitins and chitosans for the repair of wounded skin, nerve, cartilage and bone. Carbohydr Polym 2009. ; 76 : 167 – 82. [Google Scholar]