

Abstract
Kidney disease, especially when it is associated with a reduction in eGFR, can be associated with an increase in serum urate (uric acid), suggesting that hyperuricemia in subjects with kidney disease may be strictly a secondary phenomenon. Mendelian randomization studies that evaluate genetic scores regulating serum urate also have generally not found evidence that serum urate is a causal risk factor in chronic kidney disease. Nevertheless, this is countered by a large number of epidemiological, experimental, and clinical studies that suggest a potentially important role for uric acid in kidney disease and cardiovascular disease. Here we review the topic in detail. Overall, the studies strongly suggest that hyperuricemia does have an important pathogenic role that is likely driven by intracellular urate levels. An exception may be the role of extracellular uric acid in atherosclerosis and vascular calcification. One of the more striking findings on reviewing the literature is that the primary benefit of lowering serum urate in subjects with CKD is not by slowing the progression of renal disease, but rather by reducing the incidence of cardiovascular events and mortality. We recommend large scale clinical trials to determine if there is a benefit in lowering serum urate in hyperuricemic subjects in acute and chronic kidney disease and in the reduction of cardiovascular morbidity and mortality in subjects with end stage chronic kidney disease.
Keywords: hyperuricemia, acute kidney injury, chronic kidney disease, allopurinol, cardiovascular mortality
Introduction
1776 is well known as the year of independence for the United States, but it also commemorates the year that uric acid was discovered by Carl Wilhelm Scheele, a Swedish chemist. Scheele discovered that some bladder stones were made of uric acid, and while he himself had suffered from gout the year before at the young age of 32, he remained unaware that he had discovered the cause of his own disease. Almost 75 years later, Alfred Baring Garrod published his paper linking uric acid with gout and kidney disease [1]. For the rest of the 19th century, uric acid was viewed not only as a cause of gout, but also as an important cause of chronic kidney disease (CKD), arteriolosclerosis, and hypertension [2–4]. Alexander Haig even suggested that it may be involved in other conditions, including migraine, preeclampsia and diabetes [3].
In the early twentieth century, studies on uric acid were relatively few, since uric acid was difficult to measure and effective therapies were not available. Breakthroughs in chemical assays occurred in the 1920s, allowing for uric acid to be studied in more detail [5]. This led to the discovery that serum uric acid levels could vary widely in the population, and tended to be higher in countries with advanced technologies and western diets [6]. Indeed, over the course of the century mean serum uric acid (serum urate) levels tended to rise throughout the world, with mean serum urate levels today averaging around 6 mg/dl in men and 4.8 mg/dl in women [7]. In addition, hyperuricemia was defined as > 7.0 mg/dl in men and >6.0 mg/dl in women, in part because of the observation that uric acid crystal saturation occurs at a concentration of 6.8 mg/dl of uric acid in water. In addition, because uric acid circulates as a sodium urate, circulating uric acid is usually referred to as serum urate, while crystalline uric acid is referred to as uric acid.
While the emphasis on serum urate as a risk factor for gout was well established in the mid-twentieth century, investigators also recognized that both gout and hyperuricemia had a strong association with cardiovascular (CV) disease, as well as other comorbidities including hypertension, stroke, and heart failure [8,9]. A major study analyzing residents from Framingham, Connecticut, found that gout was an independent risk factor for CV disease in men [10]. Another study suggested that patients with gout were at a higher risk for developing end stage renal disease. In one study, nearly half of patients with gout had reduced kidney function and on autopsy nearly 95 percent had kidney injury, userum uratelly associated with microvascular disease, glomerulosclerosis, tubulointerstitial fibrosis, and medullary uric acid crystalline deposits [11]. By 1975 the entity of “gouty nephropathy” was considered one of the major causes of CKD [12].
Between the 1970s and 1990s a reappraisal of the role of uric acid in cardiometabolic and renal diseases was performed. One of the striking findings was the inconsistency of elevated serum urate levels and risk for CV disease, as some studies, including a repeat study by the Framingham Study group, could not show that hyperuricemia was an independent risk factor for CV mortality [13]. In addition, several investigators suggested that ‘gouty nephropathy’ might not be due to uric acid crystal deposition in the renal parenchyma, but rather be a consequence of coexisting renal disease from hypertension, diabetes or aging [14]. As a result, uric acid was no longer considered an independent risk factor for cardiovascular or renal disease, and instead was viewed as being important only in acute kidney injury (AKI) from tumor lysis syndrome, or as a cause of kidney stones [15,16]. Serum urate levels were removed from the routine chemistry (SMAC) panel, and the treatment of asymptomatic hyperuricemia was highly discouraged.
In this brief review, we will revisit the role of serum urate and uric acid crystal deposition in acute and CKD, with an emphasis not only on the role of uric acid in renal injury, but also in coincident conditions, such as systemic inflammation, insulin resistance, vascular calcification, and CV events and mortality. In general, the overall data suggests that serum urate very likely has a contributory role in subjects with acute and CKD, and that it should be viewed as a potential remediable risk factor.
Urate Metabolism in Subjects with Kidney Disease
Uric acid is the endproduct of purine degradation, and is generated from the breakdown of nucleic acids (RNA and DNA) and ATP. Uric acid can also be synthesized from amino acid precursors. Certain dietary factors can stimulate uric acid production, including purine-rich meats (especially seafood), beer (due to the alcohol and yeast content), and fructose (including fruits, honey, sugar and high fructose corn syrup) [17,18]. Other stimuli for uric acid production include ischemia, heat stress and conditions associated with rapid cell turnover (e.g. tumor lysis syndrome). Uric acid is also generated intracellularly under conditions in which aldose reductase is induced, such as with high glycemic carbohydrates, salty foods, and dehydration [19–21]. In the latter conditions, serum urate levels may not increase despite increased intracellular urate generation [20].
In most mammals, the uric acid that is generated is metabolized by urate oxidase (also known as uricase), which is ubiquitously expressed but which has the highest concentrations in the liver or kidney depending on the species. However, humans and the great apes lost uricase due to a mutation in the Miocene, and a separate mutation affected the lesser apes [22,23]. The parallel loss of uricase suggests that the loss of uricase resulted in a survival advantage at that time. Several theories have been proposed to explain the survival advantage, including potential roles of uric acid as an antioxidant to improve longevity [24], reaction time and intelligence [25]. In addition, there are postulates that uric acid might have helped to support blood pressure, fat stores and survival during a prolonged period of intermittent starvation that occurred during that time [26].
In the absence of uricase, only small amounts of uric acid are degraded, usually in response to an oxidant (generating allantoin and triuret from superoxide anion and peroxynitrite, respectively) or 6-aminouracil (from reacting with nitric oxide) [27,28]. The degradation of urate through these pathways is increased in people with oxidative stress, such as from smoking, preeclampsia, and CKD [27,29,30]. However, most subjects eliminate the excess uric acid by renal or gastrointestinal excretion. Renal excretion is driven by a number of urate transporters in the proximal tubule, of which URAT1 (SLC22A12) and Glut9 (SLC2A9) appear to be dominant in the reabsorption process. In the gut both SLC2A9 and ABCG2 mediate gut excretion of uric acid where it is further degraded by bacterial uricases.
Today serum urate levels range between 1 and 3 mg/dl in most mammals that express uricase, and 3–4 mg/dl in primates that lack uricase including humans that are on native (non-western) diets[6]. For people on western diets, the serum urate level is generally 3 to 5 mg/dl in women and 4–6 mg/dl in men. Hyperuricemia is defined as > 7 mg/dl in men and >6.0 mg/dl in women. Approximately 20% of the US population are hyperuricemic, and this is the group at most risk for gout, urate nephrolithiasis, and cardiometabolic disorders.
Genetic polymorphisms in urate transporters that modulate serum urate levels can be identified in GWAS studies and can account for approximately 6 to 8 percent of the variation of serum urate, with the strongest influence resulting from polymorphisms in SCL2A9 [31]. However, within subjects on a western diet, the degree of variation of serum urate levels accounted for by genetics increases, and may account for a sizeable fraction of the variance in serum urate [32]. Nevertheless, the effect of western diet on serum urate is immense and serum urate increased during the last century in parallel with the rise in sugar intake [6].
In the setting of CKD, the fractional excretion of uric acid will increase, as will gut excretion. Nevertheless, these responses cannot compensate fully for the reduced urate excretion and serum urate levels increase. By the time dialysis is initiated, approximately 50 percent of subjects are hyperuricemic [33]. Serum urate is removed by dialysis, so serum urate blood levels can vary in dialysis subjects dependent on how well dialyzed they are. Figure 1 shows the mechanisms leading to hyperuricemia in the CKD patient.
Figure 1. Mechanisms of Hyperuricemia in the Subject with CKD.
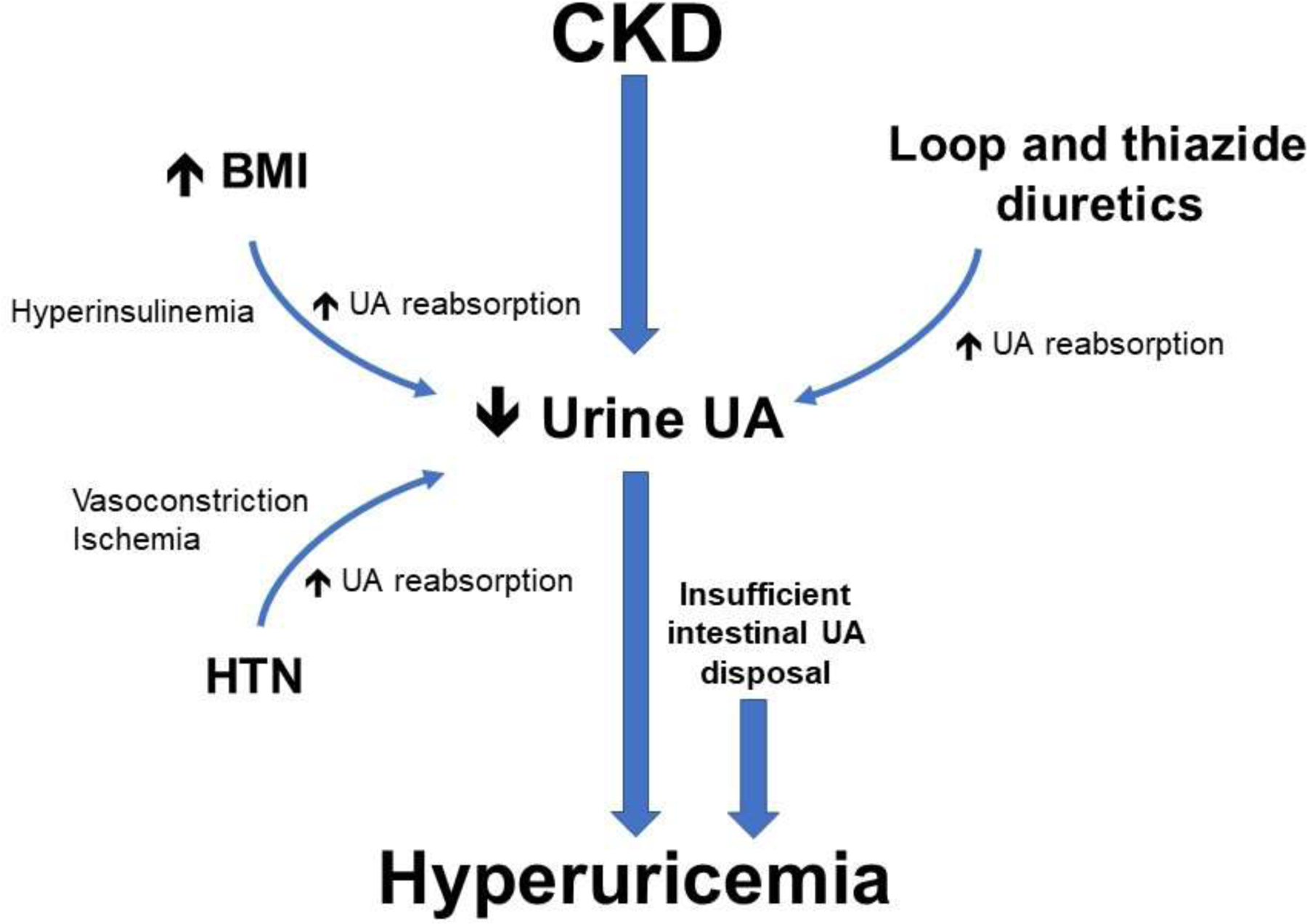
A decline in kidney function is associated with a reduction in urinary urate excretion. This may be compounded by diuretic use, and an elevated body mass index (BMI), especially when accompanied by hyperinsulinemia, can be associated with decrease uric acid excretion. Hypertension (HTN) may also reduce urate excretion, possibly related to renal vasoconstriction. While fractional excretion of renal urate excretion incrases, as well as increased compensatory excretion by the gut, these mechanisms cannot fully correct the uric acid (UA) imbalance. As a result, approximately half of subjects with CKD have hyperuricemia at the time of initiation of dialysis.
Uric acid and Acute Kidney Injury
It is well-known that uric acid causes AKI in tumor lysis syndrome by a crystal-dependent mechanism. In experimental models, intraluminal precipitation of uric acid crystals was associated with an ~50% decrease in renal blood flow and glomerular filtration rate (GFR), suggesting effects beyond a simple tubular obstructive phenomenon [34]. Sanchez-Lozada et al demonstrated that uric acid in concentrations that do not cause intraluminal crystal precipitation also reduces GFR and renal blood flow [35]. Subsequent discoveries of the physiologic actions of uric acid on the renal vasculature, tubules and inflammatory pathways substantiated the generation of the hypothesis that uric acid has a role in AKI via a crystal-independent pathway[36]. In a proof-of-concept study an experimental model of cisplatin-induced AKI, moderate hyperuricemia was associated with significantly greater proximal tubular injury (S3), parenchymal macrophage infiltration and increased expression of inflammatory mediators than the control group without intrarenal crystal deposition [37]. Uric acid lowering therapy (ULT) with urate oxidase reversed these effects.
In the evolutionary phases of AKI, at risk kidneys suffer damage followed by a decrease in GFR, kidney failure and subsequent complications. Serum urate is an independent predictor of AKI. In epidemiological studies, preoperative serum urate levels >5.5mg/dL predicted a 4 to 40-fold increased risk for postoperative AKI in a concentration-dependent manner [38,39]. Hyperuricemia has been consistently predicted to increase the risk for AKI in multiple settings including cardiac surgery [39], cisplatin associated nephrotoxicity [40], acute myeloid leukemia [41,42], radiocontrast exposure [43,44], burn injury [45] and hospitalized patients[46]. In a prospective study in cardiac surgery patients, postoperative serum urate levels exhibited significant positive linear correlations with postoperative serum creatinine and plasma neutrophil gelatinase-associated lipocalin (NGAL) and inverse correlations with kinetic estimated GFR and creatinine clearance [47]. That is, serum urate levels effectively predicted subsequent changes in conventional and novel biomarkers of AKI and was associated with decreases in GFR. The proposed mechanism whereby uric acid causes AKI involves impaired renal blood flow autoregulation related to vasoconstriction, renal hypoperfusion, ischemia/reperfusion injury and activation of the inflammatory pathways with downstream consequences.
Several interventional trials have assessed the effect of uric acid lowering therapy on AKI. In a prospective, pilot study, 26 hyperuricemic patients undergoing high-risk cardiac surgery were randomized to receive uric acid lowering therapy with urate oxidase vs. placebo. The treatment group demonstrated less structural renal injury as measured by urine NGAL. The effect size was greater in patients with more severe renal and cardiac dysfunction [48]. In another study, treatment with urate oxidase in children with advanced mature B-cell non-Hodgkin lymphoma (N=76) to prevent tumor lysis syndrome resulted in reductions of serum urate levels with an associated increase in mean GFR from 55mL/min/1.73 m2 on day –1 to 136mL/min/1.73 m2 on day 7 following treatment[49]. Prospective randomized, controlled trials in patients exposed to radiocontrast substances also demonstrated significant renal protection with allopurinol [43,50]. These results highlight the relationship between hyperuricemia, enhanced levels of renal vasoconstriction and loss of renal autoregulation leading to hypoperfusion and ischemia/reperfusion injury. Lowering serum urate reverses/attenuates these effects. Perhaps the pivotal question remains whether serum urate is a marker or causal factor in AKI. In a recent vigorous examination of this issue using the Bradford Hill criteria for causality, experimental and clinical data support the contention for a causal role for serum uric acid in AKI [51].
Figure 2 summarizes potential mechanisms by which serum urate might contribute AKI.
Figure 2. Proposed mechanism of uric acid-induced acute kidney injury.
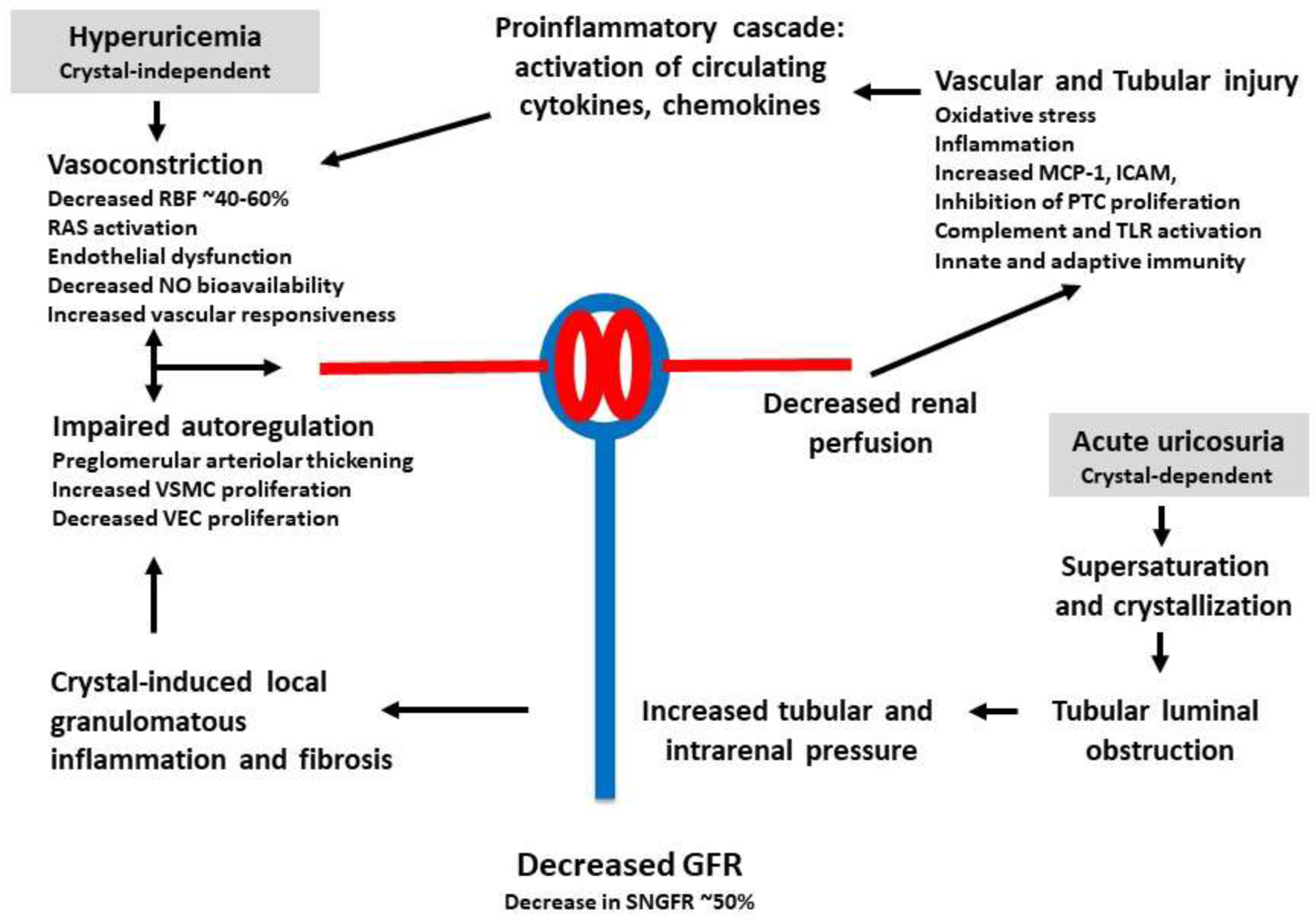
Hyperuricemia may increase the risk for AKI via both crystal-independent and crystal-dependent pathways. Hyperuricemia appears to affect renal hemodynamics, causing renal vasoconstriction, impaired autoregulation and increased glomerular pressure, while it can also affect tubular function, leading to inflammatory responses, oxidative stress, epithelial mesenchymal transition, and apoptosis. Crystalluria may also contribute to tubular injury.
Key: RBF: renal blood flow; RAS: renin-angiotensin-aldosterone system; NO: nitric oxide; VSMC: vascular smooth muscle cell; VEC: vascular endothelial cell; GFR: glomerular filtration rate; MCP-1: monocyte chemoattractant protein-1; ICAM: intercellular adhesion molecule; PTC: proximal tubular cell; TLR: Toll-like receptor.
Uric acid and Chronic Kidney Disease
Recent studies have identified hyperuricemia as a risk factor for the development of both diabetic and nondiabetic CKD [52,53]. Indeed, the presence of hyperuricemia appears to increase the risk for developing CKD by two to ten-fold, and the risk is greater in women [54]. Some studies have suggested the risk for developing CKD is greater for hyperuricemic individuals than in subjects with mild albuminuria [54]. While an occasional study has also not been able to document an increased risk for CKD in hyperuricemic subjects, [55] most studies support that hyperuricemia is a risk factor for developing CKD independent of the presence of hypertension, insulin resistance, or dyslipidemia [52,53,56]. However, hyperuricemia is not as strong as a risk factor for renal progression in subjects with preexisting CKD, and often cannot be shown to be independent of other factors in these subjects [57].
One potential mechanism by which hyperuricemia could increase the risk for CKD is via diet, as both fructose-containing sugars and purine-rich foods can raise serum urate levels. Sugary beverages have been associated with an increased risk for incident CKD in several [58–61] but not all studies [62,63], and this may be dose related since the risk is primarily seen in subjects who drank one or more sodas per day. Beer intake, which is especially strong at raising serum urate levels due to its combined yeast and alcohol intake, was also associated with an increased risk for CKD in the Jackson Heart Study [61]. Interestingly, one study also identified diet soft drinks as carrying a risk for CKD, that seemed to be independent of fructose, salt or phosphorus intake.[64]
Experimental studies have also documented that hyperuricemia can induce CKD [65]. For example, chronic hyperuricemia associated with uricase inhibition can induce a decrease in GFR associated with elevated glomerular pressure due to inadequate autoregulation [35]. This is associated with the development of arteriolosclerosis, glomerular hypertrophy, glomerulosclerosis, and tubulointerstitial disease in the absence of uric acid crystal deposition [65,66]. The mechanism is mediated by the activation of the renin angiotensin system, inhibition of endothelial nitric oxide availability, and stimulation of oxidative stress [67–69]. Uric acid also causes a phenotypic transformation of tubular cells to a mesenchymal phenotype, and can stimulate the production of prorenin, inflammatory pathways with NFκB and chemokine production, oxidative stress and apoptosis [70–75]. Similarly, uric acid can also induce oxidative stress in endothelial cells associated with mitochondrial dysfunction and insulin resistance [76,77]. Lowering uric acid with xanthine oxidase inhibitors, uricosurics and probiotics which enhance uric acid degradation can protect against kidney injury, as can agents that block the renin angiotensin system, block oxidative stress, or stimulate endothelial nitric oxide production[66–69,78,79].
In contrast, Mendelian randomization studies based on using genetic scores from gene polymorphisms affecting serum urate levels in the general population have failed to show a relationship of serum urate with CKD, while the association with gout is strong [31,80,81], with one exception [82]. While these studies have often been interpreted to show that serum urate does not have a causal role in CKD, there are other potential explanations. For example, experimental studies show that intracellular urate is likely driving the biological and renal effects, which is distinct from extracellular urate which has a primary role in driving soft tissue uric acid crystal deposition (i.e. gout). Second, while serum urate normally predicts intracellular urate levels, this may not be true in conditions in which intracellular urate formation is the primary factor driving intracellular levels, such as may occur with sugar-based diets or in settings where fructose is produced inside the cell (such as with salty food or high glycemic carbohydrates) [19,20,83]. Indeed, there is the interesting finding that extracellular urate may reduce expression of urate transporters that acts to reduce relative uptake of urate [84]. This could suggest that conditions associated with increased xanthine oxidase activity that directly elevate intracellular urate may have more biologic effects than passive hyperuricemia such as from reduce renal excretion. Furthermore, SLC2A9 gene polymorphisms account for a majority of the genetic score, but this transporter is unusual since it has divergent effects depending on the organ it is expressed in (e.g. intestinal knockout increases serum urate while renal knockout lowers serum urate)[85–87]. In addition, its effects on intracellular urate levels is not known. Hence, while the Mendelian studies do suggest caution in interpreting the epidemiological and experimental studies, they should not be taken as definitive evidence that uric acid is not important in the development and/or progression of CKD.
There have also been numerous small clinical trials assessing whether lowering serum urate levels may be helpful in slowing the progression of CKD in hyperuricemic individuals, and meta-analyses of these trials have suggested that there is indeed a modest benefit [88,89]. Nevertheless, the National Kidney Foundation (NKF) held a symposium a few years ago to review the evidence and noted that these clinical trials were often suboptimal in design including being underpowered, lacking a placebo control, and/or being too short in duration [53]. However, NKF did feel that there was enough evidence suggesting a possible beneficial effect should better quality controlled trials be performed.
Our group has recently reviewed the clinical trials from another perspective, since some negative studies had minimal or no progression of renal disease in the control group. We realized that if the control group does not show a decline in renal function, then a clinical trial would be unlikely to show a benefit in slowing kidney disease. When we reanalyzed the 23 studies that had been reported in the literature, we found that the decreasing serum urate levels was protective in those studies in which renal disease progressed. We concluded that the data suggested that hyperuricemic individuals who have progressively worsening CKD should be considered for uric acid lowering therapy [90].
More recently two clinical trials were presented at the American Society of Nephrology that countered this conclusion. The PERL study was a large placebo controlled study to investigate if allopurinol could decrease renal decline in type 1 diabetic patients with progressive CKD. The study was negative, but the patients were not hyperuricemic, had relatively stable renal function (CKD stage 2), were normotensive, and had only minimal proteinuria (mean 40ug/d albuminuria) [91]. The two-year CKD Fix study randomized normouricemic and hyperuricemic subjects with allopurinol or placebo treatments. Mean serum urate levels were in the hyperuricemic range, and no benefit was shown despite significant progression of CKD in the control group [92]. Unfortunately, 30% of patients dropped out of this study and given it was an intention to treat analysis, there were many patients who were not on allopurinol and were classified as being treated. Thus, neither study directly tested the hypothesis that lowering serum urate levels in hyperuricemic patients with CKD is renoprotective, and both suffered from high dropout rates and inclusion of normouricemic subjects. However, the studies do suggest that indiscriminant treatment of CKD with allopurinol as a means to reduce renal progression is not warranted.
Role of Uric acid in Cardiac Events Occurring in Subjects with CKD
While subjects with CKD are at risk for progression of their kidney disease, the greatest cause of morbidity and mortality are CV events. Once subjects start dialysis, mortality risks are extremely high and mostly related to sudden cardiac death. Interestingly, in subjects with CKD, the effects of lowering cholesterol with statin therapy seems blunted compared to the rest of the population. Thus, a search for other remediable therapies is needed.
Studies in the General Population.
It is well known that hyperuricemia is strongly associated with increased CV events in the non-CKD population, especially for sudden cardiac death and CV mortality. While some studies suggest hyperuricemia is an independent risk factor for cardiac events and mortality, others studies have not, and meta-analyses have yielded mixed results [93,94]. Furthermore, Mendelian randomization studies that evaluate the risk of a genetic score favoring hyperuricemia have generally failed to show a relationship with CV events [31,81]. However, two Mendelian randomization studies have identified a relationship of genetic uric acid score with CV disease, with one linking the genetic score with sudden cardiac death [95], and the other with diabetic vascular disease [96].
One possible explanation for the differing outcomes from the epidemiological studies is that there is an innate assumption with multivariable analysis that the variables need to be independent of each other to be true risk factors, but this is not true if the variables assessed are causally linked [97]. Likewise, the Mendelian randomization studies were evaluating genetic scores that influence serum urate levels, while most of the biological effects of uric acid appear to be driven by an intracellular mechanism which induces oxidative stress, endothelial dysfunction and activation of the renin angiotensin system [53].
This would suggest that the best approach to determining whether uric acid may have an independent role in CV disease would be to perform trials to determine if lowering uric acid, and especially intracellular uric acid, can provide benefit. This might be best approached by agents that block uric acid formation, such as by xanthine oxidase inhibitors. For example, hyperuricemia is known to predict cardiac death in subjects who have a history of stroke [98], and clinical trials have reported that allopurinol given to subjects with a history of stroke resulted in improved systemic and central blood pressure, reduced carotid intimal thickening [99], improved the augmentation index [100], and reduced signs of systemic inflammation [101]. Randomized clinical trials with allopurinol have been performed in subjects undergoing coronary artery bypass and have reported less cardiac injury during bypass[102] with reduced CV mortality [103]. A placebo controlled study also reported that allopurinol could reduce angina frequency and improve exercise tolerance in subjects with stable angina [104].
Studies of population registries have also provided evidence that the use of allopurinol is dose dependently associated with a a reduced risk for stroke and CV events in hypertensive subjects [105]. Other population based studies have found that allopurinol use is associated with a reduced risk for ischemic stroke and myocardial infarction in the elderly [106,107] and reduced risk for stroke or myocardial infarction in subjects with gout or diabetes [108]. In contrast, studies have not been able to show a benefit of lowering serum urate levels on congestive heart failure associated with systolic dysfunction [109]. Nevertheless, the evidence for potential CV benefit with treatment by xanthine oxidase inhibitors has led to a large ongoing study to determine if allopurinol can reduce CV events in subjects with ischemic heart disease [110].
Studies in Chronic Kidney Disease.
The presence of CKD, especially stage 3 or higher, is associated with significant CV risk [111]. A number of clinical trials have suggested that lowering serum urate levels with allopurinol can be associated with marked improvement in CV risk in people with CKD. For example, one study comparing a 9 month treatment of allopurinol with placebo in hyperuricemic subjects with CKD documented an improvement in endothelial function, augmentation index, and left ventricular hypertrophy (by cardiac MI) in subjects receiving allopurinol [112]. Three smaller clinical trials (and meta-analyses) evaluating the effect of lowering serum urate levels in CKD subjects all noted a consistent 60 percent reduction in CV events compared to the control group [88,113–115]. In another analysis of a large registry of hypertensive subjects with CKD, the use of allopurinol was associated with a nearly 65% reduction in CV events and total mortality over an 18 month period [116].
Studies in Hemodialysis Patients.
In contrast to the general observation that identify subjects with hyperuricemia to be at increased risk for CV morbidity and mortality, subjects on hemodialysis often show a J-curve relationship between serum urate levels and mortality, with the highest rate usually in the lowest quartile of serum urate values [33,117–119]. Interestingly, because uric acid excretion is reduced in the setting of end stage kidney disease, the serum urate levels in the lowest quartiles fall largely within the normal range of serum urate for the general population. Several studies suggest this “reverse epidemiological pattern” in hemodialysis patients may be a reflection that sicker hemodialysis patients often have worse nutrition which can result in lower serum albumin and serum urate, since both are modulated heavily by food intake and general nutrition [33,120,121]. Indeed, when subjects with CKD are transitioned to dialysis, the risk for CV events on dialysis is greatest for those who were hyperuricemic before they initiated dialysis [122].
The use of allopurinol in hemodialysis patients has not been studied extensively. In one small cross-over study, hyperuricemic subjects on hemodialysis were administered allopurinol 100 mg daily and had a significant decrease in systolic BP (> that 20 mm Hg) after 12 weeks [123]. In contrast, a small study in Japan reported that treatment with febuxostat decreased oxidative stress and improved endothelial function but did not improve BP in hyperuricemic dialysis patents [124]. Other studies have investigated the potential for allopurinol to reduce CV morbidity and mortality, based primarily on a retrospective analysis of dialysis registries. In one large study of over 220,000 hemodialysis patients in Japan, the use of urate lowering therapy was associated with a 35% reduction in CV mortality that remained significant after controlling for multiple variables (15% reduction) [120]. Similarly, in the Dialysis Outcomes and Practice Patterns Study performed in Japan, a 35% reduction in all-cause mortality was observed with allopurinol use, and after multivariate analysis using over 20 variables the significance was lost, except in the group that had no prior CV disease in which the protection remained around 50% [125].
Potential Roles of Uric acid in Atherosclerotic Plaques and Vascular Calcification.
The relatively dramatic reduction in CV events and mortality observed with lowering serum urate levels in subjects with CKD or end stage renal disease (ESRD) suggest a beneficial effect that may include reduction in both intracellular and extracellular urate. [126]. Urate is known to deposit in atherosclerotic plaque [127–129] and it is possible that extracellular urate might have a role in the atherosclerotic plaque formation with intra-arterial urate deposition serving as a nidus for calcium aggregation similar to its role in calcium nephrolithiasis [130]. In addition, the proinflammatory effects of urate deposition may promote progression of atherogenesis. Recently, several studies performing dual energy computed tomography (DECT) scans for vascular assessment, have demonstrated that approximately 85% of patients with gout have urate deposition in their vasculature which colocalize with sites of vascular calcification (122). Hyperuricemia is also known to be a powerful predictor of vascular calcification in subjects without renal disease, including those with type 1 diabetes. [131,132] Further studies are needed to investigate this potential mechanism of injury. Potential roles for urate deposition in development and progression of atherosclerosis and vascular calcification are shown in Figure 3.
Figure 3. Potential Mechanisms for Uric acid Involvement in Atherosclerosis, Vascular Calcification, and Cardiovascular Disease.
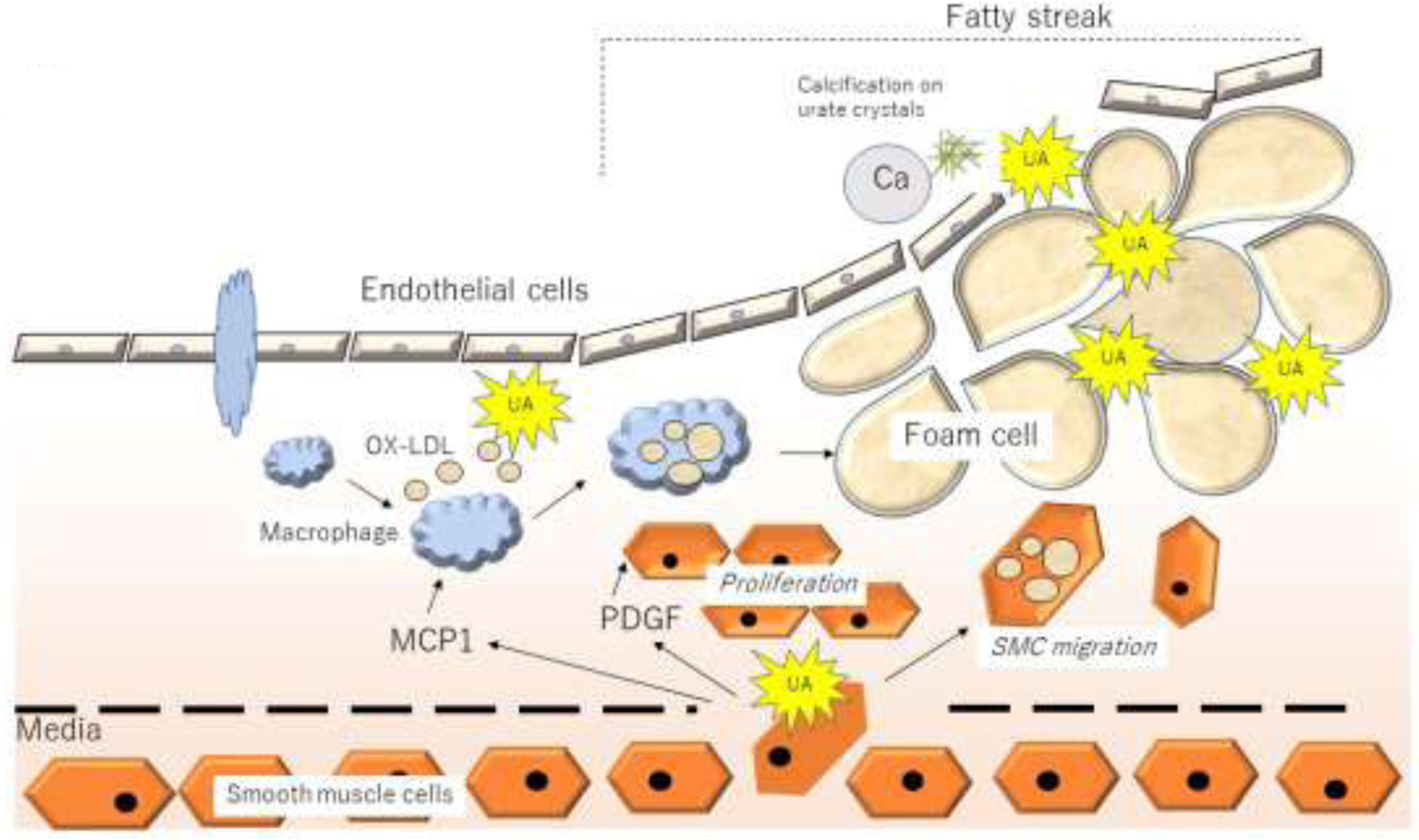
Uric acid may be generated in endothelial cells, causing local vascular smooth muscle cell proliferation and migration via the release of PDGF, and inflammation by the release of MCP-1, with facilitation of oxidation of LDL. Urate may then be deposited in plaque, in which crystals may act as a nidus for calcium deposition and vascular calcification potentially resulting in accelerated atherosclerosis. Urate deposits may also serve as a nidus for local intravascular inflammation that may stimulate atherogenesis and/or lead to adjacent inflammatory plaque
Safety Concerns.
Allopurinol can be associated with a Stevens-Johnson like syndrome that is almost exclusively observed in subjects bearing the HLA B*58 genotype, and while the frequency is <1 % in Caucasians, it approaches 2 to 4 % in African Americans and 6 to 8 % in Asians, especially the Han Chinese. Genotyping is therefore recommended in Asians and possibly African Americans populations prior to commencing allopurinol therapy. The CARES Study compared febuxostat and allopurinol in subjects at CV risk. This study observed an increased in CV mortality in the febuxostat group, especially sudden cardiac death( 2.7% in the febuxostat group vs 1.8% in the allopurinol group).[133] However, this was an intention to treat analysis and there was a 57% dropout. A more detailed analysis of this study showed that many of the CV events actually occurred following the withdrawal of the drugs, with an 18-fold increased CV event rate in the first month of cessation. [134] This suggests that there may be a rebound event upon stopping urate lowering therapies, perhaps related to the rise in uric acid and its potential to activate the renin angiotensin system [135]. Most studies do not suggest febuxostat increases the risk for CV events.[136,137] However, the CARES study does suggest that xanthine oxidase inhibitors may need to be weaned slowly in subjects at CV risk, perhaps in the presence of inhibition of the renin angiotensin system.
Uricosurics are not preferred therapies in patients with renal disease because of the risk of uricosuria, kidney stones, and possibly AKI [138]. However, when combined with xanthine oxidase inhibitors, they appear to have a much safer profile. Recombinant and pegylated uricases are primarily limited by being an intravenous therapy and the possible development of antidrug antibodies which may cause allergic reactions and reductions in efficacy. They would not be expected to affect intracellular urate production, although chronically low serum urate levels induced by recombinant uricases might still be effective at reducing intracellular urate levels.
Summary
In summary, it is well known that a reduced eGFR can result in retention of serum urate and therefore increase the risk for hyperuricemia, but there is also compelling evidence that hyperuricemia can predict the development of acute and CKD. There is relatively strong data suggesting hyperuricemia may play a role in both contrast- and ischemia-associated AKI. There is also evidence that hyperuricemia may have a role in CKD progression. While the PERL and CKD-Fix trials suggest that indiscriminant lowering of serum urate may not slow progression of CKD, several other studies found that restricting treatment to hyperuricemic individuals slows progression of kidney damage. In both acute and CKD it is likely that the beneficial effects of uric acid lowering therapy may be related to suppression of intracellular uric acid and its stimulation of inflammatory and oxidative stress pathways. Moreover, there is increasing evidence that treatments to lower serum uric acid may reduce the incidence of CV events and mortality in patients with CKD. We suggest that large-scale clinical trials are needed to address the potential CV benefit of lowering serum urate levels in subjects with chronic renal failure.
Acknowledgments
Funding.
Supported in part by NIH grants R01 DK108408 (Johnson) and R01 DK108859 (Lanaspa).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest: Dr TN and RJJ have equity with XORTX therapeutics that is making novel xanthine oxidase inhibitors, and LGL, MAL and RJJ have equity with Colorado Research Partners LLC which is making inhibitors to fructose metabolism. All other authors declare no conflicts of interest. AK is an employee of Horizon Therapeutics and has equity in Horizon Therapeutics.
References
- 1.Garrod AB: Observations on the blood and urine of gout, rheumatism and Bright’s disease Medical Chirurgical Transaction 1848;31:83–. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johnson G: On the Diseases of the Kidney. London, John W Parker and Son, 1852. [Google Scholar]
- 3.Haig A: Uric acid as a Factor in the Causation of Disease. A contribution to the pathology of high arterial tension, headache, epilepsy, mental depression, gout, rheumatism, diabetes, Bright’s disease, and other disorders., ed First. London, J & A Churchill, 1892. [Google Scholar]
- 4.Davis NC: The cardiovascular and renal relations and manifestations of gout. JAMA 1897;29:261–262. [Google Scholar]
- 5.Fishberg AM: The interpretation of increased blood uric acid in hypertension. Arch Intern Med 1924;34:503–507. [Google Scholar]
- 6.Johnson RJ, Titte S, Cade JR, Rideout BA, Oliver WJ: Uric acid, evolution and primitive cultures. Semin Nephrol 2005;25:3–8. [DOI] [PubMed] [Google Scholar]
- 7.Chen-Xu M, Yokose C, Rai SK, Pillinger MH, Choi HK: Contemporary Prevalence of Gout and Hyperuricemia in the United States and Decadal Trends: The National Health and Nutrition Examination Survey, 2007–2016. Arthritis Rheumatol 2019;71:991–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gertler MM, Garn SM, Levine SA: Serum uric acid in relation to age and physique in health and in coronary heart disease. Ann Intern Med 1951;34:1421–1431. [DOI] [PubMed] [Google Scholar]
- 9.Breckenridge A: Hypertension and hyperuricaemia. Lancet 1966;1:15–18. [DOI] [PubMed] [Google Scholar]
- 10.Abbott RD, Brand FN, Kannel WB, Castelli WP: Gout and coronary heart disease: the Framingham Study. J Clin Epidemiol 1988;41:237–242. [DOI] [PubMed] [Google Scholar]
- 11.Talbott JH, Terplan KL: The kidney in gout. Medicine (Baltimore) 1960;39:405–467. [PubMed] [Google Scholar]
- 12.Bluestone R, Waisman J, Klinenberg JR: The gouty kidney. Semin Arthritis Rheum 1977;7:97–113. [DOI] [PubMed] [Google Scholar]
- 13.Culleton BF, Larson MG, Kannel WB, Levy D: Serum uric acid and risk for cardiovascular disease and death: the Framingham Heart Study. Ann Intern Med 1999;131:7–13. [DOI] [PubMed] [Google Scholar]
- 14.Yu TF, Berger L: Impaired renal function gout: its association with hypertensive vascular disease and intrinsic renal disease. Am J Med 1982;72:95–100. [DOI] [PubMed] [Google Scholar]
- 15.Beck LH: Requiem for gouty nephropathy. Kidney Int 1986;30:280–287. [DOI] [PubMed] [Google Scholar]
- 16.Vaccarino V, Krumholz HM: Risk factors for cardiovascular disease: one down, many more to evaluate. Ann Intern Med 1999;131:62–63. [DOI] [PubMed] [Google Scholar]
- 17.Choi HK, Atkinson K, Karlson EW, Willett W, Curhan G: Purine-rich foods, dairy and protein intake, and the risk of gout in men. N Engl J Med 2004;350:1093–1103. [DOI] [PubMed] [Google Scholar]
- 18.Choi JW, Ford ES, Gao X, Choi HK: Sugar-sweetened soft drinks, diet soft drinks, and serum uric acid level: The third national health and nutrition examination survey. Arthritis Rheum 2007;59:109–116. [DOI] [PubMed] [Google Scholar]
- 19.Lanaspa MA, Ishimoto T, Li N, Cicerchi C, Orlicky DJ, Ruzycki P, Rivard C, Inaba S, Roncal-Jimenez CA, Bales ES, Diggle CP, Asipu A, Petrash JM, Kosugi T, Maruyama S, Sanchez-Lozada LG, McManaman JL, Bonthron DT, Sautin YY, Johnson RJ: Endogenous fructose production and metabolism in the liver contributes to the development of metabolic syndrome. Nat Commun 2013;4:2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lanaspa MA, Kuwabara M, Andres-Hernando A, Li N, Cicerchi C, Jensen T, Orlicky DJ, Roncal-Jimenez CA, Ishimoto T, Nakagawa T, Rodriguez-Iturbe B, MacLean PS, Johnson RJ: High salt intake causes leptin resistance and obesity in mice by stimulating endogenous fructose production and metabolism. Proc Natl Acad Sci U S A 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roncal Jimenez CA, Ishimoto T, Lanaspa MA, Rivard CJ, Nakagawa T, Ejaz AA, Cicerchi C, Inaba S, Le M, Miyazaki M, Glaser J, Correa-Rotter R, Gonzalez MA, Aragon A, Wesseling C, Sanchez-Lozada LG, Johnson RJ: Fructokinase activity mediates dehydration-induced renal injury. Kidney Int 2014;86:294–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kratzer JT, Lanaspa MA, Murphy MN, Cicerchi C, Graves CL, Tipton PA, Ortlund EA, Johnson RJ, Gaucher EA: Evolutionary history and metabolic insights of ancient mammalian uricases. Proc Natl Acad Sci U S A 2014;111:3763–3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oda M, Satta Y, Takenaka O, Takahata N: Loss of urate oxidase activity in hominoids and its evolutionary implications. Mol Biol Evol 2002;19:640–653. [DOI] [PubMed] [Google Scholar]
- 24.Ames BN, Cathcart R, Schwiers E, Hochstein P: Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: a hypothesis. Proc Natl Acad Sci U S A 1981;78:6858–6862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Orowan E: The origin of man. Nature 1955;175:683–684. [DOI] [PubMed] [Google Scholar]
- 26.Johnson RJ, Andrews P: Fructose, Uricase, and the Back-to-Africa Hypothesis. Evol Anthropol 2010;19 250–257. [Google Scholar]
- 27.Gersch C, Palii SP, Imaram W, Kim KM, Karumanchi SA, Angerhofer A, Johnson RJ, Henderson GN: Reactions of peroxynitrite with uric acid: formation of reactive intermediates, alkylated products and triuret, and in vivo production of triuret under conditions of oxidative stress. Nucleosides Nucleotides Nucleic Acids 2009;28:118–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gersch C, Palii SP, Kim KM, Angerhofer A, Johnson RJ, Henderson GN: Inactivation of nitric oxide by uric acid. Nucleosides Nucleotides Nucleic Acids 2008;27:967–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim KM, Henderson GN, Frye RF, Galloway CD, Brown NJ, Segal MS, Imaram W, Angerhofer A, Johnson RJ: Simultaneous determination of uric acid metabolites allantoin, 6-aminouracil, and triuret in human urine using liquid chromatography-mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 2009;877:65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kand’ar R, Zakova P, Muzakova V: Monitoring of antioxidant properties of uric acid in humans for a consideration measuring of levels of allantoin in plasma by liquid chromatography. Clin Chim Acta 2006;365:249–256. [DOI] [PubMed] [Google Scholar]
- 31.Tin A, Marten J, Halperin Kuhns VL, Li Y, Wuttke M, Kirsten H, Sieber KB, Qiu C, Gorski M, Yu Z, Giri A, Sveinbjornsson G, Li M, Chu AY, Hoppmann A, O’Connor LJ, Prins B, Nutile T, Noce D, Akiyama M, Cocca M, Ghasemi S, van der Most PJ, Horn K, Xu Y, Fuchsberger C, Sedaghat S, Afaq S, Amin N, Arnlov J, Bakker SJL, Bansal N, Baptista D, Bergmann S, Biggs ML, Biino G, Boerwinkle E, Bottinger EP, Boutin TS, Brumat M, Burkhardt R, Campana E, Campbell A, Campbell H, Carroll RJ, Catamo E, Chambers JC, Ciullo M, Concas MP, Coresh J, Corre T, Cusi D, Felicita SC, de Borst MH, De Grandi A, de Mutsert R, de Vries APJ, Delgado G, Demirkan A, Devuyst O, Dittrich K, Eckardt KU, Ehret G, Endlich K, Evans MK, Gansevoort RT, Gasparini P, Giedraitis V, Gieger C, Girotto G, Gogele M, Gordon SD, Gudbjartsson DF, Gudnason V, German Chronic Kidney Disease S, Haller T, Hamet P, Harris TB, Hayward C, Hicks AA, Hofer E, Holm H, Huang W, Hutri-Kahonen N, Hwang SJ, Ikram MA, Lewis RM, Ingelsson E, Jakobsdottir J, Jonsdottir I, Jonsson H, Joshi PK, Josyula NS, Jung B, Kahonen M, Kamatani Y, Kanai M, Kerr SM, Kiess W, Kleber ME, Koenig W, Kooner JS, Korner A, Kovacs P, Kramer BK, Kronenberg F, Kubo M, Kuhnel B, La Bianca M, Lange LA, Lehne B, Lehtimaki T, Lifelines Cohort S, Liu J, Loeffler M, Loos RJF, Lyytikainen LP, Magi R, Mahajan A, Martin NG, Marz W, Mascalzoni D, Matsuda K, Meisinger C, Meitinger T, Metspalu A, Milaneschi Y, Program VAMV, O’Donnell CJ, Wilson OD, Gaziano JM, Mishra PP, Mohlke KL, Mononen N, Montgomery GW, Mook-Kanamori DO, Muller-Nurasyid M, Nadkarni GN, Nalls MA, Nauck M, Nikus K, Ning B, Nolte IM, Noordam R, O’Connell JR, Olafsson I, Padmanabhan S, Penninx B, Perls T, Peters A, Pirastu M, Pirastu N, Pistis G, Polasek O, Ponte B, Porteous DJ, Poulain T, Preuss MH, Rabelink TJ, Raffield LM, Raitakari OT, Rettig R, Rheinberger M, Rice KM, Rizzi F, Robino A, Rudan I, Krajcoviechova A, Cifkova R, Rueedi R, Ruggiero D, Ryan KA, Saba Y, Salvi E, Schmidt H, Schmidt R, Shaffer CM, Smith AV, Smith BH, Spracklen CN, Strauch K, Stumvoll M, Sulem P, Tajuddin SM, Teren A, Thiery J, Thio CHL, Thorsteinsdottir U, Toniolo D, Tonjes A, Tremblay J, Uitterlinden AG, Vaccargiu S, van der Harst P, van Duijn CM, Verweij N, Volker U, Vollenweider P, Waeber G, Waldenberger M, Whitfield JB, Wild SH, Wilson JF, Yang Q, Zhang W, Zonderman AB, Bochud M, Wilson JG, Pendergrass SA, Ho K, Parsa A, Pramstaller PP, Psaty BM, Boger CA, Snieder H, Butterworth AS, Okada Y, Edwards TL, Stefansson K, Susztak K, Scholz M, Heid IM, Hung AM, Teumer A, Pattaro C, Woodward OM, Vitart V, Kottgen A: Target genes, variants, tissues and transcriptional pathways influencing human serum urate levels. Nat Genet 2019;51:1459–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Major TJ, Topless RK, Dalbeth N, Merriman TR: Evaluation of the diet wide contribution to serum urate levels: meta-analysis of population based cohorts. BMJ 2018;363:k3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee SM, Lee AL, Winters TJ, Tam E, Jaleel M, Stenvinkel P, Johnson RJ: Low serum uric acid level is a risk factor for death in incident hemodialysis patients. Am J Nephrol 2009;29:79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spencer HW, Yarger WE, Robinson RR: Alterations of renal function during dietary-induced hyperuricemia in the rat. Kidney Int 1976;9:489–500. [DOI] [PubMed] [Google Scholar]
- 35.Sanchez-Lozada LG, Tapia E, Santamaria J, Avila-Casado C, Soto V, Nepomuceno T, Rodriguez-Iturbe B, Johnson RJ, Herrera-Acosta J: Mild hyperuricemia induces vasoconstriction and maintains glomerular hypertension in normal and remnant kidney rats. Kidney Int 2005;67:237–247. [DOI] [PubMed] [Google Scholar]
- 36.Sanchez-Lozada LG, Tapia E, Rodriguez-Iturbe B, Johnson RJ, Herrera-Acosta J: Hemodynamics of hyperuricemia. Semin Nephrol 2005;25:19–24. [DOI] [PubMed] [Google Scholar]
- 37.Roncal CA, Mu W, Croker B, Reungjui S, Ouyang X, Tabah-Fisch I, Johnson RJ, Ejaz AA: Effect of elevated serum uric acid on cisplatin-induced acute renal failure. Am J Physiol Renal Physiol 2007;292:F116–122. [DOI] [PubMed] [Google Scholar]
- 38.Ejaz AA, Beaver TM, Shimada M, Sood P, Lingegowda V, Schold JD, Kim T, Johnson RJ: Uric acid: a novel risk factor for acute kidney injury in high-risk cardiac surgery patients? Am J Nephrol 2009;30:425–429. [DOI] [PubMed] [Google Scholar]
- 39.Lapsia V, Johnson RJ, Dass B, Shimada M, Kambhampati G, Ejaz NI, Arif AA, Ejaz AA: Elevated uric acid increases the risk for acute kidney injury. Am J Med 2012;125:302 e309–317. [DOI] [PubMed] [Google Scholar]
- 40.Nanji AA, Stewart DJ, Mikhael NZ: Hyperuricemia and hypoalbuminemia predispose to cisplatin-induced nephrotoxicity. Cancer Chemother Pharmacol 1986;17:274–276. [DOI] [PubMed] [Google Scholar]
- 41.Koratala A, Singhania G, Alquadan KF, Shimada M, Johnson RJ, Ejaz AA: Serum Uric Acid Exhibits Inverse Relationship with Estimated Glomerular Filtration Rate. Nephron 2016;134:231–237. [DOI] [PubMed] [Google Scholar]
- 42.Ejaz AA, Pourafshar N, Mohandas R, Smallwood BA, Johnson RJ, Hsu JW: Uric acid and the prediction models of tumor lysis syndrome in AML. PLoS One 2015;10:e0119497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xin W, Lin Z, Zhang T, Jia S: Effects of allopurinol pretreatment on the risk of contrast-induced acute kidney injury in patients undergoing percutaneous coronary intervention: A meta-analysis of randomized controlled trials. Clin Nephrol 2020;93:24–33. [DOI] [PubMed] [Google Scholar]
- 44.Kanbay M, Solak Y, Afsar B, Nistor I, Aslan G, Caglayan OH, Aykanat A, Donciu MD, Lanaspa MA, Ejaz AA, Johnson RJ, Covic A: Serum Uric Acid and Risk for Acute Kidney Injury Following Contrast. Angiology 2017;68:132–144. [DOI] [PubMed] [Google Scholar]
- 45.Liang J, Zhang P, Hu X, Zhi L: Elevated serum uric acid after injury correlates with the early acute kidney in severe burns. Burns 2015;41:1724–1731. [DOI] [PubMed] [Google Scholar]
- 46.Greenberg KI, McAdams-DeMarco MA, Kottgen A, Appel LJ, Coresh J, Grams ME: Plasma Urate and Risk of a Hospital Stay with AKI: The Atherosclerosis Risk in Communities Study. Clin J Am Soc Nephrol 2015;10:776–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ejaz AA, Alquadan KF, Dass B, Shimada M, Kanbay M, Johnson RJ: Effects of Serum Uric Acid on Estimated GFR in Cardiac Surgery Patients: A Pilot Study. Am J Nephrol 2015;42:402–409. [DOI] [PubMed] [Google Scholar]
- 48.Ejaz AA, Dass B, Lingegowda V, Shimada M, Beaver TM, Ejaz NI, Abouhamze AS, Johnson RJ: Effect of uric acid lowering therapy on the prevention of acute kidney injury in cardiovascular surgery. Int Urol Nephrol 2013;45:449–458. [DOI] [PubMed] [Google Scholar]
- 49.Galardy PJ, Hochberg J, Perkins SL, Harrison L, Goldman S, Cairo MS: Rasburicase in the prevention of laboratory/clinical tumour lysis syndrome in children with advanced mature B-NHL: a Children’s Oncology Group Report. Br J Haematol 2013;163:365–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kumar A, Bhawani G, Kumari N, Murthy KS, Lalwani V, Raju Ch N: Comparative study of renal protective effects of allopurinol and N-acetyl-cysteine on contrast induced nephropathy in patients undergoing cardiac catheterization. J Clin Diagn Res 2014;8:HC03–07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ejaz AA, Johnson RJ, Shimada M, Mohandas R, Alquadan KF, Beaver TM, Lapsia V, Dass B: The Role of Uric Acid in Acute Kidney Injury. Nephron 2019;142:275–283. [DOI] [PubMed] [Google Scholar]
- 52.Li L, Yang C, Zhao Y, Zeng X, Liu F, Fu P: Is hyperuricemia an independent risk factor for new-onset chronic kidney disease?: A systematic review and meta-analysis based on observational cohort studies. BMC Nephrol 2014;15:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Johnson RJ, Bakris GL, Borghi C, Chonchol MB, Feldman D, Lanaspa MA, Merriman TR, Moe OW, Mount DB, Sanchez Lozada LG, Stahl E, Weiner DE, Chertow GM: Hyperuricemia, Acute and Chronic Kidney Disease, Hypertension, and Cardiovascular Disease: Report of a Scientific Workshop Organized by the National Kidney Foundation. Am J Kidney Dis 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Iseki K, Ikemiya Y, Inoue T, Iseki C, Kinjo K, Takishita S: Significance of hyperuricemia as a risk factor for developing ESRD in a screened cohort. Am J Kidney Dis 2004;44:642–650. [PubMed] [Google Scholar]
- 55.Chonchol M, Shlipak MG, Katz R, Sarnak MJ, Newman AB, Siscovick DS, Kestenbaum B, Carney JK, Fried LF: Relationship of uric acid with progression of kidney disease. Am J Kidney Dis 2007;50:239–247. [DOI] [PubMed] [Google Scholar]
- 56.Kuwabara M, Niwa K, Hisatome I, Nakagawa T, Roncal-Jimenez CA, Andres-Hernando A, Bjornstad P, Jensen T, Sato Y, Milagres T, Garcia G, Ohno M, Lanaspa MA, Johnson RJ: Asymptomatic Hyperuricemia Without Comorbidities Predicts Cardiometabolic Diseases: Five-Year Japanese Cohort Study. Hypertension 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Madero M, Sarnak MJ, Wang X, Greene T, Beck GJ, Kusek JW, Collins AJ, Levey AS, Menon V: Uric acid and long-term outcomes in CKD. Am J Kidney Dis 2009;53:796–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Saldana TM, Basso O, Darden R, Sandler DP: Carbonated beverages and chronic kidney disease. Epidemiology 2007;18:501–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shoham DA, Durazo-Arvizu R, Kramer H, Luke A, Vupputuri S, Kshirsagar A, Cooper RS: Sugary soda consumption and albuminuria: results from the National Health and Nutrition Examination Survey, 1999–2004. PLoS ONE 2008;3:e3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bomback AS, Derebail VK, Shoham DA, Anderson CA, Steffen LM, Rosamond WD, Kshirsagar AV: Sugar-sweetened soda consumption, hyperuricemia, and kidney disease. Kidney Int 2010;77:609–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rebholz CM, Young BA, Katz R, Tucker KL, Carithers TC, Norwood AF, Correa A: Patterns of Beverages Consumed and Risk of Incident Kidney Disease. Clin J Am Soc Nephrol 2019;14:49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lin J, Curhan GC: Associations of sugar and artificially sweetened soda with albuminuria and kidney function decline in women. Clin J Am Soc Nephrol 2011;6:160–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bomback AS, Katz R, He K, Shoham DA, Burke GL, Klemmer PJ: Sugar-sweetened beverage consumption and the progression of chronic kidney disease in the Multi-Ethnic Study of Atherosclerosis (MESA). Am J Clin Nutr 2009;90:1172–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rebholz CM, Grams ME, Steffen LM, Crews DC, Anderson CA, Bazzano LA, Coresh J, Appel LJ: Diet Soda Consumption and Risk of Incident End Stage Renal Disease. Clin J Am Soc Nephrol 2017;12:79–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nakagawa T, Mazzali M, Kang DH, Kanellis J, Watanabe S, Sanchez-Lozada LG, Rodriguez-Iturbe B, Herrera-Acosta J, Johnson RJ: Hyperuricemia causes glomerular hypertrophy in the rat. Am J Nephrol 2003;23:2–7. [DOI] [PubMed] [Google Scholar]
- 66.Mazzali M, Hughes J, Kim YG, Jefferson JA, Kang DH, Gordon KL, Lan HY, Kivlighn S, Johnson RJ: Elevated uric acid increases blood pressure in the rat by a novel crystal-independent mechanism. Hypertension 2001;38:1101–1106. [DOI] [PubMed] [Google Scholar]
- 67.Mazzali M, Kanellis J, Han L, Feng L, Xia YY, Chen Q, Kang DH, Gordon KL, Watanabe S, Nakagawa T, Lan HY, Johnson RJ: Hyperuricemia induces a primary renal arteriolopathy in rats by a blood pressure-independent mechanism. Am J Physiol Renal Physiol 2002;282:F991–997. [DOI] [PubMed] [Google Scholar]
- 68.Sanchez-Lozada LG, Soto V, Tapia E, Avila-Casado C, Sautin YY, Nakagawa T, Franco M, Rodriguez-Iturbe B, Johnson RJ: Role of oxidative stress in the renal abnormalities induced by experimental hyperuricemia. Am J Physiol Renal Physiol 2008;295:F1134–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sanchez-Lozada LG, Tapia E, Lopez-Molina R, Nepomuceno T, Soto V, Avila-Casado C, Nakagawa T, Johnson RJ, Herrera-Acosta J, Franco M: Effects of acute and chronic L-arginine treatment in experimental hyperuricemia. Am J Physiol Renal Physiol 2007;292:F1238–1244. [DOI] [PubMed] [Google Scholar]
- 70.Ryu ES, Kim MJ, Shin HS, Jang YH, Choi HS, Jo I, Johnson RJ, Kang DH: Uric acid-induced phenotypic transition of renal tubular cells as a novel mechanism of chronic kidney disease. Am J Physiol Renal Physiol 2013;304:F471–480. [DOI] [PubMed] [Google Scholar]
- 71.Han HJ, Lim MJ, Lee YJ, Lee JH, Yang IS, Taub M: Uric acid inhibits renal proximal tubule cell proliferation via at least two signaling pathways involving PKC, MAPK, cPLA2, and NF-kappaB. Am J Physiol Renal Physiol 2007;292:F373–381. [DOI] [PubMed] [Google Scholar]
- 72.Verzola D, Ratto E, Villaggio B, Parodi EL, Pontremoli R, Garibotto G, Viazzi F: Uric acid promotes apoptosis in human proximal tubule cells by oxidative stress and the activation of NADPH oxidase NOX 4. PLoS One 2014;9:e115210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xiao J, Zhang XL, Fu C, Han R, Chen W, Lu Y, Ye Z: Soluble uric acid increases NALP3 inflammasome and interleukin-1beta expression in human primary renal proximal tubule epithelial cells through the Toll-like receptor 4-mediated pathway. Int J Mol Med 2015;35:1347–1354. [DOI] [PubMed] [Google Scholar]
- 74.Zhou Y, Fang L, Jiang L, Wen P, Cao H, He W, Dai C, Yang J: Uric acid induces renal inflammation via activating tubular NF-kappaB signaling pathway. PLoS One 2012;7:e39738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xu C, Lu A, Lu X, Zhang L, Fang H, Zhou L, Yang T: Activation of Renal (Pro)Renin Receptor Contributes to High Fructose-Induced Salt Sensitivity. Hypertension 2017;69:339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yu MA, Sanchez-Lozada LG, Johnson RJ, Kang DH: Oxidative stress with an activation of the renin-angiotensin system in human vascular endothelial cells as a novel mechanism of uric acid-induced endothelial dysfunction. J Hypertens 2010;28:1234–1242. [PubMed] [Google Scholar]
- 77.Sanchez-Lozada LG, Lanaspa MA, Cristobal-Garcia M, Garcia-Arroyo F, Soto V, Cruz-Robles D, Nakagawa T, Yu MA, Kang DH, Johnson RJ: Uric Acid-Induced Endothelial Dysfunction Is Associated with Mitochondrial Alterations and Decreased Intracellular ATP Concentrations. Nephron Exp Nephrol 2012;121:e71–e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sanchez-Lozada LG, Tapia E, Soto V, Avila-Casado C, Franco M, Zhao L, Johnson RJ: Treatment with the xanthine oxidase inhibitor febuxostat lowers uric acid and alleviates systemic and glomerular hypertension in experimental hyperuricaemia. Nephrol Dial Transplant 2008;23:1179–1185. [DOI] [PubMed] [Google Scholar]
- 79.García-Arroyo FE, Gonzaga G, Muñoz-Hernández I, Blas-Marron M, Silverio O, Tapia E, Soto V, Ranganathan N, Ranganathan P, Vyas U, Irvin A, Ir D, Robertson CE, Frank DN, Johnson RJ, Sánchez-Lozada LG: Probiotic supplements prevented oxonic acid-induced hyperuricemia and renal damage. PLoS One 2018:in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jordan DM, Choi HK, Verbanck M, Topless R, Won HH, Nadkarni G, Merriman TR, Do R: No causal effects of serum urate levels on the risk of chronic kidney disease: A Mendelian randomization study. PLoS Med 2019;16:e1002725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yang Q, Kottgen A, Dehghan A, Smith AV, Glazer NL, Chen MH, Chasman DI, Aspelund T, Eiriksdottir G, Harris TB, Launer L, Nalls M, Hernandez D, Arking DE, Boerwinkle E, Grove ML, Li M, Linda Kao WH, Chonchol M, Haritunians T, Li G, Lumley T, Psaty BM, Shlipak M, Hwang SJ, Larson MG, O’Donnell CJ, Upadhyay A, van Duijn CM, Hofman A, Rivadeneira F, Stricker B, Uitterlinden AG, Pare G, Parker AN, Ridker PM, Siscovick DS, Gudnason V, Witteman JC, Fox CS, Coresh J: Multiple genetic loci influence serum urate levels and their relationship with gout and cardiovascular disease risk factors. Circ Cardiovasc Genet 2010;3:523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ge JY, Ji Y, Zhu ZY, Li X: Genetically Elevated Serum Uric Acid and Renal Function in an Apparently Healthy Population. Urol Int 2019:1–6. [DOI] [PubMed] [Google Scholar]
- 83.Kim KM, Henderson GN, Ouyang X, Frye RF, Sautin YY, Feig DI, Johnson RJ: A sensitive and specific liquid chromatography-tandem mass spectrometry method for the determination of intracellular and extracellular uric acid. J Chromatogr B Analyt Technol Biomed Life Sci 2009;877:2032–2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ma Q, Honarpisheh M, Li C, Sellmayr M, Lindenmeyer M, Bohland C, Romagnani P, Anders HJ, Steiger S: Soluble Uric Acid Is an Intrinsic Negative Regulator of Monocyte Activation in Monosodium Urate Crystal-Induced Tissue Inflammation. J Immunol 2020;205:789–800. [DOI] [PubMed] [Google Scholar]
- 85.DeBosch BJ, Kluth O, Fujiwara H, Schurmann A, Moley K: Early-onset metabolic syndrome in mice lacking the intestinal uric acid transporter SLC2A9. Nat Commun 2014;5:4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Preitner F, Bonny O, Laverriere A, Rotman S, Firsov D, Da Costa A, Metref S, Thorens B: Glut9 is a major regulator of urate homeostasis and its genetic inactivation induces hyperuricosuria and urate nephropathy. Proc Natl Acad Sci U S A 2009;106:15501–15506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Preitner F, Pimentel A, Metref S, Berthonneche C, Sarre A, Moret C, Rotman S, Centeno G, Firsov D, Thorens B: No development of hypertension in the hyperuricemic liver-Glut9 knockout mouse. Kidney Int 2015;87:940–947. [DOI] [PubMed] [Google Scholar]
- 88.Su X, Xu B, Yan B, Qiao X, Wang L: Effects of uric acid-lowering therapy in patients with chronic kidney disease: A meta-analysis. PLoS One 2017;12:e0187550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Liu X, Zhai T, Ma R, Luo C, Wang H, Liu L: Effects of uric acid-lowering therapy on the progression of chronic kidney disease: a systematic review and meta-analysis. Ren Fail 2018;40:289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sato Y, Feig DI, Stack AG, Kang DH, Lanaspa MA, Ejaz AA, Sanchez-Lozada LG, Kuwabara M, Borghi C, Johnson RJ: The case for uric acid-lowering treatment in patients with hyperuricaemia and CKD. Nat Rev Nephrol 2019;15:767–775. [DOI] [PubMed] [Google Scholar]
- 91.Doria A, Galecki AT, Spino C, Pop-Busui R, Cherney DZ, Lingvay I, Parsa A, Rossing P, Sigal RJ, Afkarian M, Aronson R, Caramori ML, Crandall JP, de Boer IH, Elliott TG, Goldfine AB, Haw JS, Hirsch IB, Karger AB, Maahs DM, McGill JB, Molitch ME, Perkins BA, Polsky S, Pragnell M, Robiner WN, Rosas SE, Senior P, Tuttle KR, Umpierrez GE, Wallia A, Weinstock RS, Wu C, Mauer M, Group PS: Serum Urate Lowering with Allopurinol and Kidney Function in Type 1 Diabetes. N Engl J Med 2020;382:2493–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Badve SV, Pascoe EM, Tiku A, Boudville N, Brown FG, Cass A, Clarke P, Dalbeth N, Day RO, de Zoysa JR, Douglas B, Faull R, Harris DC, Hawley CM, Jones GRD, Kanellis J, Palmer SC, Perkovic V, Rangan GK, Reidlinger D, Robison L, Walker RJ, Walters G, Johnson DW, Investigators C-FS: Effects of Allopurinol on the Progression of Chronic Kidney Disease. N Engl J Med 2020;382:2504–2513. [DOI] [PubMed] [Google Scholar]
- 93.Kim SY, Guevara JP, Kim KM, Choi HK, Heitjan DF, Albert DA: Hyperuricemia and coronary heart disease: a systematic review and meta-analysis. Arthritis Care Res (Hoboken) 2010;62:170–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wheeler JG, Juzwishin KD, Eiriksdottir G, Gudnason V, Danesh J: Serum uric acid and coronary heart disease in 9,458 incident cases and 155,084 controls: prospective study and meta-analysis. PLoS Med 2005;2:e76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kleber ME, Delgado G, Grammer TB, Silbernagel G, Huang J, Kramer BK, Ritz E, Marz W: Uric Acid and Cardiovascular Events: A Mendelian Randomization Study. J Am Soc Nephrol 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yan D, Wang J, Jiang F, Zhang R, Wang T, Wang S, Peng D, He Z, Chen H, Bao Y, Hu C, Jia W: A causal relationship between uric acid and diabetic macrovascular disease in Chinese type 2 diabetes patients: A Mendelian randomization analysis. Int J Cardiol 2016;214:194–199. [DOI] [PubMed] [Google Scholar]
- 97.Johnson RJ: Finding the truth: multivariable analysis and the assassination of Abraham Lincoln. J R Coll Physicians Edinb 2018;48:153–154. [DOI] [PubMed] [Google Scholar]
- 98.Wong KY, MacWalter RS, Fraser HW, Crombie I, Ogston SA, Struthers AD: Urate predicts subsequent cardiac death in stroke survivors. Eur Heart J 2002;23:788–793. [DOI] [PubMed] [Google Scholar]
- 99.Higgins P, Walters MR, Murray HM, McArthur K, McConnachie A, Lees KR, Dawson J: Allopurinol reduces brachial and central blood pressure, and carotid intima-media thickness progression after ischaemic stroke and transient ischaemic attack: a randomised controlled trial. Heart 2014;100:1085–1092. [DOI] [PubMed] [Google Scholar]
- 100.Khan F, George J, Wong K, McSwiggan S, Struthers AD, Belch JJ: Allopurinol treatment reduces arterial wave reflection in stroke survivors. Cardiovasc Ther 2008;26:247–252. [DOI] [PubMed] [Google Scholar]
- 101.Muir SW, Harrow C, Dawson J, Lees KR, Weir CJ, Sattar N, Walters MR: Allopurinol use yields potentially beneficial effects on inflammatory indices in those with recent ischemic stroke: a randomized, double-blind, placebo-controlled trial. Stroke 2008;39:3303–3307. [DOI] [PubMed] [Google Scholar]
- 102.Tabayashi K, Suzuki Y, Nagamine S, Ito Y, Sekino Y, Mohri H: A clinical trial of allopurinol (Zyloric) for myocardial protection. J Thorac Cardiovasc Surg 1991;101:713–718. [PubMed] [Google Scholar]
- 103.Johnson WD, Kayser KL, Brenowitz JB, Saedi SF: A randomized controlled trial of allopurinol in coronary bypass surgery. Am Heart J 1991;121:20–24. [DOI] [PubMed] [Google Scholar]
- 104.Noman A, Ang DS, Ogston S, Lang CC, Struthers AD: Effect of high-dose allopurinol on exercise in patients with chronic stable angina: a randomised, placebo controlled crossover trial. Lancet 2010;375:2161–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.MacIsaac RL, Salatzki J, Higgins P, Walters MR, Padmanabhan S, Dominiczak AF, Touyz RM, Dawson J: Allopurinol and Cardiovascular Outcomes in Adults With Hypertension. Hypertension 2016;67:535–540. [DOI] [PubMed] [Google Scholar]
- 106.Singh JA, Yu S: Allopurinol and the risk of stroke in older adults receiving medicare. BMC Neurol 2016;16:164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Singh JA, Yu S: Allopurinol reduces the risk of myocardial infarction (MI) in the elderly: a study of Medicare claims. Arthritis Res Ther 2016;18:209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Singh JA, Ramachandaran R, Yu S, Curtis JR: Allopurinol use and the risk of acute cardiovascular events in patients with gout and diabetes. BMC Cardiovasc Disord 2017;17:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hare JM, Mangal B, Brown J, Fisher C Jr., Freudenberger R, Colucci WS, Mann DL, Liu P, Givertz MM, Schwarz RP: Impact of oxypurinol in patients with symptomatic heart failure. Results of the OPT-CHF study. J Am Coll Cardiol 2008;51:2301–2309. [DOI] [PubMed] [Google Scholar]
- 110.Mackenzie IS, Ford I, Walker A, Hawkey C, Begg A, Avery A, Taggar J, Wei L, Struthers AD, MacDonald TM, group A-Hs: Multicentre, prospective, randomised, open-label, blinded end point trial of the efficacy of allopurinol therapy in improving cardiovascular outcomes in patients with ischaemic heart disease: protocol of the ALL-HEART study. BMJ Open 2016;6:e013774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Foley RN, Parfrey PS, Sarnak MJ: Epidemiology of cardiovascular disease in chronic renal disease. J Am Soc Nephrol 1998;9:S16–23. [PubMed] [Google Scholar]
- 112.Kao MP, Ang DS, Gandy SJ, Nadir MA, Houston JG, Lang CC, Struthers AD: Allopurinol benefits left ventricular mass and endothelial dysfunction in chronic kidney disease. J Am Soc Nephrol 2011;22:1382–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Goicoechea M, Garcia de Vinuesa S, Verdalles U, Verde E, Macias N, Santos A, Perez de Jose A, Cedeno S, Linares T, Luno J: Allopurinol and Progression of CKD and Cardiovascular Events: Long-term Follow-up of a Randomized Clinical Trial. Am J Kidney Dis 2015 [DOI] [PubMed] [Google Scholar]
- 114.Sircar D, Chatterjee S, Waikhom R, Golay V, Raychaudhury A, Chatterjee S, Pandey R: Efficacy of Febuxostat for Slowing the GFR Decline in Patients With CKD and Asymptomatic Hyperuricemia: A 6-Month, Double-Blind, Randomized, Placebo-Controlled Trial. Am J Kidney Dis 2015;66:945–950. [DOI] [PubMed] [Google Scholar]
- 115.Tuta L, Stanigut A: Allopurinol therapy for hyperuricemia reduces inflammation and progression of renal disease in moderate chronic kidney disease. Nephrol Dial Transplant 2014;29:iii118. [Google Scholar]
- 116.Terawaki H, Nakayama M, Miyazawa E, Murata Y, Nakayama K, Matsushima M, Miyazaki M, Sato H, Sato M, Sato T, Taguma Y, Ito S: Effect of allopurinol on cardiovascular incidence among hypertensive nephropathy patients: the Gonryo study. Clin Exp Nephrol 2012 [DOI] [PubMed] [Google Scholar]
- 117.Suliman ME, Johnson RJ, Garcia-Lopez E, Qureshi AR, Molinaei H, Carrero JJ, Heimburger O, Barany P, Axelsson J, Lindholm B, Stenvinkel P: J-shaped mortality relationship for uric acid in CKD. Am J Kidney Dis 2006;48:761–771. [DOI] [PubMed] [Google Scholar]
- 118.Hsu SP, Pai MF, Peng YS, Chiang CK, Ho TI, Hung KY: Serum uric acid levels show a ‘J-shaped’ association with all-cause mortality in haemodialysis patients. Nephrol Dial Transplant 2004;19:457–462. [DOI] [PubMed] [Google Scholar]
- 119.Latif W, Karaboyas A, Tong L, Winchester JF, Arrington CJ, Pisoni RL, Marshall MR, Kleophas W, Levin NW, Sen A, Robinson BM, Saran R: Uric acid levels and all-cause and cardiovascular mortality in the hemodialysis population. Clin J Am Soc Nephrol 2011;6:2470–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sugano N, Maruyama Y, Kidoguchi S, Ohno I, Wada A, Shigematsu T, Masakane I, Yokoo T: Effect of hyperuricemia and treatment for hyperuricemia in Japanese hemodialysis patients: A cohort study. PLoS One 2019;14:e0217859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Beberashvili I, Erlich A, Azar A, Sinuani I, Feldman L, Gorelik O, Stav K, Efrati S: Longitudinal Study of Serum Uric Acid, Nutritional Status, and Mortality in Maintenance Hemodialysis Patients. Clin J Am Soc Nephrol 2016;11:1015–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Petreski T, Ekart R, Hojs R, Bevc S: Asymptomatic hyperuricemia and cardiovascular mortality in patients with chronic kidney disease who progress to hemodialysis. Int Urol Nephrol 2019;51:1013–1018. [DOI] [PubMed] [Google Scholar]
- 123.Jalalzadeh M, Nurcheshmeh Z, Mohammadi R, Mousavinasab N, Ghadiani MH: The effect of allopurinol on lowering blood pressure in hemodialysis patients with hyperuricemia. J Res Med Sci 2012;17:1039–1046. [PMC free article] [PubMed] [Google Scholar]
- 124.Tsuruta Y, Kikuchi K, Tsuruta Y, Sasaki Y, Moriyama T, Itabashi M, Takei T, Uchida K, Akiba T, Tsuchiya K, Nitta K: Febuxostat improves endothelial function in hemodialysis patients with hyperuricemia: A randomized controlled study. Hemodial Int 2015;19:514–520. [DOI] [PubMed] [Google Scholar]
- 125.Tsuruta Y, Nitta K, Akizawa T, Fukuhara S, Saito A, Karaboyas A, Li Y, Port FK, Robinson BM, Pisoni RL, Akiba T: Association between allopurinol and mortality among Japanese hemodialysis patients: results from the DOPPS. Int Urol Nephrol 2014;46:1833–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Klauser AS, Halpern EJ, Strobl S, Gruber J, Feuchtner G, Bellmann-Weiler R, Weiss G, Stofferin H, Jaschke W: Dual-Energy Computed Tomography Detection of Cardiovascular Monosodium Urate Deposits in Patients With Gout. JAMA Cardiol 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Felici C, Ciari I, Terzuoli L, Porcelli B, Setacci C, Giubbolini M, Marinello E: Purine catabolism in advanced carotid artery plaque. Nucleosides Nucleotides Nucleic Acids 2006;25:1291–1294. [DOI] [PubMed] [Google Scholar]
- 128.Suarna C, Dean RT, May J, Stocker R: Human atherosclerotic plaque contains both oxidized lipids and relatively large amounts of alpha-tocopherol and ascorbate. Arterioscler Thromb Vasc Biol 1995;15:1616–1624. [DOI] [PubMed] [Google Scholar]
- 129.Patetsios P, Song M, Shutze WP, Pappas C, Rodino W, Ramirez JA, Panetta TF: Identification of uric acid and xanthine oxidase in atherosclerotic plaque. Am J Cardiol 2001;88:188–191, A186. [DOI] [PubMed] [Google Scholar]
- 130.Coe FL, Raisen L: Allopurinol treatment of uric-acid disorders in calcium-stone formers. Lancet 1973;1:129–131. [DOI] [PubMed] [Google Scholar]
- 131.Rodrigues TC, Maahs DM, Johnson RJ, Jalal DI, Kinney GL, Rivard C, Rewers M, Snell-Bergeon JK: Serum uric acid predicts progression of subclinical coronary atherosclerosis in individuals without renal disease. Diabetes Care 2010;33:2471–2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bjornstad P, Maahs DM, Rivard CJ, Pyle L, Rewers M, Johnson RJ, Snell-Bergeon JK: Serum uric acid predicts vascular complications in adults with type 1 diabetes: the coronary artery calcification in type 1 diabetes study. Acta Diabetol 2014;51:783–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.White WB, Saag KG, Becker MA, Borer JS, Gorelick PB, Whelton A, Hunt B, Castillo M, Gunawardhana L, Investigators C: Cardiovascular Safety of Febuxostat or Allopurinol in Patients with Gout. N Engl J Med 2018;378:1200–1210. [DOI] [PubMed] [Google Scholar]
- 134.Johnson TA, Kamatani N, Kuwabara M: Xanthine Oxidase Inhibitor Withdrawal Syndrome? Comment on the Article by Choi et al. Arthritis Rheumatol 2019;71:1966–1967. [DOI] [PubMed] [Google Scholar]
- 135.Talaat KM, El-Sheikh AR: The Effect of Mild Hyperuricemia on Urinary Transforming Growth Factor Beta and the Progression of Chronic Kidney Disease. Am J Nephrol 2007;27:435–440. [DOI] [PubMed] [Google Scholar]
- 136.Zhang M, Solomon DH, Desai RJ, Kang EH, Liu J, Neogi T, Kim SC: Assessment of Cardiovascular Risk in Older Patients With Gout Initiating Febuxostat Versus Allopurinol. Circulation 2018;138:1116–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kojima S, Matsui K, Hiramitsu S, Hisatome I, Waki M, Uchiyama K, Yokota N, Tokutake E, Wakasa Y, Jinnouchi H, Kakuda H, Hayashi T, Kawai N, Mori H, Sugawara M, Ohya Y, Kimura K, Saito Y, Ogawa H: Febuxostat for Cerebral and CaRdiorenovascular Events PrEvEntion StuDy. Eur Heart J 2019;40:1778–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Sanchez-Nino MD, Zheng-Lin B, Valino-Rivas L, Sanz AB, Ramos AM, Luno J, Goicoechea M, Ortiz A: Lesinurad: what the nephrologist should know. Clin Kidney J 2017;10:679–687. [DOI] [PMC free article] [PubMed] [Google Scholar]