

Abstract
Objective
This study aimed to investigate mitochondrial changes and the mitochondrial antiviral‐signaling protein (MAVS)‐type I interferon (IFN1) signaling pathway in the muscles of anti‐melanoma differentiation gene 5(MDA5) dermatomyositis (DM) patients.
Methods
Eleven anti‐MDA5 DM and ten antibody‐negative DM patients were included. Muscle biopsies were performed in all patients. Muscle pathology and mitochondrial morphology in particular were compared between two groups. The expression of MDA5, MAVS, interferon (IFN) regulatory factor 7, and IFN‐stimulated gene 15, which are components of the MAVS‐IFN1 signaling pathway, was measured in muscle specimen. The correlation between MAVS expression in muscles and disease phenotypes and muscle pathology were analyzed.
Results
Anti‐MDA5 DM showed a significantly lower incidence of the characteristic DM pathology (P < 0.05) than antibody‐negative DM, including perifascicular fiber atrophy, inflammation, and vasculopathy. Mitochondrial abnormalities in anti‐MDA5 patients revealed a high incidence of (8/11,72.7%) and different pattern from that in antibody‐negative DM. MDA5, MAVS, IFN regulatory factor 7, and IFN stimulated gene 15 expression levels in the muscles of anti‐MDA5 DM patients were higher than those of the controls (P < 0.05) but lower than those of antibody‐negative DM patients (P < 0.05). The MAVS levels negatively correlated with manual muscle test 8 scores (r = 0.701, P = 0.016).
Conclusions
Compared to antibody‐negative DM, we presented a different distribution of the mitochondrial pathology and less severe morphology in anti‐MDA5 DM. We also revealed the enhanced but less intensive MAVS‐IFN1 signaling pathway activity in muscles of anti‐MDA5 DM. Such disparity suggested the potentially different mechanism of muscle injury in two DM groups.
Introduction
Dermatomyositis (DM) is a group of idiopathic inflammatory myopathies characterized by skin rash and skeletal myopathy. The typical histopathologic features of DM muscle pathology are the infiltration of perimysial lymphocytes and macrophages, perifascicular atrophy, as well as loss of capillaries. 1 Decreased cytochrome c oxidase (COX) stain and mitochondrial DNA depletion consistent with mitochondrial dysfunction is mostly revealed in myofibers within perifascicular area. 2 Overexpression of mitochondrial apoptosis molecules or gasdermin E‐dependent mitochondrial pyroptotic pathway might be pathogenesis for perifascicular atrophy in DM. 3
A number of myositis‐specific autoantibodies (MSA) have been identified in DM, including autoantibodies to melanoma differentiation‐associated protein 5 (MDA5), the nuclear helicase protein Mi‐2 region, nuclear matrix protein 2(NXP2), transcriptional intermediary factor 1γ(TIF1γ), and the small ubiquitin‐like modifier‐activating enzyme(SAE). 4 DM with specific autoantibodies usually presents quite distinct clinical features. Anti‐MDA5 DM, originally described in a cohort of Japanese patients in 2009, 5 accounts for 7% of juvenile myositis and 1%–30% of adult myositis. 6 Anti‐MDA5 typically is observed in hypomyopathic or amyopathic DM with a high frequency of cutaneous ulcerations and interstitial lung disease (ILD). 7 Anti‐MDA5 DM patients also demonstrate a distinct morphological pattern from other types of DM. The muscle pathology has been described as mild or unspecific changes in most cases. 1
MDA5 also known as interferon‐induced helicase‐1, is a member of the retinoic acid‐inducible gene I‐like helicase family of proteins, which are essential initiators of the innate immune response by sensing viral dsRNA in the cytoplasm upon infection and induce the oligomerization of mitochondrial antiviral‐signaling protein (MAVS). 8 MAVS interacts with mitochondrial proteins, enhancing mitochondrial fusion and inducing mitochondrial autophagy. 9 MAVS oligomerises to recruit and stimulate signaling proteins, leading to the activation of interferon (IFN) regulatory factor (IRF)3 and IRF7 and the expression of IFN‐stimulated genes (ISGs). 10 A study by Allenbach revealed the upregulation of six ISGs in the muscles of both anti‐MDA5 and classic DM patients. 1 However, mitochondrial damage in the muscles of anti‐MDA5 DM patients has not yet been profiled. We expected more dramatic and primary mitochondrial damage in anti‐MDA5 DM, which may be related to the disruption of MAVS‐IFN1 signaling. Therefore, in the present study, we microscopically and ultra‐microscopically evaluated mitochondrial pathology and quantified regulatory factors in the MAVS‐IFN1 pathway in skeletal muscles from anti‐MDA5 DM patients with the aim of clarifying the roles of mitochondrial abnormality and the MAVS‐IFN1 pathway in the disease mechanism.
Materials and Methods
Patients
This was a retrospective and observational cohort study. Of 2390 patients with suspected neuromuscular disorders who underwent muscle biopsy at Peking University First Hospital from January 2016 to September 2019, 227 patients were diagnosed with idiopathic inflammatory myopathies according to the European Neuromuscular Center criteria. 11 A total of 56 patients were diagnosed with DM, of which 11 adult anti‐MDA5 DM (1 definite DM, 7 probable DM, 3 amyopathic DM) and 10 antibody‐negative DM (9 definite DM, 1 probable DM) patients were included. Five age‐and sex‐matched individuals with normal muscle biopsy as no disease control subjects. Two double‐positive (anti‐Ro52) patients were classified into the anti‐MDA5 group since they showed the typical clinical anti‐MDA5 DM phenotype. All clinical materials used in this study were obtained for diagnostic purposes with written informed consent. The definition of disease duration in our study was from onset to biopsy. ILD was diagnosed according to the consensus classification of idiopathic interstitial pneumonias. Rapid progressive ILD (RPILD) was defined as a worsening of radiologic interstitial changes with progressive dyspnea and hypoxemia within one month after the onset of respiratory symptoms as previously reported. 12 Muscular weakness was evaluated according to manual muscle test 8 scores (MMT8), with the maximum score being 80. Disease activity was evaluated using Myositis Disease Activity Assessment Tool (MDAAT). Disease activity measure and muscle biopsy were performed at the same time. Functional outcome was assessed using the modified Rankin Scale during follow‐up of one to three years. The study was approved by the ethics committee of Peking University First Hospital [approval no.:2019181].
Antibody detection
Patients’ serum was stored at –80°C. Immunoblots to identify anti‐MDA5 antibody and all the other MSAs and myositis‐associated antibodies (MAAs) were performed according to standard methods (Euroline Autoimmune Inflammatory Myopathies 16 Ag, IgG; Euroimmun). MSAs included anti‐Mi‐2, anti‐TIF1γ, anti‐NXP2, anti‐MDA5, anti‐SAE, anti‐ signal recognition particle, anti‐EJ, anti‐Jo‐1, anti‐OJ, anti‐PL‐12, and anti‐PL‐7. MAAs included anti‐Ku, anti‐PM‐Scl100, anti‐PM‐Scl75, and anti‐RO‐52. On the basis of signal intensity, the results were classified as negative (0), weak‐positive (+), positive (++), or strong‐positive (+++). Patients with weak‐positive results and a typical clinical phenotype were included in this study.
Muscle biopsy
Open muscle biopsies were performed in all patients after consent forms were obtained. Here, we should clarify that six patients (three anti‐MDA5 and three antibody‐negative DM) had received a short course and low dose prednisone therapy (all <60 mg, oral) before the muscle biopsy. Muscle biopsies were obtained from quadriceps femoris (pt7 and 8) or biceps brachii (pt1–6 and 9–11, and control subjects). Muscle biopsies were assessed by two independent reviewers (WZ and YY). Both of them were experienced in interpretation of muscle biopsies and muscle immunoanalysis. The reviewers were blinded to the underlying MSAs of the patients. The muscle specimens were frozen in isopentane, cooled in liquid nitrogen, and stored at –80°C. The thickness of cryo sections for regular staining was 8 μm and for the immunohistochemistry staining was 6 μm. Serial frozen sections were stained with hematoxylin and eosin, modified Gomori trichrome, periodic acid‐Schiff, and oil red O and co‐stained for the detection of adenosine triphosphate enzyme (pH 4.5 and 10.8), nicotinamide adenine dinucleotide tetrazolium reductase (NADH‐TR), nonspecific esterase (NSE), succinate dehydrogenase (SDH), and COX‐SDH. Mitochondrial abnormality was defined as the presence of more than five COX‐negative fibers per frozen section as previously reported. 13 The prominent COX abnormality was defined as the ratio of COX‐deficient fibers to the total number of fibers per frozen section was over 30% and the distribution was diffuse. The sections were immunohistochemically stained with primary antibodies against human CD3, CD4, CD8, CD20, CD68, major histocompatibility complex class I (MHC‐I), membrane attack complex (MAC) and dystrophin N, C, R, dysferlin and α, β, γ‐sarcoglycans. A second specimen was fixed in 3% glutaraldehyde, post‐fixed in 1% osmium tetroxide, dehydrated with acetone, and embedded in Epon812. Ultrathin sections were contrasted with uranyl acetate and lead citrate and observed under an electron microscope.
Western blotting
The expression levels of IFN1‐signaling pathway proteins, including MDA5 (117 kD), MAVS (57 kD and 75 kD), IRF7 (54 kD), and ISG15 (18 kD), were analysed by western blotting in eleven anti‐MDA5 DM patients in comparison with eight antibody‐negative DM patients and five age‐and sex‐matched healthy control subjects. Skeletal muscle protein was extracted using a protein extraction kit (GPP1814, GenePool). The proteins were mixed with SDS‐PAGE loading buffer (5×) (GPP1820, GenePool) and boiled for 10 min; aliquots of the supernatants (40 μg/lane) were fractionated by electrophoresis using a 10% SDS acrylamide gel and electro‐transferred to PVDF membranes. The membranes were blocked with 5% milk‐blocking buffer (GPP1819, GenePool) and subsequently incubated with anti‐MDA5 (ab79055, Abcam), anti‐MAVS (ab31334, Abcam), anti‐IRF7 (ab115352, Abcam), anti‐ISG15 (ab131119, Abcam), and anti‐actin (ab8226, Abcam) antibodies at 4°C overnight. Following extensive washing with tris‐buffered saline mixed with Tween‐20 (TBS‐T), the blots were incubated with a secondary antibody (HRP‐labeled goat anti‐mouse IgG (ab6789, Abcam) or HRP‐labeled goat anti‐rabbit IgG (ab6721, Abcam)) at 20°C for 60 min. After rinsing briefly with TBS‐T, immune‐reactive protein bands were visualized using a chemiluminescence‐based detection kit (GPP1824, GenePool) according to the manufacturer’s instructions. The signals were quantified by densitometry using Quantity One v.4.6.2 software. Protein expression levels were quantified from four blots and normalized to the loading control (actin) signal.
Statistical analysis
Statistical analysis was performed using SPSS for Windows version 25.0 (IBM Corp, Armonk, NY, USA) and Prism 8.0 software (GraphPad Software). Categorical variables are presented as percentages and were compared using a Fisher’s exact test. Quantitative variables are presented as the means ± standard deviations and compared using the Student’s t‐test or Mann–Whitney test. For correlations analysis, Pearson’s test and Spearman's test were used. Two‐sided tests were used, and P < 0.05 was considered significant.
Results
Patient information
In total, 11 anti‐MDA5 DM patients (two males and nine females) were recruited into this study. The mean age at disease onset was 52.1 ± 8.7 (range, 34–66) years, and the mean disease duration was 4.1 ± 3.1 (1–12) months. All patients had cutaneous involvement, including cutaneous ulceration in 8/11 (72.7%) patients, Gottron’s sign in 9/11 (81.8%) patients, V rash in 7/11 (63.6%) patients, heliotrope periorbital oedema in 6/11 (54.5%) patients, mechanic’s hand in 5/11 (45.5%) patients, and shawl sign in 5/11 (45.5%) patients. All patients experienced ILD, and two presented RPILD. Proximal muscle weakness appeared in 8/11 (72.7%) patients; four of these patients also showed distal muscle weakness. Serum creatine kinase (CK) was mildly elevated in 5/11 (45.5%) patients (174–646 IU/L, normal: 25–170 IU/L). Liver dysfunction was present in 4/11 (36.4%) patients, despite relatively low creatine kinase levels as previously reported. 14 Systemic lupus erythematosus, cryoglobulinemia with peripheral neuropathy, and Sjögren’s syndrome appeared in three patient each. Serum antinuclear antibody was positive in 6/11 (54.5%) patients and anti‐Ro52 antibody in 3/11 (27.3%) patients. A high erythrocyte sedimentation rate was found in 7/11 (63.6%) patients. All patients had received at least one immunosuppressive therapy (glucocorticoids, tacrolimus, cyclosporine A, or cyclophosphamide) at follow‐ups. Clinical symptoms improved in 8/11 (72.7%) patients. Two patients (pt3 and pt8) presented with RPILD and died of respiratory failure 4 months after disease onset and two patients (pt9 and pt11) died of respiratory failure 1 year after disease onset. Detailed clinical and laboratory characteristics are summarized in Table S1. The 10 antibody‐negative DM patients including three males and seven females were studies as control. The mean age at disease onset was 53.1 ± 14.5 (range, 28–72) years, and the mean disease duration was 5.9 ± 4.6 (range, 1–14) months. Detailed information is summarized in Table S2. Skin ulcers were more frequent in anti‐MDA5 patients, whereas skin ulcer was absent in antibody‐negative DM group. In addition, all anti‐MDA5 patients experienced ILD. Only 20% of antibody‐negative DM patients presented ILD. CK levels were lower and MMT8 scores were significantly higher in anti‐MDA5 DM patients compared with those in antibody‐negative DM patients (P < 0.05, respectively) (Table 1).
Table 1.
Clinical characteristics between anti‐MDA5 and antibody‐negative DM groups.
| Criterion | Anti‐MDA5 | Antibody negative DM | P‐value |
|---|---|---|---|
| Female, n (%) | 9 (82) | 7 (70) | 0.635 |
| Age of onset, years | 52.1 ± 8.7 | 53.1 ± 14.5 | 0.847 |
| Disease duration, months | 4.1 ± 3.1 | 5.9 ± 4.6 | 0.296 |
| Skin ulcers, n (%) | 8 (73) | 0 | 0.001 * |
| Interstitial lung disease, n (%) | 11 (100) | 2 (20) | <0.001 * |
| MMT8 scores | 71.3 ± 6.1 | 63.2 ± 8.4 | 0.020 * |
| CK level, IU/L | 185.2 ± 209.1 | 2149.1 ± 2677.4 | 0.036 * |
Bold indicates the data that had significant difference after pairwise comparison.
MMT, manual muscle test; CK, creatine kinase.
P < 0.05.
Morphological characteristics
Among all 11 muscle specimens from anti‐MDA5 DM patients, only 1 (9.1%) showed perifascicular fiber atrophy (PFA) and 1 had regional perimysial oedema. Focal perivascular infiltration appeared in 2/11 (18.2%) patients. All patients had scattered atrophic muscle fibers. Scattered, necrotic, or regenerated muscle fibers were observed in 6/11 (54.5%) patients (Fig. 1A), with no microinfarctions. Myofibers with decreased NADH‐TR activity were observed in 3/11 (27.3%) patients; one of these patients exhibited fibers with a “moth‐eaten” appearance. Dark‐stained endomysial capillaries or perimysial vessels appeared in 8/11 (72.7%) patients on NSE staining. Macrophages were prominent, whereas T and B cells were relatively rare; CD3+ T cells were observed in 3/11 (27.3%) patients, CD4+ T cells in 4/11 (36.5%) patients, CD8+T cells in 2/11 (18.2%) patients, and CD20+B cells in 1/11 (9.1%) patients. All patients showed diffuse MHC‐I upregulation under the sarcolemma (Fig. 1B), and two showed enhanced staining in the perifascicular regions. Slight MAC deposition on the endomysial capillaries was observed in 6/11 (54.5%) patients (Fig. 1C); two of these patients also showed deposition in necrotic myofibers.
Figure 1.
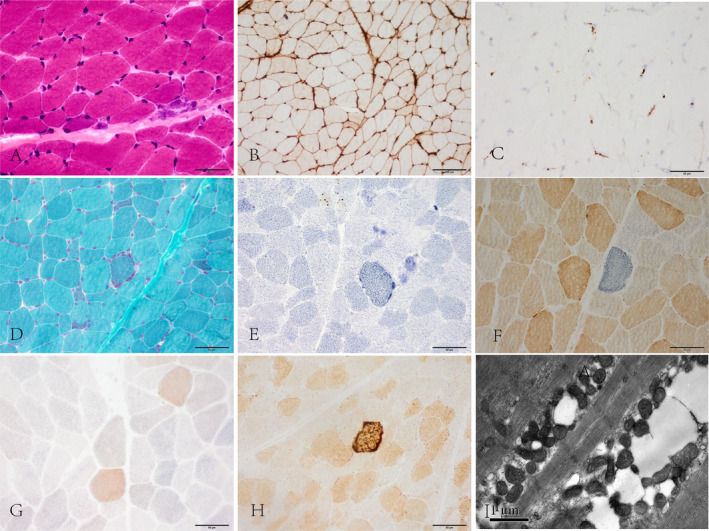
(A) Scattered atrophy, regenerated muscle fibers. HE staining. (B) Diffuse sarcolemmal MHC‐I positive immunoreactivity. (C) Mild membrane attack complex deposition on endomysial capillaries. (D) Atypical ragged red fibers appeared by modified Gomori trichrome staining. (E) SDH hyper‐reactive fibers. (F) COX‐negative fibers. (G) Numerous COX‐deficient fibers, not restricted to the perifascicular region. SDH‐COX co‐staining. (H) COX hyper‐reactive fibers. Original magnification, ×40. (I) Mitochondrial proliferation and abnormal aggregation among myofibrils. Electron microscopy. Scale bar are 1 μm. MHC‐I, major histocompatibility complex class‐I; COX, cytochrome oxidase C; SDH, succinate dehydrogenase.
Next, we compared the histology of anti‐MDA5 DM patients and antibody‐negative DM patients (Table 2). The main characteristics of DM pathology, including muscle ischaemia (perimysial oedema, 9.1% vs. 80%; PFA, 9.1% vs. 90%; decreased NADH‐TR activity, 27.3% vs. 100%), perivascular inflammation (18.2% vs. 100%), inflammatory cell infiltrates (CD3+, 27.3% vs. 90%; CD4+, 36.5% vs. 9%; CD8+, 18.2% vs.70%; CD20+, 9.1% vs. 60%), MHC‐I expression with reinforcement in perifascicular regions (18.2% vs. 90%), and MAC deposition (18.2% vs. 80%), were significantly lower in anti‐MDA5 DM patients than in antibody‐negative DM patients (P < 0.05).
Table 2.
Comparison of histopathological features of DM patients with anti‐MDA5 Ab and Ab negative.
| Histopathological features | Anti‐MDA5DM (n = 11) | Ab negative DM (n = 10) | P‐value | |
|---|---|---|---|---|
| Myofiber pathology | Necrotic/Regenerated muscle fibres, n (%) | 6 (54.5) | 9 (90) | 0.149 |
| Atrophic muscle fibres, n (%) | 11 (100) | 10 (100) | NA | |
| Regional perimysial edema, n (%) | 1 (9.1) | 8 (80) | 0.002 * | |
| Perifascicular fiber atrophy, n (%) | 1 (9.1) | 9 (90) | <0.001 * | |
| NADH‐TR activity decreasing fibers, n (%) | 3 (27.3) | 10 (100) | 0.001 * | |
| Vasculopathy | NSE hyperchromatic capillaries, n (%) | 8 (72.7) | 10 (100) | 0.214 |
| Inflammation | Perivascular inflammation, n (%) | 2 (18.2) | 10 (100) | <0.001 * |
| CD3+ T cells infiltrate, n (%) | 3 (27.3) | 9 (90) | 0.008 * | |
| CD4+ T cells infiltrate, n (%) | 4 (36.5) | 9 (90) | 0.024 * | |
| CD8+ T cells infiltrate, n (%) | 2 (18.2) | 7 (70) | 0.030 * | |
| CD20+ B cells infiltrate, n (%) | 1 (9.1) | 6 (60) | 0.024 * | |
| Immune related changes | MHC‐I expression | |||
| Diffuse in muscle fiber membrane, n (%) | 11 (100) | 10 (100) | NA | |
| enhancement in perifascicular region, n (%) | 2 (18.2) | 9 (90) | 0.002 * | |
| MAC‐positive myofiber, n (%) | 2 (18.2) | 8 (80) | 0.009 * | |
| MAC‐positive capillaries, n (%) | 6 (54.5) | 7 (70) | 0.659 | |
| Mitochondrial pathology | COX‐deficient fibers, n (%) | 8 (72.7) | 9 (90) | 0.586 |
| Diffuse COX decreasing fibers, n (%) | 3 (27.3) | 5 (50) | 0.387 | |
| COX‐deficient in perifascicular region, n (%) | 0 (0) | 5 (50) | 0.012 * | |
| Ragged red fibres, n (%) | 2 (18.2) | 3 (30) | 0.635 |
Bold indicates the data that had significant difference after pairwise comparison.
Ab, antibody; NADH‐TR, nicotinamide adenine dinucleotide tetrazolium reductase; MHC‐I, major histocompatibility complex class‐I; MAC, membrane attack complex; COX, cytochrome oxidase C; NSE, nonspecific esterase.
P < 0.05.
Mitochondrial pathology
Eight out of 11 (72.7%) patients showed mitochondrial abnormalities. Modified Gomori trichrome staining revealed a few atypical ragged red fibers in three patients (Fig. 1D). Scattered SDH hyper‐reactive fibers (Fig. 1E) were present in 8/11 (72.7%) patients. Strongly SDH‐reactive blood vessels were not found in all specimens. Eight out of 11 (72.7%) patients had scattered COX‐negative/SDH hyper‐reactive fibers (Fig. 1F) and the ratio of COX negative/SDH positive fibers to the number of total fibers per frozen section was between 0.15% and 0.75%; Three of these patients (pt5, pt6, and pt10) had numerous muscle fibers with decreased COX staining, without SDH stain hyperintensity across the fascicle (Fig. 1G), occupying up to 30% of the total myofibers. Scattered COX hyper‐reactive fibers were observed in 7/11 (63.6%) patients (Fig. 1H) and the ratio was between 0.05% and 0.55%. Nine out of 10 (90%) antibody‐negative DM patients showed mitochondrial abnormalities. Diffuse COX‐deficient fibers without SDH stain hyperintensity, mainly in the perifascicular region, were observed in 5/10 (50%) antibody‐negative DM biopsies. However, diffuse COX‐deficient fibers in the perifascicular region were not observed in any of the anti‐MDA5 DM patients (P = 0.012).
Electron microscopy of the biopsies of anti‐MDA5 DM patients revealed numerous mitochondria and abnormal mitochondrial aggregation under the sarcolemma or between the myofibrils with normal morphology. The cristae were generally regular and devoid of typical crystalloid inclusions (Fig. 1I).
MAVS‐IFN1 signalling pathway activity
The expression levels of MDA5, MAVS, IRF7, and ISG15 in the skeletal muscle of the anti‐MDA5 DM patients were significantly higher than those in the no disease controls (P = 0.003, P = 0.003, P = 0.023, and P < 0.001, respectively) (Fig. 2A and B) but lower than those in the eight antibody‐negative DM patients (P = 0.028, P = 0.011, P = 0.013, and P = 0.002, respectively) (Fig. 2C and D). Data was presented as mean ± standard deviation in Table S3.
Figure 2.
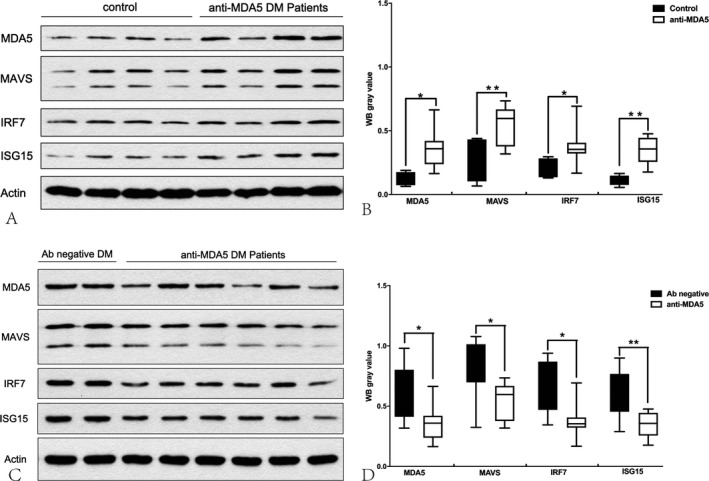
Protein expression levels of MDA5, MAVS, IRF7, and ISG15 in the skeletal muscle of the anti‐MDA5 patients were significantly higher than those in the healthy controls (A, B) but lower than those in the antibody‐negative DM patients (C, D). Protein expression levels were quantified by densitometric analysis. Actin bands were used as loading control. Data are presented as mean ± SD. Statistical analysis was performed by Student’s t‐test. *P < 0.05, **P < 0.01.
Correlation between MAVS and disease activity
Correlation analysis was used to test the influence of MDA5, MAVS, IRF7, and ISG15 on muscle strength, disease activity and functional outcome in anti‐MDA5 DM patients. The statistical results revealed that both the MAVS levels and ISG15 levels negatively correlated with MMT8 scores (P = 0.016 and P = 0.018, respectively) (Fig. 3A and B), and same results were observed in antibody‐negative DM group, both the MAVS levels and ISG15 levels negatively correlated with MMT8 scores (P = 0.033 and P = 0.031, respectively). Only the MAVS levels positively correlated with MDAAT scores (r = 0.672, P = 0.024) (Fig. 3C). Nevertheless, there was no correlation between MAVS levels and modified Rankin Scale scores. Lastly, there was no significant relationship between the other markers (MDA5, IRF7) and MMT8 or MDAAT scores, either (all P > 0.05) (Table S4).
Figure 3.
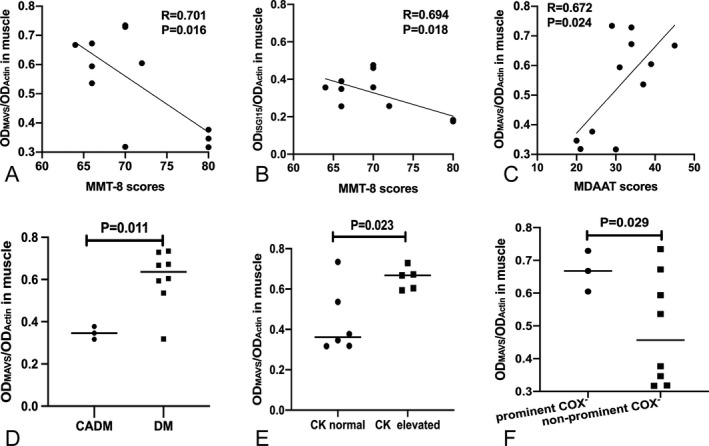
Correlations between MAVS levels and MMT8 scores, MDAAT scores (A, C). Correlations between ISG15 levels and MMT8 scores (B). The MAVS levels in muscle were compared between various phenotypes of DM and CADM subgroups, CK elevated and CK normal subgroups (D, E), The MAVS levels in muscle were compared between prominent and non‐prominent COX‐deficient fibers subgroups (F). P‐values are for Pearson’s correlation test (A, B, C). Student’s t‐test was used for the comparison (D, E, F). Horizontal lines show the mean levels of MAVS in each group. The levels of MAVS from western blot method were normalized with actin levels. MMT, muscle and manual muscle test; MDAAT, myositis disease activity assessment tool.
Comparison of MAVS in subgroups
Above all, only the MAVS significantly correlated with disease activity. Thus, this part focused on comparison of MAVS levels in subgroups. Higher levels of MAVS were detected in DM subgroup and CK elevated subgroup than in CADM subgroup (P = 0.011) and CK normal subgroup (P = 0.023) (Fig. 3D and E). Besides, the MAVS levels were significantly higher (P = 0.029) in the three patients with numerous COX‐deficient fibers than the remaining eight patients (Fig. 3F). Moreover there was a tendency of MAVS expressions increased in the patients with RPILD, PFA and MAC deposition, when compared to the patients without RPILD, PFA, or MAC deposition, respectively. Due to the small sample size, we could not draw any strong conclusions from subgroup analysis.
Discussion
There are only a few reports documenting the morphological findings of muscle specimens in adult 1 , 13 , 15 and juvenile 16 , 17 anti‐MDA5 DM patients. Our findings supported that the muscle pathology of adult anti‐MDA5 DM is distinct from that of classic DM. 1 Our series did not show prominent PFA, MAC deposition on the endomysial capillaries or perimysial inflammation, all of which are typical findings of classic DM. It suggested that immune attacks in the skeletal muscles of anti‐MDA5 DM are not so intensive or destructive as in other tissue 18 or in other types of DM. 13 , 19 The skeletal muscle presents high metabolic activities making it vulnerable to mitochondria damage which could be induced by the stress from ischemia or inflammation as usually found in classic DM. Interestingly, the abnormal mitochondria in the area of perifascicular region was much less to the antibody‐negative group whereas the scattered COX‐deficient fibers, not restricted to the perifascicular region were confined to anti‐MDA5 DM and broadly distributed COX‐deficient fibers across the specimen were observed in three anti‐MDA5 DM patients. Similar topographic distribution of COX negative fibers was also frequently observed in anti‐synthetase syndrome patients. 2 Therefore, we speculated the distinct mechanism of mitochondrial injury between anti‐MDA5 DM and antibody‐negative DM. MDA5 is one of the viral RNA sensors that induce MAVS oligomerization. 20 MAVS, is a mitochondria‐associated protein and the overexpression of MAVS could specifically induce mitochondrial fragmentation, mitochondrial turnover and mitochondrial autophagy, and increase the levels of mitochondria‐derived reactive oxygen species (ROS) production. 21 Allenbach et al. observed that only anti‐MDA5 DM showed numerous nitric oxide synthase 2‐positive muscle fibers with sarcoplasmic, not classic DM, also indicating the different pattern of mitochondrial changes. 1 This discrepancy might be mainly due to the differences of MSAs. Pinal‐Fernandez et al. demonstrated the prevalence of histological features varied among DM with different MSAs. Mi‐2+ patients had more primary inflammation, NXP2+ patients had less primary inflammation and TIF1γ+ patients had more mitochondrial dysfunction. 13 Nguyen et al. also reported the MSA specific tissue injury and speculated that there might be tissue specific receptors for the different types of MSA. 22
IFN appears to play a key role in the pathogenesis of idiopathic inflammatory myopathies, especially in DM. 23 An up‐regulated IFN signature has been observed in skeletal muscle, skin samples, and peripheral blood mononuclear cells of anti‐MDA5 DM. 1 , 24 Our study showed the components of the IFN1 signaling pathway (MDA5, MAVS, IRF7, and ISG15), were overexpressed at the protein level, suggesting that the MAVS‐IFN1 pathway is broadly involved in the muscle pathology of anti‐MDA5 DM. In systemic lupus erythematosus, high levels of IFN1 are also associated with mitochondrial dysfunctions, including enhanced mitochondrial ROS production, and that are reduced by MAVS silencing and ROS inhibition. 25 , 26 Therefore, the aberrant MAVS‐IFN1 signaling pathway activation might have partially contributed to the mitochondrial abnormalities in anti‐MDA5 DM. Moreover we found IFN1 signaling pathway activity lower than those in the antibody‐negative DM patients, which was parallel to the less severe morphological findings in anti‐MDA5 DM. Allenbach et al. also reported the expression of IFN‐stimulated genes was up‐regulated in muscle but significantly lower than classic DM patients, which further supported our foundings. 1 Conversely, the IFN1 signature levels in peripheral blood mononuclear cells were higher of anti‐MDA5 DM patients than that of antibody‐negative DM patients. 24 This discrepancy may be mainly due to differences in tissue or organ susceptibility to the attacks via anti‐MDA5 antibody regulated pathway. Besides, this discrepancy may correlate with the clinical phenotypes that differentially affect skin or muscle in DM patient. 23 We found MAVS‐IFN signaling pathway correlated with disease activity and severity in muscular aspects but not with mRS, suggesting muscle involvement not helpful to predict the whole disease prognosis. In addition, MAVS was found to express higher in samples with diffusively distributed COX deficient myofibers. It possibly be explained by the notable interacts of MAVS with the complex IV subunit COX5B, and proteins enhancing mitochondrial fusion. 10
In conclusion, anti‐MDA5 DM in our series showed relative mild morphology but distinctive pattern of the mitochondrial pathology. We also showed the overexpressed MAVS‐IFN1 signaling pathway in the skeletal muscle of the anti‐MDA5 DM patients but lower than those in the antibody‐negative DM patients. Such disparity might reflect the different pathogenesis among DMs with various MSAs. We speculate that MAVS‐IFN1pathway may have contributed to the anti‐MDA5 DM muscle pathology and phenotypes to some extent. Due to the small sample size and the lack of further quantitative measure of mitochondrial function in muscle fibers in the present study, additional investigation is still required to elucidate the precise mechanism underlying mitochondrial abnormality.
Conflicts of Interest
None.
Authors’ Contributions
YJ, WZ, and YY contributed to the study concept and design, analysis, and interpretation of the data. YL, YZ, YZ, MY, and JD contributed to the study concept and design, carried out the experiments, and acquisition of the data. HH, ZW, WZ, and YY contributed to the clinical diagnosis and biopsy of DM patients. YJ completed the statistical analysis and manuscript editing. WZ and YY contributed to the critical revision of the manuscript. The final manuscript was read and approved by all authors.
Data Sharing
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Supporting information
Table S1. Clinical characteristics of 11 adult anti‐MDA5 DM patients.
Table S2. Clinical characteristics of 10 antibody negative DM patients.
Table S3. Data of expression levels of MDA5, MAVS, IRF7, ISG15 in anti‐MDA5 DM patients and controls.
Table S4. Correlation analysis between MDA5, MAVS, IRF7, and ISG15 levels and muscle strength and disease activity in anti‐MDA5 DM patients.
Acknowledgments
The authors thank all the patients for their participation in this study. The authors also thank J. Liu, Q. Wang, and Y. Zuo for their technical assistance.
Funding Information
No funding information provided.
References
- 1. Allenbach Y, Leroux G, Suarez‐Calvet X, et al. Dermatomyositis with or without anti‐melanoma differentiation‐associated gene 5 antibodies: common interferon signature but distinct NOS2 expression. Am J Pathol 2016;186:691–700. [DOI] [PubMed] [Google Scholar]
- 2. Cerbelli B, Pisano A, Colafrancesco S, et al. Anti‐aminoacyl‐tRNA synthetase‐related myositis and dermatomyositis: clues for differential diagnosis on muscle biopsy. Virchows Arch 2018;472:477–487. [DOI] [PubMed] [Google Scholar]
- 3. Liu M, Li L, Dai T, et al. Gasdermine E‐dependent mitochondrial pyroptotic pathway in dermatomyositis: a possible mechanism of perifascicular atrophy. J Neuropathol Exp Neurol 2020;79:551–561. [DOI] [PubMed] [Google Scholar]
- 4. Wolstencroft PW, Fiorentino DF. Dermatomyositis clinical and pathological phenotypes associated with myositis‐specific autoantibodies. Curr Rheumatol Rep 2018;20:28. [DOI] [PubMed] [Google Scholar]
- 5. Sato S, Hoshino K, Satoh T, et al. RNA helicase encoded by melanoma differentiation‐associated gene 5 is a major autoantigen in patients with clinically amyopathic dermatomyositis: association with rapidly progressive interstitial lung disease. Arthritis Rheum 2009;60:2193–2200. [DOI] [PubMed] [Google Scholar]
- 6. McHugh NJ, Tansley SL. Autoantibodies in myositis. Nat Rev Rheumatol 2018;14:290–302. [DOI] [PubMed] [Google Scholar]
- 7. Moghadam‐Kia S, Oddis CV, Aggarwal R. Anti‐MDA5 antibody spectrum in western world. Curr Rheumatol Rep 2018;20:78. [DOI] [PubMed] [Google Scholar]
- 8. Nakahama T, Kato Y, Kim JI, et al. ADAR1‐mediated RNA editing is required for thymic self‐tolerance and inhibition of autoimmunity. EMBO Rep 2018;19:e46303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. He X, Zhu Y, Zhang Y, et al. RNF34 functions in immunity and selective mitophagy by targeting MAVS for autophagic degradation. EMBO J 2019;38:e100978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Meyer A, Laverny G, Bernardi L, et al. Mitochondria: an organelle of bacterial origin controlling inflammation. Front Immunol 2018;9:536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hoogendijk JE, Amato AA, Lecky BR, et al. 119th ENMC international workshop: trial design in adult idiopathic inflammatory myopathies, with the exception of inclusion body myositis, 10–12 October 2003, Naarden, The Netherlands. Neuromuscul Disord 2004;14:337–345. [DOI] [PubMed] [Google Scholar]
- 12. Labrador‐Horrillo M, Martinez MA, Selva‐O'Callaghan A, et al. Anti‐MDA5 antibodies in a large mediterranean population of adults with dermatomyositis. J Immunol Res 2014;2014:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pinal‐Fernandez I, Casciola‐Rosen LA, Christopher‐Stine L, et al. The prevalence of individual histopathologic features varies according to autoantibody status in muscle biopsies from patients with dermatomyositis. J Rheumatol 2015;42:1448–1454. [PMC free article] [PubMed] [Google Scholar]
- 14. Nagashima T, Kamata Y, Iwamoto M, et al. Liver dysfunction in anti‐melanoma differentiation‐associated gene 5 antibody‐positive patients with dermatomyositis. Rheumatol Int 2019;39:901–909. [DOI] [PubMed] [Google Scholar]
- 15. Liu Y, Zheng Y, Gang Q, et al. Perimysial microarteriopathy in dermatomyositis with anti‐nuclear matrix protein‐2 antibodies. Eur J Neurol 2020;27:514–521. [DOI] [PubMed] [Google Scholar]
- 16. Tansley SL, Betteridge ZE, Gunawardena H, et al. Anti‐MDA5 autoantibodies in juvenile dermatomyositis identify a distinct clinical phenotype: a prospective cohort study. Arthritis Res Ther 2014;16:R138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yasin SA, Schutz PW, Deakin CT, et al. Histological heterogeneity in a large clinical cohort of juvenile idiopathic inflammatory myopathy: analysis by myositis autoantibody and pathological features. Neuropathol Appl Neurobiol 2019;45:495–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Suzuki A, Kondoh Y, Taniguchi H, et al. Lung histopathological pattern in a survivor with rapidly progressive interstitial lung disease and anti‐melanoma differentiation‐associated gene 5 antibody‐positive clinically amyopathic dermatomyositis. Respir Med Case Rep 2016;19:5–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tanboon J, Nishino I. Classification of idiopathic inflammatory myopathies: pathology perspectives. Curr Opin Neurol 2019;32:704–714. [DOI] [PubMed] [Google Scholar]
- 20. Song N, Qi Q, Cao R, et al. MAVS O‐GlcNAcylation Is essential for host antiviral immunity against lethal RNA viruses. Cell Rep 2019;28:2386–2396.e5. [DOI] [PubMed] [Google Scholar]
- 21. Sun X, Sun L, Zhao Y, et al. MAVS maintains mitochondrial homeostasis via autophagy. Cell Discov 2016;2:16024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nguyen M, Do V, Yell PC, et al. Distinct tissue injury patterns in juvenile dermatomyositis auto‐antibody subgroups. Acta Neuropathol Commun 2020;8:125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gallay L, Mouchiroud G, Chazaud B. Interferon‐signature in idiopathic inflammatory myopathies. Curr Opin Rheumatol 2019;31:634–642. [DOI] [PubMed] [Google Scholar]
- 24. Zhang SH, Zhao Y, Xie QB, et al. Aberrant activation of type I interferon system may contribute to the pathogenesis of anti‐MDA5 dermatomyositis. Br J Dermatol 2018;180:1090–1098. [DOI] [PubMed] [Google Scholar]
- 25. Gao L, Bird AK, Meednu N, et al. Bone marrow‐derived mesenchymal stem cells from patients with systemic lupus erythematosus have a senescence‐associated secretory phenotype mediated by a mitochondrial antiviral signaling protein‐interferon‐beta feedback loop. Arthritis Rheumatol 2017;69:1623–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Buskiewicz IA, Montgomery T, Yasewicz EC, et al. Reactive oxygen species induce virus‐independent MAVS oligomerization in systemic lupus erythematosus. Sci Signal 2016;9:ra115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Clinical characteristics of 11 adult anti‐MDA5 DM patients.
Table S2. Clinical characteristics of 10 antibody negative DM patients.
Table S3. Data of expression levels of MDA5, MAVS, IRF7, ISG15 in anti‐MDA5 DM patients and controls.
Table S4. Correlation analysis between MDA5, MAVS, IRF7, and ISG15 levels and muscle strength and disease activity in anti‐MDA5 DM patients.