

Abstract
Pharmacogenetics studies how genetic variation leads to variability in drug response. Guidelines for selecting the right drug and right dose for patients based on their genetics are clinically effective, but are widely unused. For some drugs, the normal clinical decision making process may lead to the optimal dose of a drug that minimizes side effects and maximizes effectiveness. Without measurements of genotype, physicians and patients may adjust dosage in a manner that reflects the underlying genetics. The emergence of genetic data linked to longitudinal clinical data in large biobanks offers an opportunity to confirm known pharmacogenetic interactions as well as discover novel associations by investigating outcomes from normal clinical practice. Here we use the UK Biobank to search for pharmacogenetic interactions among 200 drugs and 9 genes among 200,000 participants. We identify associations between pharmacogene phenotypes and drug maintenance dose as well as differential drug response phenotypes. We find support for several known drug-gene associations as well as novel pharmacogenetic interactions.
Keywords: Pharmacogenetics, Pharmacogenomics, Statistical Analysis, Biobank, UK Biobank
1. Introduction
Pharmacogenetics promises to revolutionize patient care by offering personalized drug selection and dosage based on an individual’s genetics1. Variations in the genes that encode proteins involved in drug pharmacokinetics and pharmacodynamics are known to lead to interindividual heterogeneity in drug response and can greatly affect clinical outcome. Dosage guidelines have been developed by organizations such as the Clinical Implementation of Pharmacogenetic Consortium (CPIC; cpicpgx.org) to aid physicians in incorporating pharmacogenetics into their practice, however the adoption of pharmacogenetics by practicing physicians has not lived up to the optimism in the field2,3.
Doctor’s may not directly be using pharmacogenetics to inform practice, but genetics influences how patients respond to drugs nonetheless. Some drugs, such as warfarin, have a narrow therapeutic index and blood concentration of the drug must be frequently measured to ensure patient safety4. The ultimate dose at which the patient achieves the appropriate, stable blood concentration of the drug is the maintenance dose. For warfarin, this dose is strongly influenced by genetic factors such including variations in the metabolizing enzymes CYP2C9 and CYP4F2, as well as the drug target VK0RC1.
In other instances genetic variation may lead patients to be at higher risk for side effects. The frequently prescribed drug simvastatin has well known pharmacogenetic interactions with SLCO1B1 that can lead to simvastatin-induced myopathy5. While this is a rare side effect, individuals with poor functioning SLCO1B1 are at higher risk for simvastatin-induced myopathy. CPIC guidelines for simvastatin recommend that individuals with poor functioning SLCO1B1 take a reduced simvastatin dose or a different drug altogether.
Numerous pharmacogenetic drug-gene relationships have been discovered, but most pharmacogenetic studies are small and narrowly focused. The use of electronic health record and biobank scale data as a means for pharmacogenetic discovery and validation of known relationships has been proposed, but until recently databases linking clinical data with genetic data for a large number of patients were unavailable1,6. Biobanks offer an opportunity to retrospectively assess known drug-gene relationships in a clinical setting as well as offer the opportunity to discover new drug-gene associations. Biobanks and electronic health records have been used to perform targeted association studies between genomics and response to individual drugs7 as well as characterize frequency of pharmacogenetic alleles in populations8,9, but studies of drug response across a large number of drugs have not yet been performed.
The UK Biobank has been widely used to perform genome-wide association studies on a wide variety of traits, but it also includes primary care data from the United Kingdom’s National Health System10. This dataset offers longitudinal, structured clinical data for more than 220,000 participants that includes diagnoses, laboratory tests, and prescription data. This dataset offers a unique opportunity to identify associations between drug response phenotypes and genetics. Here we present a retrospective pharmacogenetic analysis linking drug exposure for 200 drugs to clinical outcome using the UK Biobank primary care data. We focus on two types of clinical outcomes of interest: maintenance dose and differential drug response.
2. Methods
2.1. Pharmacogenetic Allele Calling
We investigated drug-gene relationships for nine important pharmacogenes in the UK Biobank for 222,114 participants using primary care data from the National Health System, provided by the UK Biobank10. The pharmacogenetic alleles used in this study were derived from a previously reported procedure, described here in brief8. We used imputed genotypes from the Axiom Biobank Array released by the UK Biobank11. We included nine genes in our analysis: CYP2B6, CYP2C19, CYP2C9, CYP2D6, CYP3A5, CYP4F2, SLCO1B1, TPMT, and UGT1A1. The proteins encoded by these genes play critical roles in drug pharmacokinetics and each is included in a CPIC dosing guideline for a drug. We assigned pharmacogenetic phenotypes for each gene using PGxPOP, a tool designed for high throughput mapping of pharmacogenetic alleles and phenotypes (https://github.com/PharmGKB/PGxPOP). The analysis was limited to individuals of European descent. This included participants who self reported as European and were confirmed as European using principal component analysis.
2.2. Drug Dosage Association with Pharmacogenetics
Drugs used in this study were derived from the PharmGKB curated drug list (https://www.pharmgkb.org/downloads, drugs.zip)12. For each drug, we extracted prescription information from the UK Biobank primary care prescription data by matching the drug name and brand names in the prescription data. Dosage information and drug quantity was extracted using regular expressions that searched within the drug description. We excluded combination therapies from the analysis.
We calculated maintenance dose by determining the average milligrams of drug per day for the last five prescriptions of each drug. This was done by calculating the total milligrams of drug administered for a single prescription divided by the number of days until the next prescription. We then averaged the milligrams of drug per day over the five most recent prescriptions. Prescriptions with a quantity outside two standard deviations from the mean quantity across all participants for that drug were excluded. Subjects were required to receive a minimum of five prescriptions to be included in the analysis. We required drugs to have a minimum of 50 subjects with a maintenance dose to be included in the analysis.
We divided the analysis of maintenance dose associations into three groups of drug-gene pairs. First, we investigated the relationship between drug-gene pairs that have an existing CPIC guideline. This indicates a strong level of evidence of a relationship between a drug and a gene. Second, we investigated drug-gene pairs which have some level of evidence in PharmGKB, but no existing CPIC guideline. These pairs still have some prior evidence indicating an association, but not enough to develop a dosage guideline. Third, we investigated all other drug-gene pairs where an interaction is indicated in DrugBank13. These pairs have no prior evidence of a pharmacogenetic association. Data was grouped within each gene by predicted phenotype. For example, for CYP2C9 participants were put into bins by metabolizer class (normal metabolizers (NM), intermediate metabolizers (IM), and poor metabolizers (PM). Phenotype groups with less than ten participants for a drug are excluded from analysis.
Association between maintenance dose and pharmacogenetic phenotypes was tested for 200 drugs using two types of non-parametric statistical association tests. We used both a Kruskal-Wallis one-way analysis of variance and Jonckheere-Terpstra trend tests to test for associations between each drug and gene pair. Both types of tests are necessary to detect various relationships between dosage and genetics. First, the Kruskal-Wallis test was used to identify any pharmacogenetic phenotype (e.g. CYP2C9 PMs) that have a significant difference in the dosage from other metabolizer classes. Second, Jonckheere-Terpstra tests for an ordered relationship in ranked groups. This is a natural fit for pharmacogenetic phenotypes since there is an inherent order in function which may lead to a linear relationship with dosage (e.g. NM > IM > PM). Resulting p-values are adjusted using a Bonferroni correction. We used a covariate-adjusted dose as the response variable for each test. To do this we fit a linear regression model to the dosage using several covariates: age (at time of last prescription), sex, BMI, genotyping array, and the first for principal components of a principal component analysis (PCA) using genotype data (UK Biobank Data-Field 22009).
We tested the impact of the intronic CYP2C19 variant rs3814637 on warfarin dose. We used a two-sided Jonckheere-Terpstra test on the allele dosage against the warfarin maintenance dose. Allele dosage was determined as the sum of the alternate alleles for rs3814637.
2.3. Differential Drug Response Phenotype Association
In a separate analysis, we tested the relationship between pharmacogenes and drug response for all drugs using diagnosis codes in primary care data. We sought to identify pharmacogenomic phenotypes that would lead to a differential drug response phenotype, for example, instances where poor metabolizers have an increased risk of developing some side effect compared to normal metabolizers. For each drug included in the dosage analysis we identified all diagnoses in the primary care data in the year following the first exposure to the drug. Diagnosis codes in the primary care data are provided as Read Codes (version 2 and version 3), we mapped the Read Codes to ICD-10 codes including only the first three digits (the chapter and first two numerals). ICD-10 codes from chapters V, W, X, Y, and Z were excluded from analysis. Codes were required to have at least 100 events per drug to be included in the analysis. Diagnosis codes may represent the primary disease indication for the drug, side effects, comorbidities, or other unrelated events.
We used logistic regression to test the association between gene phenotypes and ICD-10 code incidence for each drug. This was set up using a binary indicator as the response variable and a one-hot encoding of gene phenotype. We included age (at time of first prescription), sex, genotyping array, and the first four principal components from a genotype PCA as covariates.
We evaluated three tiers of drug-gene relationship, as in the maintenance dose analysis. Drug-gene pairs with CPIC guidelines, drug-gene pairs with any level of evidence in PharmGKB but no CPIC guideline, and an exploratory analysis. For the exploratory analysis of side effect relationships we limited our search to drugs known to interact with CYP2C9, CYP2C19, and CYP2D6, as indicated by DrugBank. These genes were selected because they are promiscuous metabolizing enzymes with well defined pharmacogenetics.
3. Results
The pharmacogenetic analyses presented here included a total of 201,498 participants, after removing 20,615 participants not of European descent. More than 57 million prescriptions are contained within the primary care data, an average of 262 prescriptions per participant. Our initial drug list included 3,358 drugs. Of this, 200 were found in the UK Biobank prescription data with sufficient counts to be included in subsequent analysis.
3.1. Drug Dosage Association with Pharmacogenetics
We sought to evaluate methods for testing the relationship between maintenance dose and pharmacogenes at a biobank scale. We performed this analysis using three groups of drug-gene pairs. Of the drugs with CPIC guidance for any of the nine genes queried, there were 24 that had the minimum of 50 participants for whom a maintenance dose could be calculated. We find that nine of the drug-gene pairs have a significant difference in the dosage across gene phenotypes (Kruskal-Wallis or Jonckeere-Terpstra p < 0.05, Table 1). We do not adjust for multiple tests because these are known relationships not discoveries. Warfarin and CYP2C9 phenotypes had the most significant relationship (p ≅ 0, Jonckeere-Terpstra). The remaining twenty drug-gene pairs did not have a significant relationship between maintenance dose and gene phenotype.
Table 1.
Drug-gene dose relationship results. Drug-gene pairs are presented in three groups: drugs with CPIC guidelines, without guidelines but PharmGKB evidence, and novel associations. Level of Evidence represents the maximum level of evidence for the drug-gene relationship in PharmGKB.
| Group | Drug | Gene | Level of Evidence | # Samples | Test | p-value |
|---|---|---|---|---|---|---|
| CPIC guidance | warfarin | CYP2C9 | 1A | 6,409 | JT | 0.00E+00 |
| phenytoin | CYP2C9 | 1A | 459 | KW | 1.04E-05 | |
| azathioprine | TPMT | 1A | 799 | KW | 9.13E-03 | |
| imipramine | CYP2C19 | 2A | 348 | JT | 1.10E-23 | |
| lansoprazole | CYP2C19 | 2A | 2,793 | JT | 2.52E-02 | |
| pantoprazole | CYP2C19 | 3 | 114 | JT | 2.56E-02 | |
| simvastatin | SLCO1B1 | 1A | 34,611 | KW | 3.52E-02 | |
| warfarin | CYP4F2 | 1A | 4,559 | KW | 3.69E-02 | |
| paroxetine | CYP2D6 | 1A | 2,804 | KW | 4.22E-02 | |
| No guidance | warfarin | CYP2C19 | 3 | 6,410 | KW | 2.22E-14 |
| nicotine | CYP2B6 | 3 | 391 | JT | 6.38E-04 | |
| Novel associations | cyclosporine | CYP2C19 | NA | 166 | JT | 1.87E-05* |
| rabeprazole | CYP2C9 | NA | 223 | JT | 4.55E-05* | |
p-values with a * are significant at p <= 8.6 x 10−6, bonfenoni adjusted.
Test indicates which type of test achieved the p-value shown (JT=Jonckheere-Terpstra, KW=Kraskal Wallis). Only results with a standard enor less than 0.2 are included.
We then investigated association between maintenance dose and gene phenotype for drug-gene pairs with any level of evidence in PharmGKB but no CPIC guideline. We found two drug-gene pairs with a p-value less than 0.05 for either the Kruskal-Wallis test or Joncheere-Terpstra trend test (Table 1). The most significant was the Kruskal-Wallis test for warfarin and CYP2C19 phenotype. Investigating the dose relationship with phenotype reveals that CYP2C19 normal metabolizers have a decreased maintenance dose compared to the other CYP2C19 metabolizer classes (Figure 1, second row, first column). We followed up on this finding by interrogating the association between rs3814637 and warfarin maintenance dose.
Figure 1.
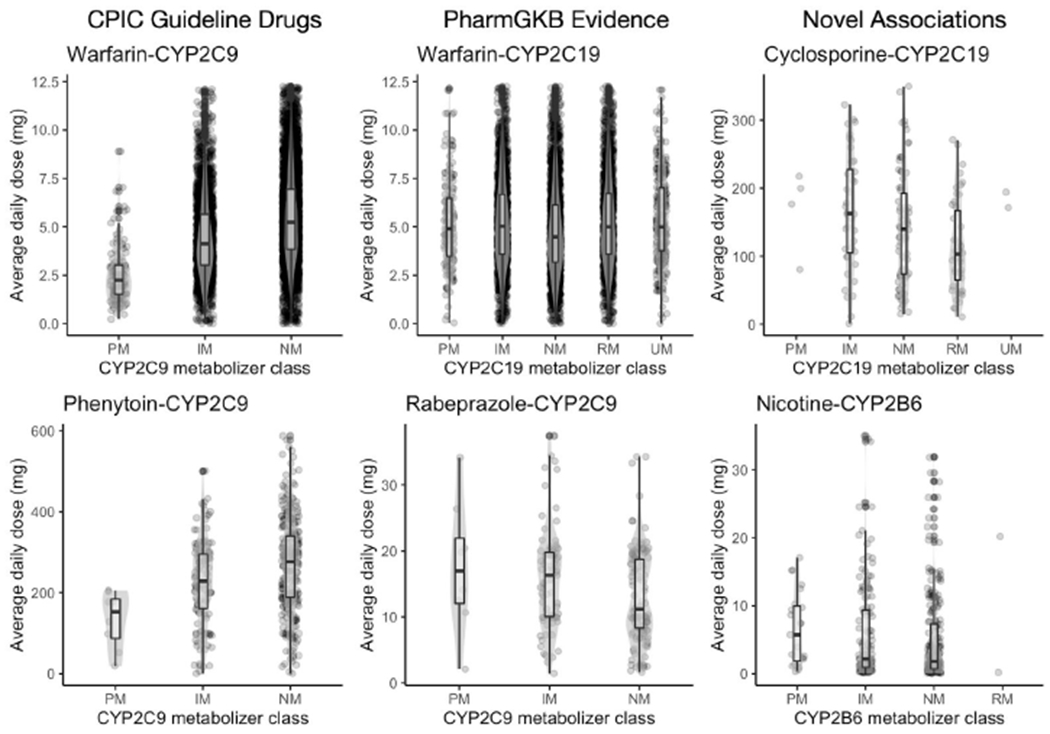
Box plots of maintenance dose for most significant drug-gene pairs. The top two most significant pairs are shown for each group (columns). Enzyme metabolizer classes are represented along the x-axis and the distribution of maintenance dose along the y-axis.
The intronic variant rs3814637 within CYP2C19 has been previously reported to be associated with warfarin response14–16. This variant is contained within several CYP2C19 star alleles: CYP2C19*1.004, CYP2C19*1.005, and CYP2C19*15.001, all of which are normal functioning alleles. We observed that normal metabolizers had an average daily dose of 4.8 mg (compared to 5.3 mg for the other metabolizer classes). We then tested the association between rs3814637 and warfarin maintenance dose. We find a significant relationship between rs3814637 dosage and warfarin maintenance dose (p <= 1.0 x 10−46, two-sided Jonckheere-Terpstra, Fig. 2).
Fig. 2.
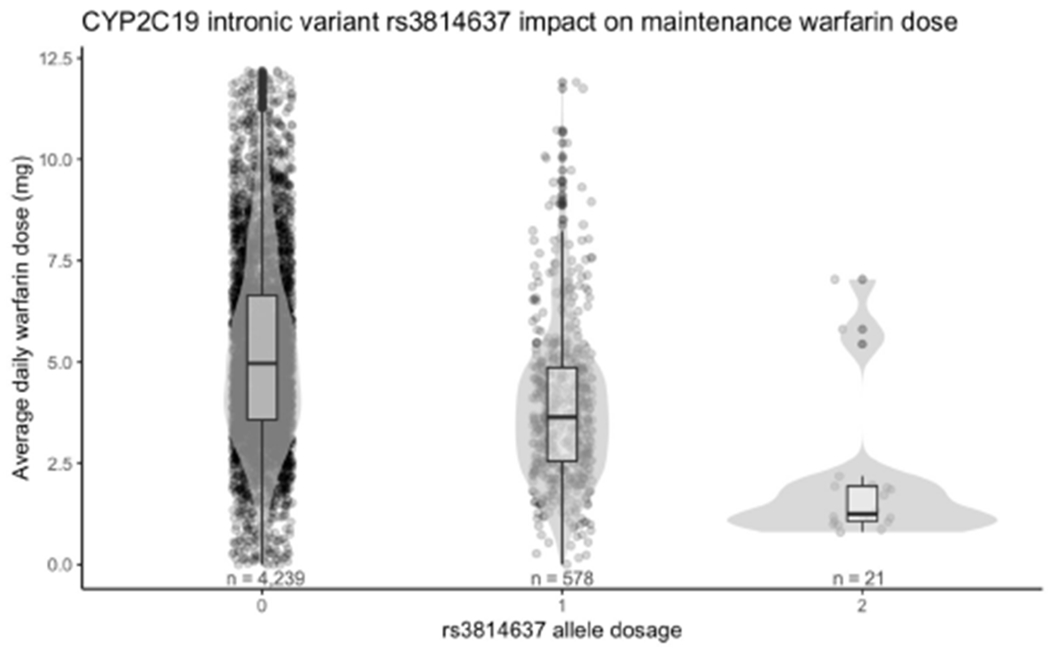
CYP2C19 intronic variant rs3814637 has a strong influence on warfarin maintenance dose. The x-axis indicates the alternate allele dosage. The y-axis is the maintenance dose.
We then analyzed the relationship between maintenance dose and gene phenotype for drug-gene pairs that had no previous indication of a pharmacogenetic relationship but are known to interact. We tested 581 drug-gene pairs and found two significant relationships between dose and gene phenotype: cyclosporine and CYP2C19, and nicotine and CYP2B6 (p < 8.6 x 10−6, Jonckheere-Terpstra, bonferroni adjusted, table 3: Novel associations).
3.2. Differential Drug Response Phenotype Association
We investigated the degree to which adverse drug reactions related to pharmacogenetics could be discovered by performing a statistical analysis of pharmacogene phenotypes and coded medical events within a one year window following the first administration of a drug. We again evaluated three drug-gene groups starting with drug-gene pairs with CPIC guidelines (Table 2, CPIC Guidance Group). The most significant side effect is a decreased incidence of herpes zoster diagnoses among CYP2C19 intermediate metabolizers (p <= 8.76 x 10−5).
Table 2.
Drug-gene side effect relationship results. Associations are presented in three groups: drug-gene pairs with CPIC guidelines, pairs with no guidelines but evidence in PharmGKB, and novel associations. Phenotype is the gene phenotype (IM: Intennediate Metabolizer, PM: Poor Metabolizer, RM: Rapid Metabolizer, UM: Ultrarapid Metabolizer, IF: Increased Function, PF: Poor Function). Odds ratio is the odds ratio relative to nonnal metabolizer or nonnal function alleles.
| Group | Drug | Gene | Level of Evidence | Phenotype | ICD-10 | Code definition | Odds ratio | p-value |
|---|---|---|---|---|---|---|---|---|
| CPIC Guidance | citalopram | CYP2C19 | 1A | IM | B02 | Herpes zoster | 0.53 | 8.76E-05 |
| simvastatin | SLCO1B1 | 1A | IF | M65 | Synovitis and tenosynovitis | 1.82 | 1.42E-04 | |
| amitriptyline | CYP2C19 | 1A | RM | R53 | Malaise and fatigue | 1.55 | 1.74E-04 | |
| amitriptyline | CYP2C19 | 1A | UM | J30 | Vasomotor and allergic rhinitis | 1.94 | 2.75E-04 | |
| codeine | CYP2D6 | 1A | PM | A52 | Late syphilis | 1.78 | 3.30E-04 | |
| ibuprofen | CYP2C9 | 1A | PM | E13 | Other specified diabetes mellitus | 2.00 | 4.90E-04 | |
| clopidogrel | CYP2C19 | 1A | RM | B08 | Viral infections characterized by skin and mucous membrane lesions | 0.59 | 5.17E-04 | |
| tamoxifen | CYP2D6 | 1A | IM | C50 | Malignant neoplasm of breast | 0.62 | 6.98E-04 | |
| simvastatin | SLCO1B1 | 1A | PF | M79 | Unspecified soft tissue disorders | 1.49 | 7.46E-04 | |
| simvastatin | SLCO1B1 | 1A | DF | M65 | Synovitis and tenosynovitis | 1.79 | 7.75E-04 | |
| No Guidance | citalopram | CYP2D6 | 3 | IM | J45 | Asthma | 1.44 | 9.13E-05 |
| citalopram | CYP2D6 | 3 | IM | 150 | Heart failure | 1.56 | 1.12E-04 | |
| simvastatin | CYP2C9 | 3 | PM | J01 | Acute sinusitis | 1.74 | 1.56E-04 | |
| citalopram | CYP2D6 | 3 | IM | J64 | Unspecified pneumoconiosis | 1.56 | 5.74E-04 | |
| propranolol | CYP2D6 | 4 | IM | O86 | Other puerperal infections | 1.85 | 6.38E-04 | |
| Novel associations | diazepam | CYP2C9 | NA | PM | M19 | Osteoarthritis | 2.33 | 4.52E-06* |
| zopiclone | CYP2C9 | NA | IM | H91 | Unspecified hearing loss | 2.20 | 1.73E-05 | |
| loratadine | CYP2D6 | NA | IM | M16 | Osteoarthritis of hip | 1.98 | 1.20E-04 | |
| tramadol | CYP2B6 | NA | PM | H61 | Disorders of external ear | 1.95 | 1.86E-04 | |
| quinine | SLCO1B1 | NA | IF | N39 | Disorders of urinary system | 1.95 | 1.87E-04 | |
indicates significance with Bonfenoni adjusted p-value threshold of 1.0 x 10−5.
Only results with a standard error less than 0.2 are included.
Next we looked to see if there are any differential drug response phenotypes enriched among drug-gene pairs with any level of evidence but no CPIC guideline. The top five results are shown in Table 2 under “No Guidance”. We find several phenotypes enriched among CYP2D6 intermediate metabolizers taking citalopram, including respiratory issues and heart failure. We also find an increased risk of sinus infections among CYP2C9 poor metabolizers on simvastatin, and an increased risk of puerperal infections among CYP2D6 intermediate metabolizers on propranolol.
We interrogated all other drugs known to be metabolized by CYP2C9, CYP2C19, or CYP2D6 for differential drug response phenotypes. This resulted in 4,806 independent association tests across 81 drugs. After multiple hypothesis corrections one side effect was significantly associated with a drug-gene pair: increased incidence of osteoarthritis in CYP2C9 poor metabolizers after taking diazepam. We show the top five results from the exploratory analysis in Table 2.
4. Discussion
Biobanks offer a powerful solution for enabling the study of relationships between drugs and genes. Large datasets linking genetic and longitudinal clinical data are becoming more broadly available and allow interrogation of the relationship between drug response and pharmacogenetic phenotypes. Here we derived drug phenotypes in the form of maintenance dose and differential drug response phenotypes for more than 200,000 participants across 200 drugs in the UK Biobank and tested their association with well established pharmacogenetic phenotypes for nine genes.
Pharmacogenetic testing is not yet common practice, but for some drugs the standard clinical procedures used to determine maintenance dose are influenced by genetics. We find evidence to support existing pharmacogenetic associations with maintenance dose. Among 24 drugs with CPIC guidance in our study we find evidence for a genetic influence on maintenance dose for nine drugs. For the remaining pairs with guidance, it is possible we are not likely to observe an association with maintenance dose because efficacy is difficult to measure or side effects are rare. Among drugs with any prior evidence of a pharmacogenetic relationship but no CPIC dosage guideline we find that maintenance dose supports the association for two drug-gene pairs. Most notably, carriers of the CYP2C19 intronic variant rs3814637 have a significantly decreased warfarin maintenance dose. The causal mechanism through which this effect occurs is unclear, and this variant itself may not be causal, rather in linkage disequilibrium with a causal variant. In GTEx, rs3814637 is associated with increased expression of CYP2C9 (the gene typically associated with warfarin response) in several tissues, although importantly not in liver. There is a gap in the amount of warfarin dosing variability that can be explained by genetics among individuals of African descent17. rs3814637 has nearly twice the allele frequency in the African population as it does in the European population (11.6% vs 6.7%)18. Although this study focuses on Europeans, this variant may explain some of the missing heritability of warfarin response among Africans, but further study is needed to confirm this relationship.
We discovered potential novel pharmacogenetic associations with maintenance dose for two drugs: cyclosporine with CYP2C19, and nicotine with CYP2B6. Both drugs are known to be metabolized by their respective associated enzymes, however there is no prior literature evidence suggesting a pharmacogenetic relationship. For both drugs, we find a decreasing association between dose and metabolizer class of their associated enzymes, where individuals with higher rates of metabolism tend to be on lower doses.
Our analysis of differential drug response phenotypes reveals associations with side effects among drug-gene pairs. This analysis is limited due to the large number of tests requiring a strict multiple hypothesis testing threshold, but produces interesting hypotheses. At first glance many of the differential phenotype associations seem unlikely, but literature evidence exists for many of the findings. For example, the most significant association among drugs with CPIC guidelines was a decreased incidence of herpes zoster among CYP2C19 intermediate metabolizers compared to CYP2C19 normal metabolizers treated with citalopram. However, two previous studies have demonstrated that SSRIs can lead to increased resistance to herpes19,20. CYP2C19 intermediate metabolizers have an increased blood concentration of citalopram and may have an increased resistance to a herpes infection. We also find CYP2C19 rapid metabolizers on clopidogrel have a decreased risk of viral skin lesions compared to CYP2C19 normal metabolizers. There is evidence that clopidogrel may inhibit viral clearance21. It may be possible that CYP2C19 rapid metabolizers have a lower concentration of clopidogrel and therefore the degree to which they are able to fight off viral infections is higher than that of CYP2C19 normal metabolizers. The most significant association is between CYP2C9 poor metabolizers on diazepam having an increased incidence of osteoarthritis. There is no literature that suggests osteoarthritis may be a side effect of diazepam, although there are studies that suggest diazepam could be used to treat pain as a result of rheumatoid arthritis. Without further evidence we cannot say whether this relationship results from pharmacogenetics and not a correlation with the drug indication or a statistical artifact.
This work has several limitations. First, we use pharmacogenetic alleles called from data imputed from genotyping arrays. We previously reported limitations in accuracy of the ability to accurately call alleles in several pharmacogenes from imputed data, notably in CYP2D68. The lack of structural variants in the dataset in addition to the inability to call rare variants may lead to inaccurate prediction of CYP2D6 phenotypes. Second, we broadly apply our maintenance dose algorithm to drugs in the UK Biobank. While this is effective for some drugs, better clinical end points may provide an improved representation of patient response. For example, a dose response curve may provide more fine grained insight into individual response and yield better insight into the genetics of drug response. It is challenging to broadly define response across drugs from numerous classes with varying indications and therapeutic indices. Even a single drug can be used for different indications and may require different doses to treat each indication. Additionally, this approach will miss patients who take a drug once and experience side effects that lead them to immediately switch drugs. No catch-all definition will suffice, but maintenance dose does reveal insight into patient response. Third, the data we used to define drug usage is in the form of prescription orders. We do not know whether the prescriptions were filled or if the patient took the drug as prescribed. Finally, we do not provide any clinical validation of the predictions presented here; further followup is needed.
Biobanks are an immense resource that allow for pharmacogenetic association testing at an unprecedented scale. Longitudinal clinical data is critical to be able to define drug response phenotypes in order to accurately assess patient response to treatment and ultimately test genetic associations. As access to biobanks continue to expand and more data is available, the ability to perform pharmacogenetic studies at large scale will increase. We believe that these resources offer a promising avenue for discovery and will further advance the field of pharmacogenetics.
5. Acknowledgments
This research has been conducted using the UK Biobank Resource under Application Number 33722. We thank all the participants in the UK Biobank study. Most of the computing for this project was performed on the Sherlock cluster. We would like to thank Stanford University, the PharmGKB resource (NIH HG010615), and the Stanford Research Computing Center for providing the computational resources that contributed to these research results. Thank you to Adam Lavertu who helped develop the ideas that led to this work. The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used for the analyses described in this manuscript were obtained from the GTEx Portal on 10/01/20.
G.M. is supported by the Big Data to Knowledge (BD2K) from the National Institutes of Health (T32 LM012409). R.B.A is supported by NIH/National Institute of General Medical Sciences PharmGKB resource (U24HG010615) and NIH GM102365. RBA is supported by the Chan Zuckerberg Biohub.
Contributor Information
Gregory McInnes, Biomedical Informatics, Stanford University, 450 Serra Mall, Stanford, CA 94305, United States of America.
Russ B Altman, Departments of Bioengineering, Genetics, Medicine, Biomedical Data Science, Stanford University, 450 Serra Mall, Stanford, CA 94305, United States of America.
References
- 1.Lavertu A et al. Pharmacogenomics and big genomic data: from lab to clinic and back again. Hum. Mol. Genet 27, R72–R78 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Relling MV & Klein TE CPIC: Clinical Pharmacogenetics Implementation Consortium of the Pharmacogenomics Research Network. Clin. Pharmacol. Ther 89, 464–467 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Krebs K & Milani L Translating pharmacogenomics into clinical decisions: do not let the perfect be the enemy of the good. Hum. Genomics 13, 39 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.International Warfarin Pharmacogenetics Consortium et al. Estimation of the warfarin dose with clinical and pharmacogenetic data. N. Engl. J. Med 360, 753–764 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramsey LB et al. The clinical pharmacogenetics implementation consortium guideline for SLCO1B1 and simvastatin-induced myopathy: 2014 update. Clin. Pharmacol. Ther 96, 423–428 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wilke RA et al. The emerging role of electronic medical records in pharmacogenomics. Clin. Pharmacol. Ther 89, 379–386 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wei W-Q et al. Characterization of statin dose response in electronic medical records. Clin. Pharmacol. Ther 95, 331–338 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McInnes G et al. Pharmacogenetics at scale: An analysis of the UK Biobank. 2020.05.30.125583 (2020) doi: 10.1101/2020.05.30.125583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reisberg S et al. Translating genotype data of 44,000 biobank participants into clinical pharmacogenetic recommendations: challenges and solutions. Genet. Med (2018) doi: 10.1038/s41436-018-0337-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sudlow C et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 12, e1001779 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bycroft C et al. Genome-wide genetic data on ~500,000 UK Biobank participants. 166298 (2017) doi: 10.1101/166298. [DOI] [Google Scholar]
- 12.Whirl-Carrillo M et al. Pharmacogenomics knowledge for personalized medicine. Clin. Pharmacol. Ther 92, 414–417 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wishart DS et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 46, D1074–D1082 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lane S et al. The population pharmacokinetics of R- and S-warfarin: effect of genetic and clinical factors. Br. J. Clin. Pharmacol 73, 66–76 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liang Y et al. Association of genetic polymorphisms with warfarin dose requirements in Chinese patients. Genet. Test. Mol. Biomarkers 17, 932–936 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jorgensen AL et al. Genetic and environmental factors determining clinical outcomes and cost of warfarin therapy: a prospective study. Pharmacogenet. Genomics 19, 800–812 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Perera MA et al. Genetic variants associated with warfarin dose in African-American individuals: a genome-wide association study. Lancet 382, 790–796 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karczewski KJ et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Irwin MR et al. Major depressive disorder and immunity to varicella-zoster virus in the elderly. Brain Behav. Immun 25, 759–766 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Irwin MR et al. Varicella zoster virus-specific immune responses to a herpes zoster vaccine in elderly recipients with major depression and the impact of antidepressant medications. Clin. Infect. Dis 56, 1085–1093 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iannacone M, Sitia G, Narvaiza T, Ruggeri ZM & Guidotti LG Antiplatelet drug therapy moderates immune-mediated liver disease and inhibits viral clearance in mice infected with a replication-deficient adenovirus. Clin. Vaccine Immunol 14, 1532–1535 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]