

Abstract
Immune response dynamics in coronavirus disease 2019 (COVID-19) and their severe manifestations have largely been studied in circulation. Here, we examined the relationship between immune processes in the respiratory tract and circulation through longitudinal phenotypic, transcriptomic, and cytokine profiling of paired airway and blood samples from patients with severe COVID-19 relative to heathy controls. In COVID-19 airways, T cells exhibited activated, tissue-resident, and protective profiles; higher T cell frequencies correlated with survival and younger age. Myeloid cells in COVID-19 airways featured hyperinflammatory signatures, and higher frequencies of these cells correlated with mortality and older age. In COVID-19 blood, aberrant CD163+ monocytes predominated over conventional monocytes, and were found in corresponding airway samples and in damaged alveoli. High levels of myeloid chemoattractants in airways suggest recruitment of these cells through a CCL2-CCR2 chemokine axis. Our findings provide insights into immune processes driving COVID-19 lung pathology with therapeutic implications for targeting inflammation in the respiratory tract.
Keywords: coronavirus disease 2019, COVID-19, lung immunity, ARDS, tissue resident memory T cells, macrophages, monocytes, single-cell RNA sequencing, cytokines, chemokines
Graphical abstract
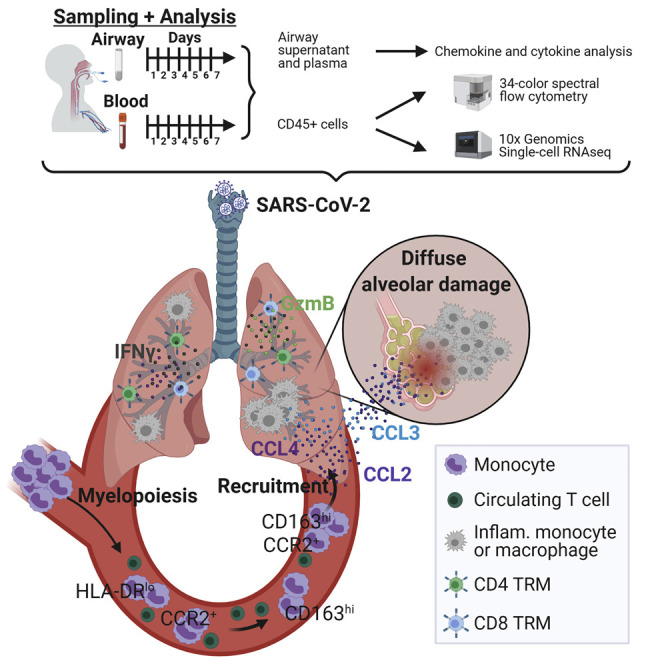
Through longitudinal profiling of paired airways and blood from patients with severe COVID-19, Szabo et al. reveal airway immune responses that correlate with age and outcome. They further identify coordinate roles for T and myeloid cells in the respiratory tract and circulation in perpetuating lung pathology and disease pathogenesis.
Introduction
The respiratory virus severe acute respiratory syndrome-coronavirus-2 (SARS-CoV-2) has resulted in devastating effects to the world’s population due to morbidity and mortality of coronavirus disease 2019 (COVID-19), as well as the life-altering measures implemented to mitigate spread. While the majority of infected individuals (>90%) develop a self-limiting disease and recover, ∼5%–10% of individuals develop severe respiratory disease marked by lung infiltrates and reduced oxygen saturation, which can progress to acute respiratory distress syndrome (ARDS), multi-organ failure, and death (Wu and McGoogan, 2020). To date, well over 2 million individuals have succumbed to COVID-19, including >500,000 in the United States alone. Risk factors for severe COVID-19 include older age and comorbidities such as obesity, diabetes, and hypertension, although younger and previously healthy individuals are also susceptible (Cummings et al., 2020; Davies et al., 2020). For individuals who recover from self-limiting illness, the immune system acts in a coordinated fashion to clear the virus and establish virus-specific immunity (Rydyznski Moderbacher et al., 2020). However, the role of the immune response in the pathogenesis of severe COVID-19 remains unclear, and understanding this phenomenon is urgently required to develop new treatment and prevention strategies.
Immune responses to respiratory virus infection are focused in the lung, where innate immune responses are initiated by tissue-resident alveolar macrophages as well as infiltrating monocytes and granulocytes (Yoo et al., 2013). The initial production of pro-inflammatory cytokines in the lung can precipitate cytokine storms in severe respiratory infections (Teijaro et al., 2011b). Adaptive immune responses mediated by CD4+ and CD8+ T cells involve mobilization of effector T cells to the lung for mediating the clearance of infected cells in situ (Kohlmeier and Woodland, 2009). A proportion of these lung effector T cells develop into tissue-resident memory T cells (TRMs), which are retained in the lung and can mediate rapid protective responses upon viral challenge (Teijaro et al., 2011a; Turner et al., 2014; Turner and Farber, 2014; Wu et al., 2014). In mouse models of SARS-CoV-1 infection, CD4+ TRM-like cells in the airways are required for protection (Zhao et al., 2016). In adult lungs, TRMs are the predominant T cell subset and persist in stable frequencies throughout many decades of life (Kumar et al., 2018; Thome et al., 2014), suggesting a crucial role in protection to respiratory pathogens. The role of resident immune cells, including alveolar macrophages and lung TRM in protection against SARS-CoV-2 infection, and their function in the pathogenesis of severe COVID-19 have yet to be elucidated.
Studies of the immune response to SARS-CoV-2 have examined innate and adaptive immune cells as well as soluble mediators in the circulation of infected individuals, revealing elevated pro-inflammatory cytokines (Hadjadj et al., 2020; Laing et al., 2020) and alterations in immune cell composition (Lucas et al., 2020; Mathew et al., 2020). Robust virus-specific adaptive immune responses have also been identified in the blood of individuals during acute infection and following recovery, including virus-specific CD4+ and CD8+ T cells (Grifoni et al., 2020; Thieme et al., 2020; Weiskopf et al., 2020), and neutralizing antibodies specific for viral proteins (Long et al., 2020; Ni et al., 2020). How these systemic immune responses relate to innate and adaptive immunity in the respiratory tract and their contribution to disease is unclear and difficult to assess.
Here, we sought to define the dynamic immune processes involved in the pathogenesis of severe COVID-19 by high-dimensional phenotypic, transcriptomic, and functional profiling of immune responses in paired airway and blood samples obtained longitudinally from patients with severe COVID-19. This analysis, in conjunction with airway and blood samples from uninfected controls, suggests key functional roles for airway T cells, monocytes, and macrophages associated with disease outcome. T cells in COVID-19 airways exhibited activated and tissue-resident signatures as well as functionally protective profiles, and their frequencies in airways (but not in blood) correlated with younger age and survival. Conversely, airway monocytes and macrophages exhibited hyperinflammatory signatures producing chemokines such as CCL2 and CCL3, while the blood contained aberrant CD163hi and HLA-DRlo monocyte populations expressing the chemokine receptor CCR2. Accordingly, infiltrating myeloid cells in COVID-19 airways likely derive from recruitment from circulation, and their frequency was associated with older age and mortality. Our results provide evidence for myeloid cell-driven lung inflammation in severe COVID-19 and reveal potential targets for reducing lung damage by inhibiting airway-specific inflammatory processes.
Results
Obtaining paired airway and blood samples from severe COVID-19 patients and controls
During the height of the pandemic in New York City, between April and June 2020, we enrolled patients with severe COVID-19 from adult and pediatric intensive care units at New York Presbyterian Hospital (see Method details for enrollment criteria). All of the enrolled patients exhibited clinical features of ARDS requiring mechanical ventilator support and intubation. We obtained daily paired airway and blood samples longitudinally from these patients starting at 24–36 h after intubation, and continuing for up to 10 days during their hospitalization (average 6–7 sample days per patient; sample numbers varied due to patient extubation or death during enrollment). Patients represented a broad age range (14–84 years old) and 8/15 (53%) died during enrollment or soon after (Table S1). Enrolled COVID-19 patients exhibited similar clinical features of disease severity and ARDS independent of outcome as assessed by markers of inflammation, neutrophil levels, Sequential Organ Failure Assessment (SOFA) score (Jones et al., 2009), and PaO2:FiO2 (P:F) ratios (Ranieri et al., 2012). Comorbidities were similar between deceased patients and those who survived, while the median age of survivors was significantly younger than those who succumbed (39 versus 72 years old). Treatments including steroids, anti-interleukin-6 (IL-6) therapy, and remdesivir were given comparably to both groups and were not associated with mortality (Table S1). All of the patients developed high levels of SARS-CoV-2-specific neutralizing antibodies as measured in plasma (Weisberg et al., 2021).
Airway samples were obtained using a saline wash of the endotracheal tube performed daily as part of clinical care, which we have previously shown contains respiratory immune cell populations (Connors et al., 2016, 2018). A total of 141 blood and airway cell preparations from COVID-19 patients were analyzed by high-dimensional spectral flow cytometry in comparison to airway samples obtained from washes of healthy control lungs (Snyder et al., 2019) and blood samples from healthy volunteers (n = 5; see Method details). Airway supernatants and blood plasma at early and late time points were assayed for cytokine and chemokine protein content, and successive samples from four COVID-19 patients were profiled by single-cell RNA sequencing (scRNA-seq) (Figure 1 A; Tables S2 and S3).
Figure 1.

Immune cell composition in airways and blood compartments of COVID-19 patients compared to healthy controls
(A) Schematic diagram showing assays performed on COVID-19 patient airway and blood samples for this study.
(B) Principal-component analysis (PCA) of all COVID-19 samples based on mean marker expression colored by site (left), condition (center), and subjects (right).
(C) UMAP embedding of flow cytometry results from all airway and blood samples combined colored by major cell lineage (top panel), and separated by tissue site in COVID-19 and healthy donor samples (bottom 4 panels) (COVID-19 airway: n = 69, COVID-19 blood: n = 83, healthy airway: n = 5, healthy blood: n = 5).
(D) Boxplots showing the frequency of major immune cell lineages of total CD45+CD66b− cells in COVID-19 and healthy airway (left) and blood (right) samples. Each dot in the boxplot represents an individual patient sample. Statistical significance was calculated using 1-way ANOVA, followed by Tukey’s honestly significant difference (HSD) post-test indicated by∗p ≤ 0.05; ∗∗p ≤ 0.01; and ∗∗∗p ≤ 0.001.
(E) Boxplots showing the frequency of each major cell lineage of total CD45+CD66b− cells in airway (top) and blood (bottom) samples collected longitudinally for COVID-19 and healthy subjects. Color of boxes corresponds to lineage, and each dot is an individual patient sample.
Distinct immune cell composition in the airways and blood of severe COVID-19 patients
Mononuclear cells in airway and blood samples were isolated from severe COVID-19 patients and uninfected healthy controls (see Method details), stained using a 34-marker panel containing antibodies specific for major lineage determinants and markers for differentiation, tissue residence, activation, and function (see Key resources table), and analyzed by spectral flow cytometry (gating strategy for mononuclear cells shown in Figure S1). Principal-component analysis (PCA) of mean marker expression for each sample segregated airway and blood samples along PC2, but not by individual (Figure 1B). While healthy airway samples intermixed with COVID-19 airway samples, healthy blood segregated from COVID-19 blood samples along PC1 (Figure 1B; Table S4). Uniform manifold approximation and projection (UMAP) embeddings of flow cytometry data from all COVID-19 and control samples showed distinct separation of the major lineages (exclusive of neutrophils) into myeloid cells, CD4+ T cells, CD8+ T cells, B cells, and innate lymphoid cells (ILCs) that were predominantly natural killer (NK) cells (Figures 1C and S1B).
Quantitative comparison of immune cell frequencies between severe COVID-19 and healthy samples for each site revealed disease- and site-associated variations. Airways exhibited similar immune cell compositions between COVID-19 and controls, with predominant myeloid cells, lower frequencies of T cells, and negligible B cells and ILCs. By contrast, blood samples demonstrated multiple differences in immune cell composition between COVID-19 and controls, including decreases in CD4+ and CD8+ T cell frequencies and substantial increases in myeloid cell frequencies (Figure 1D), consistent with previous studies (Lucas et al., 2020). Our longitudinal sampling from COVID-19 patients demonstrated that the distinct immune cell compositions in airways and blood were stable across time (Figure 1E). These findings indicate the compartmentalized distribution of immune cells in the airways during homeostasis and severe COVID-19, along with disease-driven changes in overall immune cell composition more highly manifested in blood.
Age-related alterations in airway immune cell subsets are associated with survival from severe COVID-19
Age is a strong independent risk factor for death in COVID-19 (Cummings et al., 2020; Garibaldi et al., 2021). We investigated whether the overall immune cell composition in airways and blood (average of all longitudinal samples obtained for each patient) was altered with age and/or associated with patient outcome. There were statistically significant correlations of age with immune cell frequencies in airways but not with those in blood (Figures 2 A and S2A). In particular, there was a strong positive correlation of airway myeloid cells with age (Spearman’s ρ = 0.65), while airway total and CD4+ T cell frequencies were inversely correlated with age (Spearman’s ρ = −0.56 and −0.69, respectively) (Figure 2A). Notably, mortality from severe COVID-19 was exclusive to older patients in the cohort (Figure 2A).
Figure 2.

Longitudinal assessments of immune cell composition and association with age and outcome
(A) Correlation of immune cell frequencies in the airways (left) and blood (right) with age. Each dot represents the mean immune cell frequency for each patient from all time points, and color denotes patient outcome: survived (blue), deceased (red). Statistical significance was calculated by Spearman correlation (indicated by ρ), with p value shown in each graph.
(B) Daily frequencies of immune cells in the airways (left) and blood (right) for each patient over time stratified by deceased and survived. Solid red and blue lines show mean cell lineage frequency for deceased and survived patients, respectively. Area under the curve (AUC) normalized for number of sampling days was calculated for each patient, and mean AUC is shown in each graph. Statistical significance was calculated by 1-tailed Mann-Whitney U tests (see Method details) for the AUC with Benjamini-Hochberg correction for multiple comparisons and is denoted by ∗p ≤ 0.05.
(C) k-means trajectory clustering analysis of clinical and immune cell frequencies with outcome. Left: representative trajectories of P:F ratio, SOFA score, airway myeloid cells, and airway CD4+ T cells for all patients used for k-means clustering and classification. The true outcome of each patient is denoted by red (deceased) or blue (survived) lines, and inferred clustering by k-means is denoted by solid (deceased) or dashed (survived) lines. Correct clustering denoted by red solid lines and blue dashed lines. Right: patient outcome classification performance of longitudinal k-means clustering for different combinations of immune cell trajectories and clinical parameters (SOFA score and P:F ratio). The percentage of patient outcomes successfully classified as deceased or survived is shown for each parameter measured in airways (gray) or blood (black). Dotted red line indicates classification performance by the P:F ratio and SOFA score.
Our acquisition of longitudinal data enabled the assessment of immune cell and clinical trajectories for their potential associations with disease outcome. Using an area under the curve (AUC) calculation, we found differences in the immune cell frequencies of both airway T cells and myeloid cells, as well as blood myeloid cells of patients who died versus those who survived (Figure 2B). The frequencies of other immune cells in airways (B cells, ILC) and blood (T cells, B cells, and ILCs) did not differ between these groups (Figures 2B and S2A). We further investigated whether the longitudinal trajectories of immune cell composition in the airways or blood were predictive of outcome, relative to corresponding clinical measurements of disease severity (SOFA scores and P:F ratio). We used a k-means clustering approach (see Method details) to classify the temporal dynamics of immune cell frequencies and clinical measurements (Figure 2C). The clinical parameters (SOFA score and P:F ratio) and immune cell frequencies in the blood did not predict outcome (∼50% success rate), whereas both T and myeloid cell frequencies in the airways were predictive of outcome, with a 75% success rate (Figure 2C). These results show that the frequencies of T and myeloid cells in the airways were associated with age and outcome, suggesting specific roles for these cell populations in COVID-19 disease pathogenesis.
Tissue-resident memory T cells predominate in airways and become activated in severe COVID-19
The subset composition of airway and blood T cells in severe COVID-19 versus healthy controls was examined using computational analysis of the high-dimensional flow cytometry data. Multiple markers of T cell differentiation were used to distinguish CD4+ and CD8+ naive (CD45RA+CCR7+) and memory subsets (CD45RA−CCR7−/+CD95+), TRMs (CD69+CD103+/−), regulatory T cells (Tregs; CD4+CD25+CD127−Foxp3+), along with markers of activation (human leukocyte antigen-DR isotype [HLA-DR], programmed cell death protein 1 [PD-1]), co-stimulation (CD27, CD28), senescence and terminal differentiation (CD57, killer cell lectin-like receptor subfamily G member 1 [KLRG1]), and γδ T cells (TCRGD) (Figure 3 A). We used UMAP embeddings to visualize the expression of these multiple markers by the combined airway and blood T cell dataset, showing segregation of the CD4+ lineage on the left, CD8+ on the right, and airway T cells segregating at the top of each embedding (Figure S3A). There was biased expression of CD69, CD103, PD-1, and HLA-DR in the airways, while CCR7, CD45RA, and CD127 expression was more prevalent in the blood (Figure S3A).
Figure 3.

Airway T cells in COVID-19 are dominated by TRM and activated phenotypes
(A) Heatmap displaying expression of markers within PhenoGraph-generated, hierarchical T cell clusters. A total of 24 PhenoGraph clusters were collapsed into 13 definable T cell subsets indicated along the top. Heatmap data are colored by row normalized value for each marker.
(B) UMAP embeddings of T cell subsets (as defined in A) in the blood and airways of COVID-19 patients and healthy controls (first and third rows). Pie charts indicating relative proportions of defined T cell subsets in airways and blood of COVID-19 patients and healthy controls (second and fourth rows).
(C) T cell subset frequencies in airway and blood samples from COVID-19 patients (n = 13) and healthy controls (n = 5). Boxplots show the frequency of the indicated T cell subset for each patient (average of all time points per patient) or healthy controls. Statistical significance was calculated using a 1-way ANOVA with Tukey’s test for multiple comparisons and indicated by ∗p ≤ 0.05; ∗∗p ≤ 0.01; and ∗∗∗p ≤ 0.001.
(D) Expression of T cell activation markers HLA-DR and PD-1 on T cells (total CD3+ cells) from blood and airways of COVID-19 patients and healthy controls. Left: contour plots showing mean expression on HLA-DR and PD-1 on T cells from each cohort, with airway contours colored in blue and blood colored in red. Right: frequency of T cells expressing HLA-DR and PD-1 in the airways and blood for each COVID-19 patient (n = 13; averaged over all time points) or healthy controls (n = 5). Statistical significance was calculated using a 1-way ANOVA with Tukey’s test for multiple comparisons and indicated by ∗∗∗p ≤ 0.001.
To analyze specific subsets of T cells, PhenoGraph clustering based on marker expression yielded 24 clusters, which were coalesced into 13 denoting biological subsets, functional, and/or activation states in the airways and blood (Figure 3A). Compositional analysis shown by UMAP embeddings, corresponding pie charts depicting frequency of each subset in the aggregated data (Figure 3B), and quantification of individual compiled and paired data (Figures 3C and S3B) demonstrated site- and disease-associated alterations in T cell subsets induced by severe COVID-19. T cells in COVID-19 airways were predominantly CD4+ and CD8+ TRMs, as in healthy airways, but showed elevated proportions of activated TRM and TEM (effector memory) subsets expressing HLA-DR, PD-1, and reduced levels of CD127 (Figures 3A–3C). Severe COVID-19 airways also contained reduced Treg frequencies compared to healthy airways (Figures 3B and 3C) and overall increased frequencies of circulating TEM and TCM (central memory) cells in the aggregated analysis (Figure 3B). These results suggest that T cells are being activated in situ as well as recruited to the respiratory tract.
By contrast, T cell subset proportions in COVID-19 blood were similar to those of healthy controls, with comparable frequencies of naive, TCM, and TEM populations for both CD4+ and CD8+ lineages, although some COVID-19 patients showed an increase in blood CD4+ TEM cells (Figures 3B and 3C). Both COVID-19 and healthy blood lacked measurable TRM cells, which were compartmentalized exclusively in the airways, consistent with previous studies showing TRM in airways and lungs but not blood during homeostasis (Kumar et al., 2017; Sathaliyawala et al., 2013; Snyder et al., 2019; Thome et al., 2014). Importantly, activation markers (HLA-DR, PD-1) were highly expressed by T cells specifically in COVID-19 airways, compared to the low level of expression by T cells in COVID-19 blood and in both control sites (Figure 3D). These analyses indicate that activated T cells responding to the ongoing infection in severe COVID-19 were localized to the airways and are not present at high levels in circulation, a finding that is consistent with airway but not blood T cells associated with improved outcome.
Dysregulation of myeloid cells in the airways and blood of severe COVID-19
We applied similar high-dimensional analysis to identify subsets of myeloid populations in aggregated airway and blood samples from severe COVID-19 patients and healthy controls. PhenoGraph clustering of myeloid marker expression identified 21 clusters, which were coalesced into 9 clusters based on phenotypic similarity to classical and intermediate monocyte subsets (based on CD14 and CD16 expression) (Kapellos et al., 2019) and macrophage subsets based on CD163 expression (Bharat et al., 2016), along with key differentially expressed markers HLA-DR, CD86, and CD11c (Figures 4 A and S4A). Data were visualized by UMAP and pie charts (Figure 4B), as with T cells in Figure 3.
Figure 4.

Dysregulated myeloid cell subsets in the blood and airways of COVID-19 patients
(A) Heatmap displaying expression of markers within PhenoGraph-generated, hierarchical myeloid cell clusters. A total of 21 PhenoGraph clusters were collapsed into 9 definable subsets indicated above the heatmap. Heatmap data are colored by value normalized to that of T cell expression as an internal negative control for each sample.
(B) UMAP embedding of 9 myeloid cell subsets in the airways (left) and blood (right) of COVID-19 (top) and healthy controls (bottom), with colors denoting the specific subset as defined in (A) (first and third rows). Pie charts indicating relative proportions of defined myeloid cell subsets in the airways and blood of COVID-19 patients and healthy controls (second and fourth rows).
(C) Boxplots showing compiled frequency of each myeloid subset displayed as an average of all time points collected for COVID-19 samples (CA, COVID-19 airway; CB, COVID-19 blood; HA, healthy airway; HB, healthy blood).
(D–F) Expression of myeloid markers in airways and blood shown as contour plots of expression of indicated markers by myeloid cells in airway (blue contours) and blood (red contours) samples by condition (healthy or COVID-19). Boxplots (to the right or below the contour plots) indicate the percentage of cells within each condition and site that were positive for specific markers. Statistical significance was calculated using a 1-way ANOVA followed by a Tukey HSD and indicated by ∗p ≤ 0.05; ∗∗p ≤ 0.01; and ∗∗∗p ≤ 0.001.
Severe COVID-19 airways and blood showed profound and significant alterations in the compartmentalization, distribution, and phenotypes of myeloid cell subsets compared to healthy controls (Figures 4B and 4C). In healthy controls, the myeloid populations in airways and blood were distinct; airways predominately contained CD163hi macrophages, with a minor CD163lo subset, as well as monocyte populations (CD163hi and intermediate subset) not found in blood (Figures 4B and 4C). Healthy blood contained a preponderance of HLA-DRhi classical monocytes (typically ∼80%), with dendritic cells (DCs) comprising the remaining myeloid cells (Figures 4B and 4C). By contrast, COVID-19 airways exhibited substantial phenotypic overlap with blood myeloid subsets, suggesting infiltration from blood to airways. In COVID-19 airways, conventional CD163hi macrophages (blue) were present, along with an increased frequency of CD163loHLA-DRlo (green) and CD86hiHLA-DRhi cells (yellow) relative to control airways, as well as different monocyte subsets also seen in blood (Figures 4B and 4C). In COVID-19 blood, there were stark differences in monocyte phenotypes, including a predominance of CD163hi and HLA-DRlo monocytes that are not significantly present in healthy blood and additional monocytic subsets that were shared between COVID-19 airways and blood (Figures 4B and 4C). This analysis showed major aberrations in the distribution, phenotype, and composition of myeloid cells in the airways and blood of severe COVID-19 patients.
To further investigate COVID-19-associated features of myeloid cells for each site, we analyzed the coordinate expression of antigen presentation-related molecules HLA-DR and CD86, functional myeloid markers CD64 and CD163, and CD14 and CD16 expression, which define monocyte subsets (Geissmann et al., 2003). While HLA-DR was highly expressed by myeloid cells in COVID-19 and healthy airways, myeloid cells in the blood showed a profound reduction in HLA-DR expression relative to healthy controls, reflecting recent findings and suggesting a dysregulated or immature phenotype (Figure 4D) (Schulte-Schrepping et al., 2020). Conversely, airway myeloid cells in COVID-19 patients exhibited CD163 expression that was similar to levels expressed by CD163hiHLA-DRlo monocytes aberrantly present in blood, which was at a lower level compared to healthy airway CD163hi macrophages (Figure 4E). Lastly, COVID-19 airways exhibited a striking reduction in CD16-expressing monocytes, denoting an intermediate phenotype (CD14+CD16+) compared to healthy controls (Figure 4F). Moreover, all COVID-19 blood monocyte subsets showed a reduction in HLA-DRhi cells relative to their respective healthy controls (Figure S4B). Thus, our results indicate dysregulated monocytes in the blood, alterations of airway macrophages, and evidence for the infiltration of aberrant monocyte populations into the airways, a finding that is consistent with increased airway myeloid cells associated with worse outcomes.
Single-cell transcriptome profiling reveals biased expression of activated and inflammatory signatures in airway immune cells
To dissect the functional states of T and myeloid cells in severe COVID-19, we applied transcriptome profiling by scRNA-seq to daily paired airway and blood samples from 4 patients (total of 126,564 cells; Table S3). This scRNA-seq profiling identified immune cell compositions that were consistent with flow cytometry results, along with additional airway cell types such as epithelial cells and ionocytes (Figures S5A, S5B, S6A, and S6B). Coordinate analysis of the transcriptomic profile of T and myeloid cells between airways and blood revealed how the immune processes in the respiratory tract were connected to those in circulation.
For T cells, scRNA-seq revealed distinct transcriptional profiles expressed by airways compared to circulating T cells, as depicted in UMAP of the aggregated data and heatmaps of differentially expressed genes stratified by individual sample (Figures 5A and 5B). TRM signature genes CXCR6 and ITGA1 were uniquely expressed by airway T cells (Figure 5A; Table S5), which is consistent with our previous scRNA-seq analysis of human TRM cells in airways and lungs (Snyder et al., 2019; Szabo et al., 2019). Naive and TCM cells distinguished by SELL expression were highly enriched in blood, while CCL5 expression indicated that TEM cells were found in both sites (Figure 5A). The top differentially expressed genes between airways and blood revealed that T cells from the airways exhibited a gene signature associated with TRM and tissue T cells (Kumar et al., 2017; Szabo et al., 2019), including the upregulated expression of CXCR6, ITGA1, PDCD1, LGALS, LAG3, and RBPJ compared to blood T cells (Figure 5B; Table S5). Importantly, airway T cells also upregulated expression of multiple genes encoding key cytokines and chemokines, including IFNG, CCL2, and CCL4, compared to those in blood (Figure 5B), which is consistent with an activated and pro-inflammatory state. By contrast, blood T cells exhibited a higher expression of genes associated with quiescence (TCF7, LEF1) (Choi et al., 2015) and lymphoid homing (SELL) (Figure 5B). These results demonstrated the compartmentalization of functionally activated TRM populations exhibiting the signatures of protective T cells in the respiratory tract of severe COVID-19 patients. These findings are also consistent with increased T cells in the airways correlating to better outcomes.
Figure 5.

Inflammatory gene signatures of T cells and myeloid cells are enriched in the airways of COVID-19 patients
T cells, monocytes, and macrophages from the blood and airways of COVID-19 patients were analyzed by scRNA-seq (see Method details).
(A) Separate UMAP embeddings showing gene expression from total T cells obtained from airways and blood of paired samples from 4 patients. UMAP embeddings show sample site origin, subject, and indicated gene expression (based on log2(CPM+1)).
(B) Heatmap showing major differentially expressed genes in airway compared to blood T cells from each individual patient and time point. Data are colored by row Z scored log2(CPM+1) for each sample.
(C) UMAP embeddings of total monocytes and macrophages obtained from airways and blood from 4 COVID-19 patients. UMAP embeddings show sample site origin, patient, and selected gene expression displayed as log2(CPM+1)/maximum.
(D) Heatmap of subset-defining genes, homing receptors, and key inflammatory molecules for monocytes and macrophages in airways and blood from each patient sample. The heatmap shows genes that are not differentially expressed between airways and blood (CD14-FCGR3A) and genes are consistently differentially expressed (ITGAV-TREM2).
Transcriptome analysis of myeloid cells revealed overlapping lineage markers between airways and blood, but elevated expression of inflammatory gene signatures in the airways that were consistent across individuals (Figure 5; Table S6). There was similar expression of lineage-defining genes (CD14, FCGR3A, CD68, CD163) by myeloid cells in airways and blood (Figures 5C and 5D). The expression of tissue macrophage markers MARCO and MRC1 (CD206) (Bharat et al., 2016) and the integrin ITGAV (vitronectin receptor) were specific to airway myeloid cells, while blood myeloid cells expressed transcripts for chemokine, homing, and egress receptors (CX3CR1, CCR2, SELL, S1PR4) (Figures 5C and 5D). For genes associated with myeloid cell function, airway myeloid cells exhibited elevated levels of transcripts for multiple inflammatory mediators, including chemokines for the recruitment of monocytes and macrophages (CCL2, CCL3, CCL4), lymphocytes (CCL18, CCL20, CCL23), neutrophils (CXCL3, CXCL5), complement components (C3, C1QB, C1QC), and matrix metalloproteinases (MMP9, MMP14) implicated in tissue damage in ARDS (Hendrix and Kheradmand, 2017) (Figure 5D). Together, these results demonstrate that airway myeloid cells exhibit a hyperinflammatory state in the expression of multiple cytokines and chemokines for the recruitment of immune cells. In particular, the airway-specific expression of CCL2 transcripts along with the biased expression of its receptor CCR2 by blood monocytes reveals a potential mechanism for the observed infiltration of blood-derived monocytes into the airways in severe COVID-19.
Interferon (IFN)-regulated genes are associated with innate antiviral immunity and may be dysregulated in COVID-19 (Hadjadj et al., 2020). However, there was a negligible expression of genes encoding types I, II, and III IFNs from myeloid cells, epithelial cells, or T cells (Figure S6B), consistent with the lack of SARS-CoV-2 viral sequences (see Method details) in scRNA-seq data from 4 patients. However, transcripts associated with multiple IFN-regulated gene families (i.e., ISG, IFI, IFIT, IRF, MX, and OAS) were detected in both the airway and blood myeloid cells, as well as airway epithelial and T cells (Figure S6B), suggesting a persisting antiviral state in immune cells across compartments. While the expression of IFN-regulated genes may also be triggered by IFNG expressed by airway T cells (Figures 5A and 5B), our scRNA-seq analysis indicates that major inflammatory pathways are propagated by airway myeloid cells and may include the high-level production of chemokines and cytokines for cellular recruitment and promoting tissue damage in situ.
Inflammatory mediators are highly elevated in the airways during severe COVID-19
To investigate whether the transcript expression of inflammatory markers correlated to secreted proteins, we measured cytokine and chemokine levels in airway supernatants and plasma samples from each COVID-19 patient (Table S2), using a microfluidic multiplexed secretome proteomic platform (Farhadian et al., 2020). Overall, we found marked differences in cytokine and chemokine content in the airways compared to plasma as seen in paired samples and at early (day 1) and later (days 3–7) time points (Figures 6 A, S7A, and S7B). Analytes elevated in airways compared to blood include the chemoattractants monocyte chemoattractant protein-1 (MCP-1 [CCL2]), macrophage inflammatory protein-1α (MIP-1α [CCL3]), and MIP-1β (CCL4), and T cell-associated cytokines granzyme B, IL-7, and tumor necrosis factor β (TNF-β) (Figures 6A, 6B, and S7B). By contrast, in the blood CCL2 (MCP-1), CCL3 (MIP-1α), granzyme B, TNF-β, and IL-7 were undetectable, while CCL4 (MIP-1β) was present at variable levels across patients (Figures 6A and 6B). Both blood and airways contained low and/or variable levels of molecules associated with T cell effector function (perforin, IFN-γ, IL-17, and IL-2), additional innate cytokines (IL-6 and IL-8), and TGF-β (Figures 6A, 6B, and S7B). These results show the production of pro-inflammatory chemokines and cytokines in the airway, with only a subset of these detected in the blood.
Figure 6.

COVID-19 airways contain highly elevated levels of myeloid and T cell-derived cytokines
(A) Pairwise comparison of cytokine levels averaged across both time points in airway wash and blood plasma samples collected from 15 patients. Significance indicated as ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001.
(B) Heatmap showing log10(mean X+1) pg/mL cytokine levels averaged across both time points in airway (left) and blood plasma (right) samples for each patient.
(C) Transcript levels for cytokine expression by major cell lineages identified by scRNA-seq for each patient samples indicated by color. Heatmap shows log2(mean CPM+1) gene expression.
To define the cellular origin of the soluble mediators detected in each compartment, we analyzed transcript expression for each of the molecules in Figure 6B by scRNA-seq. Overall, transcript expression of prominent cytokines/chemokines largely correlated to the protein data; airway myeloid cells expressed high levels of CCL2, CCL3, and CCL4 transcripts corresponding to the high levels of the respective proteins in airways, while blood myeloid cells expressed lower or undetectable levels of these transcripts (Figure 6C). Airway and blood myeloid cells also expressed CXCL8 and TGFB1, consistent with the protein data (Figure 6C). In the airways, T cells expressed GZMB, CXCL8, CCL4, PRF, IFNG, and TGFB1 transcripts, which were expressed by blood T cells at lower or variable levels (Figure 6C). Airway epithelial cells expressed predominantly CXCL8 transcripts, as well as lower levels of transcripts for IL-7 and several chemokines (Figure 6C). Overall, these results demonstrate a highly inflammatory environment in the airways of severe COVID-19 patients and establish a chemokine gradient for recruiting immune cells from the circulation to the site of infection, particularly for the monocyte chemoattractants CCL2 and CCL3.
Accumulation of monocytes and macrophages in the lungs of severe COVID-19 patients
We hypothesized that the production of monocyte chemoattractants in the airways may recruit dysregulated blood monocytes into the lung. We therefore examined T and myeloid cells in lung autopsy samples from COVID-19 patients with diffuse alveolar damage, the main pathological finding in COVID-19 ARDS (De Michele et al., 2020), relative to lungs from uninfected organ donor controls (Carpenter et al., 2018) (Table S7). In uninfected lungs, CD4+ and CD8+ T cells were clustered around the airway epithelium, while CD163+ monocytes and macrophages as well as neutrophils (MMP9+) were dispersed in the parenchyma (Figure 7 A, top left). In COVID-19 lungs, there was a marked increase in CD163+ myeloid cells aggregated in the alveolar spaces, critical sites for oxygenation. Also detectable were CD8+ T cells producing granzyme B, consistent with a cytotoxic response (Figure 7A). Quantitative analysis of the lung imaging data showed significant increases in the frequency and density of CD163+ monocytes and macrophages in COVID-19 versus uninfected lungs, while numbers and density of lymphocytes (CD4+ T cells, CD8+ T cells, and B cells) and neutrophils were similar between COVID-19 and control lungs (Figure 7B). However, myeloid cells in COVID-19 airways and blood did not express genes associated with proliferation (Ki67, TOP2A, UBE2C) (Figure 7C). These findings, together with our phenotypic, transcriptomic, and functional analysis of myeloid cells in airways and blood (Figures 4, 5, and 6), strongly implicate myeloid cell recruitment as a major mechanism perpetuating the inflammation and pathogenesis of severe COVID-19.
Figure 7.

COVID-19 lung autopsies exhibit specific and extensive accumulation of monocyte and macrophages relative to control lungs
(A) Lung sections obtained from non-diseased organ donors and autopsy specimens from COVID-19 patients with diffuse alveolar damage were stained with indicated antibodies and analyzed using Vectra. Representative images show staining for CD19 (B cell), CD4 or CD8 (T cells), CD163 (monocytes and macrophages), MMP9 (neutrophil), and granzyme B (cytotoxicity) in the lungs of uninfected controls (left) and COVID-19 patients (right).
(B) Quantitation of immune cell subsets in (A) for uninfected organ donor lungs (n = 3) and COVID-19 lungs (n = 5) as a frequency of total lung cells (top) or density (bottom; cells per mm2 cellular area) using InForm software. Statistical significance calculated by paired t test and indicated by ∗p ≤ 0.05.
(C) UMAP embedding showing expression of genes associated with proliferation by scRNA-seq in monocyte and macrophages derived from airways and blood, as in Figure 5.
Discussion
During the SARS-CoV-2 pandemic, the restoration of normal life is impeded first and foremost by the most severe COVID-19 cases, including debilitating ARDS and its high mortality. For respiratory viruses, protective immune responses as well as immunopathology occur in the lung and respiratory tract. While numerous studies demonstrate characteristic features of innate and adaptive immunity to SARS-CoV-2 infection that are detectable in blood (Kuri-Cervantes et al., 2020; Lucas et al., 2020; Mathew et al., 2020; Rydyznski Moderbacher et al., 2020; Schulte-Schrepping et al., 2020), the role of these COVID-19-specific immune alterations in disease pathogenesis is unclear. Several studies have assessed cellular composition within the respiratory environment in bronchoalveolar lavage samples and lung autopsies (Chua et al., 2020; Damiani et al., 2021; Liao et al., 2020; Veras et al., 2020). However, an understanding of the dynamic immune processes in the respiratory tract in the context of circulating immune cell populations is required to dissect mechanisms of disease pathogenesis.
In this study, we obtained paired respiratory and blood samples from patients with severe COVID-19 longitudinally during the course of intensive care hospitalization. We used high-dimensional profiling by spectral flow cytometry and scRNA-seq, along with multiplex cytokine quantification and immunofluorescence imaging to characterize airway and systemic immune responses in COVID-19 compared to healthy control samples. Our results suggest functional protective signatures for T cells in the respiratory tract, consistent with their associations with younger age and survival from severe COVID-19. Conversely, we identify a key role for airway myeloid cells, primarily macrophages and monocytes, in driving and perpetuating immune cell recruitment and lung inflammation, consistent with their associations with older age and mortality. We further identify a potential role for CCL2 in promoting the recruitment of dysregulated monocytes through a chemotactic axis that could serve as a therapeutic target for reducing lung inflammation and damage in severe COVID-19.
A striking feature of T cells in COVID-19 was their biased activation and functional profiles in airways. T cells in COVID-19 airways were predominantly tissue resident, bearing TRM phenotypes and gene expression profiles similar to control airways. TRM in COVID-19 airways exhibited activation profiles, including surface phenotypes (HLA-DRhiPD-1hiCD127lo) and upregulated expression of transcripts for effector molecules such as perforin, GZMB, and IFNG, features of protective T cell responses in viral infections. The presence of activated TRM in the airways of COVID-19 patients and not in control airways further indicates a virus-directed response. In mouse influenza infection, the presence of activated lung TRM in situ to influenza infection correlates with virus-specific responses (Paik and Farber, 2021; Turner et al., 2014), further supporting the idea that in situ activation is a surrogate for antiviral protective responses. Our results also suggest that T cell activation, as assessed in circulation during active COVID-19 (Mathew et al., 2020; Takahashi et al., 2020), may not reflect T cell responses in the respiratory tract.
Myeloid cells in the airways and blood of COVID-19 patients exhibited profound alterations in phenotype, function, and compartmentalization. Notably, COVID-19 airways contained tissue macrophages with inflammatory signatures expressing multiple genes encoding pro-inflammatory mediators such as CCL2, CCL3, CCL4, CXCL8, MMP, and complement components. Excessive levels of CCL2 (MCP-1), CCL3 (MIP-1α), and CCL4 (MIP-1β) proteins were detected in the airways, but not in blood, further supporting a role of airway myeloid cells in perpetuating inflammatory responses in severe COVID-19. This phenotypic and functional profile of COVID-19 airway monocytes and macrophages shares features with human macrophages in ARDS due to non-infectious causes, including the production of CCL2 and CXCL8, as well as the induction of MMPs and complement (Aggarwal et al., 2014; Morrell et al., 2019). Our results indicate that COVID-19 ARDS shares some key features with ARDS resulting from other infectious or non-infectious causes.
Our coordinate analysis of airway and blood myeloid cells and soluble mediators suggests an analogous role for airway macrophages driving lung damage in COVID-19 ARDS through the recruitment of circulating monocytes. We show that blood monocytes in severe COVID-19 express increased levels of CCR2 transcripts and aberrant CD163hiHLA-DRlo/CD86lo phenotypes compared to healthy blood monocytes. Reduced HLA-DR expression indicative of immature or dysregulated monocytes has been identified in blood myeloid cells in severe COVID-19 (Schulte-Schrepping et al., 2020; Silvin et al., 2020) and may derive from inflammation-induced mobilization from the bone marrow, called emergency myelopoiesis (Schultze et al., 2019; Shi et al., 2011; Venet et al., 2020). We show that monocyte chemoattractants (CCL2, CCL3) are specifically produced in the respiratory tract, while blood monocytes express the cognate receptor CCR2, suggesting recruitment along a CCL2 gradient as found in ARDS models (Aggarwal et al., 2014). Immunofluorescence imaging of lungs from severe COVID-19 patients also shows a striking accumulation of CD163-expressing monocytes and macrophages in the alveolar spaces of the lungs, a key site for blood gas exchange, suggesting their involvement in diffuse alveolar damage observed in COVID-19 pathology (De Michele et al., 2020). Our findings are further consistent with studies of influenza-infected individuals that identified inflammatory monocytes in severe disease (Cole et al., 2017) and increased inflammatory chemokines in the airways, which correlated with airway monocyte infiltration and disease severity (Oshansky et al., 2014).
Our results defining airway immune responses in COVID-19 and their relation to the corresponding immune reactants in blood have profound implications for treating and preventing disease. Treatments targeting systemic inflammation, either globally with steroids or specifically with cytokine blockade, have shown variable efficacy in severe COVID-19 (Della-Torre et al., 2020; Furlow, 2020). Our results suggest that targeting airway-derived cytokines such as CCL2 through CCR2 antagonists or other airway-specific mediators may be more effective in reducing lung damage or even promoting recovery from ARDS in severe COVID-19. Importantly, preventing CCL2-mediated monocyte recruitment to the lungs was an effective strategy to reduce severity in mouse models of influenza infection (Lin et al., 2008, 2011), suggesting a generalized mechanism for respiratory virus-induced lung injury.
Conversely, airway T cells exhibit protective functional signatures and increased proportions are associated with younger age and survival. Interestingly, monitoring airway T cell frequencies was a more robust predictor of outcome than the SOFA score and P:F ratio, which are two of the most commonly used severity-of-illness measures and strongest predictors of mortality in ARDS (Ranieri et al., 2012; Vincent et al., 1998). While these results require validation in larger cohort studies, airway T cell measurements could be a useful biomarker to monitor patients. In addition, promoting lung-localized immune responses could be beneficial in vaccines. The current approved SARS-CoV-2 vaccines target the generation of neutralizing antibodies and are robust strategies for establishing sterilizing immunity (Jeyanathan et al., 2020); however, respiratory targeting could be considered for individuals who are unable to develop effective antibody responses. These cohorts may include the immunocompromised or the elderly, or this strategy could be used as a booster for those at risk of infection due to frequent interactions with others through their living or work situations. A recent pre-clinical study demonstrated that intranasal administration of a recombinant SARS-CoV-2 vaccine promoted lung TRM generation and protection from viral challenge in mice (Hassan et al., 2020).
In summary, our study provides a dynamic view of ongoing respiratory and systemic immunity in severe COVID-19, revealing key roles for airway immune cells in promoting protection and driving lung inflammation. These findings have important implications for how we monitor, treat, and protect from this pandemic and future infectious challenges to the respiratory tract.
Limitations of study
Limitations of this study include a small cohort size and short sampling window (up to 10 days after intubation) during a course of prolonged acute illness. Due to the small sample size and the older age of patients who died, we were not able to separate the role of age and immune cell frequencies with outcomes. However, the strong inverse correlation that we observed between age and lower T cell frequencies suggests that there are age-related changes in airway immunity that may explain the higher mortality observed in older adults with severe COVID-19. Moreover, we only studied respiratory and blood responses in severe COVID-19 and not in more mild manifestations of the disease, and it will be interesting in future studies to incorporate upper respiratory sampling to assess immunity during a resolved infection.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Human HLA-DR BUV395 | BD | G46-6; Cat #564040; RRID:AB_2738558 |
| Anti-Human CD16 BUV496 | BD | 3G8; Cat #612944; RRID:AB_2870224 |
| Anti-Human CD163 BUV563 | BD OptiBuild | GHI/61; Cat #741402; RRID:AB_2870894 |
| Anti-Human CD33 BUV615 | BD OptiBuild | WM53; Cat #751275; RRID:AB_2875289 |
| Anti-Human PD-1 BUV661 | BD OptiBuild | EH12.1; Cat #750260: RRID:AB_2874457 |
| Anti-Human CD56 BUV737 | BD | NCAM16.2; Cat #612766; RRID:AB_2813880 |
| Anti-Human CD64 BUV805 | BD OptiBuild | 10.1; Cat #742023; RRID:AB_2871319 |
| Anti-Human CCR7 BV421 | BioLegend | G043H7; #353208; RRID:AB_11203894 |
| Anti-Human CD86 SB436 | eBioscience | IT2.2; Cat #62-0869-42; RRID:AB_2637395 |
| Anti-Human CD28 eFluor 450 | eBioscience | CD28.2; Cat #48-0289-42; RRID:AB_11151697 |
| Anti-Human CD8 BV480 | BD | RPA-T8; Cat #566121; RRID:AB_2739523 |
| Anti-Human CD20 Pacific Orange | Invitrogen | HI47; Cat #MHCD2030; RRID:AB_10375578 |
| Anti-Human CD3 BV510 | BioLegend | UCHT1; Cat #300448; RRID:AB_2563468 |
| Anti-Human CD45RA BV570 | BioLegend | HI100; Cat #304132; RRID:AB_2563813 |
| Anti-Human CD25 BV605 | BioLegend | BC96; Cat #302632; RRID:AB_11218989 |
| Anti-Human CD27 BV650 | BioLegend | O323; Cat #302828; RRID:AB_2562096 |
| Anti-Human CD69 BV711 | BioLegend | FN50; Cat #310944; RRID:AB_2566466 |
| Anti-Human CXCR5 BV750 | BD OptiBuild | RF8B2; Cat #747111; RRID:AB_2871862 |
| Anti-Human CD335 BV785 | BioLegend | 9E2: Cat #331946; RRID:AB_2810509 |
| Anti-Human CD103 BB515 | BD | Ber-ACT8; Cat #564578; RRID:AB_2738852 |
| Anti-Human CD66b FITC | BioLegend | G10F5; Cat #305104; RRID:AB_314496 |
| Anti-Human CD14 Spark Blue 550 | BioLegend | 63D3; Cat #367148; RRID:AB_2832724 |
| Anti-Human CD45 PerCP | BioLegend | HI30; Cat #304026; RRID:AB_893337 |
| Anti-Human CD57 PerCP-Cy5.5 | BioLegend | HNK-1; Cat #359622; RRID:AB_2565930 |
| Anti-Human TCR gamma/delta PerCP-eFluor 710 | eBioscience | B1.1; Cat #46-9959-42; RRID:AB_2573926 |
| Anti-Human CD138 PE | BioLegend | MI15; Cat #356504; RRID:AB_2561878 |
| Anti-Human CD4 YG584 | Cytek | SK3; Cat #R7-20041; RRID:AB_2885083 |
| Anti-Human CD123 PE-CF594 | BioLegend | 6H6; Cat #306034; RRID:AB_2566450 |
| Anti-Human CD95 PE-Cy5 | BioLegend | DX2; Cat #305610; RRID:AB_314548 |
| Anti-Human CD11c PE-Cy7 | BioLegend | 3.9; Cat #301608; RRID:AB_389351 |
| Anti-Human CD19 Spark NIR 685 | BioLegend | HIB19; Cat #302270; RRID:AB_2832581 |
| Anti-Human CD127 APC-R700 | BD | HIL-7R-M21; Cat #565185; RRID:AB_2739099 |
| Anti-Human KLRG1 APC/Fire 750 | BioLegend | SA231A2; Cat #367718; RRID:AB_2687392 |
| Anti-Human FoxP3 Alexa Fluor 647 | BioLegend | 259D; Cat #320214; RRID:AB_492984 |
| Anti-human CD19 | Leica | BT51E; Cat #CD19-163-L-CE |
| Anti-human CD8 | Leica | 4B11; Cat #CD8-4B11-L-CE |
| Anti-human CD163 | Leica | 10D6; Cat #CD163-L-CE |
| Anti-human CD4 | Abcam | EPR6855; Cat #ab133616; RRID:AB_2750883 |
| Anti-human GzmB | Leica | 11F1; Cat #GRAN-B-L-CE |
| Anti-human MMP9 | Santa Cruz Biotechnology | 2C3; Cat #sc-21733; RRID:AB_627959 |
| Anti-human CD66b Biotin | BioLegend | G10F5; Cat #305120; RRID:AB_2566608 |
| Anti-human CD235b Biotin | BioLegend | HIR2; Cat #306618; RRID:AB_2565773 |
| Chemicals, peptides, and recombinant proteins | ||
| LIVE/DEAD Fixable Blue Dead Cell Stain | Thermo Fisher Scientific | Cat #L23105 |
| DAPI | BioLegend | Cat #422801 |
| True-Stain Monocyte Blocker | BioLegend | Cat #426102 |
| TrueStain FcX | BioLegend | Cat #422302 |
| RPMI 1640 | Corning | Cat #10-040-CM |
| Ficoll-Paque PLUS | GE | Cat #17-1440-03 |
| RosetteSep Granulocyte Depletion Cocktail | STEMCELL Technologies | Cat #15624 |
| Benzonase | Millipore Sigma | Cat #E1014-5KU |
| DPBS | Corning | Cat #20-030-CV |
| EDTA | Corning | Cat #46-034-CI |
| Foxp3 / Transcription Factor Staining Buffer Set | eBioscience | Cat #00-5523-00 |
| 10% Zinc Formalin | Anatech | Cat # 170 |
| Vectashield Hard Set mounting media | Vector Labs | Cat #H1600 |
| Opal 7-Color IHC Kits | Akoya Biosciences | Cat #NEL811001KT |
| Critical commercial assays | ||
| Chromium Next GEM Single Cell 3′ Reagent kit v3.1 | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/library-prep |
| Deposited data | ||
| scRNA-seq Data | This article | https://www.covid19cellatlas.org/index.patient.html |
| Tables S5, S6, and S8 | This article | http://dx.doi.org/10.17632/fm7v43jrbk.4 |
| Software and algorithms | ||
| Python version 3.7 | Python (2020) | https://www.python.org/downloads/release/python-370/ |
| Anaconda version 4 | Anaconda (2020) | https://www.anaconda.com/products/individual |
| Rstudio version 1.2.1335 | RStudio (2019) | https://www.rstudio.com |
| R version 3.5 | R Foundation for Statistical Computing (2017) | https://www.R-project.org |
| inform Version 2.3 | Perkin Elmer, Akoya Biosceinces | https://www.akoyabio.com/phenoptics/software/inform-tissue-finder/ |
| FlowJo V 10.7 software | Tree Star | https://www.flowjo.com/ |
| Other | ||
| Dead Cell Removal Microbeads | Miltenyi Biotec | Cat #130-090-101 |
| BioMag Plus Streptavidin | Bangs Labs | Cat #BP628 |
Resource availability
Lead contact
Further information and requests for reagents should be directed to and will be fulfilled by lead author Donna L. Farber (df2396@cumc.columbia.edu)
Materials availability
This study did not generate new unique reagents.
Data and code availability
The scRNA-seq data for each sample including count matrices, normalized counts, metadata, cell annotations, and UMAP embeddings are available on the COVID-19 Cell Atlas along with interactive visualizations (https://www.covid19cellatlas.org/index.patient.html). Code for clustering of scRNA-seq data analysis is available at www.github.com/simslab/cluster_diffex2018. Additional Supplemental Items are available from Mendeley Data at http://dx.doi.org/10.17632/fm7v43jrbk.4
Experimental model and subject details
Human samples
We recruited patients from CUIMC/NYP and Morgan Stanley Children’s Hospital of NY with severe COVID-19 and ARDS (n = 15) who tested positive for SARS-CoV-2 by polymerase chain reaction (PCR) from nasopharyngeal swabs (Tables S1 and S2). Blood and airway sampling began within 24-36 hours for all patients. ARDS was defined by clinical consensus criteria including infiltrates on chest radiograph and a PaO2/FiO2 (P/F) ratio of less than 300, or pediatric criteria equivalent (Khemani et al., 2015; Ranieri et al., 2012). Exclusion criteria included patients with cancer, transplant recipients, immunodeficiencies, and existing bacterial infection. Sequential Organ Failure Assessment (SOFA) scores were calculated on all hospitalized patients using previously validated adult and pediatric score tools to provide additional clinical insight into patient disease severity (Matics and Sanchez-Pinto, 2017; Singer et al., 2016; Vasilevskis et al., 2016). All patients and samples in this study were enrolled on protocols approved by the Institutional Review Board at CUIMC. Due to the limitations placed on direct contact with infected patients and a need to conserve personal protective equipment, verbal informed consent was obtained from surrogates of critically ill COVID-19-ARDS patients.
Paired airway and blood samples were obtained from COVID-19 patients enrolled as above daily or every second day up to 7-10 days; the number of samples obtained varied due to patient death or extubation. Airway samples were collected from saline washes of the endotracheal tube during routine clinical care and blood samples were obtained by venous puncture. Healthy blood was obtained from 5 adult volunteers 31-57 years.
Control, uninfected lung tissues were obtained from deceased organ donors (who died > 2 years before the pandemic) as part of organ acquisition for clinical transplantation through an approved protocol and material transfer agreement with LiveOnNY as described previously (Carpenter et al., 2018; Dogra et al., 2020). Donors were free of cancer, chronic diseases, seronegative for hepatitis B, C, and HIV, and negative for SARS-CoV-2 by PCR (Table S7). Use of organ donor tissues does not qualify as “human subjects” research, as confirmed by the Columbia University IRB as tissue samples were obtained from brain-dead (deceased) individuals.
Method details
Processing of blood samples and isolation of PBMCs from COVID-19 patients
Whole blood collected in heparinized vacutainers was centrifuged at 400 x g for 10 min at room temperature (RT) to isolate plasma, which was then stored at −80 °C for subsequent analysis. PBMCs were isolated using Ficoll-Paque PLUS (GE) density gradient centrifugation in a Biosafety Level 2+ facility. To remove neutrophils, blood was incubated with RosetteSep Granulocyte Depletion Cocktail (StemCell Technologies, Inc.), diluted 1:3 in room temperature DPBS (Corning), layered over Ficoll-Paque PLUS in 50mL conical tubes, and centrifuged for 20 min at 1,200 x g. The PBMC layer was isolated according to the manufacturer’s instructions. Cells were washed twice with DPBS before counting with the automated NucleoCounter NC-3000 cell counter (ChemoMetec).
Processing of airway samples and isolation of airway MNCs from COVID-19 patients
Both the cellular content and supernatants were collected from airway samples. To collect airway supernatants, DPBS was added 1:1 directly to airway samples and centrifuged at 400 x g for 10 min at RT. The resulting supernatants were stored at −80 °C for subsequent analysis. To isolate airway MNCs, samples were treated with Benzonase (Millipore Sigma), purified through 100 μm filters, and centrifuged on a density gradient using Ficoll-Paque PLUS. The MNC layer was isolated according to the manufacturer’s instructions. Cells were washed twice with DPBS before counting with the automated NucleoCounter NC-3000 cell counter.
Cell preparation for scRNA-seq, library generation and sequencing
Airway and blood MNC populations were isolated as above, and the remaining neutrophils and red blood cells were removed by incubating samples with biotinylated anti-CD66b and anti-CD235ab antibodies, and depleting antibody-bound cells with streptavidin-coated magnetic beads (Bangs Labs). Dead cells were subsequently removed using the Dead Cell Removal kit (Miltenyi Biotec). The Next GEM Chromium Controller (10x Genomics) and Chromium Next GEM Single Cell 3′ Reagent kit v3.1 (10x Genomics) was used for co-encapsulation and scRNA-seq library construction as per manufacturer’s suggested protocols. Libraries were sequenced on an Illumina NovaSeq 6000, targeting ∼300M raw reads per sample (∼60,000 raw reads per cell). Sample details and number of cells sequenced in each are shown in Table S3.
Isolation of airway washes from non-diseased lungs
Uninfected lungs were obtained from deceased organ donors as described above. Airway washes were obtained by flushing out the major airway with 60 mL saline as described (Snyder et al., 2019). Cells were pelleted by centrifugation, resuspended in DPBS and stained with antibodies for flow cytometry.
High-dimensional flow cytometry
For high parameter analysis using the Cytek Aurora panel, 5x106 cells from each site were stained in 5 mL U-bottom tubes in the dark using antibody panels as described in the Key resources table. Briefly, cells were first washed with DPBS, re-suspended with LIVE/DEAD Fixable Blue Dead Cell stain (Thermo Fisher Scientific) and incubated at RT in the dark for 30 min. Cells were washed with cold FACS-buffer (DPBS + 2% FBS + 0.1 mM EDTA) and re-suspended with human TrueStain FcX and True-Stain Monocyte Blocker (BioLegend) and incubated in the dark for 10 minutes. Next, cells were incubated with a cell-surface marker staining cocktail as described in Key resources table at 4°C for 30 min. Stained cells were washed twice with cold FACS-buffer to remove unbound antibodies. For intracellular staining, cells were fixed, permeabilized and stained with intracellular antibodies (Key resources table) using a Foxp3 Transcription Factor Staining Buffer Set (eBioscience) as per manufacturer’s suggested protocols. Cells were incubated in the dark for 30 minutes with intracellular antibodies and washed twice. Data was collected on 5-laser Cytek Aurora flow cytometer (Cytek Bio).
Highly-multiplexed CodePlex chip secretome proteomics
Cryopreserved tracheal washes and plasma were thawed at room temperature for 30-60 minutes and mixed well by pipetting up and down prior to loading. An aliquot of 5.5 μL of each sample was pipetted into each macrochambers of a CodePlex chip pre-patterned with a complete copy of a 23-plex antibody array. 2% BSA/PBS was used as background control. The chip was then loaded into an IsoLight automation system and various proteins were measured by fluorescence ELISA and analyzed by the IsoSpeak software using the IsoPlexis Human Adaptive Immune Panel: GM-CSF, Granzyme B, IFN-γ, IL-10, IL-13, IL-15, IL-17A, IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IP-10, MCP-1, MIP-1α, MIP-1β, Perforin, sCD137, TGF-β1, TNF-α, TNF-β.
Multispectral staining and imaging of lung tissue
Representative samples of lung tissue 0.5–1.0 cm in thickness were obtained from organ donors and autopsy cases of individuals diagnosed with COVID-19 and found on post-mortem exam to have pathological findings consistent with diffuse alveolar damage (Table S7). Uninfected lung sections were obtained from deceased organ donors who were brain dead due to trauma > 2 years prior to the pandemic. None of the donors used for the control staining in Figure 7 showed pathological evidence of lung damage or diffuse alveolar damage.
Samples were fixed in 10% formalin (Anatech Ltd.) for 48 hours prior to dehydration and embedding in paraffin. These lung samples were sectioned at 5-mm thickness and stained using 7-color multispectral Opal reagents (Akoya Biosciences) as previously described (Gartrell et al., 2018; Weisberg et al., 2019). The multiplex panel included DAPI for nuclear counterstaining, CD4 (1:150 dilution), CD8 (1:600 dilution), CD163 (1:200 dilution), granzyme B (GzmB) (1:200 dilution), CD19 (1:50 dilution), MMP9 (1:900 dilution). Single controls and an unstained slide were stained with each group of slides. After staining, the sections were mounted in Vectashield Hard Set mounting media (Vector Labs, Cat#H1600) and stored at 4°C for up to 48 hours prior to image acquisition. Multispectral imaging and acquisition at 20x magnification (numerical aperture 0.75) was performed using the integrated Vectra 3 automated quantitative pathology imaging system (PerkinElmer) as previously described (Weisberg et al., 2019). Images were analyzed using inForm software (PerkinElmer/Akoya Biosciences). Representative areas (10-30) from each donor were chosen for quantitative analysis.
Flow cytometry analysis
Flow cytometry data was pre-gated to exclude any doublets, dead cells and CD66b+ granulocytes using FlowJo v 10.7 (Tree Star) (Figure S1). Cleaned data was exported as .fcs files with compensated parameters and analyzed further and visualized using a Python (v3.7) (Python Software Foundation. Python Language Reference, version 2.7.) computational pipeline. In brief, first the data was filtered to remove any noise using quantile gates; events that fell below 0.01% of marker expression intensity were removed from the sample. Following initial filtering, data from COVID-19 and healthy samples was merged after subsetting 60,000 events from each COVID-19 airway and blood sample, 120,000 events from each healthy blood sample, and all events from healthy airway samples. Any sample with fewer than 1000 events was removed from further analysis. The merged dataset was transformed using arcsinh function from Python numpy library (van der Walt et al., 2011) after manually adjusting the cofactor for each marker. Following normalization, the dataset was normalized on a 0-1 feature scale for each marker using MinMaxScaler function from Python scikit-learn library (Pedregosa et al., 2011). The cleaned, transformed and scaled dataset was used to run the first round of Uniform Manifold Approximation and Projection (UMAP) (McInnes et al., 2018) dimensionality reduction to remove any residual granulocyte contamination identified as clusters of CD45loCD66b+ cells. The resulting dataset was used for downstream analysis.
We ran PCA analysis at sample level using mean expression of markers in each sample, PCA loadings provided in Table S3. Next, we ran UMAP dimensionality reduction (k = 60) on this dataset using 10 lineage-defining markers (CD14, CD16, CD19, CD3, CD33, CD4, CD8, CD64, CD56, CD335). The data were projected in 2-dimensions using UMAP embeddings and clusters of major immune cell types (CD4 and CD8 T cells, B cells, NK/ILC and Myeloid cells) were identified based on expression of lineage defining markers (Figure S2A). Frequency of the major lineages was calculated for each donor sample.
For immune cell lineage and age analyses (Figures 2A and S2A), the average frequency values across all time points for each patient was calculated and plotted relative to age, showing spearman’s rho values with corresponding p value. For longitudinal analyses of immune cell frequencies between deceased and surviving patients (Figure 2B), area-under-the-curve (AUC) was calculated for each patient and divided by the number of sampling days (omitting COV007 who was sampled for one day only). Statistical significance between deceased and survived groups was calculated based on AUC using a one-tailed Mann-Whitney U test, testing the specific hypothesis that increased protective responses (T cells) are associated with positive outcomes (D < S) and increased pro-inflammatory responses (myeloid cells) would be associated with negative outcomes (D > S). We applied a Benjamini-Hochberg correction for multiple comparisons.
For lineage specific analysis (T cells, myeloid cells), we ran UMAP dimensionality reduction and subsequent Phenograph clustering (Levine et al., 2015) on each lineage specific dataset using cell subset defining markers selected based on literature review. Markers used for T cell UMAP are shown in Figure S4A. Myeloid cell markers are shown in Figure S5A. Major cell subset clusters were identified and functionally similar subsets were coalesced and manually annotated. Heatmaps were generated for average marker expression in each of the annotated clusters. Data are presented as row normalized expression of marker across all clusters. All graphs were generated using the Python matplotlib and seaborn libraries (Hunter, 2007).
Classifying patient outcomes using longitudinal K-means clustering
Patients were partitioned into two groups using a longitudinal K-means algorithm applied to the trajectories of the frequencies of myeloid, B cell, CD4 and CD8 T cell and ILCs in blood or airways in addition to the trajectories of SOFA score and P/F ratio. The proximity of two patients’ trajectories was defined using the sum of the squared Euclidean distances between their subset frequencies at each location at each time point, after normalizing each subset frequency across all patients and time points. The clustering outcome was robust to this definition of distance, giving identical results when performed using log- or logit-transformed frequencies. Classification performance was defined as the percentage of patients that were assigned to the correct outcome cluster (deceased or survived). We compared the abilities of different immune cell subsets to distinguish patient outcome by repeating the clustering analysis on different combinations of trajectories. Greater classification performance indicated increased power to identify patient outcomes. Clustering analyses were conducted in R version 3.5.3 using the kml3d package, version 2.4.2, and the results were visualized using the ggplot2 package.
Processing of scRNA-seq data
We used kallisto v0.46.2 in “BUS” mode to pseudo-align the raw reads for each sample to a merged human GRCh38 (Ensembl 93)/SARS-CoV-2 transcriptome (Bray et al., 2016; Kim et al., 2020; Melsted et al., 2019a; Melsted et al., 2019b). To correct for index swapping, which can occur on the Illumina NovaSeq 6000, we applied the algorithm of Griffiths et al. (2018) to the equivalence classes obtained from kallisto pseudo-alignment. We generated a raw count matrix from the swap-corrected BUS file using bustools v0.40.0 (Melsted et al., 2019b), filtered using the EmptyDrops algorithm (Lun et al., 2019), and removed all cells with mitochondrial pseudo-alignment rates > 20% or counts per gene greater than two standard deviations above the mean for each sample.
scRNA-seq cell annotation
We merged the scRNA-seq data from all of the airway samples and identified likely markers of specific subpopulations using the previously described drop-out score method for finding genes that are detected in fewer cells than expected given their expression level (Levitin et al., 2019; Szabo et al., 2019). Next, we computed a cell-by-cell Spearman’s rank correlation matrix using these putative marker genes. Using this matrix, we constructed a k-nearest neighbor’s graph (k = 20) as input for Louvain community detection as implemented in Phenograph (Levine et al., 2015). To associate the resulting clusters with major cell populations in the airway, we examined the statistical enrichment of the following marker genes in each cluster using the binomial test as described in Shekhar et al. (2016): T cells (CD3D, TRAC, TRBC1, TRBC2, TRDC, TRGC1, TRGC2), NK cells (NCAM1), myeloid cells (CD14, FCGR3A, CD163), epithelial/club/goblet cells (EPCAM, SCGB1A1, MUC5B, KRT78), ionocytes (CFTR), neutrophils (CD16B), plasma cells (CD19, JCHAIN), B cells (CD19, MS4A1), platelets (ITGA2B, PF4), mast cells (KIT), dendritic cells (FCER1A, CD1C) and red blood cells (HBA1, HBA2, HBB).We identified clusters as likely multiplets based on co-expression of multiple marker sets (e.g., clusters enriched in both CD14 and CD3D were marked as likely T cell / myeloid cell multiplets). All of the cells in these clusters were marked as multiplets. UMAP embeddings for all cells from each patient across all time points for airways and blood are shown in Figure S5.
In the main text, we present focused analyses on myeloid cells, T cells, and epithelial/club/goblet cells from the airway. To further refine our annotation, we re-clustered the cells annotated as each of these three cell types separately using the methods described above. We then re-analyzed the enrichment of cell type-specific markers in the resulting new clusters. As expected, this focused re-analysis of each of these three major populations identified additional putative multiplet clusters and cells that we likely mis-clustered in the initial merged analysis. We conducted two rounds of re-clustering for each of these three major cell types to produce a refined annotation. The top of Figure S6A shows a gene expression heatmap for key marker genes in the merged airway dataset colored by patient and cell type annotation. We repeated the above procedure for the merged blood scRNA-seq profiles including a focused re-clustering analysis of the cells that we originally annotated as myeloid and T cells for refinement. The bottom of Figure S6A shows a gene expression heatmap for key marker genes in the merged blood dataset colored by patient and cell type annotation.
scRNA-seq visualization and differential expression analysis
We generated merged UMAP embeddings for the blood and airway T cells (Figure 5A) and the blood and airway myeloid cells (Figure 5C). In each case, we first identified genes that were likely to contaminate either the myeloid or T cell profiles in either the blood or airway to avoid including them in any of our downstream clustering, visualization, or differential expression analysis. We conducted pairwise differential expression analysis between all of the cells annotated as a cell type-of-interest (e.g., myeloid) and each group of cells with a different annotation for the blood and airway from each patient separately. For each pairwise comparison, we randomly subsampled the two groups of cells to the same cell number. Next, we randomly subsampled the molecular counts for cells in the two groups such that they have the same average number of counts per cell. We then generated a merged count matrix for the two groups and applied the pooled normalization technique from the scran package of Lun et al. (2016) using the computeSumFactors function. Finally, we conducted a gene-by-gene, non-parametric differential expression analysis using the Mann-Whitney U-test as implemented with the function mannwhitneyu from the Python package scipy. We corrected the resulting p values for false discovery using the Benjamini-Hochberg Procedure with the function multipletests from the Python package statsmodels. Using the results of pairwise differential expression analysis, we generated a blacklist of genes for a given cell type by taking any gene with at least 10-fold enrichment in a different cell type with FDR < 0.001 in at least two patients. We removed all genes with any enrichment in the cell type-of-interest with FDR < 0.001 in any patient to avoid eliminating patient-specific markers of the cell type-of-interest. The final blacklists for blood and airway myeloid and T cells appear in Table S8.
For both myeloid and T cells, we took all of the cells in the dataset that we had annotated as each of these two cell types and used the drop-out score method described above to generate a list of putative, highly variable marker genes for each patient. Next, we generated a merged count matrix across all patients for a given cell type, which we normalized using the pooling method of Lun et al. (2016) as described above. We then generated a log-normalized submatrix (log2(counts per million +1)) containing the union of the marker gene sets identified for each patient after removing genes on the airway and blood blacklists for the cell type-of-interest. Using the PCA function in the Python package scikit-learn, we decomposed this submatrix into its principal components. We used the 10 principal components with the largest eigenvalues as input to the scRNA-seq batch correction algorithm Harmony (Korsunsky et al., 2019). We made the function HarmonyMatrix aware of only the first 10 principal components and the patient identifiers for each cell. Finally, we computed a two-dimensional embedding using the Python implementation of the Uniform Manifold Approximation and Projection (UMAP) algorithm (McInnes et al., 2018) and the Pearson correlation matrix of the Harmony-corrected principal components. These embeddings appear in Figure 5.
For the differential expression analysis between blood and airway myeloid cells and between blood and airway T cells, we used the Mann-Whitney U-test approach described above. We removed genes on the blacklists described above for each cell type prior to subsampling, normalization, and statistical testing. We also restricted this analysis to protein-coding genes and removed all T cell receptor and immunoglobulin variable regions. We performed differential expression separately on each pair of matched airway and blood samples (there are 12 patient time points for which we have matched samples). Stringent criteria were used to select the differentially expressed genes displayed in the heatmaps in Figure 5. For the myeloid cell heatmap in Figure 5, a gene had to be differentially expressed with a fold-change of at least 4 in either direction and FDR < 0.001 in at least 9 of the 12 matched sample pairs. For the T cell heatmap in Figure 5, we applied the first two criteria, but required them in only 6 of the 12 matched sample pairs. Results for all of the pairwise differential expression analyses comparing airway and blood T and myeloid cells can be found in Tables S5 and S6, respectively.
Analysis and visualization of cytokine data
Non-log transformed cytokine expression data from both time points was averaged for airway and plasma from each patient and used for paired-site analysis. Cytokine expression data from early and late time points was log10 normalized and visualized as boxplots overlaid with individual data points. The log10 normalized data was averaged across both time points and used to generate heatmaps for cytokine expression across individual patient samples. Graphs were generated using the Python matplotlib and seaborn libraries (Hunter, 2007).
Lung tissue imaging analysis
Tissue segmentation was performed using inForm software on 10-30 representative fields (Version 2.3, PerkinElmer/Akoya Biosciences). Immune cell constituents within each tissue segment were defined by the DAPI nuclear counterstain to define the nucleus of each cell, with each associated membrane detected via presence of a specific stain (CD3, CD19, CD4, GzmB, MMP9 and/or CD163). Cell segmentation was adjusted as previously described to accurately locate all cells and minimize nuclear hypersegmentation and hyposegmentation (Weisberg et al., 2019). Cells were then phenotyped by training the phenotyping algorithm of inForm software, identifying: monocytes and macrophages (CD163+ magenta cells), T cells (CD4+ cyan cells and CD8+ orange cells), B cells (CD19+ yellow cells), neutrophils (MMP9+). The cell segmentation data summary provided densities and numbers of each cell type in the lung tissue segments and the full cell segmentation data file provided the X and Y coordinates of each phenotyped cell.
Quantification and statistical analysis
Differences in means between two sample groups were compared using two-tailed t tests or two-tailed Mann-Whitney U tests, except when testing specific directed hypotheses (one-tailed). Pearson correlations were used to correlate age and immune cell frequencies. Multiple group comparisons were done using ANOVA followed by Tukey’s HSD post-test. For comparing paired airway to blood values we used paired two tailed t tests. All statistical tests were run using custom scripts based on Python sciPy library (Virtanen et al., 2020) or R (version 3.5.3.). P-values below 0.05 were considered as statistically significant. For all figures ∗∗∗ denotes p value < 0.001, ∗∗ denotes p value < 0.01, and ∗ denotes p value < 0.05.
Acknowledgments
We wish to express our gratitude to the medical ICU nurse champions Cora Garcellano, Tenzin Drukdak, Harriet Avila Raymundo, Lori Wagner, and Ricky Lee, who led the efforts to obtain patient samples for the severe COVID-19 patients; to Evelyn Hernandez and Lorena Gomez for their roles as clinical coordinators; and to the nurses and clinical staff in the Pediatric Intensive Care Unit of MSCHONY. We acknowledge the dedication, commitment, and sacrifice of the other nurses, providers, and personnel who helped care for these patients during the COVID-19 crisis. We acknowledge the suffering and loss of our COVID-19 patients and of their families and our community. We gratefully acknowledge the generosity of the donor families and the exceptional efforts of LiveOnNY transplant coordinators and staff for the organ donor lungs. This work was supported by NIH grants AI128949 and AI06697 awarded to D.L.F., a Chan Zuckerberg Initiative COVID-19 grant to D.L.F. and P.A. Sims, and AI093870 and AI150680 awarded to A.J.Y. P.D. was supported by a CRI-Irvington Postdoctoral Fellowship, and P.A. Szabo was supported by a Canadian Institutes of Health Research Fellowship. T.J.C. is supported by NIH K23 A1141686, and S.P.W. is supported by NIH K08 DK122130. Research reported in this publication was performed in the Human Immune Monitoring Core, the Columbia Single Cell Analysis Core, and the Sulzberger Columbia Genome Center, which are supported by NCI cancer center support grant P30CA013696. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank Eldad Hod for the use of his laboratory for sample processing, and Carly Ziegler and Dr. Alex Shalek of MIT for sharing their merged human SARS-CoV-2 genome and transcriptome annotation.
Author contributions
P.A. Szabo, S.B.W., J.I.G., and P.D. processed samples, designed and optimized high-dimensional flow cytometry panels, analyzed data, made figures, and wrote the manuscript. P.A. Szabo and S.B.W. processed samples for scRNA-seq profiling and encapsulation using 10X Chromium; I.K. prepared and sequenced the 10X libraries. P.D. designed the Python pipeline for flow cytometry data. M.R.B. monitored and consented the intensive care unit (ICU) patients, oversaw the clinical data analysis, and collected samples. T.J.C. obtained and maintained the institutional review board (IRB) protocols, consented patients, and processed samples. M.M.L.P., R.M., E.I., and M.C. obtained and processed the patient samples. S.E.M., C.P., and A.J.Y. statistically analyzed the longitudinal data and performed the k-means clustering analysis. J.D.-P. captured and analyzed the patient data. J.Z., M.S., and S.M. performed the cytokine analysis of airway supernatants and blood plasma. S.P.W. planned, designed, and analyzed the lung autopsy imaging experiments. A.S. provided the lung autopsy samples and associated data. A.K. performed the immunohistochemistry of lung autopsies. P.A. Sims planned the scRNA-seq experiments, analyzed the data, and wrote and edited the manuscript. D.L.F. oversaw the compliance, planned the experiments, coordinated the sample acquisition and data acquisition/analysis, analyzed the data, and wrote and edited the paper.
Declaration of interests
J.Z., M.S., and S.M. have competing interests with IsoPlexis. The remaining authors declare no competing interests.
Published: March 11, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.immuni.2021.03.005.
Supplemental information
References
- Aggarwal N.R., King L.S., D’Alessio F.R. Diverse macrophage populations mediate acute lung inflammation and resolution. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014;306:L709–L725. doi: 10.1152/ajplung.00341.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharat A., Bhorade S.M., Morales-Nebreda L., McQuattie-Pimentel A.C., Soberanes S., Ridge K., DeCamp M.M., Mestan K.K., Perlman H., Budinger G.R., Misharin A.V. Flow Cytometry Reveals Similarities Between Lung Macrophages in Humans and Mice. Am. J. Respir. Cell Mol. Biol. 2016;54:147–149. doi: 10.1165/rcmb.2015-0147LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray N.L., Pimentel H., Melsted P., Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016;34:525–527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- Carpenter D.J., Granot T., Matsuoka N., Senda T., Kumar B.V., Thome J.J.C., Gordon C.L., Miron M., Weiner J., Connors T., et al. Human immunology studies using organ donors: impact of clinical variations on immune parameters in tissues and circulation. Am. J. Transplant. 2018;18:74–88. doi: 10.1111/ajt.14434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y.S., Gullicksrud J.A., Xing S., Zeng Z., Shan Q., Li F., Love P.E., Peng W., Xue H.H., Crotty S. LEF-1 and TCF-1 orchestrate T(FH) differentiation by regulating differentiation circuits upstream of the transcriptional repressor Bcl6. Nat. Immunol. 2015;16:980–990. doi: 10.1038/ni.3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua R.L., Lukassen S., Trump S., Hennig B.P., Wendisch D., Pott F., Debnath O., Thürmann L., Kurth F., Völker M.T., et al. COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 2020;38:970–979. doi: 10.1038/s41587-020-0602-4. [DOI] [PubMed] [Google Scholar]
- Cole S.L., Dunning J., Kok W.L., Benam K.H., Benlahrech A., Repapi E., Martinez F.O., Drumright L., Powell T.J., Bennett M., et al. MOSAIC Investigators M1-like monocytes are a major immunological determinant of severity in previously healthy adults with life-threatening influenza. JCI Insight. 2017;2:e91868. doi: 10.1172/jci.insight.91868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connors T.J., Baird J.S., Yopes M.C., Zens K.D., Pethe K., Ravindranath T.M., Ho S.H., Farber D.L. Developmental Regulation of Effector and Resident Memory T Cell Generation during Pediatric Viral Respiratory Tract Infection. J. Immunol. 2018;201:432–439. doi: 10.4049/jimmunol.1800396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connors T.J., Ravindranath T.M., Bickham K.L., Gordon C.L., Zhang F., Levin B., Baird J.S., Farber D.L. Airway CD8(+) T Cells Are Associated with Lung Injury during Infant Viral Respiratory Tract Infection. Am. J. Respir. Cell Mol. Biol. 2016;54:822–830. doi: 10.1165/rcmb.2015-0297OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings M.J., Baldwin M.R., Abrams D., Jacobson S.D., Meyer B.J., Balough E.M., Aaron J.G., Claassen J., Rabbani L.E., Hastie J., et al. Epidemiology, clinical course, and outcomes of critically ill adults with COVID-19 in New York City: a prospective cohort study. Lancet. 2020;395:1763–1770. doi: 10.1016/S0140-6736(20)31189-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damiani S., Fiorentino M., De Palma A., Foschini M.P., Lazzarotto T., Gabrielli L., Viale P.L., Attard L., Riefolo M., D’Errico A. Pathological post-mortem findings in lungs infected with SARS-CoV-2. J. Pathol. 2021;253:31–40. doi: 10.1002/path.5549. [DOI] [PubMed] [Google Scholar]
- Davies N.G., Klepac P., Liu Y., Prem K., Jit M., Eggo R.M., CMMID COVID-19 working group Age-dependent effects in the transmission and control of COVID-19 epidemics. Nat. Med. 2020;26:1205–1211. doi: 10.1038/s41591-020-0962-9. [DOI] [PubMed] [Google Scholar]
- De Michele S., Sun Y., Yilmaz M.M., Katsyv I., Salvatore M., Dzierba A.L., Marboe C.C., Brodie D., Patel N.M., Garcia C.K., Saqi A. Forty Postmortem Examinations in COVID-19 Patients. Am. J. Clin. Pathol. 2020;154:748–760. doi: 10.1093/ajcp/aqaa156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Della-Torre E., Campochiaro C., Cavalli G., De Luca G., Napolitano A., La Marca S., Boffini N., Da Prat V., Di Terlizzi G., Lanzillotta M., et al. SARI-RAF Study Group. SARI-RAF Study Group Members Interleukin-6 blockade with sarilumab in severe COVID-19 pneumonia with systemic hyperinflammation: an open-label cohort study. Ann. Rheum. Dis. 2020;79:1277–1285. doi: 10.1136/annrheumdis-2020-218122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogra P., Rancan C., Ma W., Toth M., Senda T., Carpenter D.J., Kubota M., Matsumoto R., Thapa P., Szabo P.A., et al. Tissue Determinants of Human NK Cell Development, Function, and Residence. Cell. 2020;180:749–763.e13. doi: 10.1016/j.cell.2020.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farhadian S., Glick L.R., Vogels C.B.F., Thomas J., Chiarella J., Casanovas-Massana A., Zhou J., Odio C., Vijayakumar P., Geng B., et al. Acute encephalopathy with elevated CSF inflammatory markers as the initial presentation of COVID-19. BMC Neurol. 2020;20:248. doi: 10.1186/s12883-020-01812-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furlow B. COVACTA trial raises questions about tocilizumab’s benefit in COVID-19. Lancet Rheumatol. 2020;2:e592. doi: 10.1016/S2665-9913(20)30313-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garibaldi B.T., Fiksel J., Muschelli J., Robinson M.L., Rouhizadeh M., Perin J., Schumock G., Nagy P., Gray J.H., Malapati H., et al. Patient Trajectories Among Persons Hospitalized for COVID-19: A Cohort Study. Ann. Intern. Med. 2021;174:33–41. doi: 10.7326/M20-3905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartrell R.D., Marks D.K., Hart T.D., Li G., Davari D.R., Wu A., Blake Z., Lu Y., Askin K.N., Monod A., et al. Quantitative Analysis of Immune Infiltrates in Primary Melanoma. Cancer Immunol. Res. 2018;6:481–493. doi: 10.1158/2326-6066.CIR-17-0360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissmann F., Jung S., Littman D.R. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- Griffiths J.A., Richard A.C., Bach K., Lun A.T.L., Marioni J.C. Detection and removal of barcode swapping in single-cell RNA-seq data. Nat. Commun. 2018;9:2667. doi: 10.1038/s41467-018-05083-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grifoni A., Weiskopf D., Ramirez S.I., Mateus J., Dan J.M., Moderbacher C.R., Rawlings S.A., Sutherland A., Premkumar L., Jadi R.S., et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell. 2020;181:1489–1501.e15. doi: 10.1016/j.cell.2020.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjadj J., Yatim N., Barnabei L., Corneau A., Boussier J., Smith N., Péré H., Charbit B., Bondet V., Chenevier-Gobeaux C., et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science. 2020;369:718–724. doi: 10.1126/science.abc6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan A.O., Kafai N.M., Dmitriev I.P., Fox J.M., Smith B.K., Harvey I.B., Chen R.E., Winkler E.S., Wessel A.W., Case J.B., et al. A Single-Dose Intranasal ChAd Vaccine Protects Upper and Lower Respiratory Tracts against SARS-CoV-2. Cell. 2020;183:169–184.e13. doi: 10.1016/j.cell.2020.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrix A.Y., Kheradmand F. The Role of Matrix Metalloproteinases in Development, Repair, and Destruction of the Lungs. Prog. Mol. Biol. Transl. Sci. 2017;148:1–29. doi: 10.1016/bs.pmbts.2017.04.004. [DOI] [PubMed] [Google Scholar]
- Hunter J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007;9:90–95. [Google Scholar]
- Jeyanathan M., Afkhami S., Smaill F., Miller M.S., Lichty B.D., Xing Z. Immunological considerations for COVID-19 vaccine strategies. Nat. Rev. Immunol. 2020;20:615–632. doi: 10.1038/s41577-020-00434-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones A.E., Trzeciak S., Kline J.A. The Sequential Organ Failure Assessment score for predicting outcome in patients with severe sepsis and evidence of hypoperfusion at the time of emergency department presentation. Crit. Care Med. 2009;37:1649–1654. doi: 10.1097/CCM.0b013e31819def97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapellos T.S., Bonaguro L., Gemünd I., Reusch N., Saglam A., Hinkley E.R., Schultze J.L. Human Monocyte Subsets and Phenotypes in Major Chronic Inflammatory Diseases. Front. Immunol. 2019;10:2035. doi: 10.3389/fimmu.2019.02035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khemani R.G., Smith L.S., Zimmerman J.J., Erickson S., Pediatric Acute Lung Injury Consensus Conference Group Pediatric acute respiratory distress syndrome: definition, incidence, and epidemiology: proceedings from the Pediatric Acute Lung Injury Consensus Conference. Pediatr. Crit. Care Med. 2015;16(5, Suppl 1):S23–S40. doi: 10.1097/PCC.0000000000000432. [DOI] [PubMed] [Google Scholar]
- Kim D., Lee J.Y., Yang J.S., Kim J.W., Kim V.N., Chang H. The Architecture of SARS-CoV-2 Transcriptome. Cell. 2020;181:914–921.e10. doi: 10.1016/j.cell.2020.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlmeier J.E., Woodland D.L. Immunity to respiratory viruses. Annu. Rev. Immunol. 2009;27:61–82. doi: 10.1146/annurev.immunol.021908.132625. [DOI] [PubMed] [Google Scholar]
- Korsunsky I., Millard N., Fan J., Slowikowski K., Zhang F., Wei K., Baglaenko Y., Brenner M., Loh P.R., Raychaudhuri S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods. 2019;16:1289–1296. doi: 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar B.V., Connors T.J., Farber D.L. Human T Cell Development, Localization, and Function throughout Life. Immunity. 2018;48:202–213. doi: 10.1016/j.immuni.2018.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar B.V., Ma W., Miron M., Granot T., Guyer R.S., Carpenter D.J., Senda T., Sun X., Ho S.H., Lerner H., et al. Human Tissue-Resident Memory T Cells Are Defined by Core Transcriptional and Functional Signatures in Lymphoid and Mucosal Sites. Cell Rep. 2017;20:2921–2934. doi: 10.1016/j.celrep.2017.08.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuri-Cervantes L., Pampena M.B., Meng W., Rosenfeld A.M., Ittner C.A.G., Weisman A.R., Agyekum R.S., Mathew D., Baxter A.E., Vella L.A., et al. Comprehensive mapping of immune perturbations associated with severe COVID-19. Sci. Immunol. 2020;5:eabd7114. doi: 10.1126/sciimmunol.abd7114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laing A.G., Lorenc A., Del Molino Del Barrio I., Das A., Fish M., Monin L., Muñoz-Ruiz M., McKenzie D.R., Hayday T.S., Francos-Quijorna I., et al. A dynamic COVID-19 immune signature includes associations with poor prognosis. Nat. Med. 2020;26:1623–1635. doi: 10.1038/s41591-020-1038-6. [DOI] [PubMed] [Google Scholar]
- Levine J.H., Simonds E.F., Bendall S.C., Davis K.L., Amir A.D., Tadmor M.D., Litvin O., Fienberg H.G., Jager A., Zunder E.R., et al. Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis. Cell. 2015;162:184–197. doi: 10.1016/j.cell.2015.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitin H.M., Yuan J., Cheng Y.L., Ruiz F.J.R., Bush E.C., Bruce J.N., Canoll P., Iavarone A., Lasorella A., Blei D.M., Sims P.A. De novo gene signature identification from single-cell RNA-seq with hierarchical Poisson factorization. Mol. Syst. Biol. 2019;15:e8557. doi: 10.15252/msb.20188557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao M., Liu Y., Yuan J., Wen Y., Xu G., Zhao J., Cheng L., Li J., Wang X., Wang F., et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020;26:842–844. doi: 10.1038/s41591-020-0901-9. [DOI] [PubMed] [Google Scholar]
- Lin K.L., Suzuki Y., Nakano H., Ramsburg E., Gunn M.D. CCR2+ monocyte-derived dendritic cells and exudate macrophages produce influenza-induced pulmonary immune pathology and mortality. J. Immunol. 2008;180:2562–2572. doi: 10.4049/jimmunol.180.4.2562. [DOI] [PubMed] [Google Scholar]
- Lin K.L., Sweeney S., Kang B.D., Ramsburg E., Gunn M.D. CCR2-antagonist prophylaxis reduces pulmonary immune pathology and markedly improves survival during influenza infection. J. Immunol. 2011;186:508–515. doi: 10.4049/jimmunol.1001002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long Q.X., Liu B.Z., Deng H.J., Wu G.C., Deng K., Chen Y.K., Liao P., Qiu J.F., Lin Y., Cai X.F., et al. Antibody responses to SARS-CoV-2 in patients with COVID-19. Nat. Med. 2020;26:845–848. doi: 10.1038/s41591-020-0897-1. [DOI] [PubMed] [Google Scholar]
- Lucas C., Wong P., Klein J., Castro T.B.R., Silva J., Sundaram M., Ellingson M.K., Mao T., Oh J.E., Israelow B., et al. Yale IMPACT Team Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature. 2020;584:463–469. doi: 10.1038/s41586-020-2588-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun A.T., Bach K., Marioni J.C. Pooling across cells to normalize single-cell RNA sequencing data with many zero counts. Genome Biol. 2016;17:75. doi: 10.1186/s13059-016-0947-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun A.T.L., Riesenfeld S., Andrews T., Dao T.P., Gomes T., Marioni J.C., Participants in the 1st Human Cell Atlas Jamboree EmptyDrops: distinguishing cells from empty droplets in droplet-based single-cell RNA sequencing data. Genome Biol. 2019;20:63. doi: 10.1186/s13059-019-1662-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew D., Giles J.R., Baxter A.E., Oldridge D.A., Greenplate A.R., Wu J.E., Alanio C., Kuri-Cervantes L., Pampena M.B., D’Andrea K., et al. UPenn COVID Processing Unit Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science. 2020;369:eabc8511. doi: 10.1126/science.abc8511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matics T.J., Sanchez-Pinto L.N. Adaptation and Validation of a Pediatric Sequential Organ Failure Assessment Score and Evaluation of the Sepsis-3 Definitions in Critically Ill Children. JAMA Pediatr. 2017;171:e172352. doi: 10.1001/jamapediatrics.2017.2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInnes L., Healy J., Saul N., Grossberger L. UMAP: Uniform Manifold Approximation and Projection. J. Open Source Softw. 2018;3:861. [Google Scholar]
- Melsted P., Booeshaghi A.S., Gao F., Beltrame E., Lu L., Hjorleifsson K.E., Gehring J., Pachter L. Modular and efficient pre-processing of single-cell RNA-seq. bioRxiv. 2019:673285. [Google Scholar]
- Melsted P., Ntranos V., Pachter L. The barcode, UMI, set format and BUStools. Bioinformatics. 2019;35:4472–4473. doi: 10.1093/bioinformatics/btz279. [DOI] [PubMed] [Google Scholar]
- Morrell E.D., Bhatraju P.K., Mikacenic C.R., Radella F., 2nd, Manicone A.M., Stapleton R.D., Wurfel M.M., Gharib S.A. Alveolar Macrophage Transcriptional Programs Are Associated with Outcomes in Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2019;200:732–741. doi: 10.1164/rccm.201807-1381OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni L., Ye F., Cheng M.L., Feng Y., Deng Y.Q., Zhao H., Wei P., Ge J., Gou M., Li X., et al. Detection of SARS-CoV-2-Specific Humoral and Cellular Immunity in COVID-19 Convalescent Individuals. Immunity. 2020;52:971–977.e3. doi: 10.1016/j.immuni.2020.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshansky C.M., Gartland A.J., Wong S.S., Jeevan T., Wang D., Roddam P.L., Caniza M.A., Hertz T., Devincenzo J.P., Webby R.J., Thomas P.G. Mucosal immune responses predict clinical outcomes during influenza infection independently of age and viral load. Am. J. Respir. Crit. Care Med. 2014;189:449–462. doi: 10.1164/rccm.201309-1616OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik D.H., Farber D.L. Influenza infection fortifies local lymph nodes to promote lung-resident heterosubtypic immunity. J. Exp. Med. 2021;218:e20200218. doi: 10.1084/jem.20200218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedregosa F., Varoquaux G., Gramfort A., Michel V., Thirion B., Grisel O., Blondel M., Prettenhofer P., Weiss R., Dubourg V., et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011;12:2825–2830. [Google Scholar]
- Ranieri V.M., Rubenfeld G.D., Thompson B.T., Ferguson N.D., Caldwell E., Fan E., Camporota L., Slutsky A.S., ARDS Definition Task Force Acute respiratory distress syndrome: the Berlin Definition. JAMA. 2012;307:2526–2533. doi: 10.1001/jama.2012.5669. [DOI] [PubMed] [Google Scholar]
- Rydyznski Moderbacher C., Ramirez S.I., Dan J.M., Grifoni A., Hastie K.M., Weiskopf D., Belanger S., Abbott R.K., Kim C., Choi J., et al. Antigen-Specific Adaptive Immunity to SARS-CoV-2 in Acute COVID-19 and Associations with Age and Disease Severity. Cell. 2020;183:996–1012.e19. doi: 10.1016/j.cell.2020.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathaliyawala T., Kubota M., Yudanin N., Turner D., Camp P., Thome J.J., Bickham K.L., Lerner H., Goldstein M., Sykes M., et al. Distribution and compartmentalization of human circulating and tissue-resident memory T cell subsets. Immunity. 2013;38:187–197. doi: 10.1016/j.immuni.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte-Schrepping J., Reusch N., Paclik D., Baßler K., Schlickeiser S., Zhang B., Krämer B., Krammer T., Brumhard S., Bonaguro L., et al. Deutsche COVID-19 OMICS Initiative (DeCOI) Severe COVID-19 Is Marked by a Dysregulated Myeloid Cell Compartment. Cell. 2020;182:1419–1440.e23. doi: 10.1016/j.cell.2020.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultze J.L., Mass E., Schlitzer A. Emerging Principles in Myelopoiesis at Homeostasis and during Infection and Inflammation. Immunity. 2019;50:288–301. doi: 10.1016/j.immuni.2019.01.019. [DOI] [PubMed] [Google Scholar]
- Shekhar K., Lapan S.W., Whitney I.E., Tran N.M., Macosko E.Z., Kowalczyk M., Adiconis X., Levin J.Z., Nemesh J., Goldman M., et al. Comprehensive Classification of Retinal Bipolar Neurons by Single-Cell Transcriptomics. Cell. 2016;166:1308–1323.e30. doi: 10.1016/j.cell.2016.07.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C., Jia T., Mendez-Ferrer S., Hohl T.M., Serbina N.V., Lipuma L., Leiner I., Li M.O., Frenette P.S., Pamer E.G. Bone marrow mesenchymal stem and progenitor cells induce monocyte emigration in response to circulating toll-like receptor ligands. Immunity. 2011;34:590–601. doi: 10.1016/j.immuni.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvin A., Chapuis N., Dunsmore G., Goubet A.G., Dubuisson A., Derosa L., Almire C., Hénon C., Kosmider O., Droin N., et al. Elevated Calprotectin and Abnormal Myeloid Cell Subsets Discriminate Severe from Mild COVID-19. Cell. 2020;182:1401–1418.e18. doi: 10.1016/j.cell.2020.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer M., Deutschman C.S., Seymour C.W., Shankar-Hari M., Annane D., Bauer M., Bellomo R., Bernard G.R., Chiche J.D., Coopersmith C.M., et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3) JAMA. 2016;315:801–810. doi: 10.1001/jama.2016.0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder M.E., Finlayson M.O., Connors T.J., Dogra P., Senda T., Bush E., Carpenter D., Marboe C., Benvenuto L., Shah L., et al. Generation and persistence of human tissue-resident memory T cells in lung transplantation. Sci. Immunol. 2019;4:eaav5581. doi: 10.1126/sciimmunol.aav5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo P.A., Levitin H.M., Miron M., Snyder M.E., Senda T., Yuan J., Cheng Y.L., Bush E.C., Dogra P., Thapa P., et al. Single-cell transcriptomics of human T cells reveals tissue and activation signatures in health and disease. Nat. Commun. 2019;10:4706. doi: 10.1038/s41467-019-12464-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T., Ellingson M.K., Wong P., Israelow B., Lucas C., Klein J., Silva J., Mao T., Oh J.E., Tokuyama M., et al. Yale IMPACT Research Team Sex differences in immune responses that underlie COVID-19 disease outcomes. Nature. 2020;588:315–320. doi: 10.1038/s41586-020-2700-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teijaro J.R., Turner D., Pham Q., Wherry E.J., Lefrançois L., Farber D.L. Cutting edge: tissue-retentive lung memory CD4 T cells mediate optimal protection to respiratory virus infection. J. Immunol. 2011;187:5510–5514. doi: 10.4049/jimmunol.1102243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teijaro J.R., Walsh K.B., Cahalan S., Fremgen D.M., Roberts E., Scott F., Martinborough E., Peach R., Oldstone M.B., Rosen H. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell. 2011;146:980–991. doi: 10.1016/j.cell.2011.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thieme C.J., Anft M., Paniskaki K., Blazquez-Navarro A., Doevelaar A., Seibert F.S., Hoelzer B., Konik M.J., Brenner T., Tempfer C., et al. Robust T cell response towards spike, membrane, and nucleocapsid SARS-CoV-2 proteins is not associated with recovery in critical COVID-19 patients. Cell Rep. Med. 2020;1:100092. doi: 10.1016/j.xcrm.2020.100092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thome J.J., Yudanin N., Ohmura Y., Kubota M., Grinshpun B., Sathaliyawala T., Kato T., Lerner H., Shen Y., Farber D.L. Spatial map of human T cell compartmentalization and maintenance over decades of life. Cell. 2014;159:814–828. doi: 10.1016/j.cell.2014.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner D.L., Bickham K.L., Thome J.J., Kim C.Y., D’Ovidio F., Wherry E.J., Farber D.L. Lung niches for the generation and maintenance of tissue-resident memory T cells. Mucosal Immunol. 2014;7:501–510. doi: 10.1038/mi.2013.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner D.L., Farber D.L. Mucosal resident memory CD4 T cells in protection and immunopathology. Front. Immunol. 2014;5:331. doi: 10.3389/fimmu.2014.00331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Walt S., Colbert S.C., Varoquaux G. The NumPy Array: A Structure for Efficient Numerical Computation. Comput. Sci. Eng. 2011;13:22–30. [Google Scholar]
- Vasilevskis E.E., Pandharipande P.P., Graves A.J., Shintani A., Tsuruta R., Ely E.W., Girard T.D. Validity of a Modified Sequential Organ Failure Assessment Score Using the Richmond Agitation-Sedation Scale. Crit. Care Med. 2016;44:138–146. doi: 10.1097/CCM.0000000000001375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venet F., Demaret J., Gossez M., Monneret G. Myeloid cells in sepsis-acquired immunodeficiency. Ann. N Y Acad. Sci. 2020 doi: 10.1111/nyas.14333. [DOI] [PubMed] [Google Scholar]
- Veras F.P., Pontelli M.C., Silva C.M., Toller-Kawahisa J.E., de Lima M., Nascimento D.C., Schneider A.H., Caetité D., Tavares L.A., Paiva I.M., et al. SARS-CoV-2-triggered neutrophil extracellular traps mediate COVID-19 pathology. J. Exp. Med. 2020;217:e29291129. doi: 10.1084/jem.20201129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent J.L., de Mendonça A., Cantraine F., Moreno R., Takala J., Suter P.M., Sprung C.L., Colardyn F., Blecher S. Use of the SOFA score to assess the incidence of organ dysfunction/failure in intensive care units: results of a multicenter, prospective study. Working group on “sepsis-related problems” of the European Society of Intensive Care Medicine. Crit. Care Med. 1998;26:1793–1800. doi: 10.1097/00003246-199811000-00016. [DOI] [PubMed] [Google Scholar]
- Virtanen P., Gommers R., Oliphant T.E., Haberland M., Reddy T., Cournapeau D., Burovski E., Peterson P., Weckesser W., Bright J., et al. SciPy 1.0 Contributors SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods. 2020;17:261–272. doi: 10.1038/s41592-019-0686-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg S.P., Carpenter D.J., Chait M., Dogra P., Gartrell-Corrado R.D., Chen A.X., Campbell S., Liu W., Saraf P., Snyder M.E., et al. Tissue-Resident Memory T Cells Mediate Immune Homeostasis in the Human Pancreas through the PD-1/PD-L1 Pathway. Cell Rep. 2019;29:3916–3932.e5. doi: 10.1016/j.celrep.2019.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg S.P., Connors T.J., Zhu Y., Baldwin M.R., Lin W.H., Wontakal S., Szabo P.A., Wells S.B., Dogra P., Gray J., et al. Distinct antibody responses to SARS-CoV-2 in children and adults across the COVID-19 clinical spectrum. Nat. Immunol. 2021;22:25–31. doi: 10.1038/s41590-020-00826-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiskopf D., Schmitz K.S., Raadsen M.P., Grifoni A., Okba N.M.A., Endeman H., van den Akker J.P.C., Molenkamp R., Koopmans M.P.G., van Gorp E.C.M., et al. Phenotype and kinetics of SARS-CoV-2-specific T cells in COVID-19 patients with acute respiratory distress syndrome. Sci. Immunol. 2020;5:eabd2071. doi: 10.1126/sciimmunol.abd2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T., Hu Y., Lee Y.T., Bouchard K.R., Benechet A., Khanna K., Cauley L.S. Lung-resident memory CD8 T cells (TRM) are indispensable for optimal cross-protection against pulmonary virus infection. J. Leukoc. Biol. 2014;95:215–224. doi: 10.1189/jlb.0313180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z., McGoogan J.M. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72 314 Cases From the Chinese Center for Disease Control and Prevention. JAMA. 2020;323:1239–1242. doi: 10.1001/jama.2020.2648. [DOI] [PubMed] [Google Scholar]
- Yoo J.K., Kim T.S., Hufford M.M., Braciale T.J. Viral infection of the lung: host response and sequelae. J. Allergy Clin. Immunol. 2013;132:1263–1276. doi: 10.1016/j.jaci.2013.06.006. quiz 1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J., Zhao J., Mangalam A.K., Channappanavar R., Fett C., Meyerholz D.K., Agnihothram S., Baric R.S., David C.S., Perlman S. Airway Memory CD4(+) T Cells Mediate Protective Immunity against Emerging Respiratory Coronaviruses. Immunity. 2016;44:1379–1391. doi: 10.1016/j.immuni.2016.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The scRNA-seq data for each sample including count matrices, normalized counts, metadata, cell annotations, and UMAP embeddings are available on the COVID-19 Cell Atlas along with interactive visualizations (https://www.covid19cellatlas.org/index.patient.html). Code for clustering of scRNA-seq data analysis is available at www.github.com/simslab/cluster_diffex2018. Additional Supplemental Items are available from Mendeley Data at http://dx.doi.org/10.17632/fm7v43jrbk.4