

Abstract
In vitro studies with keloid fibroblasts frequently present contradictory results. This may occur because keloids present distinct genotypic and phenotypic characteristics in its different regions, such as the peripheral region in relation to the central region. We suggest an explant model for keloid fibroblasts harvesting, standardising the initial processing of keloid samples to obtain fragments from different regions, considering its biological differences, for primary cell culture. The different keloid regions were delimited and fragments were obtained using a 3‐mm diameter punch. To remove fragments from the periphery, the punch was placed in one longitudinal line extremity, respecting the lesion borders. For the central region, it was placed in the intersection of lines at the level of the largest longitudinal and transversal axes, the other fragments being removed centrifugally in relation to the first one. Primary fibroblast culture was carried out by explant. Flow cytometry analysis showed cell cycle differences between the groups, confirming its different origins and biological characteristics. In conclusion, our proposed model proved itself efficient for keloid fibroblast isolation from specific regions and cultivation. Its simplicity and ease of execution may turn it into an important tool for studying the characteristics of the different keloid‐derived fibroblasts in culture.
Keywords: Cell culture techniques, cultured cells, fibroblasts, keloid, in vitro
INTRODUCTION
Cell culture is an important tool for the study of cellular physiology without interference of local or systemic factors present in the organism. It allows detecting the mechanism of action of cellular regulators at gene expression and cell signalling levels 1, 2.
Conventional cell culture provides a reductionist view of cells in a bi‐dimensional arrangement in contrast to their normal multi‐cellular, three‐dimensional environment. This can be considered advantageous as it provides defined experimental parameters to investigate the phenotypic consequences of genetic alterations. In contrast to whole organisms, each particular cell line constitutes a phenotypically and genetically uniform population of individual cells derived from one tissue (2). On the other hand, when cells are cultured in vitro, several variables may affect the cellular phenotype, for example, contamination, confluence degree, cell–cell adhesion and seeding density (3).
When considering keloids, a benign fibroproliferative cicatricial neoplasia with unknown physiopatogenesis 4, 5, 6, and potential for autonomous growth and in vitro development, even in the absence of humoral factors 7, 8, 9, the use of in vitro models to understand their formation mechanisms becomes essential (10). The importance of such models is corroborated by the fact that keloids occur exclusively in humans 11, 12 which makes research with animals very difficult 13, 14. Recently, Butler et al. (15) developed an in vitro organotypic skin model to simulate keloid biology that can serve as a surrogate to study keloid formation without an animal model.
Clinical observations showed that different keloid regions exhibit different growth characteristics. Central regions are shrunken and soft in texture and have been generally termed by Ladin et al. (16) the ‘older parts of the keloid’.
Alterations of apoptosis and cell proliferation have been implicated in keloid aetiology. Appleton et al. (17) observed a peculiar compartmentalisation of cell apoptosis, proliferation and necrosis in keloid tissue with scanty proliferating cells in the central area of keloid and the apoptotic phenomenon more evident in the peripheral areas, thus hypothesising maturation of keloids through this pathway of cellular clearance. Ladin et al. (16) investigated p53 and bcl‐2, which referred both positivity in keloids in the hypercellular peripheral lesional areas. An inverse distribution of fas expression was showed with staining being limited to the central, more hypocellular regions. This reversed phenotype in the older areas of the keloid may prevent malignant degeneration, thus favouring normal apoptosis as evidenced by prominent fas expression (16).
In vitro studies with keloid fibroblasts frequently present contradictory results 16, 18, 19, 20, 21, 22. This may be because of the fact that keloids present distinct genotypic 18, 23 and phenotypic 22, 24 characteristics in different regions of the lesion itself, such as the peripheral region in relation to the central one, or the superficial portion in relation to the basal region, and also, if they were in clinical activity (growth, hyperaemia, pruritus and/or pain) at the moment of sample collection 10, 22.
Ladin et al. also investigated keloid fibroblasts in culture and showed that cultured keloid fibroblasts between passages 3 and 6 were consistently p53+, bcl‐2+, whereas normal human and neonatal foreskin fibroblasts were consistently p53−, bcl‐2−. However, they did not specify the region of the keloid from which fibroblasts were derived, even though their work strongly suggests that they derived from the peripheral area (16).
Luo et al. isolated and cultivated fibroblasts from the superficial, central and basal regions of keloid lesions. They examined the growth behaviour of each fibroblast fraction in short‐term and long‐term cultures and calculated the percentage of apoptotic cells. Fibroblasts obtained from the superficial and basal regions of keloid tissue showed population doubling times and saturation densities similar to normal fibroblasts. In contrast, central keloid fibroblasts showed reduced doubling times and reached higher cell densities. In long‐term culture, central keloid fibroblasts formed a stratified three‐dimensional structure, contracted the self‐produced extracellular matrix and gave rise to nodular cell aggregates, mimicking the formation of keloid tissue (25).
Giugliano et al. showed that fibroblasts derived from the central part of keloid lesions grow faster than peripheral and non keloid fibroblasts and, in long‐term cultures, became stratified assuming a three‐dimensional structure. Compared with peripheral and non keloid fibroblasts, central keloid fibroblasts presented an increased production of both interleukin‐6 and vascular endothelial growth factor (26).
Lu et al. showed that cultured fibroblasts derived from both central and peripheral parts of keloids displayed significant resistance to Fas‐mediated apoptosis. Also, their analysis of cell cycle distribution indicated that the majority of fibroblasts derived from peripheral parts of keloids were in proliferative periods of the cell cycle (G2‐S phase), whereas the majority of fibroblasts derived from keloid centres were in G0‐G1 phase. Fas and Bcl‐2 expression did not differ significantly between the groups, but p53 expression was much higher in fibroblasts derived from central parts. These findings suggested that differences in cell cycle distribution and p53 protein expression may account for the different growth characteristics of keloid peripheries and centres (22).
Taken together, these studies illustrate the importance of specifying which part of the keloid is being used, so as to evaluate its clinical status. Some discrepancies found in cell culture studies involving keloid‐derived fibroblasts 27, 28, 29, 30, 31, 32 may be explained by this lack of information concerning the origin and clinical status of the keloid cells used.
Thus, to reduce biases in studies involving keloid fibroblasts culture, it is imperative to standardise the collection of these fibroblasts, and researchers should report the details of the collection method used, the region of the keloid from which cultured fibroblasts were derived and also if keloids were in clinical activity at the moment of collection. The present study suggests an explant model for keloid fibroblasts culture, with the standardisation of the initial process of keloid samples to obtain fragments from different regions, taking into consideration its biological differences, in order to perform primary culture of keloid fibroblasts.
METHODS
Collection of keloid fragments to obtain fibroblasts
This method applies to planar, non peduncular keloids. Fresh keloids obtained at the time of surgical excision were used following informed consent and with approval from Universidade Federal de Sao Paulo's Ethical Committee.
Four non Caucasian female patients from the Plastic Surgery Division of the Universidade Federal de Sao Paulo, aged 18–36 years who had a planar, non peduncular keloid on the trunk of at least 1‐year evolution, in clinical activity (presenting one or more of the following characteristics: growth, hyperaemia, pruritus and/or pain), were surgically treated. The exclusion criteria were, briefly: keloids previously treated; patients with chronic dermatopathies, metabolic, collagen or degenerative/auto‐immune diseases; malignant neoplasms or patients submitted to systemic or topic treatment with corticosteroids. For our research purposes, the keloids measured at least 3 × 2 cm at the longitudinal and transversal axes, respectively. Keloids were excised in monobloc, in subcutaneous plane by fusiform peri‐keloidean incision, including a skin fragment in the extremities, which corresponds to the cutaneous exceeding tissue necessary for an adequate suture coaptation (Figure 1).
Figure 1.
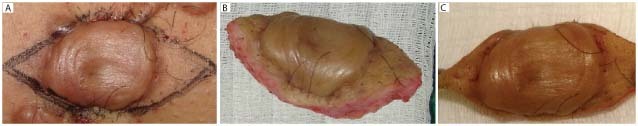
Keloid excision in monobloc by fusiform peri‐keloidean incision including a skin fragment in the extremities.
Keloid fragments used for primary fibroblast culture were obtained from the excised specimens by a circular punch of 3 mm diameter and 10 mm depth (Figure 2). Keloid adjacent skin fragments, used here for comparison purposes (control group), were obtained from the most distant point in relation to the keloid border, maintaining a minimum distance of 5 mm from the border (Figure 2A).
Figure 2.
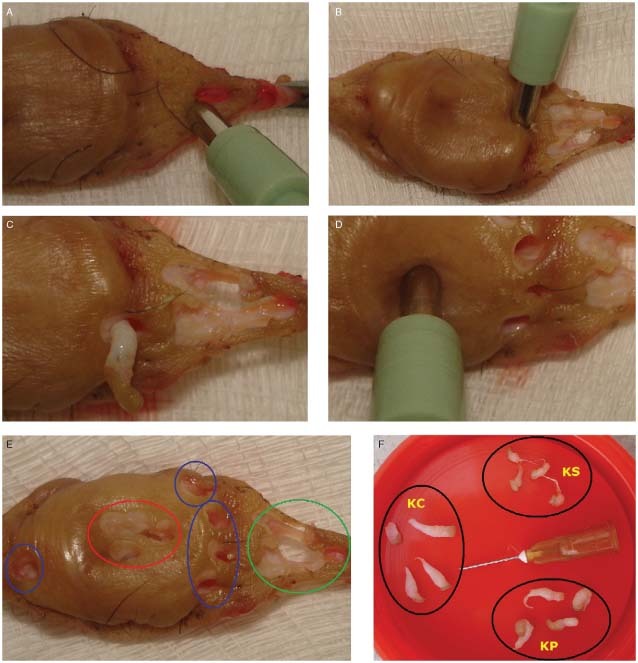
Removal of keloid fragments from its different regions using a 3‐mm circular punch, 100 mm depth. (A) Removal of fragment from the keloid adjacent skin. (B) Removal of the first fragment from the peripheral region, the punch being placed in one of the longitudinal extremities. (C) Close‐up showing the fragment obtained from the peripheral region. (D) Removal of the keloid fragment from the central region. (E) Final aspect of the surgical part showing the orifices after fragment removal. In red, fragment orifices from the keloid central region. In blue, from the peripheral region and in green, from the adjacent skin. (F) Fragments obtained from the keloid central region (KC), peripheral region (KP) and adjacent skin (KS).
The central keloid region should be marked from the right angle intersection between two lines placed at the level of the largest longitudinal axis and the largest lesion transversal axis. The keloid peripheral region corresponds to the most distant points of the central intersection within the internal limits of the lesion border.
To remove the fragments from the peripheral region, the punch was placed in one of the longitudinal line extremities (most distant point in relation to the centre) respecting the lesion border. The other fragments were removed in the same direction (clockwise) in relation to the first point (Figure 2B, C).
To remove the first fragment from the central region, the punch was positioned in the intersection of the two lines situated at the level of the largest longitudinal axis and the largest transversal axis. The other fragments were removed centrifugally in relation to the first one, clockwise (Figure 2D, E).
The amount of fragments removed depends on the size of the keloid. At least four fragments should be obtained from each region (Figure 2F).
Fragments collected from the keloid surface reach a maximum of 10 mm depth. To collect fragments from deeper keloid portions (basal region), the surgical part should be cut longitudinally. The depth from where the fragments are to be collected should be specified by the researcher.
Primary fibroblast culture and subculture
Fibroblast harvesting was carried out by explant using the method described by Keira et al. (33), with adaptations. Fragments were placed in 15 ml conic tubes and washed with 10 ml phosphate‐buffered saline (PBS; Cultilab, SP, Brazil) containing penicillin (100 Ul/ml; Gibco, Carlsbad, CA, USA) and streptomycin (100 µm/ml; Gibco) six times under vigorous agitation, changing tubes and PBS in each repetition. Fragments were incubated (37°C, 30 minutes) in 10 ml Dulbecco's modified Eagle's medium (DMEM; Cultilab). Then, fragments were transferred to 60 mm2 Petri dishes, in square areas marked by perpendicular lines made with scalpel. Plates were left semi‐opened in the laminar flow for 30 minutes, for the fragments to adhere to its surface. Then, 6 ml of DMEM 15% fetal bovine serum (FBS; Cultilab), penicillin (100 UI/ml; Gibco) and streptomycin (100 µg/ml; Gibco) were added to each plate. Plates were kept in humidified incubator (37°C, 95% O2, 5% CO2).
The culture medium was changed every 2 days, for this rate enables the maintenance of ideal pH conditions between 7·6 and 7·8 without non physiologic upheavals 34, 35. This pH stability aims a balance between cellular proliferation and cellular biosynthesis activity of the fibroblasts (36). A few days after establishing the primary culture, we could observe spindle‐like cells proliferating from the edges of the explanted tissue, regarded as culturing fibroblasts (37) (Figure 3), as reported by Ehrlich et al. (38). Fibroblast satisfactory proliferation is observed in approximately 7–14 days 35, 36, 37, 38.
Figure 3.
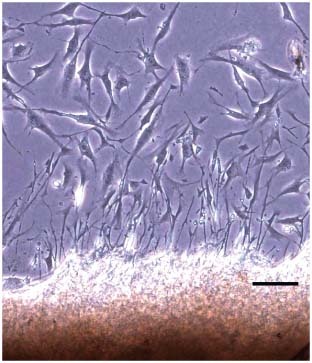
Fibroblasts proliferating from the edges of the explanted tissue to the Petri dish after 7 days in culture (42). Optical microscopy. Bar: 100 µm.
Subculturing (passage) was performed when cellular confluence reached approximately 80%. For this, the culture medium was aspirated and the keloid fragments discarded. The plate containing fibroblasts was washed with PBS, then quickly rinsed with Versene [PBS with 0·05 M ethylene diamine tetra acetic acid (EDTA); Sigma Chemical Co., Saint Louis, MO, USA] and 1 ml 0·25% trypsin with 0·02% EDTA was added. The plate was kept for 2 minutes in the incubator and taken to the microscope to confirm fibroblast detachment. Trypsin was neutralised with 3·0 ml 10% DMEM FBS and the cellular suspension centrifuged (100 g, 6 minutes). The pellet was resuspended in 10% DMEM FBS and antibiotics, and 100 000 cells were seeded in each 75 cm2 culture flask.
Cell cycle analysis by flow cytometry
Cell cycle distribution patterns of cultured fibroblasts from different keloid regions were analysed by flow cytometry at the third passage, and results are representative of four independent experiments. Cells were washed in PBS and fixed (formalin in PBS 0·4%; 30 minutes, 4°C). Thereafter, cells were washed two times in PBS and incubated in 500 µl PBS containing 0·1% saponin and 250 mg/l RNAse at 37°C, for 30 minutes, and then stained with 50 µg/ml propidium iodide (ICN, Costa Mesa, CA). Cellular DNA was analysed by FACSCalibur System (BD Biosciences, San Jose, CA, USA), and WinMDI v.2.9 software was used to determine the percentage of cells in the G0‐G1, G2‐M/S phases. A total of 10 000 events were analysed to determine the positivity percentage of cell markers and cell cycle.
Cell morphology analysis by confocal microscopy
Cells were grown to subconfluence in coverslips (in 12‐well plates), in standard medium, washed in PBS and fixed in formaldehyde in PBS (0·4%; 30 minutes), then exposed to glycine in PBS (0·1 M; 10 minutes) and twice to bovine serum albumin (BSA) in PBS (2%; 30 minutes), to reduce background interference. They were immunostained with 0·33 M AlexaFluor‐488 (green) or AlexaFluor‐594 (red) conjugated to phalloidin (Molecular Probes, Carlsbad, CA, USA), stained with DAPI (Sigma) and MitoTracker Green (Sigma) in PBS (2% BSA, 30 minutes). Thereafter, cells were washed three times in PBS for 10 minutes and mounted in slides, in solution 1:1 PBS/glycerol. Fluorescence was observed using a Zeiss Laser Scanning Confocal Microscope (LSM‐500), with the appropriate filters. Cells were analysed at the third passage, and results are representative of six independent experiments.
Statistical analysis
The results obtained were analysed using a one‐way analysis of variance followed by the Student–Newman–Keuls multiple range test. Data were analysed by GraphPad Prism v.3.0 software.
RESULTS
Analysis of cell cycle distribution
Cell cycle distribution was quantitatively measured by analysing DNA content using flow cytometry (Figure 4A). A comparison between the groups is shown as the percentage of cells in the G0‐G1 (M2) and G2‐M/S (M3) phases in each group (Figure 4B, C). Concerning the mitotic index, our data showed approximately 60% of peripheral keloid fibroblasts distributed in G2‐M/S phases (M3), whereas only about 41% of adjacent skin and central keloid fibroblasts were in those proliferative phases, with no significant differences between the last two groups. In contrast, the majority of adjacent skin and central keloid fibroblasts were distributed to the G0‐G1 phase (≅58%). Thus, there are significant differences in cell cycle distribution between peripheral keloid fibroblasts and both adjacent skin and central keloid fibroblasts (P < 0·05) (Figure 4B). On the other hand, the apoptotic index of the adjacent skin fibroblasts was significantly higher (≅2%) than those of central (≅0·9%) and peripheral (≅1%) keloid fibroblasts, with no significant differences between the last two groups (Figure 4C).
Figure 4.
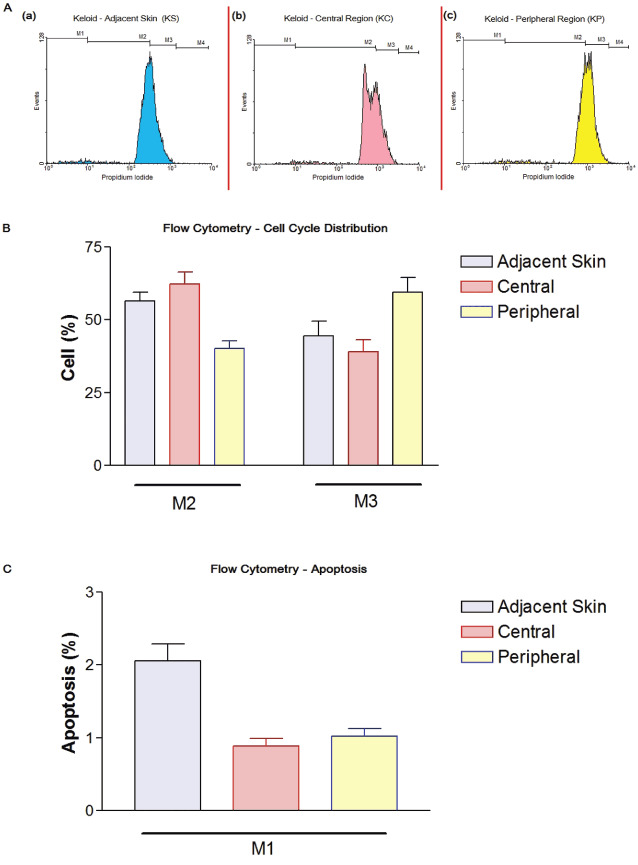
Flow cytometry. (A) Cell cycle analysis from one of the experiments showing the different profiles between the groups. M1: apoptotic cells; M2: G0‐G1 phase; M3: G2‐M/S phases; M4: post‐G2‐M. Fibroblasts from: (a) adjacent skin; (b) central region; (c) peripheral region. (B) Cell cycle quantitative distribution. Data analysed with one‐way analysis of variance (ANOVA) followed by Newman–Keuls (significance level P < 0·05). (C) Apoptotic cells. Data analysed with one‐way ANOVA followed by Newman–Keuls (significance level P < 0·05). (B and C) Values represent the mean ± SEM of at least four different experiments.
Fibroblasts morphological analysis
Morphological analysis by confocal microscopy of the fibroblasts from all three regions studied has shown preserved morphology, with absence of blebbings and homogeneous cytoskeleton distribution (Figure 5). We can observe actin filament distribution and mitochondria in the perinuclear region. The overlapped images show the organelles colocalisation and mitochondria around the nucleus (Figure 6). The organelles and cytoskeleton distribution presented normal morphology. Apoptosis classic features were not observed (5, 6).
Figure 5.
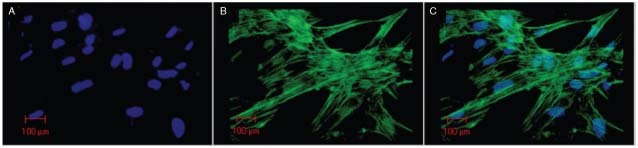
Confocal microscopy. Cultured fibroblasts from the adjacent skin. (A) Cell nuclei stained with DAPI (blue); (B) actin filaments immunostained with phalloidin/AlexaFluor‐488 (green); (C) overlapped images.
Figure 6.
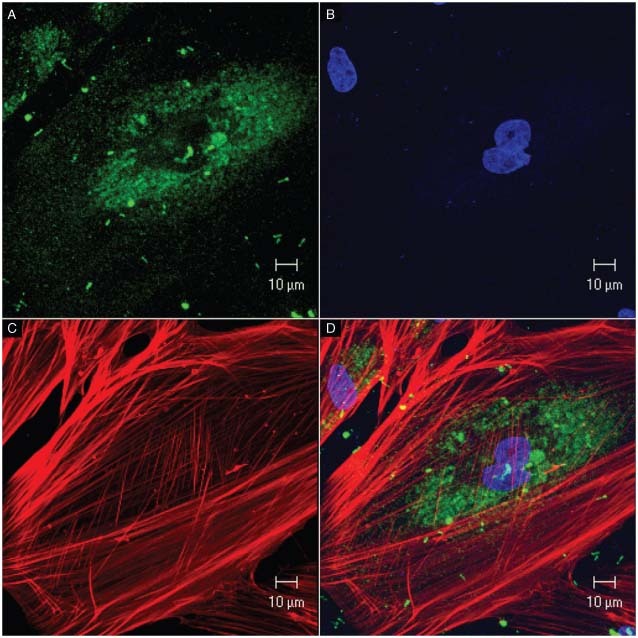
Confocal microscopy. Cultured fibroblasts from the adjacent skin. (A) Mitochondria stained with MitoTracker Green; (B) cell nuclei stained with DAPI (blue); (C) actin filaments immunostained with phalloidin/AlexaFluor‐594 (red); (D) overlapped images.
DISCUSSION AND CONCLUSION
The importance of specifying which part of the keloid lesion will be studied, so as its clinical status (active × resting keloid), have been strongly emphasised because many biological differences between fibroblasts derived from different keloid regions had been already reported 10, 16, 17, 18, 24, 25, 26, 27, 28, 39, 40. Our analysis of the cell cycle distribution indicated that 60% of peripheral keloid fibroblasts were in the proliferative periods of the cell cycle, whereas the majority of adjacent skin and central keloid fibroblasts were distributed to the G0‐G1 phase (≅58%) (Figure 4B). Also, the adjacent skin fibroblasts showed a higher apoptotic index compared with those of both central and peripheral keloid fibroblasts. This imbalance between proliferation and apoptosis may be responsible for keloid pathogenesis. Our data corroborate with the ones obtained by Lu et al. (22), which compared the cell cycle distribution of central and peripheral keloid fibroblasts.
Primary cell culture using explant techniques, when compared with techniques of enzymatic dissociation (using dispase and/or collagenase), provides an initial lower cell yield. Nevertheless, it allows better preservation of cell characteristics, avoiding significant cellular trauma, caused by the enzymatic attack, which may apply selective pressure in long‐term culture because it has already been proved that vertebrate cells are severely stressed by enzymatic dispersion 41, 42. Several chemical and mechanical isolation procedures have already been compared with optimise cell yield and minimise DNA damage by the method itself. If compared with collagenase isolation, mechanical cell dissociation gave less DNA damage (43). However, given the structural keloid characteristics (such as hardness), mechanical cell dissociation methods do not apply for keloid primary cell culture. Trypsinisation, used for subculturing (passaging), resulted in low DNA damage, similar to those obtained by mechanical dissociation (43).
Because of the potentially prejudicial characteristics of enzymatic dissociation, many researchers prefer to be cautious, choosing more conservative methods, such as the one presented here.
On the basis of our results, we present a simple, reliable and reproducible model which can also be adapted by the researcher to its specific conditions. Our proposed model proved itself efficient for fibroblast isolation from different keloid regions and its in vitro cultivation. Its simplicity and ease of execution may turn it into an important tool for studying and understanding the specific characteristics of the different keloid‐derived fibroblasts in culture.
ACKNOWLEDGEMENTS
The authors wish to thank Silvana Gaiba for her helpful suggestions. None of the authors has a financial interest in any of the products, devices or drugs mentioned in this article.
REFERENCES
- 1. McKeehan WL, Barnes D, Reid L, Stanbridge E, Murakami H, Sato GH. Frontiers in mammalian cell culture. In Vitro Cell Dev Biol 1990;26:9–23. [DOI] [PubMed] [Google Scholar]
- 2. Grimm S. The art and design of genetic screens: mammalian culture cells. Nat Rev Genet 2004;5:179–89. [DOI] [PubMed] [Google Scholar]
- 3. Dubertret L, Coulomb B. Reconstruction of human skin in culture. C R Seances Soc Biol Fil 1994;188:235–44. [PubMed] [Google Scholar]
- 4. Hochman B, Nahas FX, Sobral CS, Arias V, Locali RF, Juliano Y, Ferreira LM. Nerve fibers: a possible role in keloid pathogenesis. Br J Dermatol 2008;158:651–2. [DOI] [PubMed] [Google Scholar]
- 5. Hochman B, Locali RF, Matsuoka PK, Ferreira LM. Intralesional triamcinolone acetonide for keloid treatment: a systematic review. Aesthetic Plast Surg 2008;32:705–9. [DOI] [PubMed] [Google Scholar]
- 6. Berman B, Bieley HC. Adjunct therapies to surgical management of keloids. Dermatol Surg 1996;22:126–30. [DOI] [PubMed] [Google Scholar]
- 7. Hochman B, Ferreira LM, Vilas Bôas FC, Mariano M. Hamster (Mesocricetus auratus) cheek pouch as an experimental model to investigate human skin and keloid heterologous graft. Acta Cir Bras 2004;19:79–88. [Google Scholar]
- 8. Placik OJ, Lewis VL. Immunologic associations of keloids. Surg Gynecol Obstet 1992;175:185–93. [PubMed] [Google Scholar]
- 9. Hochman B, Vilas Bôas FC, Mariano M, Ferreira LM. Keloid heterograft in the hamster (Mesocricetus auratus) cheek pouch. Acta Cir Bras 2005;20:200–12. [DOI] [PubMed] [Google Scholar]
- 10. Lee SS, Yosipovitch G, Chan YH, Goh CL. Pruritus, pain, and small nerve fiber function in keloids: a controlled study. J Am Acad Dermatol 2004;51:1002–6. [DOI] [PubMed] [Google Scholar]
- 11. Niessen FB, Spauwen PHM, Schalkwijk J, Kon M. On the nature of hypertrophic scars and keloids: a review. Plast Reconstr Surg 1999;104:1435–58. [DOI] [PubMed] [Google Scholar]
- 12. O’Sullivan ST, O’Shaughnessy M, O’Connor TP. Aetiology and management of hypertrophic scars and keloids. Ann R Coll Surg Engl 1996;78:168–75. [PMC free article] [PubMed] [Google Scholar]
- 13. Ramos ML, Gragnani A, Ferreira LM. Is there an ideal animal model to study hypertrophic scarring? J Burn Care Res 2008;29:363–8. [DOI] [PubMed] [Google Scholar]
- 14. Ogawa R, Chin MS. Animal models of keloids and hypertrophic scars. J Burn Care Res 2008;29:1016–7. [DOI] [PubMed] [Google Scholar]
- 15. Butler PD, Ly DP, Longaker MT, Yang GP. Use of organotypic coculture to study keloid biology. Am J Surg 2008;195:144–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ladin DA, Hou Z, Patel D, McPhail M, Olson JC, Saed GM, Fivenson DP. p53 and apoptosis alterations in keloids and keloid fibroblasts. Wound Repair Regen 1998;6:28–37. [DOI] [PubMed] [Google Scholar]
- 17. Appleton I, Brown JB, Willoughby DA. Apoptosis, necrosis, and proliferation. Possible implications in the etiology of keloids. Am J Pathol 1996;149:1441–7. [PMC free article] [PubMed] [Google Scholar]
- 18. Teofoli P, Barduagni S, Ribuffo M, Campanella A, De Pita’ O, Puddu P. Expression of Bcl‐2, p53, c‐jun and c‐fos protooncogenes in keloids and hypertrophic scars. J Dermatol Sci 1999;22:31–7. [DOI] [PubMed] [Google Scholar]
- 19. Chodon T, Sugihara T, Igawa HH, Funayama E, Furukawa H. Keloid‐derived fibroblasts are refractory to Fas‐mediated apoptosis and neutralization of autocrine transforming growth factor‐beta1 can abrogate this resistance. Am J Pathol 2000;157:1661–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tanaka A, Hatoko M, Tada H, Iioka H, Niitsuma K, Miyagawa S. Expression of p53 family in scars. J Dermatol Sci 2004;34:17–24. [DOI] [PubMed] [Google Scholar]
- 21. Lu F, Gao J, Ogawa R, Hyakusoku H, Ou C. Fas‐mediated apoptotic signal transduction in keloid and hypertrophic scar. Plast Reconstr Surg 2007;119:1714–21. [DOI] [PubMed] [Google Scholar]
- 22. Lu F, Gao J, Ogawa R, Hyakusoku H, Ou C. Biological differences between fibroblasts derived from peripheral and central areas of keloid tissues. Plast Reconstr Surg 2007;120:625–30. [DOI] [PubMed] [Google Scholar]
- 23. Lu F, Gao J, Li X. The distribution of cell cycle on fibroblasts derived from the pathological scars and analysis of Fas gene mutations in keloids using polymerase chain reaction‐based single‐strand conformation polymorphism. Zhonghua Yi Xue Za Zhi 2000;80:709–12. [PubMed] [Google Scholar]
- 24. Liu Y, Gao J, Liu X, Lu F, Liu H. Correlation analysis between clinical phenotypes of keloids and polymorphism of p53 gene codon 72. Zhongguo Xiu Fu Chong Jian Wai Ke Za Zhi 2008;22:1433–6. [PubMed] [Google Scholar]
- 25. Luo S, Benathan M, Raffoul W, Panizzon RG, Egloff DV. Abnormal balance between proliferation and apoptotic cell death in fibroblasts derived from keloid lesions. Plast Reconstr Surg 2001;107:87–96. [DOI] [PubMed] [Google Scholar]
- 26. Giugliano G, Pasquali D, Notaro A, Brongo S, Nicoletti G, D’Andrea F, Bellastella A, Sinisi AA. Verapamil inhibits interleukin‐6 and vascular endothelial growth factor production in primary cultures of keloid fibroblasts. Br J Plast Surg 2003;56:804–09. [DOI] [PubMed] [Google Scholar]
- 27. Tuan TL, Hwu P, Ho W, Yiu P, Chang R, Wysocki A, Benya PD. Adenoviral overexpression and small interfering RNA suppression demonstrate that plasminogen activator inhibitor‐1 produces elevated collagen accumulation in normal and keloid fibroblasts. Am J Pathol 2008;173:1311–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xu B, Liu ZZ, Zhu GY, Yang JF, Zhao JP, Wang JC, Cai JL. Efficacy of recombinant adenovirus‐mediated double suicide gene therapy in human keloid fibroblasts. Clin Exp Dermatol 2008;33:322–8. [DOI] [PubMed] [Google Scholar]
- 29. Zhang GY, Yi CG, Li X, Zheng Y, Niu ZG, Xia W, Meng Z, Meng CY, Guo SZ. Inhibition of vascular endothelial growth factor expression in keloid fibroblasts by vector‐mediated vascular endothelial growth factor shRNA: a therapeutic potential strategy for keloid. Arch Dermatol Res 2008;300:177–84. [DOI] [PubMed] [Google Scholar]
- 30. Naim R, Naumann A, Barnes J, Sauter A, Hormann K, Merkel D, Aust W, Braun T, Bloching M. Transforming growth factor‐beta1‐antisense modulates the expression of hepatocyte growth factor/scatter factor in keloid fibroblast cell culture. Aesthetic Plast Surg 2008;32:346–52. [DOI] [PubMed] [Google Scholar]
- 31. Mukhopadhyay A, Khoo A, Cheong HH, Chan SY, Aalami O, Lim IJ, Phan TT. Targeting of Sp1 transcription factor: a novel therapeutic approach for keloids, an in vitro analysis. Exp Dermatol 2007;16:1023–31. [DOI] [PubMed] [Google Scholar]
- 32. Xia W, Kong W, Wang Z, Phan TT, Lim IJ, Longaker MT, Yang GP. Increased CCN2 transcription in keloid fibroblasts requires cooperativity between AP‐1 and SMAD binding sites. Ann Surg 2007;246:886–95. [DOI] [PubMed] [Google Scholar]
- 33. Keira SM, Ferreira LM, Gragnani A, Duarte IS, Santos, IAN. Experimental model for fibroblast culture. Acta Cir Bras 2004;19:11–6. [Google Scholar]
- 34. Booth BA, Polak KL, Uitto J. Collagen biosynthesis by human skin fibroblasts. I. Optimization of the culture conditions for synthesis of type I and type III procollagens. Biochem Biophys Acta 1980;607:145–60. [DOI] [PubMed] [Google Scholar]
- 35. Ceccarini C, Eagle H. pH as a determinant of cellular growth and contact inhibition. Proc Nat Acad Sci 1971;68:229–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nigra TP, Martin GR, Eagle H. The effect of environmental pH on collagen synthesis by cultured cells. Biochem Biophys Res Commun 1973;53:272–81. [DOI] [PubMed] [Google Scholar]
- 37. Campaner AB. TGF‐beta 1 in fibrosis formation of skin in athymic mice. PhD Thesis. São Paulo: Federal University of São Paulo 2005; 129p.
- 38. Ehrlich HP, Desmoulière A, Diegelmann RF, Cohen IK, Compton CC, Garner WL, Kapanci Y, Gabbiani G. Morphological and immunochemical differences between keloid and hypertrophic scar. Am J Pathol 1994;145:105–13. [PMC free article] [PubMed] [Google Scholar]
- 39. Desmouliere A, Redard M, Darby I, Gabbiani G. Apoptosis mediates the decrease in cellularity during the transition between granulation tissue and scar. Am J Pathol 1995;146:56–66. [PMC free article] [PubMed] [Google Scholar]
- 40. Angel P, Karin M. The role of jun, fos and the AP‐1 complex in cell proliferation and transformation. Biochem Biophys Acta 1991;1072:129–57. [DOI] [PubMed] [Google Scholar]
- 41. Dietel M, Arps H, Gerding D, Trapp M, Niendorf A. Establishment of primary cell cultures: experiences with 155 cell strains. Klin Wochenschr 1987;65:507–12. [DOI] [PubMed] [Google Scholar]
- 42. Rubin H. Cell aging in vivo and in vitro. Mech Ageing Dev 1997;98:1–35. [DOI] [PubMed] [Google Scholar]
- 43. Kosmehl T, Hallare AV, Reifferscheid G, Manz W, Braunbeck T, Hollert H. A novel contact assay for testing genotoxicity of chemicals and whole sediments in zebrafish embryos. Environ Toxicol Chem 2006;25:2097–106. [DOI] [PubMed] [Google Scholar]