

Abstract
Ti(salen) complexes catalyze the asymmetric [3 + 2] cycloaddition of cyclopropyl ketones with alkenes. While high enantioselectivities are achieved with electron-rich alkenes, electron-deficient alkenes are less selective. Herein, we describe mechanistic studies to understand the origins of catalyst and substrate trends in an effort to identify a more general catalyst. Density functional theory (DFT) calculations of the selectivity determining transition state revealed the origin of stereochemical control to be catalyst distortion, which is largely influenced by the chiral backbone and adamantyl groups on the salicylaldehyde moieties. While substitution of the adamantyl groups was detrimental to the enantioselectivity, mechanistic information guided the development of a set of eight new Ti(salen) catalysts with modified diamine backbones. These catalysts were evaluated with four electron-deficient alkenes to develop a three-parameter statistical model relating enantioselectivity to physical organic parameters. This statistical model is capable of quantitative prediction of enantioselectivity with structurally diverse alkenes. These mechanistic insights assisted the discovery of a new Ti(salen) catalyst, which substantially expanded the reaction scope and significantly improved the enantioselectivity of synthetically interesting building blocks.
Graphical Abstract
INTRODUCTION
Stereoselective catalysis of radical-based organic transformations remains a challenge in modern synthetic chemistry in part because of the high reactivity and relatively limited knowledge about reaction pathways involving radical intermediates.1 Recent advances have made possible a plethora of highly enantioselective reactions mediated by radical intermediates.2 In contrast to the rapid developments in this area, mechanistic understanding of catalytic radical-based reactivities lags behind.3 Better understanding will lead to rational design and optimization of next-generation catalysts with broader scope and improved synthetic applications.
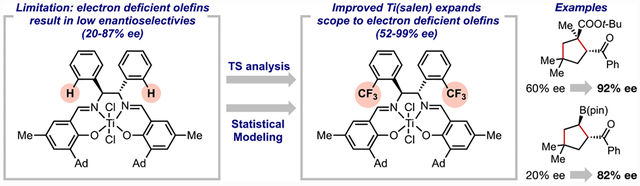
In this context, we have recently developed a new catalytic strategy that harnesses the single-electron redox activity of Ti complexes for the formal [3 + 2] cycloaddition of cyclopropyl ketones and alkenes (Figure 1).4 Notably, the use of a chiral Ti(salen) complex ((R,R)-4a) renders this transformation diastereo- and enantioselective, producing polysubstituted cyclopentanes from alkenes and cyclopropyl ketones. This development is interesting for several reasons. Synthetically, cyclopentanes are common structures in complex bioactive compounds.5 In particular, cyclopentylcarboxylic acids (cf. 3e and 3f) have been studied as structural analogs for prolines in the discovery and evaluation of peptidyl drugs.6 On a fundamental level, enantioselective radical reactions catalyzed by Ti predominantly rely on chiral cyclopentadienyl ligands,7 which are typically cumbersome to synthesize. The successful use of salen compounds, a class of modular chiral ligands, in enantioselective reactions will have significant implications in this burgeoning area of research.4b
Figure 1.

Reported Ti(salen)-catalyzed [3 + 2] cycloaddition. Electron-deficient alkenes yield products with lower enantioselectivities.
In the initial discovery, we found that Ti catalysts such as (R,R)-4a generally promoted the reaction in excellent diasteroand enantioselectivity for styrene-type substrates with electron-rich and -neutral substituents (e.g., (S,S)-3a–c). Nonetheless, reactions with electron-deficient alkenes led to much poorer stereocontrol, and the application of lower reaction temperatures was necessary to achieve reasonable levels of enantioselectivity at the expense of longer reaction times and lower effciency. Specifically, acrylates, methacrylates, phenyl vinyl sulfone, and vinyl pinacolboronate ester gave rise to products in high diastereoselectivity but low enantioselectivity (Figure 1, (S,S)-3e–h). Reactions using such Michael acceptors often afford more synthetically useful products. For example, acrylate-derived products (S,S)-3e and (S,S)-3f can be used as amino acid analogues in peptide synthesis,6 whereas vinyl pinacolboronate ester 2h provides product (S,S)-3h with a stereogenic C–B bond that could be further elaborated into other synthetically useful compounds.8
We sought to address this significant limitation of our reaction system and improve the Ti(salen) catalyst by means of probing the mechanism of stereochemical induction. Metal-salen complexes have been widely used in enantioselective catalysis in the context of small molecule9 and polymer synthesis.10 Empirically, several structural factors including the nature of the metal center,11 the interplay of steric and electronic factors garnered by the salicylaldehyde groups,12 and the conformation of the complex13 can profoundly influence the reaction enantioselectivity. In contrast to the extensive use of metal-salen complexes in asymmetric catalysis, mechanistic understanding of the high enantioselectivity imparted by these chiral catalysts remains largely underexplored with the exception of the extensive, elegant studies of the hydrolytic kinetic resolution of epoxides by Jacobsen.14 Against this backdrop, we aimed to gain further insights into the mechanism of stereochemical induction in the context of the Ti-catalyzed [3 + 2] cycloaddition reaction with the primary goal of providing general guidelines for new catalyst design to improve currently challenging substrate classes, namely electron poor alkenes. Given the rigidity of metal salen complexes, we hope that these guidelines can be translated to other enantioselective transformations catalyzed by these privileged chiral catalysts.
Thus, we report herein a combination of experimental and computational tools to interrogate the mechanism of this Ti(salen)-catalyzed [3 + 2] cycloaddition. Both the structural features of catalysts and substrates were systematically modified to probe structure-selectivity relationships. By combining theory and statistical mathematical modeling, a stereochemical model was developed that features the role of catalyst distortion as well as intermediate positioning provided by both the chiral diamine backbone and the ortho-substituents on the salicylaldehyde of the catalyst. Ultimately, the mechanistic understanding informed the design of a new catalyst, which significantly expanded and improved the scope of the reaction.
RESULTS AND DISCUSSION
A. Experimental and Computational Studies of Reaction Mechanism.
The proposed mechanism for this reaction begins with the generation of an active Ti(III) catalyst via single electron reduction by Mn (Figure 2A). Subsequently, reductive ring opening of the cyclopropyl substrate occurs regioselectively under the action of the Ti(III) catalyst to yield a tertiary carbon-centered radical I. Addition to a radical acceptor alkene then proceeds to form intermediate II, which cyclizes to generate the final product. This stepwise, single-electron mechanism is supported by a spin trapping experiment (Figure 2B).4a Nonlinear effect and diffusion NMR experiments (see Experimental SI Section 6) were conducted to rule out cooperative reactivity between multiple catalysts, which has been reported for other metal-salen systems.15 Diffusion NMR experiments revealed that the catalyst is monomeric in the ground state. This information, taken together with the lack of a nonlinear effect of the catalytic reaction, suggests that this system operates through a monomeric catalyst. With experimental evidence to support a radical mechanism and a monomeric catalyst, computational analysis was carried out to assess the energetic feasibility of the mechanism.
Figure 2.

(A) Proposed mechanism for the Ti(salen)-catalyzed [3 +2] cycloaddition of cyclopropylketones with alkenes. (B) Spin trapping experiment supports a single electron mechanism.
The proposed catalytic cycle was computed using Gaussian0916 with a model Ti catalyst with only the salen core and p-chlorostyrene 2b as the substrate to reduce the computational expense (Figure 3). Alternative mechanisms, such as a concerted reductive ring opening and radical addition, were considered but such TSs were not located. As proposed, the reaction first proceeds through a TS corresponding to reductive ring opening (TS1) to yield radical intermediate I, which occurs with a free energy of activation of 26.9 kcal/mol relative to the catalyst-ketone complex (preTS1) and the styrene at infinite separation. The addition of this radical to the olefin (TS2) is irreversible and the rate-determining step (RDS). The subsequent radical cyclization (TS3) establishes the stereogenic centers and thus is the enantio- and diastereoselectivity-determining step. With this truncated model catalyst system, the cis cyclization pathway is slightly lower in energy than that of the trans pathway.
Figure 3.

Catalytic cycle for the Ti(salen) catalyzed [3 + 2] cycloaddition calculated with a model catalyst. Energies are reported in kcal/mol. Computational method: IEFPCM(EtOAc)-M06/def2-TZVP//M06/6–31G(d)-LANL2DZ level of theory.
The achiral model catalyst allowed us to interpret the feasibility of reaction steps in the proposed mechanism, but does not explain the observed selectivities. Therefore, to interpret the origins of selectivity, we next performed computational analysis of TS3, the selectivity-determining step, with the full optimal catalyst.17
B. Computational Analysis of the Selectivity Determining Transition States with the Full Catalyst System.
For the diastereo- and enantio-determining radical cyclization step, four possible TSs are considered: trans-Re, trans-Si, cis-Re, and cis-Si, each of which leads to a stereoisomer of the cyclopentane product. Conformers in each of these TS categories were also explored and generated by performing a conformational search (see Computational SI Section IV for details). These conformers were then used as candidate structures for TS optimization using Gaussian09 with the IEFPCM(EtOAc)-M06/6–31+G(d,p)//M06/6–31G(d)-SDD level of theory.16 From this process, the lowest-energy structure for each of the four stereochemical pathways was interpreted further. The lowest-energy pathway was calculated to be trans-Re TS3 (hereon referred to as major TS3), which is lower in energy by 0.8 kcal/mol relative to cis-Si TS3 and 1.5 kcal/mol relative to trans-Si TS3 (hereon referred to as minor TS3). The cis-Re TS3 pathway was significantly higher in energy and thus was excluded from further analysis (4.2 kcal/mol higher in energy than major TS3). These results are consistent with the experimentally observed enantio- and diastereoselectivity (Figure 4A). This method provided values in agreement with experiment for two additional styrene substrates (see Computational SI Section IV for details), further validating the DFT method.
Figure 4.

(A) Major TS3 scenarios to consider for analysis of reaction of 2a with (R,R)-4a, including calculated and experimental ΔΔG‡ values. Computational method: IEFPCM(EtOAc)-M06/6–31+G(d,p)//M06/6–31G(d)-SDD level of theory. (B) Qualitative model developed for rationalizing selectivity outcomes based on intermediate positioning and catalyst distortion and (C) overlay of each catalyst TS with the lowest-energy ground state catalyst (gray) to visualize catalyst distortion in each TS3. Distortion/interaction analysis was performed using single point energies calculated with the M06/6–31+G(d,p)-SDD level of theory.
Visual inspection of all three TSs shows that a well-defined chiral pocket is created by the backbone and constrained by the large adamantyl groups. Conformational analysis of the catalyst core in the key TSs shows the general positions of the adamantyl groups are similar in the minor TS3 and cis-Si TSs but inverted for the favored major TS3. Thus, viewing the catalyst along the C2 axis allows for the creation of a quadrant diagram based on the general positions of the adamantyl and backbone phenyl substituents. In this diagram, two quadrants are categorized as “full” with the catalyst adamantyl and phenyl substituents positioned toward the intermediate and the other two quadrants are “empty” with the adamantyl and phenyl substituents positioned away (Figure 4B). The large steric profile of the adamantyl groups directs the intermediate to adopt a position that places aromatic substituents on the intermediate in proximity to the salen core. Therefore, the distinct catalyst conformations in each TS result in disparate contacts between catalyst and intermediate. Specifically, the qualitative model shows that the intermediate substituents are arranged in the TS that leads to competing enantiomers to occupy two empty and one full quadrants. As such, the impact of minimizing energetically repulsive contacts in one TS over another is not clear. Intriguingly, we noticed that the catalyst in the major TS3 adopts a similar planar conformation to that of the ground state of the catalyst (Figure 4C, left), whereas the catalyst is significantly distorted to a stepped conformation to form the minor or cis-Si TSs (Figure 4C, center and right). The differences in catalyst-intermediate arrangement and catalyst conformation prompted further assessment using distortion/interaction and noncovalent interaction (NCI) analyses to determine the stereocontrolling factors.
A distortion/interaction analysis was performed to investigate the nature and energetic cost of catalyst distortion.18 This analysis quantifies the energy required for the catalyst and intermediate to distort into the TS conformation and the interaction energies that are established for the TS to favorably form despite the distortion. Single-point calculations in the gas-phase at the M06/6–31+G(d,p)-SDD level were employed to evaluate the relative roles of distortion and interaction on the overall energy differences for the diastereomeric TSs (see Computational SI Section VI). Using this method, the overall energy differences for the TSs forming enantiomeric products (ΔΔE‡enant. = ΔE‡ Minor‑TS3 – ΔE‡ Major‑TS3) and TSs forming diastereomeric products (ΔΔE‡diaster. = ΔE‡cis‑Si‑TS3 − ΔE‡Major‑TS3) are 0.7 and 2.9 kcal/mol, respectively.19 The calculated distortion energy differences reveal that cis-Re (3.1 kcal/mol) and minor (3.2 kcal/mol) TSs exhibit similar levels of distortion in reference to the major TS. Analysis of the contributions from the catalyst and intermediate fragments for each enantiomer suggests that the differences in distortion energy arise from catalyst distortion (3.09 kcal/mol)) rather than intermediate distortion (0.08 kcal/mol). Yet, the interaction energies reveal a significant difference in interaction energies for the minor TS3 relative to major TS3 (−2.5 kcal/mol), but not cis-Si TS3 relative to major TS3 (0.2 kcal/mol). This suggests that the interactions between catalyst and intermediate are more favorable in the minor TS3 than major TS3.
Natural energy decomposition analysis (NEDA) was performed to analyze the specific contribution of different interactions to the differences in total interaction energy for the TSs (see Computational SI Section VII). Although the minor TS3 has a greater level of repulsive interactions from steric crowding relative to the major TS3, minor TS3 also features more stabilizing attractive NCIs, ultimately resulting in an overall more negative total interaction energy. Furthermore, the values from this analysis imply that the gem-dimethyl contacts with the salen backbone in minor TS3 are more energetically costly than the interactions with the 4-chlorophenyl in major TS3. NCI plots were generated, demonstrating that a network of attractive interactions between the arenes on the intermediate and salen core occur in the TSs, further confirming our analysis (see Computational SI Section VIII).20 Although the interaction energy for the minor TS3 is more stabilizing, the energetic penalty for distortion outweighs the stabilizing interactions. On the basis of these analyses, we propose that catalyst distortion is the major contributor to the observed stereoselectivities with the p-chlorostyrene substrate 2b. The notion of a preferred conformation of a metal-salen in dictating selectivity was first invoked for rationalizing selectivity in Mn(salen)-catalyzed epoxidations, but it is plausible that catalyst distortion plays a significant role in these reactions as well.9e,21
To determine if catalyst distortion was a stereocontrolling factor among a wider range of substrates, we next performed TS analysis with 2-vinylpyridine (2l). This substrate reacted to yield products in low enantioselectivity (39% ee). Since enantioselectivity, and not diastereoselecivity, was the limiting aspect of this reaction with many substrates, we focused only on TSs that form enantiomeric products. TS analysis with substrate 2l was conducted to assess if catalyst distortion was also the primary enantiocontrolling factor with poorly performing substrates. The relevant TSs (trans-Re = major TS3 and trans-Si = minor TS3) were located using the same computational method as for the p-chlorostyrene TSs. The lowest energy major TS3 pathway was calculated to be 0.8 kcal/mol lower than the minor TS3 (Figure 5, top). This corresponds well with the lower levels of enantioselectivity obtained with this substrate. Distortion/interaction analysis of the TSs revealed that unlike the p-chlorostyrene substrate 2b, catalyst (2.7 kcal/mol) and substrate distortion (1.0 kcal/mol) both contribute to the relative difference in distortion energy between the TSs. Since the distortion contribution (+3.7 kcal/mol) is much larger than the energy difference between the major and minor TSs, other factors must be contributing to the enantioselectivity outcome. Most notably, NEDA demonstrated that minor TS3 has a larger attractive energy term than major TS3. Although these attractive interactions between catalyst and intermediate lower the enantioselectivity, the overall sense of stereoinduction can be explained by catalyst distortion.
Figure 5.

TS analysis for reaction of catalyst (R,R)-4a with 2-vinylpyridine 2l as substrate (top). Distortion/interaction analysis for these TSs (bottom).
This analysis indicated that improving enantioselectivity with poorly performing substrates requires increasing catalyst distortion and/or reducing the stabilizing interactions in the minor TS3. We reasoned that modifying the catalyst structure to increase steric bulk would increase catalyst distortion.
Our stereochemical models revealed that both adamantyl groups and chiral diamine backbone substituents play critical organizational roles in the selectivity-determining TS, resulting in disparate catalyst distortion. The critical role of the adamantyl groups is supported by results from a screen of a training set of Ti(salen) catalysts with variations of the salicylaldehyde portion to probe the steric influence of the ortho substituents and electronic tuning of the metal center by the para substituent. The data set revealed large ortho substituents (t-butyl (t-Bu) or adamantyl (Ad)) on the catalyst are required to access enantioenriched products. Specifically, ortho-t-Bu catalysts yielded products with moderate enantioselectivities and ortho-Ad catalysts formed products in excellent enantio- and diastereoselectivity regardless of the para substituent on the catalyst (see Experimental SI Section 4 for details). Notably, tuning the electronics of the catalyst through the para substituent had a minimal effect on selectivity. Attempts to further increase the enantioselectivity by improving the size of the ortho-substituents beyond Ad proved unfruitful. Overall, this clearly demonstrated that varying the ortho and para substituents on the salicylaldehyde are not effective approaches for improving the catalyst, and that alternative structural modifications to the catalyst (e.g., varying the diamine backbone) needed to be considered.
C. Catalyst Backbone SAR and Statistical Modeling.
After our studies of structural modifications on the salicylaldehyde moiety reinforced the critical role of the adamantyl groups, we next explored introducing substituents on the diamine backbone of Ti(salen) to create a more compact stereochemical environment for the cyclization transition state to induce a higher degree of catalyst distortion in the minor TS. We note that such modifications are largely underexplored in the area of asymmetric catalysis by metal-salen complexes, despite the fact that the structure of the diamine backbone has been shown to affect enantioselectivity in certain reaction systems.22
As a first experimental support for the stereochemical hypothesis, we subjected a modified catalyst to the reaction in which the phenyl groups on the diamine backbone were replaced with pentafluorophenyl groups ((S,S)-4aa) (Figure 6).23 This modification perturbs the steric and electronic environment of the active site. Indeed, reaction with this catalyst resulted in a significant reduction of diastereo- and enantioselectivity (2:1 dr, 77% ee) relative to the parent catalyst (S,S)-4aa (14:1 dr, 90% ee; Figure 6). In the original optimization of this reaction with styrene, it was also observed that both diastereo- and enantioselectivity diminished when the phenyl diamine backbone was replaced with a cyclohexane-1,2-diamine backbone (from >19:1 to 14:1 dr, 97 to 69% ee).4a These results provide support that structural variation of the catalyst diamine backbone can have significant implications for stereoselectivity outcomes. Given these initial explorations did not result in a more selective catalyst, we reasoned that more subtle changes to the catalyst diamine backbone may be necessary to effectively increase catalyst distortion and reduce favorable interactions in minor TS without significantly changing the reaction pathway.
Figure 6.

Modification of the chiral backbone substituents results in diminishing of stereoselectivity.24
Accordingly, we designed a series of eight Ti(salen) catalysts that incorporate different chiral diamine backbones ((S,S)-4a–h). This series of catalysts bear an ortho substituent on each of the aryl groups on the backbone, which based on the computational models are perfectly positioned into the chiral pocket where the intermediate assembly is bound. We first evaluated these catalysts in the reaction with 1 and p-chlorostyrene 2b and observed slightly increased enantioselectivities (from −90% to between −93% and −96% ee; Figure 7). This result suggests that the catalyst modification did not lead to a detrimental effect on enantioinduction. Nevertheless, the enantioselectivities are comparable across the series of catalysts. We propose that the general lack of sensitivity to these catalyst modifications for the styrene substrate is a consequence of similar influences on the major and minor TS 3s. Essentially, comparable differences in the interplay of catalyst distortion and interaction energy result in no additional differentiation in the relative energies of these two TSs, and subsequently little influence on enantioselectivity.
Figure 7.

Graphical representation of substrate structure-selectivity trends as a function of catalyst backbone B5 Sterimol value. Each trend line represents a different olefin substrate.
In contrast, when this set of eight new Ti(salen) catalysts were explored in reactions with four electron-deficient alkene reactants, all of which performed poorly with the original optimal (S,S)-4a catalyst (Figure 7), significant enhancements were observed in enantioselectivity. Increasing the backbone steric profile results in a more confined Ti active site, thus introducing a greater degree of catalyst distortion for dictating enantioselectivity outcomes with these substrates. Initial analysis of the enantioselectivity data of the new catalyst series revealed a trend between the B5 Sterimol value of the backbone arene substituent and enantioselectivity. The size of the backbone substituent was not simply related to their efficacy, as the most selective one was neither too large nor too small. In fact, a goldilocks effect was observed in which the optimal catalyst for all four alkenes possesses the medium-sized o-trifluoromethylphenyl backbone substituents. Although the specific catalyst trends vary for the alkenes, catalysts with aryl backbones smaller or larger than this group yield inferior enantioselectivity outcomes (Figure 7). This suggests that smaller backbones result in inadequate levels of catalyst distortion for the minor TS3.25 Yet, beyond a certain size, the steric interactions and subsequent distortion begin to impact both the minor and major TSs, resulting in lower levels of enantioselectivity. Catalyst (S,S)-4e provides the optimal backbone to enhance catalyst distortion in the minor TS3, but not the major TS3. The results with p-chlorostyrene 2b are also plotted, demonstrating the general lack of sensitivity to these structural modifications of the backbone substituent. Overall, these data provided a ΔΔG‡ range of 1.5 kcal/mol, which constitutes a sufficient statistical spread to employ multivariate linear regression (MLR) statistical modeling.26
MLR statistical modeling was then used to interrogate the catalyst and substrate effects specific to these alkenes. This approach relates molecular descriptor sets for the alkenes and catalysts to enantioselectivity outcomes.26 Because the inherent enantioselectivity of the new catalysts is masked by the styrene subset in which the ee variation is small, only the substrates exhibiting variable selectivity as a function of catalyst structure were further investigated. To appropriately capture the relevant substrate features in the selectivity-determining step, we acquired parameters such as NBO charges, spin densities, and SOMO energy computationally using truncated intermediate II with a surrogate TiCl3 in place of the full Ti(salen) catalyst. This was a simple yet crucial means of describing the molecular features most relevant to the enantio-determining step. Parameters such as Sterimol values and dihedral angles were collected from the optimized catalyst (S,S)-1a–h structures. Backbone specific parameters, such as polarizability, quadrupole moment, and molecular surface area, were obtained from the relevant arene structures and employed as additional catalyst descriptors for aiding model development. Using forward stepwise linear regression in MATLAB, a three-parameter statistical model was developed, consisting of two catalyst terms, namely, polarizability and B5 Sterimol value of the arene on the diamine backbone, and one intermediate parameter, namely, the NBO charge at the carbon-centered radical in intermediate II (Figure 8, plot I). Notably, the catalyst descriptors have larger coefficients in the model than the intermediate parameter, demonstrating the critical role of the catalyst backbone in enantioselectivity outcomes and implying significant catalyst control in this reaction. This is consistent with the proposal that catalyst distortion, as opposed to catalyst-intermediate interactions, is the primary factor in dictating enantioselectivity outcomes.
Figure 8.

MLR statistical models I and II developed with 8 catalyst X 4 substrate matrix (32 data points for training set, top). Both models suggest there is significant catalyst control in enantioselectivity outcomes. Predictions for novel substrates with improved catalyst (S,S)-1e (Ar = o-CF3Ph) are labeled. All other predictions are for product (R,R)-3h with the catalyst series.
Cross-validation and external validation in which the results of all eight catalysts with a vinyl pinacolboronate ester substrate (Figure 9, 2h) were predicted (see Figure 8 for predictions), suggesting the model is robust. Furthermore, the model effectively predicted the results of three structurally diverse substrates (Figure 9, 2i, 2j, 2k) with new catalyst (S,S)-4e. The prediction for the acrylonitrile-derived product ((R,R)-3i) is less accurate, which we attribute to its substantially smaller steric profile relative to the other substrates that are more effectively modeled. All attempts at predicting results for styrene-type substrates (e.g., 2b, 2d, 2m) with the model were unsuccessful and it was not possible to develop robust models when styrene substrates were included in the training set. This is consistent with our hypothesis that the factors controlling enantioselectivity differ in the styrenyl substrate class.
Figure 9.
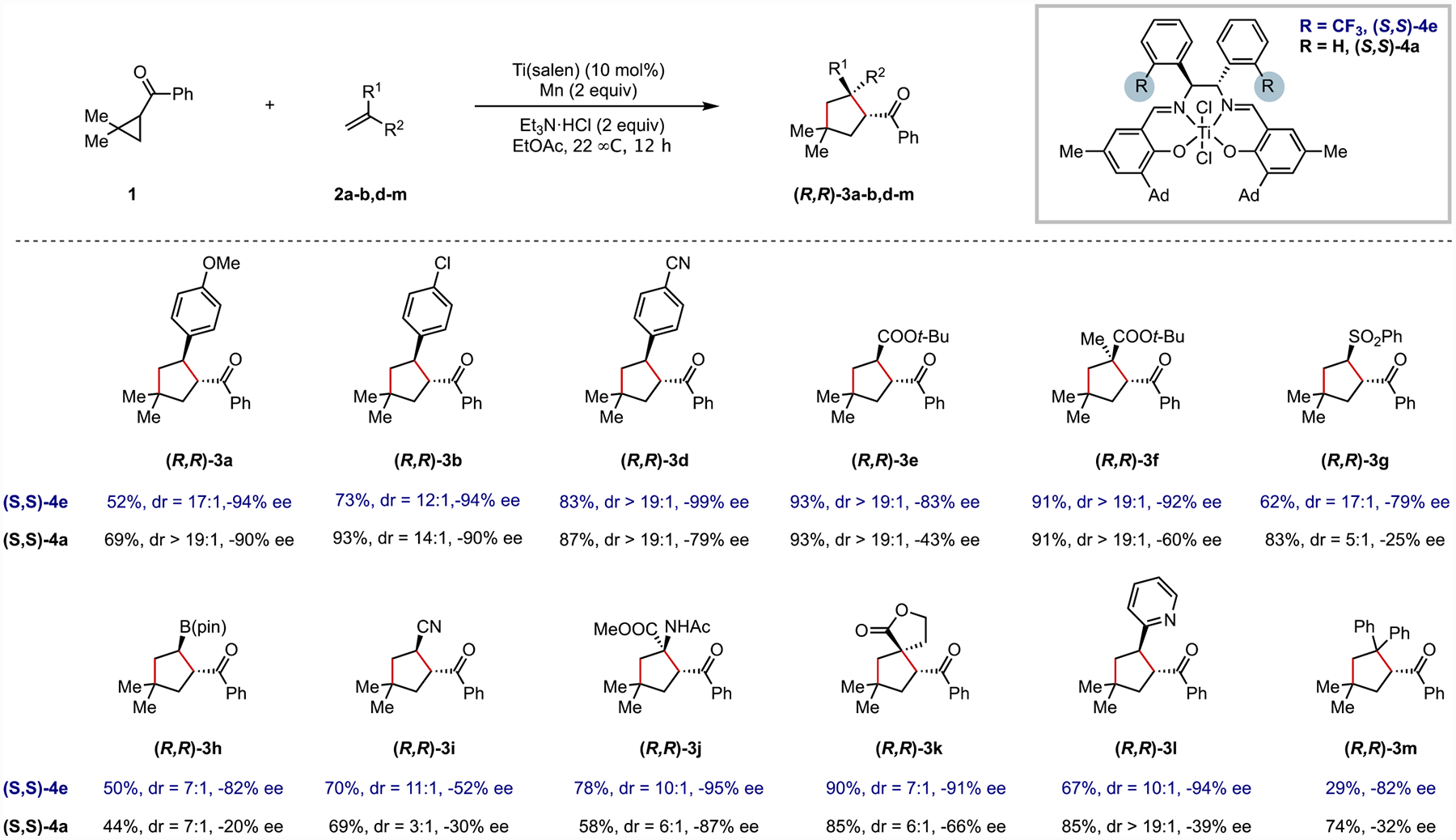
Improved performance of catalyst (S,S)-4e with previously challenging substrates.
The catalyst descriptors, polarizability and B5 Sterimol value of the aryl groups on the backbone, presumably capture the role of backbone substituents in influencing the degree of catalyst distortion (Figure 8). The two parameters are required to encapsulate the goldilocks effect of the backbone size. Intriguingly, a single parameter was sufficient at describing the diverse substrates. This is in part due to the substantial range in these NBO charge values (−0.55 up to 0.21). Yet, interpretation of this parameter in the enantio-determining TS was unclear. Thus, our efforts focused toward developing a mechanistically interpretable model.
A correlation matrix was generated for the parameters in order to identify more digestible parameters related to the NBOC2 parameter. Specifically, the spin density at the carbon centered radical (spinC2) and the Ti–O bond distance were determined as correlative with NBOC2 of the intermediate II structures (R2 = 0.65). Using these additional parameters, a four-parameter model was developed by manually altering the model to include these parameters in place of NBO (Figure 8, plot II). These two substrate parameters are interpreted as capturing the position of the TS along the reaction coordinate in the context of the Hammond postulate, in which more stabilized TSs represent more product-like TSs and result in higher levels of enantioselectivity. Although this model is more interpretable, it is less accurate in predicting out-of-sample compared to the three-parameter model. The value of these models is the ability to quickly, computationally assess how novel substrates will perform in this reaction without requiring exhaustive TS analysis.
Yet we remained curious how the new, improved catalyst provided such marked improvements in enantioselectivity. Thus, a final set of TS analysis was performed with the 2-vinylpyridine substrate 2l and the best catalyst (R,R)-4e. The lowest energy major TS3 pathway was calculated to be 5.0 kcal/mol lower in energy than the minor TS3 pathway (Figure 10). Although the energy difference is overestimated, the reproduction of experimental enantioselectivity trends, specifically that this new catalyst (S,S)-4e should promote the reaction with 2-vinylpyridine with higher levels of enantioselectivity than catalysts (S,S)-4a, demonstrates the strength of the computational analysis. Distortion/interaction analysis was performed with single-point energy calculations in gas phase with M06/6–31+G(d,p)-SDD level. This revealed that the distortion energy difference between the TS 3s was increased(6.0 kcal/mol), whereas the relative interaction energy difference was reduced (−1.6 kcal/mol). This suggests that the backbone modification was an effective avenue for both increasing catalyst distortion and reducing the stabilizing interaction energy in the minor TS3.
Figure 10.

TS analysis for reaction of catalyst (R,R)-4e with 2-vinylpyridine 2l as substrate (top). Distortion/interaction analysis for these TS 3s (bottom).
D. Substrate Scope Expansion Using New-Generation Ti(salen).
The catalyst modification informed by computational analysis led us to identify an improved Ti(salen) catalyst (S,S)-4e for the [3 + 2] cycloaddition reaction. Thus, the scope with the new Ti(salen) catalyst (S,S)-4e was substantially expanded to include various previously challenging, electron-deficient alkenes (Figure 9). Although the MLR statistical model and TS analysis suggests that the enantio-determining factors likely differ for styrene-type substrates from Michael acceptors, enantioselectivity was retained or improved for styrene-type substrates using the new catalyst ((R,R)-3a, (R,R)-3b, (R,R)-3d, and (R,R)-3m). In other words, the new catalyst constitutes a more general catalyst for a broader range of reaction partners without compromise to the original results, a particularly challenging feat in asymmetric catalysis.
With respect to electron-deficient alkenes, the precise improvement in diastereo- and enantioselectivity varied for each substrate, with the most significant improvement being for 2-vinylpyridine 2l, which constituted a 1.4 kcal/mol increase in differential free energy of activation. Notably, this improved Ti(salen) catalyst provides access to a diverse suite of synthetically useful products in high diastereo- and enantioselectivity. For example, proline analogues ((R,R)-3e–f) and a quaternary α-amino acid ((R,R)-3j) can be accessed from acrylates. Reactions with phenyl vinyl sulfone and vinyl pinacolboronate ester give rise to carbocycles with stereogenic C–S and C–B bonds (e.g., (R,R)-3g–h) that can be further transformed into other useful products. Finally, several products with quaternary stereogenic centers, including a spiro-bicyclic structure ((R,R)-3k), could be readily formed from the corresponding 1,1-disubstituted alkenes with excellent stereoselectivity.
CONCLUSIONS
The ability to control organic radical reactivity in a selective manner remains a synthetic challenge, but could provide access to unique useful structural scaffolds. In the Ti-catalyzed [3 + 2] cycloaddition reaction, through a suite of computational and experimental mechanistic studies, catalyst distortion was elucidated to dictate stereochemical outcomes. The mechanistic insights aided in our search for an improved catalyst, which substantially expanded the reaction scope and provided a collection of synthetically interesting products in high diastereo- and enantioselectivity. A catalyst–substrate matrix based on the new catalysts allowed for the development of an MLR statistical model that could predict the performance of each catalyst with a novel substrate. The predictive power of this model was demonstrated through accurate prediction of enantioselectivity outcomes for various substrates in reactions with the improved catalyst. This work demonstrates the utility of mechanistic studies in guiding catalyst optimization toward a more broadly applicable transformation. Furthermore, this development represents a rare example of metal-salen asymmetric catalysis in which the modification of the chiral diamine backbone was systematically studied to understand and improve reaction stereoselectivity. Given the broad use of metal-salen complexes in enantioselective synthesis, we anticipate that our discovery will have broader implications in transformations catalyzed by these privileged catalysts. Our future directions will focus on using the information gleaned from TS analysis to expand reactivity with different substrate classes.
Supplementary Material
ACKNOWLEDGMENTS
This work is dedicated to the occasion of Prof. Eric Jacobsen’s 60th birthday and the 30th anniversary of his initial publication on Mn(salen)-catalyzed asymmetric epoxidation (commonly known as the Jacobsen epoxidation; J. Am. Chem. Soc. 1990, 112, 2801-2803). Financial support is provided by the National Science Foundation (CHE-1763436 (M.S.S)) and the National Institute of Health (GM134088 (S.L.)). Computational resources were provided from the Center for High Performance Computing (CHPC) at the University of Utah and the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the National Science Foundation grant number ACI-1548562. SGR acknowledges the National Science Foundation for a graduate research fellowship. SGR thanks Dr. Jolene P. Reid for computational training and helpful discussions in regard to MLR modeling and TS analysis.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c07128.
Experimental procedures, and characterization data (PDF)
Computational details (PDF)
The authors declare no competing financial interest.
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.0c07128
Contributor Information
Sophia G. Robinson, Department of Chemistry, University of Utah, Salt Lake City, Utah 84112, United States.
Xiangyu Wu, Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, United States;.
Binyang Jiang, Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, United States.
Matthew S. Sigman, Department of Chemistry, University of Utah, Salt Lake City, Utah 84112, United States;.
Song Lin, Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, United States;.
REFERENCES
- (1).(a) Sibi MP; Porter NA Enantioselective Free Radical Reactions. Acc. Chem. Res 1999, 32, 163–171. [Google Scholar]; (b) Gansäuer A; Bluhm H Reagent-Controlled Transition-Metal-Catalyzed Radical Reactions. Chem. Rev 2000, 100, 2771–2788. [DOI] [PubMed] [Google Scholar]; (c) Bar G; Parsons AF Stereoselective Radical Reactions. Chem. Soc. Rev 2003, 32, 251–263. [DOI] [PubMed] [Google Scholar]; (d) Wang K; Kong W Recent Advances in Transition Metal-Catalyzed Asymmetric Radical Reactions. Chin. J. Chem 2018, 36, 247–256. [Google Scholar]; (e) Uraguchi D; Ohmatsu K; Ooi T Organic Molecular Catalysts in Radical Chemistry: Challenges Toward Selective Transformations. Molecular Technology; Wiley–VCH: Weinheim, Germany, 2019; Vol. 4, pp 163–197. [Google Scholar]
- (2).For recent examples, see the following:; (a) Streuff J; Feurer M; Bichovski P; Frey G; Gellrich G Enantioselective Titanium(III)-Catalyzed Reductive Cyclization of Ketonitriles. Angew. Chem., Int. Ed 2012, 51, 8661–8664. [DOI] [PubMed] [Google Scholar]; (b) Xu X; Zhu S; Cui X; Wojtas L; Zhang XP Cobalt (II)-catalyzed asymmetric olefin cyclopropanation with c-ketodiazoacetates. Angew. Chem., Int. Ed 2013, 52, 11857–11861. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Huo H; Shen X; Wang C; Zhang L; Röse P; Chen LA; Harms K; Marsch M; Hilt G; Meggers E Asymmetric photoredox transition-metal catalysis activated by visible light. Nature 2014, 515, 100–103. [DOI] [PubMed] [Google Scholar]; (d) Zhu R; Buchwald SL Versatile enantioselective synthesis of functionalized lactones via copper-catalyzed radical oxyfunctionalization of alkenes. J. Am. Chem. Soc 2015, 137, 8069–8077. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Zhang W; Wang F; McCann SD; Wang D; Chen P; Stahl SS; Liu G Enantioselective cyanation of benzylic C–H bonds via copper-catalyzed radical relay. Science 2016, 353, 1014–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Wang Z; Yin H; Fu GC Catalytic enantioconvergent coupling of secondary and tertiary electrophiles with olefins. Nature 2018, 563, 379–383. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Song L; Fu N; Ernst BG; Lee WH; Frederick MO; DiStasio RA Jr; Lin S Dual Electrocatalysis Enables Enantioselective Hydrocyanation of Conjugated Alkenes. Nat. Chem 2020, 12, 747–754. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Shin NY; Ryss JM; Zhang X; Miller SJ; Knowles RR Light-driven deracemization enabled by excited-state electron transfer. Science 2019, 366, 364–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).For reviews on stereocontrol principles in radical reactions, see the following:; (a) Kamigaito M; Satoh K Stereoregulation in living radical polymerization. Macromolecules 2008, 41, 269–276. [Google Scholar]; (b) Liu L; Ward RM; Schomaker JM Mechanistic Aspects and Synthetic Applications of Radical Additions to Allenes. Chem. Rev 2019, 119, 12422–12490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).(a) Hao W; Harenberg JH; Wu X; MacMillan SN; Lin S Diastereo- and enantioselective formal [3 + 2] cycloaddition of cyclopropyl ketones and alkenes via Ti-catalyzed radical redox relay. J. Am. Chem. Soc 2018, 140, 3514–3517. [DOI] [PubMed] [Google Scholar]; (b) McCallum T; Wu X; Lin S Recent Advances in Titanium Radical Redox Catalysis. J. Org. Chem 2019, 84, 14369–14380. [DOI] [PubMed] [Google Scholar]
- (5).(a) Hudlicky T; Price JD Anionic approaches to the construction of cyclopentanoids. Chem. Rev 1989, 89, 1467–1486. [Google Scholar]; (b) Heasley B Stereocontrolled preparation of fully substituted cyclopentanes: relevance to total synthesis. Eur. J. Org. Chem 2009, 2009, 1477–1489. [Google Scholar]; (c) Barrero A; Quilez del Moral J; Herrador M; Rodriguez H; Perez Morales M Cyclopentane Sesquiterpenes from Fungi: Ocurrence-Bioactivity, Biosynthesis and Chemical Synthesis. Curr. Org. Chem 2009, 13, 1164–1181. [Google Scholar]
- (6).(a) Defauw JM; Murphy MM; Jagdmann E Jr.; Hu H; Lampe JW; Hollinshead SP; Mitchell TJ; Crane HM; Heerding JN; Mendoza JS; Davis JE; Darges JW; Hubbard FR; Hall SE Synthesis and Protein Kinase C Inhibitory Activities of Acyclic Balanol Analogs That Are Highly Selective for Protein Kinase C over Protein Kinase A. J. Med. Chem 1996, 39, 5215–5227. [DOI] [PubMed] [Google Scholar]; (b) Muegge I; Podlogar BL 3D-Quantitative Structure Activity Relationships of Biphenyl Carboxylic Acid MMP-3 Inhibitors: Exploring Automated Docking as Alignment Method. Quant. Struct.-Act. Relat 2001, 20, 215–222. [Google Scholar]; (c) Stoll V; Stewart KD; Maring CJ; Muchmore S; Giranda V; Gu YY; Wang G; Chen Y; Sun M; Zhao C; Kennedy AL; Madigan DL; Xu Y; Saldivar A; Kati W; Laver G; Sowin T; Sham HL; Greer J; Kempf D Influenza Neuraminidase Inhibitors: Structure-Based Design of a Novel Inhibitor Series. Biochemistry 2003, 42, 718–727. [DOI] [PubMed] [Google Scholar]; (d) Raha K; Merz KM Large-Scale Validation of a Quantum Mechanics Based Scoring Function: Predicting the Binding Affinity and the Binding Mode of a Diverse Set of Protein-Ligand Complexes. J. Med. Chem 2005, 48, 4558–4575. [DOI] [PubMed] [Google Scholar]; (e) Hanessian S; Yun H; Hou Y; Yang G; Bayrakdarian M; Therrien E; Moitessier N; Roggo S; Veenstra S; Tintelnot-Blomley M; Rondeau J-M; Ostermeier C; Strauss A; Ramage P; Paganetti P; Neumann U; Betschart C Structure-Based Design, Synthesis, and Memapsin 2 (BACE) Inhibitory Activity of Carbocyclic and Heterocyclic Peptidomimetics. J. Med. Chem 2005, 48, 5175–5190. [DOI] [PubMed] [Google Scholar]
- (7).(a) Cesarotti E; Kagan HB; Goddard R; Krüger C Synthesis of New Ligands for Transition Metal Complexes: Menthyland Neomenthyl-Cyclopentadienes. J. Organomet. Chem 1978, 162, 297–309. [Google Scholar]; (b) Wild FRWP; Zsolnai L; Huttner G; Brintzinger HH Ansa-Metallocene Derivatives IV. Synthesis and Molecular Structures of Chiral Ansa-Titanocene Derivatives with Bridged Tetrahydroindenyl Ligands. J. Organomet. Chem 1982, 232, 233–247. [Google Scholar]; (c) Ye B; Cramer N A Tunable Class of Chiral Cp Ligands for Enantioselective Rhodium(III)-Catalyzed C-H Allylations of Benzamides. J. Am. Chem. Soc 2013, 135, 636–639. [DOI] [PubMed] [Google Scholar]
- (8).For representative examples of stereoselective transformation of organoboron compounds, see the following:; (a) Matteson DS; Man HW; Ho OC Asymmetric Synthesis of Stegobinone via Boronic Ester Chemistry. J. Am. Chem. Soc 1996, 118, 4560–4566. [Google Scholar]; (b) Imao D; Glasspoole BW; Laberge VS; Crudden CM Cross-Coupling Reactions of Chiral Secondary Organoboronic Esters with Retention of Configuration. J. Am. Chem. Soc 2009, 131, 5024–5025. [DOI] [PubMed] [Google Scholar]; (c) Ohmura T; Awano T; Suginome M Stereospecific Suzuki-Miyaura Coupling of Chiral α-(Acylamino)Benzylboronic Esters with Inversion of Configuration. J. Am. Chem. Soc 2010, 132, 13191–13193. [DOI] [PubMed] [Google Scholar]; (d) Nave S; Sonawane RP; Elford TG; Aggarwal VK Protodeboronation of Tertiary Boronic Esters: Asymmetric Synthesis of Tertiary Alkyl Stereogenic Centers. J. Am. Chem. Soc 2010, 132, 17096–17098. [DOI] [PubMed] [Google Scholar]; (e) Mlynarski SN; Karns AS; Morken JP Direct Stereospecific Amination of Alkyl and Aryl Pinacol Boronates. J. Am. Chem. Soc 2012, 134, 16449–16451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).For reviews, see the following:; (a) Larrow JF; Jacobsen EN Asymmetric Processes Catalyzed by Chiral (Salen)Metal Complexes. In Organometallics in Process Chemistry; Springer: Berlin, 2004; pp 123–152. [Google Scholar]; (b) Katsuki T Catalytic Asymmetric Oxidations Using Optically Active (Salen)Manganese(III) Complexes as Catalysts. Coord. Chem. Rev 1995, 140, 189–214. [Google Scholar]; (c) Canali L; Sherrington DC Utilisation of Homogeneous and Supported Chiral Metal (Salen) Complexes in Asymmetric Catalysis. Chem. Soc. Rev 1999, 28, 85–93. [Google Scholar]; (d) Jacobsen EN Asymmetric Catalysis of Epoxide Ring-Opening Reactions. Acc. Chem. Res 2000, 33, 421–431. [DOI] [PubMed] [Google Scholar]; (e) Katsuki T Chiral Metallosalen Complexes: Structures and Catalyst Tuning for Asymmetric Epoxidation and Cyclopropanation. Adv. Synth. Catal 2002, 344, 131–147. [Google Scholar]; (f) Bandini M; Cozzi PG; Umani-Ronchi A [Cr (Salen)] as a ‘Bridge’ between Asymmetric Catalysis, Lewis Acids and Redox Processes. Chem. Commun 2002, 9, 919–927. [DOI] [PubMed] [Google Scholar]; (g) Katsuki T Some Recent Advances in Metallosalen Chemistry. Synlett 2003, 3, 281–291. [Google Scholar]; (h) Cozzi PG Metal – Salen Schiff Base Complexes in Catalysis: Practical Aspects. Chem. Soc. Rev 2004, 33, 410–421. [DOI] [PubMed] [Google Scholar]; (i) McGarrigle EM; Gilheany DG Chromium- and Manganesesalen Promoted Epoxidation of Alkenes. Chem. Rev 2005, 105, 1563–1602. [DOI] [PubMed] [Google Scholar]; (j) Achard TRJ; Clutterbuck LA; North M Asymmetric Catalysis of Carbon–Carbon Bond-Forming Reactions Using Metal (Salen) Complexes. Synlett 2005, 12, 1828–1847. [Google Scholar]; (k) Baleizao C; Garcia H Chiral Salen Complexes: An Overview to Recoverable and Reusable Homogeneous and Heterogeneous Catalysts. Chem. Rev 2006, 106, 3987–4043. [DOI] [PubMed] [Google Scholar]; (l) Matsumoto K; Saito B; Katsuki T Asymmetric Catalysis of Metal Complexes with Non-Planar ONNO Ligands: Salen, Salalen and Salan. Chem. Commun 2007, 35, 3619–3627. [DOI] [PubMed] [Google Scholar]
- (10).For reviews, see the following:; (a) Darensbourg DJ; Mackiewicz RM; Phelps AL; Billodeaux DR Copolymerization of CO2 and Epoxides Catalyzed by Metal Salen Complexes. Acc. Chem. Res 2004, 37, 836–844. [DOI] [PubMed] [Google Scholar]; (b) Coates GW; Moore DR Discrete Metal-Based Catalysts for the Copolymerization of CO2 and Epoxides: Discovery, Reactivity, Optimization, and Mechanism. Angew. Chem., Int. Ed 2004, 43, 6618–6639. [DOI] [PubMed] [Google Scholar]; (c) Darensbourg DJ Making Plastics from Carbon Dioxide: Salen Metal Complexes as Catalysts for the Production of Polycarbonates from Epoxides and CO2. Chem. Rev 2007, 107, 2388–2410. [DOI] [PubMed] [Google Scholar]; (d) Osten KM; Mehrkhodavandi P Indium Catalysts for Ring Opening Polymerization: Exploring the Importance of Catalyst Aggregation. Acc. Chem. Res 2017, 50, 2861–2869. [DOI] [PubMed] [Google Scholar]
- (11).For representative examples, see the following:; (a) Kowalczyk R; Sidorowicz Ł; Skarzewski J Asymmetric Nitroaldol Reaction Catalyzed By a Chromium(III)–Salen System. Tetrahedron: Asymmetry 2007, 18, 2581–2586. [Google Scholar]; (b) Sibi MP; Nad S Enantioselective Radical Reactions: Stereoselective Aldol Synthesis from Cyclic Ketones. Angew. Chem., Int. Ed 2007, 46, 9231–9234. [DOI] [PubMed] [Google Scholar]; (c) Mazet C; Jacobsen EN Dinuclear {(Salen)Al} Complexes Display Expanded Scope in the Conjugate Cyanation of α,β-Unsaturated Imides. Angew. Chem., Int. Ed 2008, 47, 1762–1765. [DOI] [PubMed] [Google Scholar]; (d) Zeng X; Cao Z; Wang X; Chen L; Zhou F; Zhu F; Wang C; Zhou J Activation of Chiral (Salen)AlCl Complex by Phosphorane for Highly Enantioselective Cyanosilylation of Ketones and Enones. J. Am. Chem. Soc 2016, 138, 416–425. [DOI] [PubMed] [Google Scholar]
- (12).For representative examples, see the following:; (a) Palucki M; Finney NS; Pospisil PJ; Güler ML; Ishida T; Jacobsen EN The Mechanistic Basis for Electronic Effects on Enantioselectivity in the (salen)Mn(III)-Catalyzed Epoxidation Reaction. J. Am. Chem. Soc 1998, 120, 948–954. [Google Scholar]; (b) Bryliakov KP; Talsi EP Iron-Catalyzed Oxidation of Thioethers by Iodosylarenes: Stereoselectivity and Reaction Mechanism. Chem. - Eur. J 2007, 13, 8045–8050. [DOI] [PubMed] [Google Scholar]; (c) Kull T; Peters R Contact Ion Pair Directed Lewis Acid Catalysis: Asymmetric Synthesis of trans-Configured –Lactones. Angew. Chem., Int. Ed 2008, 47, 5461–5464. [DOI] [PubMed] [Google Scholar]; (d) Park J; Lang K; Abboud KA; Hong S Self-Assembled Dinuclear Cobalt (II)-Salen Catalyst Through Hydrogen-Bonding and Its Application to Enantioselective Nitro-Aldol (Henry) Reaction. J. Am. Chem. Soc 2008, 130, 16484–16485. [DOI] [PubMed] [Google Scholar]; (e) Kowalczyk R; Kwiatkowski P; Skarzewski J; Jurczak J Enantioselective Nitroaldol Reaction Catalyzed by Sterically Modified Salen - Chromium Complexes. J. Org. Chem 2009, 74, 753–756. [DOI] [PubMed] [Google Scholar]; (f) Koya S; Nishioka Y; Mizoguchi H; Uchida T; Katsuki T Asymmetric Epoxidation of Conjugated Olefins with Dioxygen. Angew. Chem., Int. Ed 2012, 51, 8243–8246. [DOI] [PubMed] [Google Scholar]; (g) Hutson GE; Turkmen YE; Rawal VH Salen Promoted Enantioselective Nazarov Cyclizations of Activated and Unactivated Dienones. J. Am. Chem. Soc 2013, 135, 4988–4991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).For representative examples see the following:; (a) Belokon YN; Green B; Ikonnikov NS; Larichev VS; Lokshin BV; Moscalenko MA; North M; Orizu C; Peregudov AS; Timofeeva GI Mechanistic Investigation of the Asymmetric Addition of Trimethylsilyl Cyanide to Aldehydes Catalysed by Dinuclear Chiral (Salen) Titanium Complexes. Eur. J. Org. Chem 2000, 2000, 2655–2661. [Google Scholar]; (b) Matsumoto K; Sawada Y; Saito B; Sakai K; Katsuki T Construction of Pseudo-Heterochiral and Homochiral Di-m-Oxotitanium (Schiff Base) Dimers and Enantioselective Epoxidation Using Aqueous Hydrogen Peroxide. Angew. Chem., Int. Ed 2005, 44, 4935–4939. [DOI] [PubMed] [Google Scholar]; (c) Xu Z-J; Fang R; Zhao C; Huang J-S; Li G-Y; Zhu N; Che C-M Cis-X-Bis (Carbonyl) Ruthenium-Salen Complexes: X-Ray Crystal Structures and Remarkable Catalytic Properties toward Asymmetric Intramolecular Alkene Cyclopropanation. J. Am. Chem. Soc 2009, 131, 4405–4417. [DOI] [PubMed] [Google Scholar]; (d) North M; Watson JM Asymmetric Addition of Cyanide to b -Nitroalkenes Catalysed by Chiral Salen Complexes of Titanium-(IV) and Vanadium(V). ChemCatChem 2013, 5, 2405–2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).(a) Annis DA; Jacobsen EN Polymer-Supported Chiral Co(Salen) Complexes Mechanistic Investigations in the Hydrolytic Kinetic Resolution of Terminal Epoxides. J. Am. Chem. Soc 1999, 121, 4147–4154. [Google Scholar]; (b) Nielsen LPC; Stevenson CP; Blackmond DG; Jacobsen EN Mechanistic Investigation Leads to a Synthetic Improvement in the Hydrolytic Kinetic Resolution of Terminal Epoxides. J. Am. Chem. Soc 2004, 126, 1360–1362. [DOI] [PubMed] [Google Scholar]; (c) Nielsen LPC; Zuend SJ; Ford DD; Jacobsen EN Mechanistic Basis for High Reactivity of (Salen)Co-OTs in the Hydrolytic Kinetic Resolution of Terminal Epoxides. J. Org. Chem 2012, 77, 2486–2495. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ford DD; Nielsen LPC; Zuend SJ; Musgrave CB; Jacobsen EN Mechanistic Basis for High Stereoselectivity and Broad Substrate Scope in the (Salen)Co(III)-Catalyzed Hydrolytic Kinetic Resolution. J. Am. Chem. Soc 2013, 135, 15595–15608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).(a) Hansen KB; Leighton JL; Jacobsen EN On the Mechanism of Asymmetric Nucleophilic Ring-Opening of Epoxides Catalyzed by (Salen)CrIII Complexes. J. Am. Chem. Soc 1996, 118, 10924–10925. [Google Scholar]; (b) Konsler RG; Karl J; Jacobsen EN Cooperative Asymmetric Catalysis with Dimeric Salen Complexes. J. Am. Chem. Soc 1998, 120, 10780–10781. [Google Scholar]
- (16).Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Mennucci B; Petersson GA; Nakatsuji H; Caricato M; Li X; Hratchian HP; Izmaylov AF; Bloino J; Zheng G; Sonnenberg JL; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark M; Heyd JJ; Brothers E; Kudin KN; Staroverov VN; Keith T; Kobayashi R; Normand J; Raghavachari K; Rendell A; Burant JC; Iyengar SS; Tomasi J; Cossi M; Rega N; Millam JM; Klene M; Knox JE; Cross JB; Bakken V; Adamo C; Jaramillo J; Gomperts R; Stratmann RE; Yazyev O; Austin AJ; Cammi R; Pomelli C; Ochterski JW; Martin RL; Morokuma K; Zakrzewski VG; Voth GA; Salvador P; Dannenberg JJ; Dapprich S; Daniels AD; Farkas O; Foresman JB; Ortiz JV; Cioslowski J; Fox DJ Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, 2013. [Google Scholar]
- (17). Because the selectivity-determining step occurs after the RDS, it would be difficult to experimentally study it with traditional physical organic experimental tools.
- (18).(a) Champagne PA; Houk KN Origins of Selectivity and General Model for Chiral Phosphoric Acid-Catalyzed Oxetane Desymmetrizations. J. Am. Chem. Soc 2016, 138, 12356–12359. [DOI] [PubMed] [Google Scholar]; (b) Bickelhaupt FM; Houk KN Analyzing Reaction Rates with the Distortion/Interaction-Activation Strain Model. Angew. Chem., Int. Ed 2017, 56, 10070–10086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19). A variety of levels of theory were employed for identifying the optimal method for single-point energy calculations (details tabulated in the Supporting Information). Electronic energies consistently underestimate the barrier for enantioselectivity and overestimate the energy barriers for diastereoselectivity regardless of the level of theory or inclusion of solvation model. Free energies for the TSs were all in reasonable agreement with experimental values, but this distortion/interaction analysis is performed using electronic energies because of the calculation of the separated distorted reactants. Thus, while the calculated dE values are not as accurate as the computed dG, the general trends for the substrate-catalyst combinations are analyzed to provide insight into this system, rather than focusing on the exact values.
- (20).(a) Contreras-Garcia J; Johnson E; Keinan S; Chaudret R; Piquemal J-P; Beratan D; Yang W NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. J. Chem. Theory Comput 2011, 7, 625–632. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Johnson ER; Keinan S; Mori-Sanchez P; Contreras-Garcia J; Cohen AJ; Yang W Revealing Noncovlanet Interactions. J. Am. Chem. Soc 2010, 132, 6498–6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).(a) Hashihayata T; Punniyamurthy T; Irie R; Katsuki T; Akita M; Moro-Oka Y Conformational Analysis of Cationic (R, S)-and (R, R)-(Salen)manganese Complexes Possessing Axial Chirality as a Chiral Element Based on X-ray Crystallography: An Explanation of the Effect of Apical Ligand on Enantioselection by (salen) manganese Catalyst. Tetrahedron 1999, 55, 14599–14610. [Google Scholar]; (b) Nishikori H; Ohta C; Oberlin E; Irie R; Katsuki T Mn-Salen Catalyzed Nitrene Transfer Reaction: Enantioselective Imidation of Alkyl Aryl Sulfides. Tetrahedron 1999, 55, 13937–13946. [Google Scholar]
- (22).(a) Shaw S; White JD Asymmetric Catalysis Using Chiral Salen-Metal Complexes: Recent Advances. Chem. Rev 2019, 119, 9381–9426. [DOI] [PubMed] [Google Scholar]; (b) Discolo CA; Touney EE; Pronin SV Catalytic Asymmetric Radical-Polar Crossover Hydroalkoxylation. J. Am. Chem. Soc 2019, 141, 17527–17532. [DOI] [PubMed] [Google Scholar]
- (23).(a) Wheeler SE; Houk KN Substituent Effects in the Benzene Dimer are Due to Direct Interactions of the Substituents with the Unsubstituted Benzene. J. Am. Chem. Soc 2008, 130, 10854–10855. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cockroft SL; Hunter CA; Lawson KR; Perkins J; Urch CJ Electrostatic Control of Aromatic Stacking Interactions. J. Am. Chem. Soc 2005, 127, 8594–8595. [DOI] [PubMed] [Google Scholar]
- (24). Please note that the catalysts in the original method report (J. Am. Chem. Soc. 2018, 140, 3514–3517) possess (R, R) diamine backbones, whereas catalysts with modified diamine backbones in this study were prepared with (S, S) diamine backbones. Thus, the (S, S) products formed with the (R, R) catalyst have positive (+) % ee, whereas performing the reaction with the (S, S) catalysts yields (R, R) products reported with negative (−) % ee values. All TS analysis was performed with (R, R) catalysts.
- (25). Note that for the training set series the catalyst all have the (S, S) backbone, whereas the original report of this method and all TS analysis employed the (R, R) backbone. Thus, for these reactions the major TS3 is trans-Si and the minor TS3 is trans-Re.
- (26).For recent reviews of MLR statistical modeling see the following:; (a) Reid JP; Sigman MS Comparing Quantitative Prediction Methods for the Discovery of Small-Molecule Chiral Catalysts. Nat. Rev. Chem 2018, 2, 290–305. [Google Scholar]; (b) Santiago CB; Guo J-Y; Sigman MS Predictive and Mechanistic Multivariate Linear Regression Models for Reaction Development. Chem. Sci 2018, 9, 2398–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Sigman MS; Harper KC; Bess EN; Milo A The Development of Multidimensional Analysis Tools for Asymmetric Catalysis and Beyond. Acc. Chem. Res 2016, 49, 1292–1301. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.