

Abstract
Objective
KCNQ2‐associated developmental and epileptic encephalopathies (DEE) present with seizures and developmental impairments. The relation between seizures and functional impairments in affected children and the relation of a specific genetic variant to seizure control remains unknown.
Methods
Parents of children with documented KCNQ2 variants who participated in a structured, online natural history survey provided information about seizure history, functional mobility, hand use, communication function, and feeding independence. Bivariate analyses were performed with nonparametric methods and logistic regression was used for multivariable analyses.
Results
Thirty‐nine children (20, 51% girls, median age 4.5 years, interquartile range (IQR) 1.9—19.3) had a median age of seizure onset of 1 day (IQR 1—3 days). The most common seizure types were bilateral tonic‐clonic (N = 72, 28%) and bilateral tonic (N = 13, 33%). Time since last seizure was <6 months (N = 18, 46%), 6–23 months (N = 11, 28%), and ≥24 months (N = 10 26%). Severe functional impairment was reported for mobility (62%), hand grasp (31%), feeding (59%), and communication (77%). Twenty‐eight (72%) were impaired in ≥2 domains. There were only weak and inconsistent associations between seizure recency and individual impairments or number of impairments after adjustment for other factors. The functional location of the variants within the Kv7.2 protein was not associated with seizure control.
Interpretation
Seizures in KCNQ2‐DEE are often well‐controlled, but children have severe impairments regardless. With the increased potential for precision therapies targeting the Kv7.2 channel or the KCNQ2 gene itself, identifying the most relevant and sensitive clinical endpoints will be critical to ensure successful trials of new therapies.
Introduction
KCNQ2‐developmental epilepsy and encephalopathy (DEE) is a rare neurodevelopmental brain disease estimated to affect ~100 new infants per year in the US. 1 KCNQ2 encodes the voltage‐gated potassium channel Kv7.2, variants in which were first recognized in association with a relatively “benign” form of familial neonatal epilepsy (BFNE), with often self‐limited seizures and typical development. The clinical phenotype associated with pathogenic variants in KCNQ2 was later extended to include severe neonatal‐onset encephalopathy and epilepsy, typically with tonic seizures and burst suppression on EEG. 2 , 3 Another phenotype includes onset a few months later, often presenting with infantile spasms. 2 , 4
Because of its relatively recent recognition and its extreme rareness, descriptions of KCNQ2‐associated epilepsy are based on nonsystematic, retrospectively ascertained clinical data of individual patients from across multiple sites. The current profiles based on these multicenter case series indicate a severe disorder with impairments in multiple domains. Seizures, however, while the initial presenting symptom in the neonatal period, have been described as remitting in some patients. 2
The relation between seizures and the encephalopathic features or impairments associated with KCNQ2‐DEE has been hypothesized but not established. The concept of “epileptic encephalopathy,” 5 in which seizures themselves negatively influence brain development, is a clear possibility; however, a direct effect of impaired Kv7.2 channel function, regardless of seizures, must be considered. The growing interest in precision therapies for rare genetic disorders is resulting in novel compounds being considered for evaluation in human trials. The target outcomes for such trials must be carefully selected as seizures may not be the only or even most important outcome in patients, 6 , 7 and cessation of seizures may or may not result in improvement in other important disease manifestations.
To accelerate understanding of rare DEEs and to provide an understanding of the many conditions and morbidities associated with DEEs, a natural history survey was initiated. Integral to the study methods was the inclusion of parent input into the selection of features to study, specific questions used, and appropriate response categories for those questions. A study goal was to provide a rapid means of performing surveys and eventually longitudinal studies of DEEs with systematically collected parent‐reported information as such data would approximate the type of data collected from parents in the clinical trial setting. Here we present the results of the initial KCNQ2 cohort in whom the frequency and nature of severe impairments and concerns were assessed by parent report in a systematic fashion.
To address the potentially separate roles of seizures versus the KCNQ2 disease itself, we focused on the relationship between seizure freedom and four indicators of functional impairment (mobility, hand grasp, feeding, and communication) and four clinical outcomes identified as important to parents (sleep disruption, gastrointestinal dysfunction, cortical visual impairment, and autism diagnosis or features).
Methods
In collaboration with parents representing different DEE family groups, including the KCNQ2 Cure Alliance and the Jack Pribaz Foundation, a survey was developed to assess not only seizures but also several areas of functional and developmental impairment and other concerns identified by parents. Details of the survey development were presented previously. 8 Where possible, we utilized existing instruments or modified them and incorporated parent feedback in determining response options. The survey was designed as a series of topic‐specific forms for seizures, gross and fine motor abilities, communication, feeding, behavior, sleep, gastrointestinal function, vision, hearing, and other outcomes.
Gross motor function was assessed with age‐appropriate wording from the Gross Motor Function Classification System (GMFCS) for children ≥2 years old. 9 Based on the GMFCS, gross motor ability was dichotomized as dependent (need of mobility device or assistance from a person or aid) versus independent. For children <2 years‐old, gross motor ability was dichotomized based on a parental assessment of development as moderately to severely delayed versus within expected range or mildly delayed. Fine motor skills were based on parent report of hand grasp and purposeful manipulation of objects.
Communication function was assessed with the Communication Function Classification System (CFCS) for children ≥2 years old. 10 A dichotomized variable was also created based on the CFCS to reflect difficulty communicating with unfamiliar or familiar people (dependent) versus effective communication with known and unfamiliar people (independent). For children <2 years old, communication ability was dichotomized based on a parental assessment of communication skills as moderately to severely delayed versus within expected range or mildly delayed.
Feeding ability was reflected by the use of a gastrotomy tube (none, partially fed, and exclusively fed) and a four‐point question about independence for feeding purposes (fully independent, requires some assistance or supervision, requires considerable assistance or supervision, completely dependent on someone else). This latter reflected concepts from the eating and drinking ability classification system (EDACS) 11 and was modified based on parent input. The dichotomous variable for feeding dependence was based on exclusive tube feeding or a rating by parents of needing considerable or complete help in feeding for children ≥2 years old. For children <1 year old, it was based only on the presence of a gastrotomy tube, and for children 12–23 months, it was based on the presence of a gastrotomy tube or complete dependence on someone else.
A sum of the four domains in which there was significant impairment, dependence, or delay was constructed. A full report on functional abilities in KCNQ2 and other DEE groups has been published previously. 8
Parents were asked about the frequency of sleep disturbances due to seizures and independent of seizures. For these analyses, we focused on nonseizure‐related nocturnal awakenings as reported in the past month (6–7 nights/week, 1–5 nights/week, and <1/week, including none). Checklists were provided for vision and hearing concerns. As cortical visual impairment (CVI) was specifically identified as a concern by parents, we have focused on CVI; however, all information is based strictly on parent report. Gastrointestinal symptoms, specifically constipation and dysmotility in the past month, were combined into an either‐or versus neither variable. In the survey, parents indicated whether their child carried a formal autism diagnosis or had features without a diagnosis. A variable to reflect endorsement of either option versus neither was created.
The seizure form reviewed the onset of seizures, types of different seizures experienced (including age at onset and age at last for each type), and antiseizure medications used. We specifically determined the occurrence of the most recent seizure of any type. Parents were provided formal descriptions of ten different seizure types to guide their responses. 12 Parents could provide other seizure types, not in our list with descriptions of those seizures.
Parents were asked to provide information on their children’s KCNQ2 variants, preferably a de‐identified copy of the genetic testing report although parent‐reported variants were accepted. All variants were reviewed and classified according to ACMG criteria by a licensed genetic counselor (SM). 13
Inclusion criteria for the analytic sample: Because of our focus on definitive KCNQ2‐associated disorders, only children for whom the parents provided the specific KCNQ2 variant were included in these analyses. For our primary analyses, we included children with variants classified as pathogenic or likely pathogenic as well as children with variants of unknown significance provided the parents had been tested and the variant was not inherited. We further limited analyses to children for whom information about seizures and functional abilities were provided by parents.
For analyses of seizures, our primary measure of seizure freedom was ≥6 months seizure‐free. Consequently, we planned to limit analyses to children >6 months old at the time of the interview. In a parallel set of analyses, we considered longer times seizure‐free (<6, 6–23 ≥ 24 months) as an ordinal variable in children ≥2 years old. For children ≥2 years of age, we also (secondarily) examined the multilevel, ordinal variables for the GMFCS, CFCS, and the feeding independence variable.
The survey was implemented in CLIRINX®, a novel web‐based program for facilitating multicenter, clinical studies. 14 It was disseminated to parents through the family groups. Parents were able to return and complete the forms in as many sittings as they chose. While we encouraged parents to complete all forms within about a week’s time, we accepted any information submitted. Data were collected from December 2018 through February 2020. Up to three reminders were sent if a parent had not completed all of the forms.
Data were analyzed in SAS 9.4©. Descriptive statistics were calculated with chi‐square, t‐test, and Wilcoxon rank‐sum tests, as appropriate to the data structure. Given the limited sample size, exact P‐values were used when possible. Multivariable analyses were performed with logistic regression.
Parents provided electronic, informed consent through a secure online system in CLIRINX®. All procedures were approved by the Lurie Children’s Hospital Institutional Review Board.
Results
In total, 106 participants registered in the KCNQ2 project. Of these, 50 provided their child’s genetic variant. One was excluded as the variant was a large deletion that incorporated KCNQ2 as well as EEF1A2 (associated with epilepsy and intellectual disability 15 ) and CHRNA4 (associated with an autosomal dominant form of epilepsy 16 ). One child had an inherited variant interpreted as a VUS. Of the 48 remaining, seizure information was not provided for eight, and functional impairment information was missing for one other. Analyses were performed for 39 children with documented pathogenic or likely pathogenic variant or a de novo VUS and complete phenotypic data. No child was younger than 6 months old.
The 39 participants who met all inclusion criteria, including confirmation of their KCNQ2 variants, came from the United States (N = 23), United Kingdom (N = 3), Canada (N = 2), the Netherlands (N = 3), and one from each of eight other countries. Mothers comprised 36 (92%) of responding parents. The 39 children included 20 (51%) girls. The median age of the children at the time of the survey was 4.5 years (interquartile range (IQR) 1.9 to 9.2, range 0.6 to 19.3). Twelve (31%) children were younger than 2 years old, 13 (33%) were 2–4 years old, eight (21%) were 5–9 years old, and six (15%) were ≥10 years old.
Variants
Thirty‐nine patients were reported to have 32 different variants with five variants reported for two separate patients and one variant reported for three. Two distinct variants were reported at each of the amino acid positions 256 and 281. Of the 32 variants, 17 (24 children) were pathogenic, 12 (12 children) likely pathogenic, and two (3 children) were de novo VUS by ACMG variant interpretation criteria (Table S1). Of the 32 unique variants, eight were within the intracellular domains (six C‐terminal, two linker), three within the extracellular domain (pore region), and 21 within the transmembrane domains (Fig. 1). Twenty‐seven children had confirmed de novo variants; for 12, the inheritance was unknown.
Figure 1.
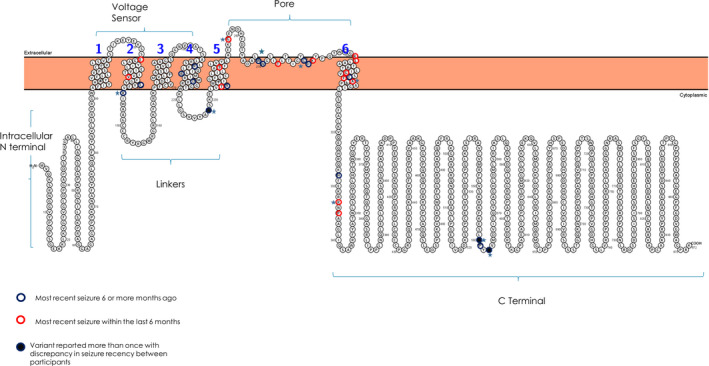
Location of variants within the Kv7.2 protein and seizure control associated with each variant.
Seizures
The median age of seizure onset, was the first day of life (IQR 1 to 3 days). Thirty‐seven children (95%) had their first seizure within the first week of life. The remaining two children had seizure onset at 1 and 6 months of age. At the time of the survey, the majority of children (N = 31, 79%) were reported to have had one or two different seizure types, four children had three types, and four children had four types of seizures. The two most common seizure types were bilateral tonic‐clonic (N = 28, 72%), followed by bilateral tonic (N = 13, 33%). All but four children had one or both of these types. Other seizure types included infantile spasms (N = 9, 23%), myoclonic (N = 6, 15%), and other focal motor seizures (N = 6, 15%). Additional seizure types occurred in only a few patients each. Age at onset for each seizure type is provided in Table 1. During the most active stage of their epilepsy, 35 (92%) of children reportedly had daily seizures. On days seizures occurred, multiple (≥3) daily seizures were reported for 37 (95%) children with 19 (49%) reporting ten or more seizures per day.
Table 1.
Seizure types for children with KCNQ2‐DEE.
| Seizure type 1 | N (%) | Median age at first in months (IQR) | Occurred in past 6 months |
|---|---|---|---|
| Bilateral tonic‐clonic | 28 (72%) | 0.06 (0.03–0.13) | 8 (29%) |
| Bilateral tonic | 13 (33%) | 0.03 (0.03–0.22) 2 | 6 (46%) |
| Bilateral clonic | 2 (5%) | 0.03 2 | 1 (50%) |
| Hemiconvulsion | 1 (3%) | 0.13 | 0 |
| Absence | 2 (5%) | 39 (18–60) | 1 (50%) |
| Behavioral Arrest | 4 (10%) | 45.5 (13.5–78) | 2 (50%) |
| Myoclonic | 6 (15%) | 1 (1–9) | 3 (50%) |
| Infantile spasms | 9 (23%) | 3 (0.07–5.0) | 1 (11%) |
| Other focal motor | 6 (15%) | 0.6 (0.1–5.0) | 2 (33%) |
No parent reported atonic seizures.
Missing one response each.
In the previous week, 32 (82%) children reportedly had no seizures. This included 21 (54%) children with no seizures in the past 6 months (remote seizures). Of the remaining, three children had seizures on 1–2 days in the previous week and three on 4–7 days. The median age in the remote seizure group (4.5 years, IQR 2.6–10.1 years) was only slightly older than in the recent seizure group (3.4 years, IQR 1.0–8.1, P = 0.62). In the remote seizure group, the median time since the last seizure was 23 months (IQR 17–55.0, range 7–125); time seizure‐free was 6–23 m (N = 11), and ≥24 m (N = 10). The age at last definite seizure was 13 months (IQR 3 to 36, range 0.5–108). There was no discernable pattern in the location of the variants with respect to moderate seizure freedom (≥6 months), and in a few instances, children with the same variant were discordant for seizure outcome (Fig. 1).
Treatment
Of 38 patients with treatment information, 31 (82%) were taking anti‐seizure medication. This included 17/18 (94%) of those <6 m seizure‐free, 9/10 (90%) 6–23 m seizure‐free, and 5/10 (50%) ≥24 m seizure‐free (P = 0.007). Parents reported current or past use of 24 different anti‐seizure medications (ASM). The most frequently reported were levetiracetam (N = 30) and phenobarbital (N = 31). The most common sodium channel blocking agent reported was oxcarbazepine (N = 17), followed by phenytoin and carbamazepine (N = 12 each), and lacosamide (N = 5). Ezogabine (retigabine), a potassium channel opener, was reported in four patients. Parents reported drug effectiveness and reasons for stopping if discontinued (Fig. [Link], [Link]). Only 2/12 (17%) children became completely seizure‐free on carbamazepine whereas the comparable numbers were 9/17 (53%) for oxcarbazepine and 5/12 (42%) for phenytoin. Only 2/4 (50%) children became seizure‐free on ezogabine. No child had undergone resective or other intracranial epilepsy surgery or device implantation (e.g., vagus nerve stimulator).
Functional abilities
All 39 children had information about levels of function for the four primary functional domains. Twenty‐four (62%) did not walk or, if <2 years old, had moderate to severe delays; 12 (31%) did not grasp objects with their hands, 23 (59%) were mostly or entirely dependent on someone else for feeding, and 30 (77%) were ineffective communicators with unfamiliar people and mostly ineffective with persons known to them. The number of children with these levels of impairments in 0, 1, 2, 3, and 4 domains was 4 (10%), 7 (18%), 10 (26%), 10 (26%), and 8 (20%). The number of domains with functional impairment was not significantly different in children <2 versus ≥2 years old (P = 0.60); however, by individual domain, children <2 years were less likely to fall in the dependent range for feeding (P = 0.03).
Gut dysmotility or constipation was reported for 28 (72%), CVI for 15 (38%), and autism diagnosis or features by 16/38 (42%) children. Night‐time awakenings not associated with seizures were noted to occur rarely (<1/ week) or never by 12/36 (34%), 1–5 nights per week by 15 (42%), and 6–7 nights per week by 9 (25%) parents.
We compared the proportion with significant functional impairment in each domain, the number of impaired functional domains, and the proportion with each of the four other parent‐prioritized clinical outcomes in those with recent (<6 months) and remote (≥6 months) seizures (Table 2, Fig. 1A). We found only modest, borderline significant associations when comparing children with recent versus remote seizures, suggesting that those with recent seizures were more likely to have impaired hand use (P = 0.05) and more nonseizure‐related night‐time awakenings (P = 0.06). For most other outcomes, there was only a few percentage‐point difference in the prevalence of functional and other outcomes in those with recent versus remote seizures.
Table 2.
Bivariate associations between time seizure‐free and significant impairment or dependence in each of four functional areas and for four morbidities specifically identified by parents.
| All Children (N = 39) 1 | Children >2 years old (N = 28) 2 | |||||
|---|---|---|---|---|---|---|
| Seizure Recency | Grouped months seizure‐free | |||||
| Domain or feature | <6 months (N = 18) | ≥6 months (N = 21) | <6 months (N = 10) | 6–23 months (N = 8) | ≥24 months (N = 10) | |
| Motor | N for children >2 y | |||||
| Independent (N = 15) | 5 (28%) | 10 (48%) | (N = 13) | 4 (40%) | 2 (25%) | 7 (70%) |
| Dependent (N = 24) | 13 (72%) | 11 (52%) | (N = 15) | 6 (60%) | 6 (75%) | 3 (30% |
| P = 0.32 | P = 0.19 | |||||
| Hands | ||||||
| Grasp (N = 27) | 9 (50%) | 18 (86%) | (N = 21) | 6 (60) | 5 (63%) | 10 (100%) |
| No grasp (N = 12) | 9 50%) | 3 (14%) | (N = 7) | 4 (40%) | 3 (37%) | 0 |
| ` | P = 0.03 | P = 0.04 | ||||
| Eating | ||||||
| Independent/PO (N = 16) | 7 (39%) | 9 (43%) | (N = 9) | 2 (20%) | 3 (37%) | 4 (40%) |
| Dependent/g‐tube (N = 23) | 11 (61%) | 12 (57%) | (N = 19) | 8 (80%) | 5 (63%) | 6 (60%) |
| P = 1.0 | P = 0.35 | |||||
| Communication | ||||||
| Independent (N = 9) | 4 (22%) | 5 (24%) | (N = 5) | 1 (10%) | 0 | 4 (60%) |
| Dependent (N = 30) | 14 (78%) | 16 (76%) | (N = 23) | 9 (90%) | 8 (100%) | 6 (40%) |
| P = 1.0 | P = 0.09 | |||||
| Constipation/dysmotility | ||||||
| No (N = 11) | 6 (33%) | 5 (24%) | (N = 8) | 4 (40%) | 4(50%) | 0 |
| Yes (N = 28) | 12 (67%) | 16 (76%) | (N = 20) | 6 (60%) | 4 (50%) | 10 (100%) |
| P = 0.72 | P = 0.05 | |||||
| Nights with nonseizure awakenings (prior month) 3 | ||||||
| <1/week or never (N = 12) | 3 (20%) | 9 (43%) | (N = 9) | 3 (37%) | 3 (37%) | 3 (30%) |
| 1–5 nights/week (N = 15) | 6 (40%) | 9 (43%) | (N = 12) | 3 (37%) | 4 (50%) | 25 (50%) |
| 6–7 nights/week (N = 9) | 6 (40%) | 3 (14%) | (N = 5) | 2 (25%) | 1 (13%) | 2 (20%) |
| P = 0.06 2 | P = 0.92 | |||||
| Cortical Visual Impairment | ||||||
| No (N = 24) | 9 (50%) | 15 (71%) | (N = 16) | 4 (40%) | 3 (37%) | 9 (90%) |
| Yes (N = 15) | 9 (50%) | 6 (29%) | (N = 12) | 6 (60%) | 5 (63%) | 1 (10%) |
| P = 0.20 | P = 0.03 | |||||
| Autism diagnosis or features | ||||||
| No (N = 22) | 10 (59%) | 12 (57%) | (N = 14) | 5 (50%) | 4 (50%) | 5 (50%) |
| Yes (N = 16) | 7 (41%) | 9 (43%) | (N = 14) | 5 (50%) | 4 (50%) | 5 (50%) |
| P = 1.0 | P = 1.0 | |||||
P‐value based on Fisher Exact test for all 2 × 2 tables.
P‐value based on Mantel–Haenszel Chi‐square test for trend on 1 degree of freedom.
Responses were missing for sleep disturbances (N = 3), and autism (N = 1).
For children older than 2 years (N = 28), the same associations were examined with the ordinal representation of time seizure‐free (<6, 6–23, ≥24 months) to test whether longer periods seizure‐free were associated with lower prevalence of impairment or morbidity (Table 2, Fig. 1B). The findings suggested less impairment with longer time seizure‐free for hand use (P = 0.04) and cortical visual impairment (P = 0.03). Constipation or gut dysmotility was more commonly reported in children who had been seizure‐free ≥2 years (100%) than in children who were seizure‐free <6 months (60%) and 6–23 months (50%, P = 0.03).
We adjusted all bivariate associations in Table 2 and Figure 1 for age at the time of study participation to ensure none of the findings might be due to older children having acquired more skills and having the opportunity to have longer periods seizure‐free (Table 3). Multivariable adjustment did not substantially alter any of the associations, nor was age significantly associated with any of the clinical outcomes studied.
Table 3.
Multivariable age‐adjusted associations between time seizure‐free and significant impairment or dependence in each of four functional areas and for four morbidities specifically identified by parents.
| ≥6 months versus <6 months seizure‐free in all children (N = 39) | Grouped time seizure‐free in children ≥2 years old 2 | |||||
|---|---|---|---|---|---|---|
| Odds ratio 3 | 95% (CI) | P‐value | Odds ratio 3 | 95% (CI) | P‐value | |
| Walking | 0.52 | (0.13, 2.12) | 0.36 | 0.57 | (0.22, 1.46) | 0.24 |
| Hand grasp | 0.18 | (0.04, 0.87) | 0.03 | 0.29 | (0.08, 1.05) | 0.06 |
| Feeding | 0.75 | (0.20, 2.83) | 0.67 | 0.68 | (0.25, 1.81) | 0.44 |
| Communication | 0.86 | (0.18, 4.01) | 0.85 | 0.33 | (0.07, 1.48) | 0.15 |
| # Domains with significant impairment | 0.43 | (0.13, 1.44) | 0.17 | 0.44 | (0.19, 1.03) | 0.06 |
| Constipation/dysmotility | 1.44 | (0.34, 6.09) | 0.62 | 3.13 | (0.88, 11.14) | 0.08 |
| Nights awake/week 1 | 0.25 | (0.06, 0.98) | 0.05 | 0.92 | (0.37, 2.31) | 0.87 |
| Cortical visual impairment | 0.36 | (0.09, 1.42) | 0.14 | 0.33 | (0.11, 0.93) | 0.04 |
| Autism diagnosis or features | 0.85 | (0.21, 3.34) | 0.81 | 0.94 | (0.38, 2.32) | 0.89 |
Abbreviation: CI, Confidence interval.
grouped as none or <1/week, 1–5/week, 6–7/week.
Grouped time seizure‐free: <6, 6–23, ≥24 months.
All estimates are adjusted for age and represent the change in the prevalence of the outcome with greater time seizure‐free.
We also considered age at last seizure versus time seizure‐free in a series of logistic regression models for children ≥2 years‐old. To avoid over‐specifying the models, current age was not included. For none of the functional or clinical outcomes, did age at last seizure approach statistical significance in bivariate or multivariable logistic regression analyses. Finally, whether or not a child was currently taking ASMs was also considered. While current ASM use had modest bivariate associations with some outcomes, it did not substantially alter any of the associations between functional impairments and seizures already described above.
For children ≥2 year old, we examined the association between time seizure‐free (<6 months, 6–23, and ≥24 months) and the full range of responses for the GMFCS, CFCS, and the feeding independence variable (Fig. 2). Both bivariate and multivariable (adjusted for age) analyses indicated at most modest associations between time seizure‐free with feeding (P = 0.06) and the CFCS (P = 0.08) and no clear association with the GMFCS (P = 0.21). Few children were assessed to be in the highest level of function or independence on any of these three measures.
Figure 2.
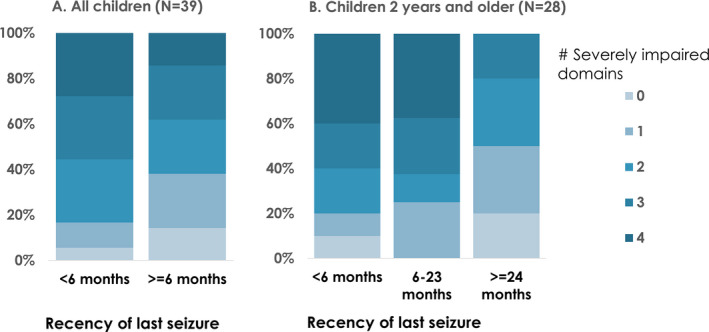
Association between seizure recency and number of domains with severe impairment. A. For all children comparing recent (<6 months) and remote (>=6 months) seizures (P = 0.14) and B. For children 2 years and older comparing seizure recency of <6, 6–23, and >=24 months (P = 0.05).
Discussion
KCNQ2‐DEE typically presents as neonatal or very early infantile‐onset epilepsy. 1 , 3 In our cohort, almost all children had onset in the first week of life. Importantly, the majority of the cohort was no longer having seizures at the time when parents completed the surveys, either because seizures were well‐controlled on medication or had resolved and children were no longer taking medication. KCNQ2‐DEE is typically associated with a severe degree of disability, as evidenced by the large proportion of children in this cohort who were dependent for mobility, feeding, hand use, and communication. The majority of the children had substantial impairments in two, three, or all four domains. Only four children had independent function in all four domains, but even these children had delays and impairments. For example, two did not use speech as a primary mode of communication and only one was described as having gross motor skills in the expected range for age.
KCNQ2 variants of children in this study are comparable to those reported in the literature, and the overall description of the cohort in terms of seizure types and pervasiveness of impairments is also comparable to what might be surmised from the literature. Our study adds a systematic assessment of the levels of impairment seen in children with documented KCNQ2‐DEE and examines the relationship between seizures and functional impairment. In fact, there were only modest and inconsistent associations between seizure freedom (of up to 2 or more years) and occurrence of severe functional impairments and of other clinical outcomes important to parents (sleep, GI symptoms, autism, and CVI).
The concept of epileptic encephalopathy 5 , 17 implies that seizure activity in the developing brain contributes to subsequent developmental impairment and disability by interfering with normal developmental function. Evidence supporting this concept largely comes from studies demonstrating that earlier onset and longer duration with uncontrolled seizures are associated with poorer developmental and cognitive scores later in childhood. 18 , 19 , 20 , 21 , 22 , 23
KCNQ2‐DEE poses an interesting contrast to the result from two large randomized trials for infantile spasms and several surgical series, which have motivated and supported the concept of “epileptic encephalopathy.” 5 , 17 In this KCNQ2 cohort, there were only modest and often inconsistent relationships between basic functional abilities and time seizure‐free. Per the epileptic encephalopathy hypothesis, earlier control of seizures should be associated with higher levels of development and function, yet we did not find seizure freedom or younger age at last seizures to be strongly associated with better outcomes. We suspect that seizures play a substantial role in daily fluctuations in a child’s level of function and that, over time, uncontrolled seizures do contribute to developmental impairments and resulting disability. The presence of seizures very early during development may disrupt normal developmental processes so profoundly that they are permanently affected. Anecdotally, some parents reported that they were sure seizures had begun in utero. On the other hand, disability in KCNQ2‐DEE, likely also reflects ongoing Kv7.2 dysfunction in the central nervous system as developmental impairments apparently persist long after cessation of seizures in many cases. This is entirely plausible as KCNQ2 (and KCNQ3) expression, which begins in the human brain in late fetal life 24 persists into adulthood in both rodent and human brain. 24 , 25
Important limitations include that all data are based on parent‐reported information. Parent and patient‐reported outcomes, however, are an important and recognized source of information for primary and secondary outcomes in randomized trials. 6 , 7 The outcome measures employed were directly drawn from measures already in use or adapted from existing measures. Our sample covered a broad age range from infancy through early adulthood. Measures of development for infants and young children, yield to measures of function and impairment as children grow older. Hence, our metrics for children younger than two years differed from those in older children. We also note that the parent‐reported functional measures are nongranular; however, data collection was systematic thus facilitating grouping of patients and comparisons based on consistently collected data.
The survey was disseminated through family group websites and Facebook pages, which may introduce ascertainment bias. Members of these groups, and those most likely to participate in such surveys, may not be representative of all families or the full spectrum of disease manifestations; the children included may represent those more severely affected.
Most children were exposed to one or more sodium blocking agent, most often oxcarbazepine. The reported information did not reflect overwhelming success of any therapy. In fact, seizure response for carbamazepine (17%) was much lower than might be expected based on a recent report of benign familial neonatal epilepsy secondary to KCNQ2 or KCNQ3 variants in which 88% of patients had cessation of seizures with treatment. 26 Importantly, our data about treatment responses did not include information about cotreatments, and attribution of drug effect would be uncertain.
Our sample size is limited. KCNQ2‐DEE is a rare disorder, so this is not unexpected. Although we had a larger group of participants, analyses were restricted to those with documented genetic variants. Having such limited numbers places acknowledged constraints on statistical power. While our results cannot rule out that seizures adversely affect the outcomes examined, they also do not suggest seizures strongly determine these outcomes. In fact, most associations examined indicated little or no difference (within a few percentage points) for outcomes in children with different levels of seizure freedom.
Seizures are the earliest, most common, and an especially distressing symptom that brings children with KCNQ2‐DEE to medical attention. Accordingly, studies of therapeutic outcomes to date have focused on seizures. The finding that many children stop having seizures very early—either because their seizures are well‐controlled with available medications or resolve on their own, poses challenges for future trials of therapeutics as, within this already very rare disease population, many children would not be eligible for trials targeting seizures as the primary outcome. The Food and Drug Administration (FDA) has issued guidance to industry for the development of therapeutics for rare diseases. 6 , 7 This guidance emphasizes the importance of having a well‐delineated natural history, identifying patient‐relevant clinical outcomes, and identifying or developing appropriate clinical outcome assessments that can measure those outcomes in a way that is sensitive to meaningful change in the context of a randomized trial.
The functional measures we used, while not granular, represent the full range of function from fully independent to dependent and are either verbatim or adapted versions of measures in standard use in the rehabilitation literature. 27 They provided a clinically meaningful summary of ability to disability spectrum and demonstrated the severe disability in all domains; nearly half of KCNQ2‐DEE patients were dependent in three or all four domains.
The parents from the KCNQ2 community have emphasized the importance of basic functions, communication, sleep, GI function, and behavior. The measures we employed highlighted the severe degree of impairment in these children but were never intended for use in monitoring change in randomized trials. For that, it is necessary to identify or develop well‐validated clinical outcome assessments that are appropriate for tracking change in people with severe levels of disability. For basic functional abilities (mobility, communication), the level of disability is so severe, that standard measures often used in the general population may be inadequate. For example, the Vineland Scales of Adaptive Behavior, a standardized, clinically valuable assessment instrument, was used in a cohort of children with SCN2A‐DEE, a different but similarly severe neurodevelopmental disorder. 28 Vineland scores were approximately 3 standard deviations below the test mean, several domains had substantial “floor effects” (over half of subjects had raw scores of 0) and, over the age span and longitudinally, the rate of skill acquisition was so slow that standardized scores actually fell over time although there was no apparent regression in performance as reflected by stable or slightly increasing raw scores. The Vineland was recently used in two trials evaluating cannabidiol. Although cannabidiol had a robust effect on seizures, and the Global Impression of Change index improved significantly in the treatment groups in both trials, there was no change in Vineland scores. 29 , 30
To support the anticipated upcoming wave of clinical trials of new, precision medicine therapeutics for the DEEs, a more detailed understanding of the levels of function and disability and validated measures to assess meaningful changes will be necessary. Seizures are a critical outcome; however, they are not the only outcome and in the case of KCNQ2‐DEE, not persistent in many children. Furthermore, our findings do not support the notion that control of clinical seizures alone is sufficient to prevent or reverse KCNQ2‐related encephalopathy. The promise of new precision therapies – including the gene‐targeted therapies – is not simply to suppress symptoms. The goal is to treat the disease itself and, in doing so, return a patient to optimal health. The extent to which this can be achieved, after early onset seizures and encephalopathy, is an important question to be addressed in formal clinical trials. To determine the value of therapies for achieving this goal, trials must include validated measures of important, nonseizure disease manifestations that are sufficiently granular for the targeted population and sensitive to meaningful change over time, especially in what is often the limited timeframe of a randomized trial. Our findings, and the measures employed, can be used to guide the design of parent‐reported clinical trial outcomes for patients with KCNQ2‐DEE. In conjunction with parent‐reported seizures, video‐EEG, data from wearable devices, 31 and other assessments of seizure burden, parent‐reported outcomes regarding developmental function as well as clinician‐administered performance measures suitable to the patients’ level of ability can serve as valuable measures to assess the efficacy of treatments, from potassium channel openers to emerging gene editing and antisense oligonucleotide approaches.
Conflict of Interest
A. T. Berg: None. S. Mahida: None. A. Poduri: None.
Figure 3.
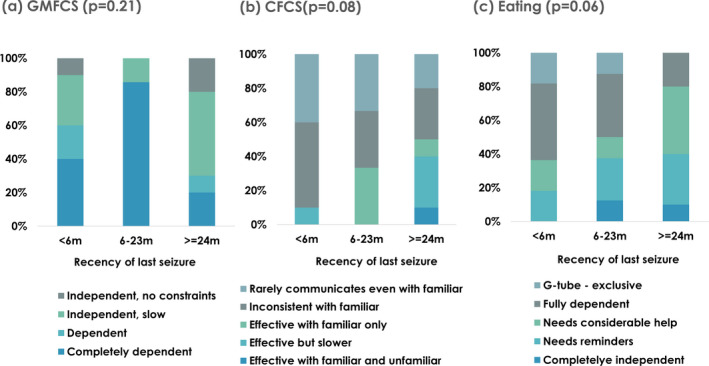
For children 2 years and older, associations between seizure recency (<6, 6–23, and >=24 months) and the full range of responses for (A) the Gross Mobility Function Classification System (GMFCS), (B) Communication Function Classification System (CFCS), and (C) Eating Independence. All P‐values are based on a Mantel–Haenszel chi‐square for trend.
Supporting information
Figure S1. Medications used in children with KCNQ2‐DEE: a. Reported effect of seizure control of medications and b. Reported reasons for stopping medications.
Table S1. KCNQ2 Variants of patients included in the study sample and their ACMG classification and location within the Kv7.1 protein.
Acknowledgments
We are very grateful to the many families who participated in the design and implementation of this study and who participated in the study itself and to Gerry Nebitt whose expertise and support made this study possible.
Funding information
The analyses for this project were funded by the Stanley Manne Children’s Research Institute and Ann & Robert H. Lurie Children’s Hospital of Chicago under the Precision Medicine Strategic Research Initiative and by a grant from the Pediatric Epilepsy Research Foundation, Dallas, TX. AP was supported by the Translational Research Program, Boston Children’s Hospital; AP and SM were supported by the KCNQ2 Alliance.
Funding Statement
This work was funded by Stanley Manne Children’s Research Institute grant ; Ann & Robert H. Lurie Children’s Hospital of Chicago grant ; Pediatric Epilepsy Research Foundation grant ; Translational Research Program, Boston Children’s Hospital grant ; KCNQ2 Alliance grant .
References
- 1. Shellhaas RA, Wusthoff CJ, Tsuchida TN, et al. Profile of neonatal epilepsies: characteristics of a prospective US cohort. Neurology 2017;89:893–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Weckhuysen S, Ivanovic V, Hendrickx R, et al. Extending the KCNQ2 encephalopathy spectrum: clinical and neuroimaging findings in 17 patients. Neurology 2013;81:1697–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Weckhuysen S, Mandelstam S, Suls A, et al. KCNQ2 encephalopathy: emerging phenotype of a neonatal epileptic encephalopathy. Ann Neurol 2012;71:15–25. [DOI] [PubMed] [Google Scholar]
- 4. Fang ZX, Zhang M, Xie LL, et al. KCNQ2 related early‐onset epileptic encephalopathies in Chinese children. J Neurol 2019;266:2224–2232. [DOI] [PubMed] [Google Scholar]
- 5. Berg AT, Cross JH. Towards a modern classification of the epilepsies? Lancet Neurol 2010;9:459–461. [DOI] [PubMed] [Google Scholar]
- 6. FDA . Workshop on natural history studies of rare diseases. 2012. Bethesda, MD: FDA. [Google Scholar]
- 7. FDA . Rare diseases: natural history studies for drug development guidance for industry. In: Adminsitration FD , ed. 2019. https://www.fda.gov/media/122425/download. [Google Scholar]
- 8. Berg AT, Gaebler‐Spira D, Wilkening G, et al. Nonseizure consequences of Dravet syndrome, KCNQ2‐DEE, KCNB1‐DEE, Lennox‐Gastaut syndrome, ESES: a functional framework. Epilepsy Behav 2020;111:107287. [DOI] [PubMed] [Google Scholar]
- 9. Morris C, Galuppi BE, Rosenbaum PL. Reliability of family report for the gross motor function classification system. Dev Med Child Neurol 2004;46:455–460. [DOI] [PubMed] [Google Scholar]
- 10. Hidecker MJ, Paneth N, Rosenbaum PL, et al. Developing and validating the communication function classification system for individuals with cerebral palsy. Dev Med Child Neurol 2011;53:704–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sellers D, Mandy A, Pennington L, et al. Development and reliability of a system to classify the eating and drinking ability of people with cerebral palsy. Dev Med Child Neurol 2014;56:245–251. [DOI] [PubMed] [Google Scholar]
- 12. Berg AT, Millichap JJ. The 2010 revised classification of seizures and epilepsy. Continuum 2013;19:571–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nesbitt G. CLIRINX [online]. https://www.clirinx.com/.
- 15. Lam WW, Millichap JJ, Soares DC, et al. Novel de novo EEF1A2 missense mutations causing epilepsy and intellectual disability. Molec Gentet Genome Med 2016;4:465–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Steinlein OK, Mulley JC, Propping P, et al. A missense mutation in the neuronal nicotinic acetylcholine receptor alpha 4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet 1995;11:201–203. [DOI] [PubMed] [Google Scholar]
- 17. Howell KB, Harvey AS, Archer JS. Epileptic encephalopathy: use and misuse of a clinically and conceptually important concept. Epilepsia 2016;57:343–347. [DOI] [PubMed] [Google Scholar]
- 18. O'Callaghan FJ, Edwards SW, Alber FD, et al. Safety and effectiveness of hormonal treatment versus hormonal treatment with vigabatrin for infantile spasms (ICISS): a randomised, multicentre, open‐label trial. Lancet Neurol 2017;16:33–42. [DOI] [PubMed] [Google Scholar]
- 19. O'Callaghan FJ, Lux AL, Darke K, et al. The effect of lead time to treatment and of age of onset on developmental outcome at 4 years in infantile spasms: evidence from the United Kingdom Infantile Spasms Study. Epilepsia 2011;52:1359–1364. [DOI] [PubMed] [Google Scholar]
- 20. Berg AT, Zelko FA, Levy SR, Testa FM. Age at onset of epilepsy, pharmacoresistance, and cognitive outcomes: a prospective cohort study. Neurology 2012;79:1384–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cormack F, Cross JH, Isaacs E, et al. The development of intellectual abilities in pediatric temporal lobe epilepsy. Epilepsia 2007;48:201–204. [DOI] [PubMed] [Google Scholar]
- 22. Jonas R, Nguyen S, Hu B, et al. Cerebral hemispherectomy: hospital course, seizure, developmental, language, and motor outcomes. Neurology 2004;62:1712–1721. [DOI] [PubMed] [Google Scholar]
- 23. Vasconcellos E, Wyllie E, Sullivan S, et al. Mental retardation in pediatric candidates for epilepsy surgery: the role of early seizure onset. Epilepsia 2001;42:268–274. [DOI] [PubMed] [Google Scholar]
- 24. Kanaumi T, Takashima S, Iwasaki H, et al. Developmental changes in KCNQ2 and KCNQ3 expression in human brain: possible contribution to the age‐dependent etiology of benign familial neonatal convulsions. Brain Dev 2008;30:362–369. [DOI] [PubMed] [Google Scholar]
- 25. Tinel N, Lauritzen I, Chouabe C, et al. The KCNQ2 potassium channel: splice variants, functional and developmental expression. Brain localization and comparison with KCNQ3. FEBS Lett 1998;438:171–176. [DOI] [PubMed] [Google Scholar]
- 26. Sands TT, Balestri M, Bellini G, et al. Rapid and safe response to low‐dose carbamazepine in neonatal epilepsy. Epilepsia 2016;57:2019–2030. [DOI] [PubMed] [Google Scholar]
- 27. Paulson A, Vargus‐Adams J. Overview of four functional classification systems commonly used in cerebral palsy. Children 2017;4. 10.3390/children4040030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Berg AT, Palac H, Wilkening G, et al. SCN2A‐developmental epilepsies and encephalopathies: challenges to trial‐readiness for non‐seizure outcomes. Epilepsia 2021;62:258–268. [DOI] [PubMed] [Google Scholar]
- 29. Devinsky O, Cross JH, Laux L, et al. Trial of cannabidiol for drug‐resistant seizures in the Dravet syndrome. N Engl J Med 2017;376:2011–2020. [DOI] [PubMed] [Google Scholar]
- 30. Devinsky O, Patel AD, Cross JH, et al. Effect of cannabidiol on drop seizures in the Lennox‐Gastaut syndrome. N Engl J Med 2018;378:1888–1897. [DOI] [PubMed] [Google Scholar]
- 31. Onorati F, Regalia G, Caborni C, et al. Multicenter clinical assessment of improved wearable multimodal convulsive seizure detectors. Epilepsia 2017;58:1870–1879. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Medications used in children with KCNQ2‐DEE: a. Reported effect of seizure control of medications and b. Reported reasons for stopping medications.
Table S1. KCNQ2 Variants of patients included in the study sample and their ACMG classification and location within the Kv7.1 protein.