

Abstract
Purpose of review
Atherosclerosis is a complicated cardiovascular disease characterized by unbalanced lipid metabolism and unresolved inflammation that occurred inside of arteries. The transcytosis of low- density lipoprotein (LDL) across the endothelium and its accumulation in the arterial wall is the initial step of atherosclerosis. Here we summarize recent research into the understanding of the regulatory mechanisms of endothelial LDL transcytosis and its relevance in the development of atherosclerosis.
Recent findings
A number of recent studies have revealed the contribution of caveolae, activin-like kinase 1 (ALK1) or scavenger receptor B1 (SR-B1) in endothelial LDL transcytosis and the progression of atherosclerosis. Caveolin-1 (Cav-1), the major protein component in caveolae, is required for the formation of caveolae and caveolae-mediated LDL uptake and transcytosis across the endothelium. SR-B1 and ALK1 directly bind LDL and facilitate the transport of LDL through the endothelial cells. The change in expression of caveolae-associated proteins and SR-B1 regulates endothelial LDL transcytosis and the pathogenesis of atherosclerosis.
Summary
Caveolae, ALK1 and SR-B1 are identified as key regulators in the LDL transcytosis across the endothelium. Endothelial LDL transcytosis might be a potential therapeutic approach to limit the initiation of early atherosclerosis and treat the atherosclerotic vascular diseases.
Keywords: low density lipoprotein, transcytosis, endothelial cell, atherosclerosis
INTRODUCTION
Atherosclerosis is a complex cardiovascular disease characterized by unbalanced lipid metabolism and unresolved inflammation that occurred inside of arteries [1, 2]. The infiltration and retention of athero-prone lipoproteins, predominately low density lipoprotein (LDL), in the vessel wall is considered as the initial step in the development of atherosclerosis [2]. Oxidative modification of the retained LDL enhances its pro-atherosclerotic potential through multiple inflammatory and immunologic mechanisms leading to increased lipid-laden foam cell formation and vascular inflammation to facilitate the progression of atherosclerosis [3]. During the progression of atherosclerosis, the flux of pro-atherogenic apoB-containing lipoproteins (LDL, VLDL and chylomicrons) and athero-protective high-density lipoprotein (HDL) into the vessel wall governs the progression of this vascular disease [4]. The transport of athero-prone LDL particles across the endothelium is critical for the initiation, progression and regression of atherosclerosis. However, the precise mechanisms governing the fluxes of lipoproteins in the vasculature are not fully understood. In this review article, we have summarized the recent findings in the field, highlighting the novel mechanisms involved in the transport of LDL into the arterial wall and its relevance in the development of atherosclerosis.
LDL TRANSCYTOSIS ACROSS ENDOTHELIUM
The LDL particles are heterogeneous lipid-protein complex with diameter ranged from 22 nm to 28 nm [5]. The transport of macromolecules across the endothelium is mediated by either paracellular or transcellular pathway. However, the paracellular transport of lipoproteins with a diameter > 6 nm through the endothelium is limited by inter-endothelial junctions including tight junctions, adherens junctions and gap junctions [6]. Numerous studies have revealed the involvement of transcytosis in the process of LDL transport across the endothelium [7–12]. Unlike the classical LDL receptor (LDLR)–dependent endocytosis pathway through which LDL is degraded by lysosomal enzymes releasing free cholesterol, intact LDL can pass through the endothelial cell (EC) layer by specific transcytosis pathways involved in caveolae, activin-like kinase 1 (ALK1) or scavenger receptor B1 (SR-B1) protein (Figure 1) [7, 8, 11]. LDLR has been reported to coordinate the LDL transcytosis in the blood-brain barrier (BBB) that avoid fusion with lysosomes [13], however, it’s unlikely to be required for endothelial LDL transcytosis in the athero-prone regions since degradation of LDLR by proprotein convertase subtilisin/kexin type 9 (PCSK9) did not influence the LDL transcytosis in EC [9]. In addition to the well-recognized mechanism of endothelial LDL transcytosis in the growth of atherosclerotic lesion, improved endothelial barrier function toward LDL permeability and degradation in the aortic wall precedes the morphological lesion regression using an anti-Apob antisense oligonucleotide in cholesterol-fed Ldlr-deficient mice [14]. Moreover, the plasma cholesterol lowering had no or less effect on the permeability of the vessel wall toward plasma proteins investigated by intravenous injection of Evans blue [14], indicating the involvement of LDL transcytosis in both the progression and regression of atherosclerosis.
Figure 1. The uptake and transport of low-density lipoprotein (LDL) in endothelial cells (ECs).
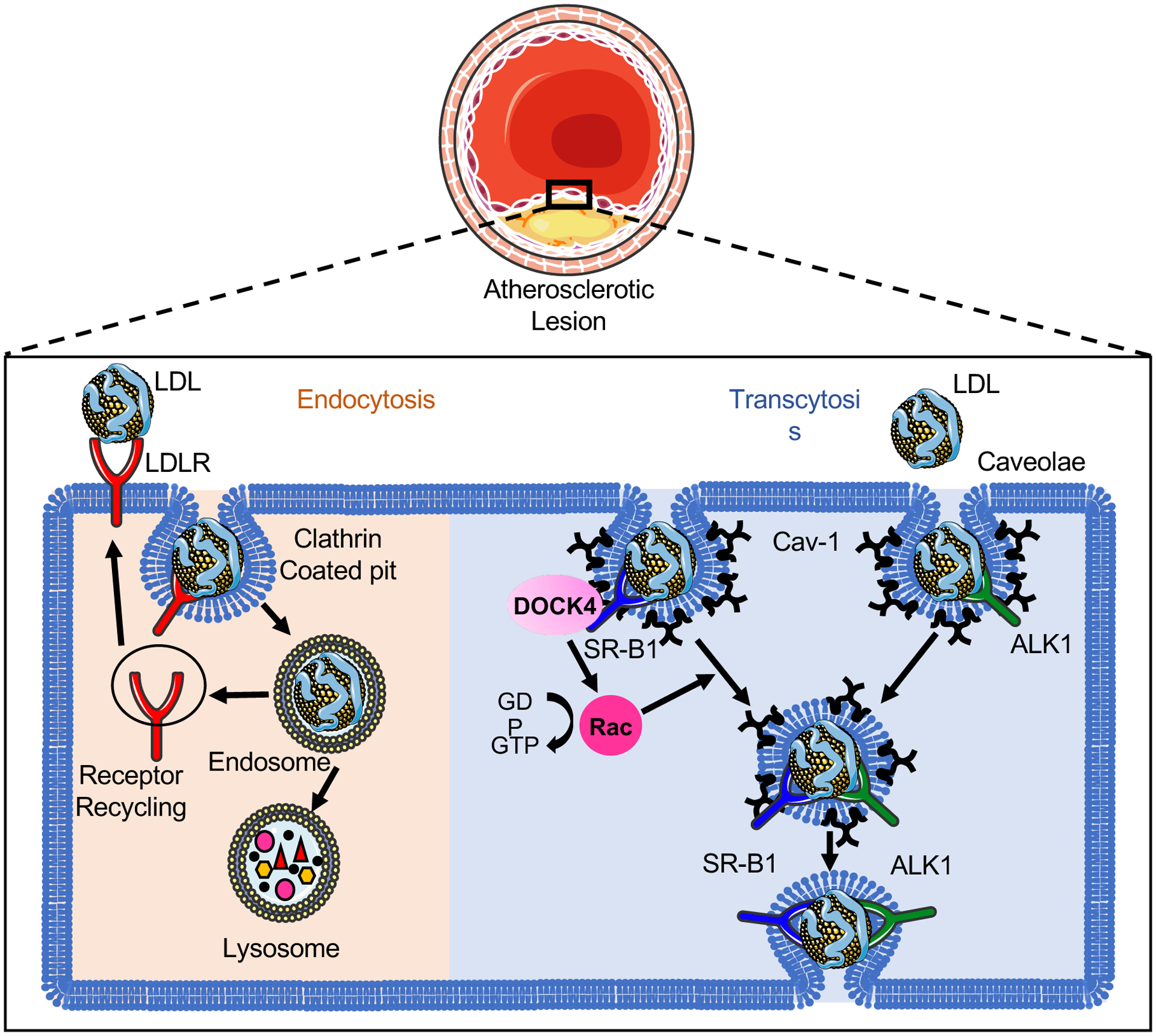
The classical LDL receptor (LDLR) pathway mediates the clustering of LDL-LDLR complex into clathrin-coated pits. The internalized coated vehicle fuses with endosomes, where the low pH in the endosome leads to the dissociation of LDLR from LDL and the recycling to the cell surface. The retained LDL particles travel further to lysosome and subsequently undergoes lysosomal degradation. Unlike the LDLR–dependent endocytosis pathway through which LDL is degraded by lysosomal enzymes releasing free cholesterol, intact LDL can transport across the endothelium by specific transcytosis pathways involved in caveolae, activin-like kinase 1 (ALK1) or scavenger receptor B1 (SR-B1) protein. Caveolin-1 (Cav-1) is the major protein component of caveolae that is required for the formation of caveolae in ECs. SR-B1 and ALK1 directly bind the LDL and locate in the caveolae. The recruitment of the guanine-nucleotide-exchange factor DOCK4 to SR-B1 facilitates the SR-B1 internalization and LDL transcytosis by coupling LDL binding to SR-B1 with Rac1 activation. The endocytosed LDL particles are then transferred to the opposite side of the cell leading to the retention of LDL in the arterial wall.
CAVEOLAE-MEDIATED LDL TRANSCYTOSIS IN EC
Caveolae is a highly uniform “bulb-shape” structure observed at the plasma membrane of specific cell types including EC, smooth muscle cell and fibroblasts [15]. In EC, caveolae is dynamic and highly motile microdomain involved in signal transduction and endocytosis [15]. Caveolin-1 (Cav-1) is the major protein component in caveolae mediating the formation of caveolae and provoking caveolae-associated functions in EC [15, 16]. Studies from our lab and others have elucidated an important role forCav-1 in LDL transcytosis and atherosclerosis [7, 10, 12, 17]. Genetic ablation of Cav-1 on Apoe−/− mice inhibited the progression of atherosclerosis by reducing LDL infiltration and macrophage accumulation into the arterial wall and increasing nitric oxide (NO) production [12]. Importantly, the specific overexpression of Cav-1 in the endothelium reversed the phenotypes observed in Cav-1/Apoe double knockout (Cav-1−/−Apoe−/−) mice [12]. Similar findings of Cav-1 in endothelial LDL transcytosis were observed in Cav-1−/− aortic rings incubated with 125I-labeled LDL ex vivo [10].
Our recent study revealed the co-localization of apoB, the main protein in LDL, and caveolae in the endothelium of the atheroprone regions of the aortic arch [7]. Absence of Cav-1 inhibited the LDL infiltration in the atheroprone regions of aortic arch both in vivo and ex vivo, while silencing of Cav-1 in human coronary aortic endothelial cells (HCAECs) markedly attenuated LDL transcytosis measured by TIRF microscopy in vitro [7]. Similar to the results observed in Apoe−/− mice, genetic deficiency of Cav-1 in Ldlr knockout mice significantly attenuated the infiltration of ApoB-containing lipoproteins and the initiation of atherosclerosis [7]. The suppression of atherosclerosis in absence of Cav-1 was independent of the increased endothelial nitric oxide synthase (eNOS) activity observed in Cav-1 deficient mice [7]. The physiological role of Cav-1 in regulating LDL transcytosis and the initiation of atherosclerotic lesions was further supported by the high-level expression of Cav-1 in the endothelium of human atherosclerotic plaques and the higher expression of Cav-1 in the athero-prone regions where disturbed flow occurs compared to athero-protected regions in the murine aortic arch (Figure 2) [7]. In addition to changes in Cav-1 expression, we also showed that the number of basolateral and intracellular caveolae were significantly increased in athero-prone areas (lower curvature of the aortic arch) compared to athero-protective areas (greater curvature of the aortic arch) (Figure 2). In contrast, the number of caveolae in the apical plasma membrane of the endothelium was significantly reduced in athero-prone areas. Together, these findings indicate that hemodynamics and mechanical forces influence caveolae number, morphology and cellular distribution in different locations of the aortic endothelium facilitating the transport of LDL into the arterial wall and the growth of atherosclerotic lesion [7].
Figure 2. Caveolae-mediated LDL transcytosis in endothelial cells (ECs) of aortic athero-prone regions.
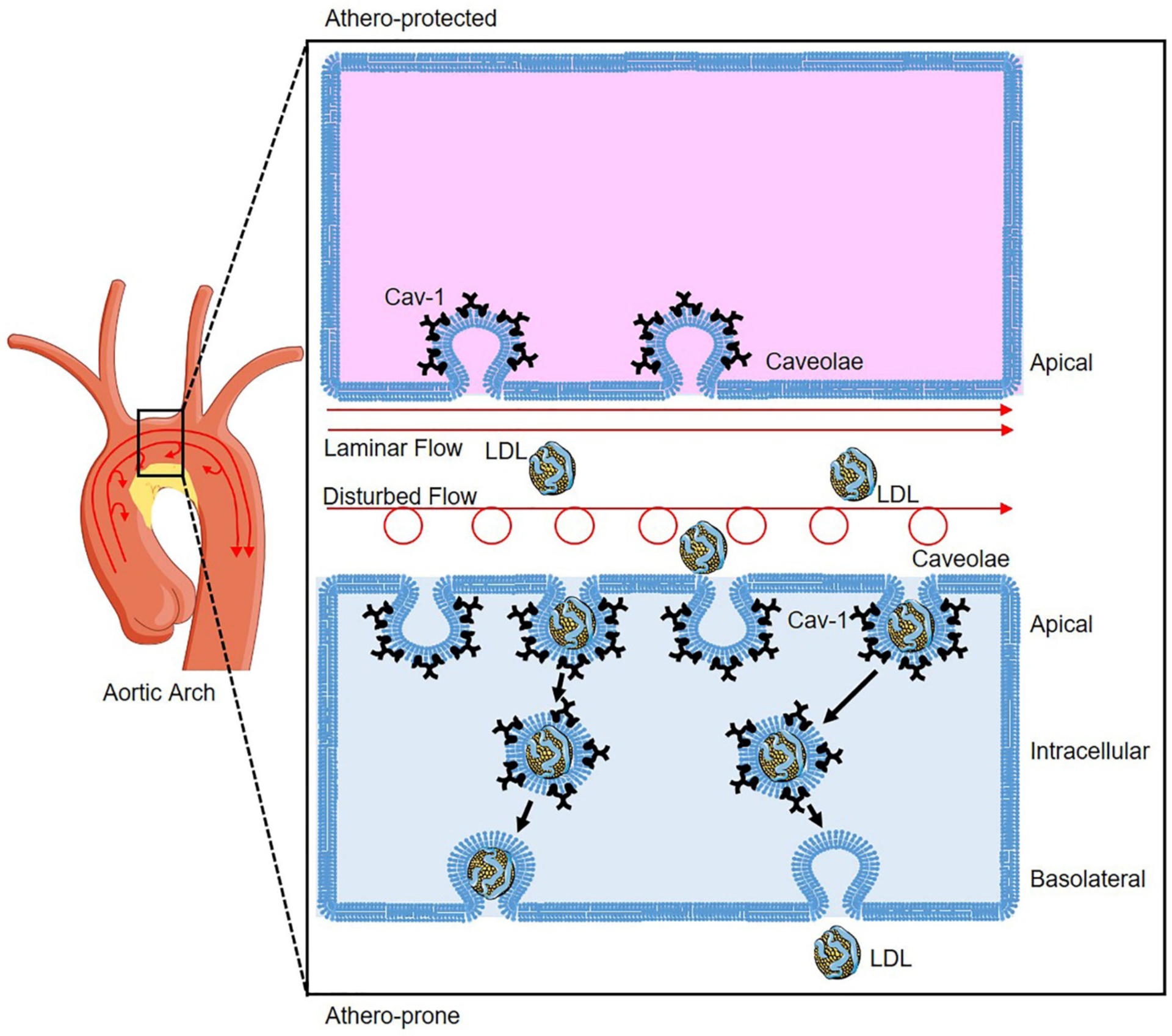
Atherosclerosis occurs preferentially at the athero-prone regions (low curvature) of disturbed flow with low shear stress, whereas laminar flow with high shear stress protects against atherosclerosis. Disturbed flow promotes higher expression of Caveolin-1 (Cav-1) in the athero-prone regions of aortic arch, which induces more caveolae formation compared to ECs of athero-protected regions where laminar flow conditions exist. In the ECs of athero-prone regions, caveolae colocalized with LDL redistribute from apical to intracellular compartment facilitating LDL transcytosis to induce LDL deposition and progression of atherosclerosis.
Recent reports have shown that intact endothelial autophagy is required to maintain the endothelial accumulation of native LDL and oxidized LDL (oxLDL) in vitro, as well as oxLDL retention in vivo [21]. Notably, we and others have uncovered the participation of caveolae in regulating autophagy activation in different cell types including ECs, adipocytes and cardiomyocytes [22–24]. Cav-1 deficiency promoted autophagy activation by enhancing autophagic flux in the aortic endothelium from athero-prone regions, by which protected against the initiation of atherosclerosis on the background of Ldlr−/− [22]. However, the pharmacological inhibition of autophagy by 3-methyladenine in Cav-1−/− mice attenuated the vascular inflammation and macrophage infiltration, but had no effect on neutral lipid accumulation in the atherosclerotic plaques [22], indicating that Cav-1-mediated autophagy might not contribute to LDL transcytosis process.
ENDOTHELIAL TRANSCYTOSIS OF LDL BY SR-B1
SR-B1 is well-known as an HDL receptor involved in reverse cholesterol transport (RCT) process by facilitating the efflux of cholesterol from peripheral macrophages and the selective uptake of cholesteryl esters from HDL into the liver [25, 26]. Besides acting as an HDL receptor, SR-B1 has been recognized as a high-affinity receptor for LDL, oxidized LDL and very low-density lipoprotein (VLDL) [27]. An early study leaded by Lee and colleagues uncovered an unexpected role for SR-B1 in endothelial LDL transcytosis in HCAECs [9]. Using total internal reflection fluorescence (TIRF) microscopy, the authors found that overexpression of SR-B1 markedly increases LDL transcytosis in ECs. Conversely, silencing the expression of SR-B1 using siRNAs attenuates LDL transport across the endothelial monolayer [9]. SR-B1-mediated LDL transcytosis was significantly inhibited by the addition of excess HDL, the canonical ligand for SR-B1, indicating the competition for the receptor between different lipoproteins [9]. Notably, SR-B1 has been found to be present in the basolateral compartment of EC [28] and facilitate the transport of excess cholesterol from peripheral cells of arterial wall followed by its return by the lymphatic system to the bloodstream by binding to HDL in the lymphatic EC [29, 30]. The competitive binding of SR-B1 with both HDL and LDL might prevent the endothelial transport of proatherogenic lipoproteins and promote the cholesterol efflux from the arterial wall to protect against atherosclerotic vascular diseases. Indeed, reduction of atherosclerosis was observed in the mice that overexpression of SR-B1 in EC, which might be attributed to greater cholesterol uptake by HDL in the basolateral compartment [28].
Most recently, the mechanisms by which SR-B1 regulates LDL transcytosis across the endothelium and development of atherosclerosis has been identified in the mice lacking SR-B1 selectively in the endothelium on an apolipoprotein E null (Apoe−/−) background [8]. SR-B1 directly bound the LDL and facilitated the transcytosis of LDL across endothelial monolayers by recruiting the guanine nucleotide exchange factor dedicator of cytokinesis 4 (DOCK4), and promoting SR-B1 internalization and LDL transcytosis by activation of Rac Family Small GTPase 1 (RAC1) [8]. These findings were further supported by the higher SR-B1 expression in athero-prone regions of mouse aorta and in human atherosclerotic arteries [8]. Remarkably, the genetic ablation of SR-B1 in ECs showed significantly reduced LDL accumulation in arterial wall and markedly less atherosclerosis [8], suggesting the important role of SR-B1 in LDL transcytosis and the progression of atherosclerosis. The identification of SR-B1 as receptor of both HDL and LDL suggests that the binding of HDL to SR-B1 might prevent the LDL transcytosis in the arterial endothelium to limit atherosclerosis progression [31]. While these findings indicates that suppression of SRB1 in EC might be beneficial to prevent atherosclerosis, global deficiency of SR-B1 results in massive atherosclerosis in mice [32], and coronary artery disease in humans [33] due to impair RCT, thus raising a concern about the therapeutic value of inhibiting SR-B1.
ALK1-MEDIATED LDL TRANSCYTOSIS IN EC
A genome-wide RNAi screening in the human EC line EA.hy926 identified ALK1 as an LDL-binding protein mediating LDL transcytosis [11]. ALK1 is a well-known transforming growth factor beta-1 (TGFβ−1) receptor highly expressed on ECs and binds bone morphogenetic proteins (BMP) −9 and −10 [34]. Silence ALK1 using siRNA in HCAECs reduced the transport of DiI-LDL from the apical to the basolateral membrane, while overexpression of ALK1 resulted in increased LDL transcytosis by TRIF imaging [11]. The regulation of ALK1 in LDL transcytosis was further confirmed by trans-well assay incubated with 125I-labelled LDL [11]. Notably, endothelial deficiency of ALK1 inhibited the LDL uptake into aortic endothelium in vivo indicating the involvement of ALK1 in the LDL uptake and transcytosis [11]. Intriguingly, ALK1-mediated LDL transcytosis was not affected by its kinase activity and the binding of other ligands such as BMP-9 [11]. However, the lethality of sustained ALK1 in adult endothelium limits the exploration of ALK1-mediated LDL transcytosis during atherogenesis [11, 35].
REGULATION OF ENDOTHELIAL LDL TRANSCYTOSIS
Caveolae are subcellular endocytic organelle that is rich in lipids and proteins including SR-B1, ALK1 and Cav-1.[36] The identification of SR-B1, ALK1 and Cav-1 as major regulators of LDL transcytosis and accumulation in the arterial wall provides the possibility targeting caveolae-associated molecules for the progression of atherosclerosis. Clinical studies have demonstrated a correlation between hyperglycemia and cardiovascular events including atherosclerosis [37, 38]. A recent study showed that high glucose suppressed the autophagic degradation of Cav-1 through AMPK-MTOR-PIK3C3 pathway, which led to the accumulation of Cav-1 in the cytosol and formation of caveolae in the cell membrane [39]. The increased level of Cav-1 and caveolae therefore facilitated the LDL transcytosis across the endothelium [39]. The mutation of Cav-1 carrying a defective LC3-interacting region (LIR) resulted in increased autophagic degradation of Cav-1 and reduced LDL transcytosis in ECs [39]. Studies from the same group revealed the effects of proinflammatory cytokine C-reactive protein (CRP) on LDL transcytosis and atherosclerosis [40, 41]. CRP treatment promoted the initiation of early atherosclerosis in diet-induced Apoe−/− mouse model through increased endothelial LDL transcytosis and the retention of LDL in human umbilical venous walls [41]. The CRP-stimulated LDL transcytosis was associated with the production of reactive oxygen species (ROS), NLRP3 inflammasome activation, and the expression of caveolae-associated proteins (Cav-1, cavin-1 and DNM2) in ECs [40, 41]. As similar with CRP, angiotensin II significantly promoted LDL transcytosis across the endothelium through the production of ROS and the expression of caveolae-associated proteins [42].
A recent study from Ghaffari et al provided first evidence that estrogen impaired the transcytosis of LDL in ECs, supporting a sex difference in LDL transcytosis [43]. The LDL transcytosis was higher in coronary ECs from men than premenopausal women, which was inhibited by estrogen [43]. Mechanically, estrogen bound to G-protein coupled estrogen receptor (GPER), instead of canonical ERα and ERβ, to repress SR-B1 expression and reduce LDL transcytosis, but had no effect on the expression of ALK1 and Cav-1 [43]. Importantly, depletion of SR-B1 by siRNA in ECs abolished the estrogen-mediated inhibition of LDL transcytosis while overexpression of SR-B1 in female ECs partially restored the sensitivity of LDL transcytosis to exogenous estrogen [43]. Li et al. reported that C1q/TNF related protein 5 (CTRP5), a secreted glycoprotein expressed predominantly in endothelium, promoted LDL transcytosis across endothelial monolayers in vitro, which might be attributed to the upregulation of LDLR related protein 1 (LRP1) and LRP1-mediated LDL transcytosis secondary to the increased 12/15-lipoxygenases pathway [44]. However, considering the endocytic uptake and degradation of LDL by LRP1 in macrophages [45], the contribution of LRP1 in endothelial LDL transcytosis need to be further investigated.
CONCLUSION
LDL transcytosis across the endothelium and its retention in the arterial wall are critical for the initiation of atherosclerosis. Caveolae, ALK1 and SR-B1 are key regulators of endothelial LDL transcytosis and change in expression of these regulators influences LDL transcytosis in ECs and subsequent progression of atherosclerosis (Figure 1). Interestingly, both SR-B1 and ALK1 directly bind LDL and locate in the caveolae, future studies are required to distinguish the specific contribution of SR-B1 and ALK1 in caveolae-mediated LDL transcytosis. Due to the technical limitation for in vivo imaging of LDL transcytosis across the endothelium, the precise mechanisms and molecular regulation of LDL transcytosis in vivo are still unclear. The technological breakthrough of in vivo imaging and the further exploration of its molecular mechanisms, LDL transcytosis might be recognized as a potential therapeutic target to limit the accumulation of LDL in the arterial wall and the development of atherosclerotic vascular diseases.
KEY POINTS.
LDL transcytosis across the endothelium followed by the retention of LDL in the arterial wall is the initial event of atherosclerosis.
Caveolae, ALK1 and SR-B1 are key regulators in endothelial LDL transcytosis and the progression of atherosclerosis.
Endothelial LDL transcytosis is regulated by hyperglycemia, proinflammatory cytokines and estrogen through the change of novel key regulators involved in this process.
Targeting endothelial LDL transcytosis might be a potential therapeutic approach to the treatment of atherosclerotic vascular diseases.
ACKNOWLEDGEMENTS
This work was supported by grants from the National Institutes of Health (R35HL135820 to CF-H), and the American Heart Association (16EIA27550005 to CF-H).
Footnotes
CONFLICTS OF INTEREST
None.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
* of special interest
** of outstanding interest
- [1].Back M, Yurdagul A Jr., Tabas I et al. Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat Rev Cardiol 2019; 16:389–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Tabas I, Williams KJ, Boren J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation 2007; 116:1832–1844. [DOI] [PubMed] [Google Scholar]
- [3].Tabas I, Bornfeldt KE. Macrophage Phenotype and Function in Different Stages of Atherosclerosis. Circ Res 2016; 118:653–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ouimet M, Barrett TJ, Fisher EA. HDL and Reverse Cholesterol Transport. Circ Res 2019; 124:1505–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Williams PT, Superko HR, Haskell WL et al. Smallest LDL particles are most strongly related to coronary disease progression in men. Arterioscler Thromb Vasc Biol 2003; 23:314–321. [DOI] [PubMed] [Google Scholar]
- [6].Komarova YA, Kruse K, Mehta D, Malik AB. Protein Interactions at Endothelial Junctions and Signaling Mechanisms Regulating Endothelial Permeability. Circ Res 2017; 120:179–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7]**.Ramirez CM, Zhang X, Bandyopadhyay C et al. Caveolin-1 Regulates Atherogenesis by Attenuating Low-Density Lipoprotein Transcytosis and Vascular Inflammation Independently of Endothelial Nitric Oxide Synthase Activation. Circulation 2019; 140:225–239. [DOI] [PMC free article] [PubMed] [Google Scholar]; Study showing different pattern of Cav-1 expression and caveolae distribution in the endothelial cells of athero-prone and athero-resistant area and revealing the mechanisms by which Cav-1 regulate atherogenesis through LDL transcytosis and vascular inflammation independent of endothelial nitric oxide synthase activation.
- [8]**.Huang L, Chambliss KL, Gao X et al. SR-B1 drives endothelial cell LDL transcytosis via DOCK4 to promote atherosclerosis. Nature 2019; 569:565–569. [DOI] [PMC free article] [PubMed] [Google Scholar]; Data demonstrating that high density lipoprotein receptor SR-B1 in endothelial cells mediates the transcytosis of LDL and its accumulation in arterial wall by recruiting DOCK4 to promote the progression of atherosclerosis.
- [9].Armstrong SM, Sugiyama MG, Fung KY et al. A novel assay uncovers an unexpected role for SR-BI in LDL transcytosis. Cardiovasc Res 2015; 108:268–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Frank PG, Pavlides S, Cheung MW et al. Role of caveolin-1 in the regulation of lipoprotein metabolism. Am J Physiol Cell Physiol 2008; 295:C242–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11]**.Kraehling JR, Chidlow JH, Rajagopal C et al. Genome-wide RNAi screen reveals ALK1 mediates LDL uptake and transcytosis in endothelial cells. Nat Commun 2016; 7:13516. [DOI] [PMC free article] [PubMed] [Google Scholar]; Evidence from genome-wide RNAi screen identifies the critical role of ALK1 in LDL transcytosis and uptake in the endothelial cells.
- [12].Fernandez-Hernando C, Yu J, Suarez Y et al. Genetic evidence supporting a critical role of endothelial caveolin-1 during the progression of atherosclerosis. Cell Metab 2009; 10:48–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Dehouck B, Fenart L, Dehouck MP et al. A new function for the LDL receptor: transcytosis of LDL across the blood-brain barrier. J Cell Biol 1997; 138:877–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bartels ED, Christoffersen C, Lindholm MW, Nielsen LB. Altered metabolism of LDL in the arterial wall precedes atherosclerosis regression. Circ Res 2015; 117:933–942. [DOI] [PubMed] [Google Scholar]
- [15]*.Parton RG. Caveolae: Structure, Function, and Relationship to Disease. Annu Rev Cell Dev Biol 2018; 34:111–136. [DOI] [PubMed] [Google Scholar]; A comprehensive, integrated review of caveolae underlying molecular composition, formation, functions and relationship to diseases.
- [16].Chidlow JH Jr., Sessa WC. Caveolae, caveolins, and cavins: complex control of cellular signalling and inflammation. Cardiovasc Res 2010; 86:219–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Fernandez-Hernando C, Yu J, Davalos A et al. Endothelial-specific overexpression of caveolin-1 accelerates atherosclerosis in apolipoprotein E-deficient mice. Am J Pathol 2010; 177:998–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18]*.Chen S, Wang X, Wang J et al. Genomic variant in CAV1 increases susceptibility to coronary artery disease and myocardial infarction. Atherosclerosis 2016; 246:148–156. [DOI] [PMC free article] [PubMed] [Google Scholar]; Case-control association studies identifies genomic variant in CAV-1 is associated with significant risk of CAD and MI in humans.
- [19].Holm H, Gudbjartsson DF, Arnar DO et al. Several common variants modulate heart rate, PR interval and QRS duration. Nat Genet 2010; 42:117–122. [DOI] [PubMed] [Google Scholar]
- [20].Ellinor PT, Lunetta KL, Albert CM et al. Meta-analysis identifies six new susceptibility loci for atrial fibrillation. Nat Genet 2012; 44:670–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Torisu K, Singh KK, Torisu T et al. Intact endothelial autophagy is required to maintain vascular lipid homeostasis. Aging Cell 2016; 15:187–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhang X, Ramirez CM, Aryal B et al. Cav-1 (Caveolin-1) Deficiency Increases Autophagy in the Endothelium and Attenuates Vascular Inflammation and Atherosclerosis. Arterioscler Thromb Vasc Biol 2020:ATVBAHA120314291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Le Lay S, Briand N, Blouin CM et al. The lipoatrophic caveolin-1 deficient mouse model reveals autophagy in mature adipocytes. Autophagy 2010; 6:754–763. [DOI] [PubMed] [Google Scholar]
- [24].Wu D, Xie F, Xiao L et al. Caveolin-1-Autophagy Pathway Mediated Cardiomyocyte Hypertrophy Induced by Apelin-13. DNA Cell Biol 2017; 36:611–618. [DOI] [PubMed] [Google Scholar]
- [25].Acton S, Rigotti A, Landschulz KT et al. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science 1996; 271:518–520. [DOI] [PubMed] [Google Scholar]
- [26].Yancey PG, de la Llera-Moya M, Swarnakar S et al. High density lipoprotein phospholipid composition is a major determinant of the bi-directional flux and net movement of cellular free cholesterol mediated by scavenger receptor BI. J Biol Chem 2000; 275:36596–36604. [DOI] [PubMed] [Google Scholar]
- [27].Calvo D, Gomez-Coronado D, Lasuncion MA, Vega MA. CLA-1 is an 85-kD plasma membrane glycoprotein that acts as a high-affinity receptor for both native (HDL, LDL, and VLDL) and modified (OxLDL and AcLDL) lipoproteins. Arterioscler Thromb Vasc Biol 1997; 17:2341–2349. [DOI] [PubMed] [Google Scholar]
- [28].Vaisman BL, Vishnyakova TG, Freeman LA et al. Endothelial Expression of Scavenger Receptor Class B, Type I Protects against Development of Atherosclerosis in Mice. Biomed Res Int 2015; 2015:607120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lim HY, Thiam CH, Yeo KP et al. Lymphatic vessels are essential for the removal of cholesterol from peripheral tissues by SR-BI-mediated transport of HDL. Cell Metab 2013; 17:671–684. [DOI] [PubMed] [Google Scholar]
- [30].Martel C, Li W, Fulp B et al. Lymphatic vasculature mediates macrophage reverse cholesterol transport in mice. J Clin Invest 2013; 123:1571–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zhang X, Fernandez-Hernando C. The Janus-faced role of SR-BI in atherosclerosis. Nature Metabolism 2019; 1:586–587. [DOI] [PubMed] [Google Scholar]
- [32].Braun A, Trigatti BL, Post MJ et al. Loss of SR-BI expression leads to the early onset of occlusive atherosclerotic coronary artery disease, spontaneous myocardial infarctions, severe cardiac dysfunction, and premature death in apolipoprotein E-deficient mice. Circ Res 2002; 90:270–276. [DOI] [PubMed] [Google Scholar]
- [33]*.Zanoni P, Khetarpal SA, Larach DB et al. Rare variant in scavenger receptor BI raises HDL cholesterol and increases risk of coronary heart disease. Science 2016; 351:1166–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]; Large population-based studies reveal a homozygote for a loss-of-function variant in SCARB1, the gene encoding SR-B1, increases plasmia HDL-C levels and risk of CHD.
- [34].David L, Mallet C, Mazerbourg S et al. Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood 2007; 109:1953–1961. [DOI] [PubMed] [Google Scholar]
- [35].Park SO, Wankhede M, Lee YJ et al. Real-time imaging of de novo arteriovenous malformation in a mouse model of hereditary hemorrhagic telangiectasia. J Clin Invest 2009; 119:3487–3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Anderson RG. The caveolae membrane system. Annu Rev Biochem 1998; 67:199–225. [DOI] [PubMed] [Google Scholar]
- [37].Chait A, Bornfeldt KE. Diabetes and atherosclerosis: is there a role for hyperglycemia? J Lipid Res 2009; 50 Suppl:S335–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Roussel R, Steg PG, Mohammedi K et al. Prevention of cardiovascular disease through reduction of glycaemic exposure in type 2 diabetes: A perspective on glucose-lowering interventions. Diabetes Obes Metab 2018; 20:238–244. [DOI] [PubMed] [Google Scholar]
- [39].Bai X, Yang X, Jia X et al. CAV1-CAVIN1-LC3B-mediated autophagy regulates high glucose-stimulated LDL transcytosis. Autophagy 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Bian F, Yang XY, Xu G et al. CRP-Induced NLRP3 Inflammasome Activation Increases LDL Transcytosis Across Endothelial Cells. Front Pharmacol 2019; 10:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Bian F, Yang X, Zhou F et al. C-reactive protein promotes atherosclerosis by increasing LDL transcytosis across endothelial cells. Br J Pharmacol 2014; 171:2671–2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Bian F, Cui J, Zheng T, Jin S. Reactive oxygen species mediate angiotensin II-induced transcytosis of low-density lipoprotein across endothelial cells. Int J Mol Med 2017; 39:629–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43]*.Ghaffari S, Naderi Nabi F, Sugiyama MG, Lee WL. Estrogen Inhibits LDL (Low-Density Lipoprotein) Transcytosis by Human Coronary Artery Endothelial Cells via GPER (G-Protein-Coupled Estrogen Receptor) and SR-BI (Scavenger Receptor Class B Type 1). Arterioscler Thromb Vasc Biol 2018; 38:2283–2294. [DOI] [PubMed] [Google Scholar]; The first evidence showing a sex difference in endothelial LDL transcytosis by regulating SR-B1 expression.
- [44].Li C, Chen JW, Liu ZH et al. CTRP5 promotes transcytosis and oxidative modification of low-density lipoprotein and the development of atherosclerosis. Atherosclerosis 2018; 278:197–209. [DOI] [PubMed] [Google Scholar]
- [45].Sakr SW, Eddy RJ, Barth H et al. The uptake and degradation of matrix-bound lipoproteins by macrophages require an intact actin Cytoskeleton, Rho family GTPases, and myosin ATPase activity. J Biol Chem 2001; 276:37649–37658. [DOI] [PubMed] [Google Scholar]