

Abstract
For the past 20 years, super‐oxidised solutions (SOSs) have been shown to be potent antimicrobials and disinfectants via oxidative damage. However, the potential toxicity of SOSs on eukaryotic cells has not been documented in vitro. This is relevant because oxygen and chlorine reactive species may possibly induce ageing and irreversible cellular dysfunctions that eventually produce cell death. The present study investigates the cytotoxicity and oxidative stress induced by a novel, pH‐neutral SOS (i.e. Microcyn™, MCN) on young, primary diploid – human dermal fibroblast (HDF) cultures. For this purpose, hydrogen peroxide (HP) was used as a positive control of oxidative damage. When these solutions were used at concentrations indicated for wound care (i.e. undiluted MCN or 880 mM HP), HP was significantly more toxic than MCN. After 5 and 30 minutes of exposure, cell viability was 38% and 5%, respectively, in 880 mM HP‐treated cells versus 75% and 70% in MCN‐treated populations, respectively. HP induced both apoptosis and necrosis, whereas MCN induced only necrosis. Genotoxic and ageing studies were then conducted at sublethal HP concentrations as previously reported in the literature. Cellular DNA and RNA were partially degraded only in HDFs exposed to 500 μM HP for 30 minutes but not in those exposed to undiluted MCN. At this same concentration, HP induced the formation of 8‐hydroxy‐2′deoxyguanosine adducts in HDFs but this effect was neither observed in control‐ nor observed in MCN‐treated cells. HDFs were further exposed to 5 μM HP or 10% MCN for 1 month. The expression of senescence‐associated‐β‐galactosidase was only significantly elevated in cells chronically exposed to 5 μM HP. Altogether, these results show that MCN is significantly less cytotoxic than antiseptic HP concentrations (i.e. 880 mM) and that, in vitro, it does not induce genotoxicity or accelerated ageing.
Keywords: Apoptosis, Cell aging, Cell viability, Human fibroblasts, Super‐oxidised water
Introduction
Super‐oxidised solutions (SOSs) are electrochemically processed aqueous solutions manufactured from pure water and salt (e.g. USP‐grade sodium chloride, NaCl). During the electrolysis process, molecules are dissociated and reactive species of chlorine and oxygen are formed. These species can potentially damage nucleic acids, proteins or lipids (1). For example, oxygen free radicals such as superoxide (•O2 −) and the hydroxyl radical (•OH) could react directly with proteins and other macromolecules to produce carbonyls (aldehydes and ketones), cross‐linking and lipid peroxidation (2). Others like hydrogen peroxide (H2O2) and hypochlorite (OCl−) are not themselves free radicals but these oxygen‐containing molecules can facilitate free‐radical formation (3).
For the past 20 years, SOSs have been shown to be potent antimicrobial agents (4). They have already been tested as disinfectants for instruments and hard inanimate surfaces 5, 6. The literature also describes applications for human beings in various clinical disorders 7, 8. A pH‐neutral SOS (Microcyn™, MCN), for example, is used as a wound care product in various countries. This is a hypotonic solution with an osmolarity of 13 mOsm and known chemical species including super‐oxidised water (99·98%), hypochlorous acid, sodium hypochlorite and sodium chloride at pH 7·2–7·8. Because of its high oxidation–reduction potential (ORP > 800), MCN efficiently kills bacteria, fungi, viruses and even spores in seconds to minutes depending on the type of test (i.e. suspension or carrier tests) (9).
The mechanism of action for MCN eradication of various micro‐organisms has not been fully defined but it involves a two‐step process: MCN alters the integrity of the bacterial cell wall – or the viral capsids – because of its high ORP. Cell lysis occurs via the osmotic pressure imbalance between the cell and the (hypotonic) MCN solution. Using contrast microscopy, bacterial swelling is observed within the first 30 seconds of exposure to MCN followed by bacterial cell rupture (10).
MCN does not induce skin sensitisation or irritation in animal studies despite its high ORP and antimicrobial activity (11). A major concern, however, has been the potential oxidative damage to human cells. Therefore, the effects of MCN on cell viability and oxidative stress in young primary diploid – human dermal fibroblast (HDF) cultures, as measured by 8‐hydroxy‐2′deoxyguanosine (8‐OHdG) adducts, nucleic acid stability and ageing process, were evaluated in this study. HDFs were thus exposed to different concentrations of MCN and hydrogen peroxide (HP). HP was used as a positive control of oxidative stress.
Materials and methods
Chemicals
All chemicals and reagents were of analytical grade and were purchased from Sigma Chemical Co. (St Louis, MO). MCN was purchased commercially. This is a hypotonic solution with an osmolarity of 13 mOsm and known chemical species including super‐oxidised water (99·98%), hypochlorous acid (0·00252%), sodium hypochlorite (0·00357%) and sodium chloride (0·0105%) at pH 7·2–7·8. The total free available chlorine in the solution ranges from 50 to 80 p.p.m. Several other reactive species are also generated in trace amounts during its production including ozone, HP and chlorine dioxide. However, the latter compounds are unstable and are not measurable in the final product.
Cell culture
HDFs were isolated from normal human neonatal foreskin. Informed consent was obtained from both parents of the child (Hospital Angeles del Pedregal, Mexico City, Mexico). The isolated tissue was washed three times in phosphate‐buffered salt solution (PBS 1×) containing 1500 U/ml of penicillin/streptomycin (Sigma Chemical Co.). Foreskin slices were incubated for 4 hours at 37°C under sterile conditions in 0·3% collagenase (CLS‐2; Worthington Biochem, Lakewood, NJ) in Dulbecco‘s modified Earl’s medium (DMEM; GIBCO, Carlsbad, CA) containing 2 mM l‐glutamine and 1500 U/ml penicillin/streptomycin/antimicotic (Sigma). After incubation, cells and tissue debris were removed by centrifugation at 800 g for 5 minutes. The pellet was re‐suspended in DMEM supplemented with 2 mM of l‐glutamine, 1500 U/ml penicillin/streptomycin/antimicotic supplemented (complete media) with 10% foetal bovine serum (FBS; GIBCO). The filtrate was seeded onto 75 cm2 tissue culture flasks (Nunc‐International, Rochester, NY). All cell cultures were kept at 37°C with 5% CO2. Twenty‐four hours later, cultures were washed three times in sterile saline solution (0·9% NaCl) for the removal of red blood cells.
Cell cycle analysis
This assay was used to evaluate the ploidy of HDF cultures. After removal of the maintenance media, proliferating cells (1 × 106) were washed three times with PBS pH 7·4 and harvested from the flasks with 0·25% trypsin–versene solution. Trypsinisation was terminated with DMEM containing 10% FBS. The cellular suspension was centrifuged at 800 g for 5 minutes. The resulting pellet was stained with propidium iodide as per the manufacturer‘s instructions (CycleTEST™ PLUS DNA Reagent Kit, BD, Franklin Lakes, NJ). Samples were analysed in a FACSCalibur Flow Cytometer (BD, Palo Alto, CA). The ModFit LT software was used to determine the various cell cycle phases (12).
Determination of the population’s doubling times
Frozen cell stocks were thawed at 37°C, washed once in complete DMEM and further plated (1 × 106 cells). At 80–90% confluence, cells were dissociated and subcultured at 5 × 105 cells in T25 culture flasks (Nunc‐International). Doubling times were calculated as follows: t d= ln(2) × (delta T)/ln(final count/initial count), where delta T is the time from plating to subculture, in hours; final count is the number of cells at subculture; and initial count is the number of cells plated at the start of the culture (13).
Viability assay
Cells growing in T25 culture flasks (Nalge; Nunc‐International) at 60–70% of confluence (5 × 105) were exposed in triplicate to either DMEM supplemented with 10% Bovine Fetal Serum (SFB) (control), 500 μM HP or 880 mM HP (3%) or different dilutions of MCN for 5 and 30 minutes. All the solutions were prepared fresh with saline solution (0·9% NaCl) for each experiment. After removal of the media, HP and MCN, cells were very carefully scraped for mechanical detachment from the plates immediately, and the cell viability was determined by flow cytometry (FACScalibur; BD) for 7‐aminoactinomycin D staining according to the manufacturer‘s instructions (BD).
Necrosis–apoptosis assay
The ratios of necrosis and apoptosis in the cells after exposure to antiseptic concentrations of HP (880 mM) and MCN (100%) were determined using annexin V–fluoroisothiocyanate (FITC) and propidium iodide according to the manufacturer’s protocol (Annexin‐V‐Fluos Staining kit; Roche, New Brunswick, NJ). These measurements were recorded simultaneously in the cells used for the cell viability assay.
Genomic DNA extraction
Total DNA was extracted from 3 × 106 control and acute treated cells. The fibroblast cultures in T75 flasks were washed once with PBS and then exposed to MCN (100%) or 500 μM of HP in saline solution (0·9% NaCl) or saline solution for the control treatment for 30 min at 37°C in 5% CO2 atmosphere. After exposure time, the solutions were retired, and new complete media were added. The DNA was extracted 3 hours later. The DNA isolation was carried out in triplicate as per the manufacturer‘s instructions for the QIAamp DNA midi kit (Qiagen™, Hilden, Germany). Total genomic DNA (600 ng) was dissolved in a mixture of 9 μl tris‐EDTA (TE) buffer and 1 μl gel loading buffer (0·25% bromophenol blue, 0·25% xylene cyanol, 30% v/v glycerol) and then loaded into a 1% agarose gel in tris acetic acid (TAE) buffer. The isolated DNA was analysed by native electrophoresis containing 0·5 mg/ml of ethidium bromide. DNA Ladder (100 pb) of Promega™ (Madison, WI) was used as size marker. To control for integrity of the DNA, the expression of the β‐globin gene was also analysed using primers and conditions already described (14).
Reverse transcriptase–polymerase chain reaction
Total RNA was obtained from 3 × 106 control and acute treated cells (30 minutes of exposure with MCN 100% and HP (500 μM) as per the manufacturer’s instructions (Qiagen™). Isolated RNAs (5 μg) were used to create complementary DNA (cDNA) libraries with the SuperScript™ First‐Strand Synthesis System (Invitrogen™, Carlsbad, CA) and the oligo(dT18) primer. Two microlitres of the resulting reverse transcriptase (RT) products were further expanded with Taq DNA polymerase (Roche). The polymerase chain reaction (PCR) mixture for amplification contained 10 mM Tris–HCl (pH 8·5), 50 mM KCl, 1·5 mM de MgCl2, 0·05 mM dithiotheratol, 0·05 mM ethylenediaminetetraacetic acid, 0·5% Tween, 0·5 Nonidet P40, 25% glycerol, 0·2 mM deoxynucleotide triphosphate, 0·2 μM of specific primers and 2·5 U of the Taq DNA polymerase in 50 μl of reaction. The housekeeping human gene glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) was analysed with the following primer sequences for obtain one 460‐pb‐sized fragment: up 5′‐ACC ACA GTC CAT GCC ATC AC‐3′ and down 5′‐TCC ACC ACC CTG TTG CTT GTA‐3′. The amplification was carried out in a thermal cycler (Genius, Techne, Staffordshire, UK) using the following program: 3 minutes at 94°C for an initial denaturation; 94, 60 and 72°C for 40 seconds each, for 30 cycles, and one final extension step at 72°C for 5 minutes. The isolated RNA and PCR products were analysed by electrophoresis on a 1·5% agarose gel that contained ethidium bromide. Contamination of genomic DNA in all the RNA samples was detected trough the RT–PCR reaction without the enzyme RT.
Cell treatment for 8OH‐guanosine measures (acute treatment)
HDF cultures used in these experiments were in passage 3 (10–15 doublings populations). A total of 5 × 106 cells were seeded in 175‐cm2 culture flasks (Nunc) with complete DMEM and incubated for 48 hours (60–70% confluence). Three groups of the 40 × 106 cells each (five flasks per group) were then formed: (1) control group, with cells exposed to DMEM without serum; (2) cells treated with a sublethal concentration of HP (500 μM) and (3) cells treated with MCN without dilution (i.e. 100% solution). Another three groups of cells were treated in the same way but kept in supplemented medium for 3 hours after the exposure to HP and MCN (recovery group) and prior to DNA extraction. The exposure was conducted with 10 ml of each experimental solution for 30 minutes at 37°C and 5% CO2. After the exposure time, the solution was removed, and complete DMEM was added.
8‐OHdG enzyme‐linked immunosorbent assay
Control and treated cells were detached from the flasks with 0·25% trypsin (w/v) and further centrifuged. The pellets were recovered and DNA isolated from 40 × 106 cells (Qiagen™) as per the manufacturer‘s instructions. All DNA samples were analysed on agarose gels prior to digestion. All samples were normalised at 100 μg of total DNA. Samples were then re‐suspended in 200 μl of 20 mM sodium acetate (pH 4·8) and digested down to nucleotides with 20 μg of nuclease P1 (US Biological, Swampscott, MA) at 37°C for 30 minutes. This step was followed by the addition of 20 μl of 1 M Tris–HCl (pH 7·4) and treatment with 1·3 U of Escherichia coli alkaline phosphatase (Roche) at 37°C for 60 minutes 15, 16. After digestion, the samples were passed through a 30 000‐molecular‐weight cut‐off filter (UltraFree MC, Millipore, Bedford, MA), and 50 μl of the digested samples were used for the ELISA BIOXITECH® 8‐OHdG‐EIA™ kit (OXIS Health Products, Inc, Portland, OR). The assay was carried out at triplicate.
Cell treatment for senescence‐associated‐β‐galactosidase determination (chronic)
Chronic oxidative stress was induced by 5 μM HP or 10% MCN (v/v) in complete DMEM. Medium exchange was carried out every 2 days. The treatment started with HDFs in passage 2, and it was continued for up to passage 12 (<40–50 doublings). Cells with the same passage doublings were compared to age‐matched control cells grown in the absence of HP or MCN.
Senescence‐associated‐β‐galactosidase staining
A total of 1 × 105 cells in the treated and control groups were seeded in six‐well plates (in triplicate for each condition). After 24 hours, cells were washed twice in cold PBS, fixed at room temperature in 2 ml of 2% formaldehyde/0·2% glutaraldehyde for 5 min, further washed and incubated with fresh staining solution (1 mg of 5‐bromo‐4‐chloro‐3‐indolyl β‐d‐galactosidase (X‐Gal)/ml, 40 mM citric acid/sodium phosphate, pH 6·0/5 mM potassium ferrocyanide/5 mM potassium ferricyanide/150 mM NaCl and 2 mM MgCl2). Plates were incubated at 37°C without CO2 for 16 hours. The number of blue (positive) cells was counted in 20 fields at 400× magnification and expressed as number of positive cells (17).
DNA ploidy of HDF cultures
HDF cultures were obtained from three different foreskins, which were pooled and cryopreserved together for the purpose of this study. Only diploid cells were used for all experiments. On cell cycle analysis, DNA diploidy was defined as the presence of a single G0–G1 peak with a CV ≤7% and a corresponding G2/M peak collected from at least 20 000 total events (Figure 1).
Figure 1.
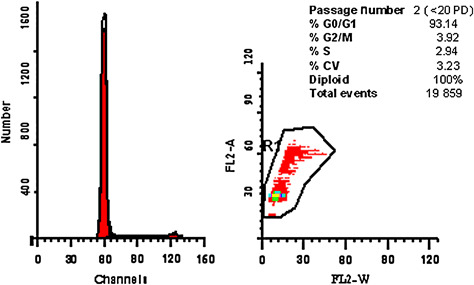
Cell cycle analysis of human dermal fibroblast (HDF) cultures isolated from human foreskin. This is a representative DNA histogram of an HDF culture at passage 2 (<20 PD) in which more than 93% of the population was on phase G0–G1 (n= 3).
Statistical analysis
Statistical analysis was performed by the T test for dependent samples. P < 0·05 was considered significant. The Tukey honest significant difference test was used for the analysis of variance between groups. The Statistica version 6·00 software was used for these analyses.
Results
Effects of MCN on the cellular viability of HDFs
Figure 2A shows the dose–response curves in HDF cultures exposed to different concentrations of MCN and HP. In this assay, cells were exposed to concentrations used for wound care (i.e. undiluted MCN and 880 nM HP) and to diluted solutions for 5 and 30 minutes. Cell viability decreased 25–30% when HDFs were exposed to undiluted MCN or 500 μM HP independent of the exposure time. At this concentration, MCN induced a statistically significant reduction in HDF’s viability in comparison to the control group (P < 0·05). In contrast, cell viability was 39% and 5% after exposure to 880 mM HP for 5 and 30 minutes, respectively (Figure 2A). MCN induced cell death through necrosis because >15% of the cells incorporated propidium iodide in the flow cytometry analysis at both exposure times (Figure 2B). MCN does not induce cell death via apoptosis because only 3% of MCN‐treated cells exposed annexin‐V on the cellular surface (Figure 2C). This percentage is similar to that measured in the control group. Finally, 880 mM HP induced necrosis/apoptosis in 20%/15% and 75%/20% of treated cells at 5 and 30 minutes of exposure, respectively.
Figure 2.
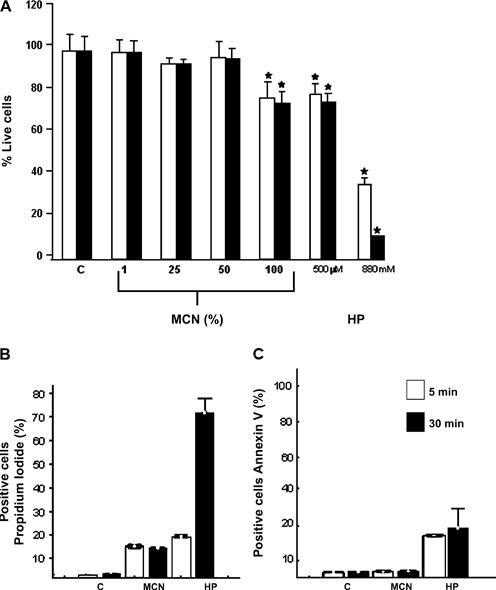
Acute effects of MCN and hydrogen peroxide (HP) on primary human dermal fibroblasts (HDFs) viability. (A) Cell viability of primary HDFs exposed to different concentrations of MCN and HP. Cells (5 × 105) were exposed to antiseptic concentrations of MCN (100%, undiluted) or HP (880 mM) as well as to diluted solutions for 5 and 30 minutes (blank and black bars, respectively). Cell viability was measured by 7‐aminoactinomycin D incorporation using flow cytometry analysis (* significant statistical difference, P < 0·05). (B) Propidium iodide staining of HDFs exposed to undiluted MCN or 880 mM HP after 5 and 30 minutes of exposure. Positive staining was evaluated by flow cytometry as a marker of necrosis. (B) Annexin V–FITC detection in HDFs exposed to either undiluted MCN or 880 mM HP after 5 and 30 minutes of exposure. Positive staining was evaluated as a marker of apoptosis. Percentage values expressed as mean ± SD (n= 3).
Effects of acute exposure of HDF to MCN on nucleic acid stability
Oxidation agents are known to cause direct damage to nucleic acids. It has also been shown that 500 μM HP is a sublethal dose that can be used as a positive control of genotoxicity in in vitro experiments (18). Figure 3A shows a native electrophoresis of genomic DNA (gDNA) samples from undiluted MCN‐ and 500 μM HP‐treated HDFs. In MCN samples, there were high‐molecular‐weight DNA fragments (>12 kb) that suggest integrity of the gDNA, whereas there was a DNA degradation smear in the HP samples. The integrity of total RNA extracted from MCN‐ and 500 μM HP‐treated cells was also analysed under denaturing conditions on agarose gels. Two discreet bands that correspond to intact ribosomal eukaryotic RNAs (i.e. 28S and 18S) were seen only in the control and MCN samples. In contrast, there was a low‐molecular‐weight smear in the HP samples because of RNA degradation (Figure 3B).
Figure 3.
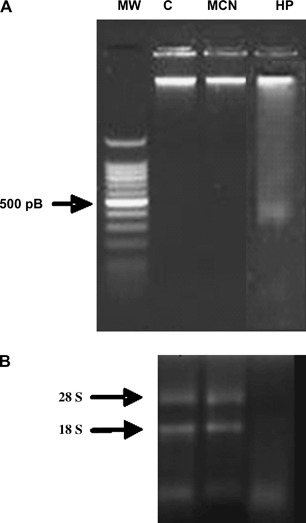
The acute treatment of human dermal fibroblasts (HDFs) with undiluted MCN did not alter DNA or RNA stability. (A) Ethidium‐bromide‐stained 1% agarose gel. The 70 ng of total gDNA was loaded for native electrophoresis of DNA extracted from non treated HDF cultures (C) and from treated cultures with undiluted MCN or 500 μM hydrogen peroxide (HP). (B). Two micrograms of total RNA samples were run on a 1·5% denaturing agarose gel stained with ethidium bromide. The 18S and 28S ribosomal RNA bands are clearly visible in the samples from control (C) and MCN‐treated cells. The degraded RNA appears as a lower molecular weight smear in the sample from HP‐treated cells.
To further analyse the integrity of nucleic acids, we amplified samples from cells treated with undiluted MCN or 500 μM HP. gDNA samples from both conditions were used as a template for PCR amplification of the human β‐globin gene. One 270 pb fragment could be amplified from all samples, even in the HP samples (Figure 4, upper panel). This is not unexpected because the gDNA degradation in HP‐treated cells was only partial. RNA samples from both conditions also underwent reverse transcription and amplification (RT–PCR) of the constitutive human GAPDH gene. This reaction is widely used as control of RT–PCR amplification because of the abundance and steady levels of this messenger in eukaryotic cells (19). In this study, RT–PCR assays used the same starting cDNA load for the amplification reaction of all samples. As shown in Figure 4 (lower panel), RNA samples obtained from cells exposed to undiluted MCN were appropriate for the amplification of this gene in the same proportion as in the control samples. In contrast, the partial or total degradation of the RNA in HP‐treated cells reduced the quantity of GAPDH transcripts and the amplification levels of the GAPDH human gene in these samples.
Figure 4.
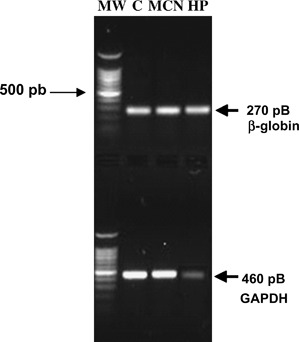
Amplification of the human β‐globin and glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) genes. Human dermal fibroblasts (HDFs) were treated with either undiluted MCN or 500 μM hydrogen peroxide (HP) for 30 minutes prior to gDNA and total RNA extraction. Upper panel: Ethidium‐bromide‐stained 1% agarose gel. DNA samples (300 ng) from control and treated HDFs were used for polymerase chain reaction (PCR) amplification of the human β‐globin gene as described in the Materials and Methods section. Lower panel: Stained 1% agarose gel showing PCR products of the human GAPDH gene. Even when the complementary DNA load used for amplification of all HDFs samples was the same for the three conditions, only the treatment of HDFs with 500 μM HP reproducibly diminished the amplification efficiency of GAPDH (n= 3).
Oxidative DNA damage and adduct formation (8‐OHdG) in HDFs
It is known that the production of 8‐OHdG adducts in a cell is a marker of oxidative damage at specific residues of the DNA molecule (20). In addition, high cellular levels of this adduct correlate with mutagenesis, carcinogenesis and cellular ageing 21, 22, 23. Figure 5 shows the levels of 8‐OHdG adducts present in DNA samples from treated HDFs with control, undiluted MCN or a sublethal concentration of HP (i.e. 500 μM) for 30 minutes. DNA extraction was then performed immediately (T0) or 3 hours (T3) after exposure to evaluate any change in the concentration of adducts. The exposure of HDFs to MCN for 30 minutes did not increase the formation of adducts in these cells in comparison to the controls at T0 and T3. In contrast, the amount of 8‐OHdG adducts observed in HDFs treated with 500 μM HP for 30 minutes increased 20 times in comparison to the control at T0. These cells were able to repair the oxidation process and partially decrease the levels of 8‐OHdG adducts after 3 hours (T3) of incubation in supplemented DMEM. Despite this recovery period, HP‐treated cells still presented five times more adducts than control‐ or MCN‐treated cells at T3.
Figure 5.
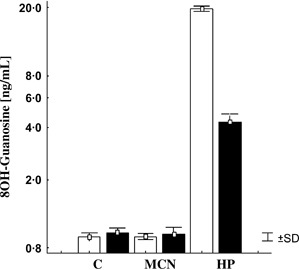
Levels of 8‐hydroxy‐2′deoxyguanosine (8‐OHdG) adducts in human dermal fibroblasts. Cells were treated with control (C), undiluted MCN or 500 μM hydrogen peroxide (HP). DNA was further extracted right after the exposure (T0, white bars) or 3 hours after the challenge period (T3, black bars). DNA was digested, and the 8‐OHdG adducts were measured by enzyme‐linked immunosorbent assay kit as per the manufacturer’s instructions. Only cells treated with a sublethal – but genotoxic – HP dose showed an increase in the levels of the 8‐OHdG adducts at T0. These adduct levels partially decreased after a 3‐hour recovery period of the cells in supplemented medium. Values (ng/ml) as mean ± SD (n= 3).
Effects of chronic exposure of HDFs to low concentrations of MCN or HP
It is known that chronic oxidative stress induced by low HP concentrations promotes premature ageing in human fibroblasts (24). In order to mimic prolonged oxidative stress in this study, primary HDF cultures were chronically exposed to low concentrations of MCN (10%) and HP (5 μM) during 20 passage doubling (PD). After 1 month of continuous exposure, the expression of the senescence‐associated‐β‐galactosidase (SA‐β‐Gal) enzyme was analysed. The expression and activity of this isoform have been previously associated with the senescence process in vivo and in vitro (25). Chronic treatment of HDFs with the sublethal HP concentration herein tested increased the SA‐β‐Gal expression in 86% of cells. In contrast, the treatment with MCN did not induce overexpression of this protein.
Discussion
It is common practice to evaluate efficacy and safety of wound cleansers by demonstrating antimicrobial activity and lack of skin irritation and sensitisation in animal models. SOSs have been shown to be effective antimicrobials with potential clinical applications 4, 7, 8, 10. Of these SOSs, a neutral‐pH solution (i.e. MCN) is currently used in wound care and does not induce skin sensitisation or irritation in animal models despite its high ORP (>800) (11). However, a major concern when using SOSs in wounds has been the potential induction of oxidative stress in HDFs. We have therefore conducted a series of in vitro experiments to explore this issue.
For this purpose, the toxicity of MCN was compared with that of another common wound care product, HP, which is also the best positive control for oxidative stress in vitro. Cell viability and oxidative stress markers were evaluated in HDFs at different concentrations of MCN and HP. Direct exposure of HDFs to either agent was the preferred route of administration for acute experiments in order to avoid inactivation with proteins found in supplemented media (26). All experiments were conducted with primary HDF cultures that contain more heterogeneous cell populations than established cell lines 27, 28. This was important in order to have a more representative cell population with different fibroblast phenotypes, thus more closely mimicking the cell population heterogeneity found in wounds. Finally, 8‐OHdG adducts and SA‐β‐Gal expression were selected as cellular markers of oxidative stress because elevation of their levels correlates with mutagenesis, carcinogenesis or cellular ageing 20, 21, 22, 23, 24.
Others have showed that the HP concentration regularly used for wound care (i.e. 3% or 880 mM) is lethal to HDFs, whereas sublethal concentrations of HP (e.g. 500 and 5 μM) are still genotoxic to these cells 24, 26. Under our experimental conditions, more than 90% of the cells died after 30 minutes of exposure to 880 mM HP. In contrast, exposure of HDFs to undiluted MCN or to 500 μM HP for 30 minutes induced cell death in 30% of the population. However, the viable HDFs exposed to undiluted MCN did not show DNA or RNA degradation or increased levels of 8‐OHdG adducts, as did those HDFs exposed to 500 μM HP. Furthermore, when HDFs were chronically exposed to an even lower concentration of HP (5 μM) or MCN (10% v/v) for 1 month, premature ageing was only induced in the HP‐treated group. These results showed that undiluted MCN is significantly less cytotoxic than commonly used, antiseptic HP concentrations (i.e. 880 mM) and that it neither induces DNA oxidation nor accelerated ageing, as do sublethal HP concentrations.
To evaluate the type of cell death induced by HP and MCN, fibroblasts underwent annexin‐V and propidium iodide staining and were further analysed by flow cytometry. As reported elsewhere 29, 30, 31, HP also induced apoptosis and necrosis in HDFs under our experimental conditions. In theory, HP induces oxidation of nucleic acids and plasma membrane, which leads to apoptosis and necrosis, respectively. In contrast, undiluted MCN induced only necrosis. Because exposure to MCN for 30 minutes did not alter DNA or RNA stability, it is possible that exposure to undiluted MCN produced only limited damage to the plasma membranes of HDFs.
The induction of DNA oxidation by sublethal concentrations of HP (500 μM), but not by undiluted MCN, is further evidence that MCN does not affect cell nuclei. At this concentration, HP caused a 20‐fold increase in levels of 8‐OHdG in HDFs. Instead, the level of DNA adducts in MCN‐treated cells remained the same as in the control cells. Because cultures of young fibroblasts (<20 PDs) had been reported to re‐establish the basal levels of 8‐OHdG in a period of 60 minutes after the removal of the oxidative agent (24), we cultured MCN‐ and HP‐treated cells in supplemented medium for 3 hours prior to a second measurement of 8‐OHdG. After this recovery period, adduct levels in HP‐treated cells were still five times higher than in control and MCN‐treated cells. These data confirm that sublethal concentrations of HP induce extensive DNA oxidation that cannot be repaired in 3 hours, whereas MCN does not induce the accumulation of DNA adducts, as originally hypothesised.
Finally, it has been shown that accelerated ageing in fibroblasts, as measured by the expression of SA‐β‐Gal, can be induced by chronic exposure of these cells to low concentrations of HP (i.e. 5 μM) 24, 25. The expression of this isoform has been associated with degenerative processes in vivo and with cellular ageing or ‘replicative senescence’ in vitro 18, 19. After 1 month of exposure to 5 μM HP, there was increased expression of SA‐β‐Gal in HDFs but not with 10% MCN. Moreover, only 5 μM HP increased the doubling times in HDFs (which is a characteristic of senescent cultures) as compared to control‐ and MCN‐treated cells (data not shown). These results are intriguing because MCN kills bacteria, fungi, viruses and spores in short periods of time, from seconds to few minutes, depending on the type of test conducted. In suspension tests, for example, a log10 reduction factor ≥8 in the level of bacteria usually occurs in 15–30 seconds of exposure to MCN (9). Apparently, the reactive species of SOS rapidly react and denature the bacterial cell wall and membrane followed by lysis (32).
We conclude that exposure of HDFs to undiluted MCN, as used in wound care, is significantly less cytotoxic than antiseptic HP concentrations (i.e. 880 mM). In addition, MCN does not induce DNA oxidation or accelerated ageing, as do sublethal HP concentrations. These results are supported by the lack of sensitisation, irritation or genotoxicity that have been described (11).
References
- 1. Beckman KB, Ames BN. The free radical theory of aging matures. Physiol Rev 1998;78:547–81. [DOI] [PubMed] [Google Scholar]
- 2. De Grey AD. Reactive oxygen species production in the mitochondrial matrix: implications for the mechanism of mitochondrial mutation accumulation. Rejuvenation Res 2005;8:13–7. [DOI] [PubMed] [Google Scholar]
- 3. Suzuki T, Masuda M, Friesen MD, Fenet B, Ohshima H. Novel products generated from 2′‐deoxyguanosine by hypochlorous acid or a myeloperoxidase – H2O2‐Cl‐system: identification of diimino‐imidazole and amino‐imidazolone nucleosides. Nucleic Acids Res 2002;30:2555–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tanaka H, Hirakata Y, Kaku M, Yoshida R, Takemura H, Mizukane R, Ishida K, Tomono K, Koga H, Kohno S, Kamihira S. Antimicrobial activity of superoxidized water. J Hosp Infect 1996;34:43–9. [DOI] [PubMed] [Google Scholar]
- 5. Park H, Hung YC, Kim C. Effectiveness of electrolyzed water as a sanitizer for treating different surfaces. J Food Prot 2002;65:1276–80. [DOI] [PubMed] [Google Scholar]
- 6. Nelson D. Newer technologies for endoscope disinfection: electrolyzed acid water and disposable‐component endoscope systems. Gastrointest Endosc Clin N Am 2000;10:319–28. [PubMed] [Google Scholar]
- 7. Sekiya S, Ohmori K, Harii K. Treatment of infectious skin defects or ulcers with electrolyzed strong acid aqueous solution. Artif Organs 1997;21:32–8. [DOI] [PubMed] [Google Scholar]
- 8. Inoue Y, Endo S, Kondo K, Ito H, Omori H, Saito K. Trial of electrolyzed strong acid aqueous solution lavage in the treatment of peritonitis and intraperitoneal abscess. Artif Organs 1997;21:28–31. [DOI] [PubMed] [Google Scholar]
- 9. Landa‐Solís C, González‐Espinosa D, Guzmán B, Snyder M, Reyes‐Terán G, Torres K, Gutiérrez AA. Microcyn a novel super‐oxidized water with neutral pH and disinfectant activity. J Hosp Infect 2005;61:291–9. [DOI] [PubMed] [Google Scholar]
- 10. Martínez‐Munive A, Menéndez‐Skertchly A, Toiber M, Hernández‐Hernández B, Padilla‐Longoria R, Quijano‐Orvananos F. Super‐oxidized water (Microcyn 60) for mesh hernioplasty in grossly contaminated fields: an experimental study. SE 163. In: American College of Surgeons 91st Annual Clinical Congress; 2005. Oct 16–20; San Francisco (CA). [Google Scholar]
- 11. Gutiérrez AA. The science behind stable, super‐oxidized water. Exploring the various applications of super‐oxidized solutions. Wounds 2006;18 (Suppl Jan):7–10. [Google Scholar]
- 12. Dressler LG, Seamer LC. Controls, standards and histogram interpretation in DNA flow cytometry. Methods Cell Biol 1994;41:241–62. [DOI] [PubMed] [Google Scholar]
- 13. Stein GH. SV‐40‐transformed human fibroblasts: evidence for cellular aging in pre‐crises cells. J Cell Physiol 1985;125:36–44. [DOI] [PubMed] [Google Scholar]
- 14. Inadome Y, Noguchi M. Selection of higher molecular weight genomic DNA for molecular diagnosis from formalin‐fixed material. Diagn Mol Pathol 2003;12:231–6. [DOI] [PubMed] [Google Scholar]
- 15. Fraga C, Motchnik P, Shigenaga M, Helbock H, Jacob RA, Ames BN. Ascorbic acid protects against endogenous oxidative DNA damage in human sperm. Proc Natl Acad Sci U S A 1991;88:11003–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shen CL, Song W, Pence BC. Interactions of selenium compounds with other antioxidants in DNA damage and apoptosis in human normal keratinocytes. Cancer Epidemiol Biomarkers Prev 2001;10:385–90. [PubMed] [Google Scholar]
- 17. Goberdhan P, Lee X, Basile G, Acosta M, Scout G, Roskelley C, Medrano E, Linskens M, Rubeli I, Pereira‐Smith O, Peacocke M, Campisi J. A biomarker that identifies senescent human cell in culture and in aging skin in vivo . Proc Natl Acad Sci U S A 1995;92:9663–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Frippiat C, Chen QM, Zdanov S, Magalhaes JP, Remacle J, Toussaint O. Subcytotoxic H2O2 stress triggers a release of transforming growth factor‐beta, which induces biomarkers of cellular senescence of human diploid fibroblast. J Biol Chem 2001;276:2531–7. [DOI] [PubMed] [Google Scholar]
- 19. Dheda K, Huggett JF, Bustin SA, Johnson MA, Rook G, Zumla A. Validation of housekeeping genes for normalizing RNA expression in real‐time PCR. Biotechniques 2004;37:112–4, 116, 118–9. [DOI] [PubMed] [Google Scholar]
- 20. Valko M, Izakovic M, Mazur M, Rhodes CJ, Telser J. Role of oxygen radicals in DNA damage and cancer incidence. Mol Cell Biochem 2004;266:37–56. [DOI] [PubMed] [Google Scholar]
- 21. Lj Marnett. Oxyradicals and DNA damage. Carcinogenesis 2000;21:361–70. [DOI] [PubMed] [Google Scholar]
- 22. Choi AM, Pignolo RJ, ApRhys CM, Cristofalo VJ, Holbrook NJ. Alteration in the molecular response to DNA damage during cellular aging of cultures fibroblast: reduced AP‐1 activation and collagenase gene expression. J Cell Physiol 1995;164:65–73. [DOI] [PubMed] [Google Scholar]
- 23. Ottender M, Lutz WK. Correlation of DNA adduct levels with tumor incidence: carcinogenic potency of DNA adducts. Mutat Res 1999;424:237–47. [DOI] [PubMed] [Google Scholar]
- 24. Wolf FI, Torsillo A, Covacci V, Fasanella S, Montanary M, Boninsegna A, Cittadini A. Oxidative DNA damage as a marker of aging in WI‐38 human fibroblasts. Exp Gerontol 2002;37:647–56. [DOI] [PubMed] [Google Scholar]
- 25. Severino J, Allen RG, Balin S, Ballin A, Cristafolo VJ. Is β‐galactosidase staining a marker of senescence in vitro and in vivo? Exp Cell Res 2000;257:162–71. [DOI] [PubMed] [Google Scholar]
- 26. Wilson JR, Mills JG, Prather ID, Dimitrijevich D. A toxicity index of skin and wound cleansers used on in vitro fibroblasts and keratinocytes. Adv Skin Wound Care 2005;18:373–8. [DOI] [PubMed] [Google Scholar]
- 27. Harley CB, Sherwood SW. Aging of cultured human skin fibroblast. Methods Mol Biol 1997;75:23–30. [DOI] [PubMed] [Google Scholar]
- 28. Jones GE, Wise CJ. Establishment, maintenance and cloning of human dermal fibroblast. Methods Mol Biol 1997;75:13–21. [DOI] [PubMed] [Google Scholar]
- 29. Naderi J, Hung M, Pandey S. Oxidative stress‐induced apoptosis in dividing fibroblast involves activation of p38 MAP kinase and over expression of Bax: resistance of quiescent cells to oxidative stress. Apoptosis 2003;8:91–100. [DOI] [PubMed] [Google Scholar]
- 30. Tanaka K, Asanuma M, Ogawa N. Molecular basis of anti‐apoptotic effect of immunophilin ligands on hydrogen peroxide‐induced apoptosis in human glioma cells. Neurochem Res 2004;29:1529–36. [DOI] [PubMed] [Google Scholar]
- 31. Arrigo AP, Firdaus WJ, Mellier G, Moulin M, Paul K, Diaz‐latoud C, Kretz‐remy C. Cytotoxic effects induced by oxidative stress in cultured mammalian cells and protection provided by Hsp27 expression. Methods 2005;35:126–38. [DOI] [PubMed] [Google Scholar]
- 32. Kiura H, Sano K, Morimatsu S, Nakano T, Morita C, Yamaguchi M, Maeda T, Katsuoka Y. Bactericidal activity of electrolyzed acid water from solution containing sodium chloride at low concentration, in comparison with that at high concentration. J Microbiol Methods 2002;49:285–93. [DOI] [PubMed] [Google Scholar]