

Abstract
Multidrug efflux pumps present a challenge to treatment of bacterial infections, making it vitally important to understand their mechanism of action. Here we investigated the nature of substrate binding within LmrP of L. lactis, a prototypical multidrug transporter of the major facilitator superfamily. We determined the crystal structure of LmrP in a ligand-bound outward-open state and observed an embedded lipid in the binding cavity of LmrP. Molecular dynamics simulations, directed mutagenesis and native mass spectrometry confirm the presence of an anionic lipid, and show its need to stabilise the observed ligand-bound structure. Mutants engineered to disrupt binding of the embedded lipid display reduced transport of some, but not all antibiotic substrates. Our results suggest that a lipid within the binding cavity could provide a malleable hydrophobic component that allows adaptation to the presence of different substrates, helping to explain the broad specificity of this protein and possibly other multidrug transporters.
Introduction
Efflux pumps can provide cells with a broad-range resistance mechanism when exposed to cytotoxic agents. By protecting cells from acute chemical stress, they create conditions that enable the emergence of long-lasting genetic adaptation. The ability of a single transporter to efficiently bind and expel structurally diverse molecules constitutes a structural conundrum that remains poorly understood. Multidrug exporters are known to transport compounds ranging from small dyes and short detergents to large antibiotics1,2. In some extreme cases (such as the mammalian P-glycoprotein), a given transporter has been described to recognise molecules as diverse as small organic compounds, lipids and even peptides3,4. Such remarkable polyspecific substrate recognition is difficult to reconcile with the traditional view of protein ligand interaction, where binding sites have evolved to provide optimal complementation for one or a few ligands. Efflux pumps employ transport mechanisms used by metabolite transporters of the MATE, MFS, SMR, RND, PACE and ABC (super)families5, albeit without the substrate specificity of classical transporters. From a structural standpoint it is particularly challenging to understand how such large differences in mass, size and chemical organisation can be accommodated by a single binding pocket. Therefore it is expected that high-resolution structures of multidrug transporters bound with various substrates would help decipher this particular enigma.
One of the better structurally characterised systems are the gram-negative bacterial RND extrusion pumps where multiple crystal structures of AcrB have been obtained by different groups, some with well-defined bound substrates6–8. These structures show that substrates locate in a complex site made of two binding pockets, separated by a switch loop, that change in size depending on the state of the protein within the transport cycle (access, binding, extrusion). While analysis of AcrB structures indicates that substrates do not bind via an induced-fit mechanism, it was proposed that the conformational plasticity of the two pockets may allow diverse modes of binding, although the molecular basis of polyspecificity remains largely unclear9,10.
The recent cryo-EM structures of both MRP1 bound to leukotriene C4 (LC4) and of human P-glycoprotein bound to taxol provide examples of ABC multidrug transporters bound to physiological ligands11,12. LC4 is bound within MRP1 through the coordinated action of two chemically distinct halves of the binding site, one positively charged binding pocket and one hydrophobic binding pocket. Within human P-glycoprotein, taxol is coordinated through non-bonded interactions and through the side chains of three binding site glutamines, although these interactions aren’t divided into the two distinct pockets observed for MRP1. Despite clear differences in the nature of substrate coordination in both proteins (which is reflected in their substrate specificities), plasticity of the binding pocket size and variation in the specific side chains involved in coordination is thought to underpin the adaptability of both of these proteins to different substrates.
The structure of MdfA, a secondary multidrug transporter of the Major Facilitator Superfamily (MFS), bound to chloramphenicol identifies key polar interactions between the substrate and an interior aspartate (D34)13. However, how the protein coordinates the rest of the ligand is not entirely clear from the structures as no other interactions (i.e. VDW contacts) are observed.
In this study, we set out to uncover the molecular basis of the polyspecific nature of LmrP, a prototypical bacterial multidrug transporter of the MFS family. LmrP is a model MFS expressed by L. lactis and has a broad spectrum of substrates, from antibiotics to DNA dyes14,15. All LmrP substrates are lipophilic, have at least one charge at neutral pH and are typically of a mass between 300 and 850 Da16, although detergents and Ca2+ ions have also been suggested as substrates14,17. Double Electron Electron resonance (DEER) studies have shown that the transporter alternates between at least two states, inward-open and outward-open, depending on the protonation states of key acidic residues, namely Asp68 and Glu32718. Owing to its extensive functional and biophysical characterisation16,19,20, LmrP is a particularly well-suited model for deciphering the fundamental principles of efflux pumps.
Results and Discussion
LmrP shows limited conformational adaptation to substrate
The ability of a given multidrug resistance transport protein (MDR) to recognise structurally diverse substrates could require conformational adaption of the protein scaffold. Using DEER spectroscopy, we have previously shown that binding of the prototypical MDR substrate Hoechst 33342 leads to stabilisation of the outward open state18. In order to explore possible structural adaptation, we here monitored the conformational dynamics of LmrP in the presence of 6 known substrates15,16 by performing DEER distance measurements using 5 spin-labelled cysteine reporter pairs. We selected 2 cysteine pairs on the extracellular side and 2 on the intracellular side that were previously shown to report on changes in the conformational equilibrium, as well as 1 pair within the transmembrane region that had been demonstrated to be sensitive to substrate binding18. We observe mostly minor modifications of distance distribution for the conformational distance reporters but significant changes for the binding reporter with all ligands, thereby confirming that the different substrates bind the transporter under such experimental conditions (Figure 1 and Supplementary Figure 1). These results suggest that large conformational changes of the protein are not required to bind ligands as different as ethidium bromide and verapamil. On the other hand, for each of the six ligands, we observed distance distributions that are fully compatible with an outward-open state (Figure 1), suggesting a common overall conformation for the high-affinity state.
Figure 1. DEER spectroscopy shows limited conformational change upon substrate binding to LmrP.
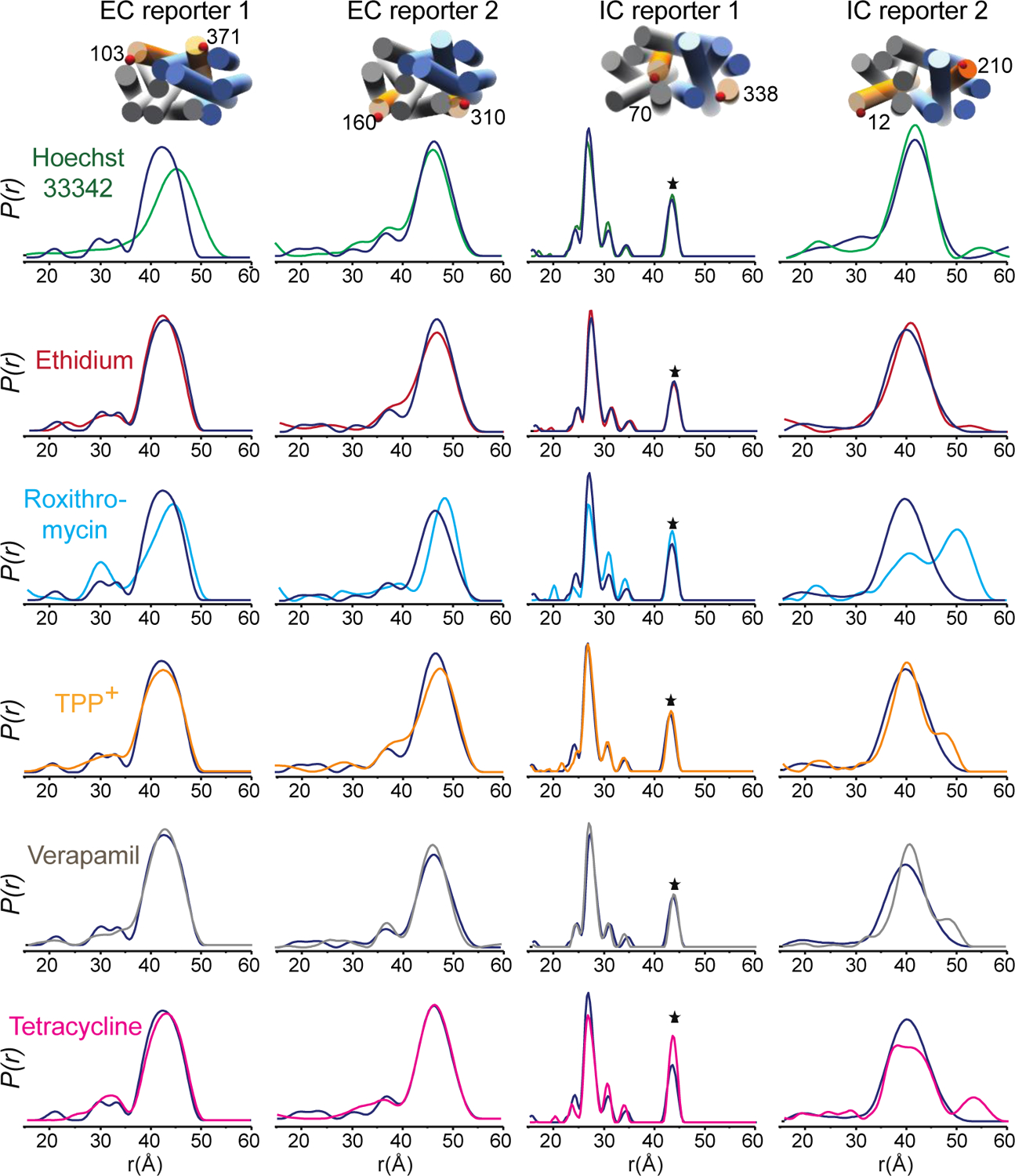
The conformational equilibrium of LmrP is compatible with the outward facing conformation in the apo state (blue) and in the presence of Hoechst 33342 (green), ethidium bromide (red), roxithromycin (cyan), TPP+ (yellow), verapamil (grey) and tetracycline (magenta), as evidenced by distance distributions observed with spin-labels located on the extracellular (EC) and intracellular (IC) termini of transmembrane helices. Distributions were normalised. Interspin distance is denoted by r, with P(r) indicating the distance probability. The * denotes the peak due to aggregation in the sample (see methods).
Crystallisation of LmrP
We sought to investigate the molecular details allowing the necessary binding pocket plasticity by solving the crystal structure of LmrP in complex with Hoechst 33342. Crystallisation of LmrP required extensive screening of purification and crystallisation conditions. In particular, precisely controlling the lipidation state of the purified protein proved essential to obtaining diffracting crystals in a reproducible manner (see Methods). Highest resolution crystals also required the removal of a 4 residue stretch within LmrP that was predicted to be flexible (according to the SERp server21). These residues (199–202) are located in the 3rd intracellular loop that connects the N-terminal and C-terminal halves of LmrP. Co-crystallisation of this LmrP mutant with Hoechst 33342 resulted in crystals diffracting up to 2.9 Å resolution. Subsequently, initial phases were obtained using crystals grown after selenomethionine replacement of the 16 native methionines, with the anomalous signal of the SeMet also used to guide model building (Supplementary Figure 2). Data collection and refinement statistics for native and anomalous datasets are given in Table 1.
Table 1. Crystallographic data and refinement statistics.
Values in parenthesis refer to the highest resolution bin. Data collection statistics for the native dataset are given prior to the use of an anisotropic resolution cut-off.
| Data collection | Native | Se peak |
|---|---|---|
| Space group | C2221 | C2221 |
| Unit cell parameters (Å) | 104.74, 139.99, 115.70 | 104.72, 143.19, 115.64 |
| Wavelength (Å) | 0.97166 | 0.97925 |
| Resolution | 47.7–2.9 | 45.0–3.8 |
| Observations | 367272 (18981) | 181358 (8425) |
| Unique observations | 19088 (1303) | 8938 (652) |
| Multiplicity | 19.2 (14.6) | 20.3 (12.9) |
| I/σ(I) | 9.1 (0.2) | 9.0 (1.4) |
| Rpim | 0.04 (2.91) | 0.068 (1.032) |
| Completeness | 99.5 (94.6) | 99.9 (100.0) |
| CC1/2 | 1 (0.23) | 0.998 (0.621) |
| Refinement | ||
| Resolution | 20.0–2.9 | |
| Rwork/Rfree | 22.4/ 23.5 | |
| R.m.s.d., bonds (Å) | 0.01 | |
| R.ms.d., angles (°) | 1.23 | |
| No. of atoms | ||
| Protein | 2981 | |
| Hoechst | 34 | |
| Lipids | 72 | |
| Detergent / Glycerol / Water | 79 | |
| Mean B-factors (Å2) | ||
| Protein | 82.3 | |
| Hoechst | 74.5 | |
| Lipids | 103.9 | |
| Detergent / Glycerol / Water | 106.48 | |
| Ramachandran | ||
| Favoured (%) | 96 | |
| Allowed (%) | 4 |
Architecture of LmrP
LmrP follows the canonical MFS fold of 12 transmembrane α-helices (TM) arranged in two pseudo-symmetrical bundles of 6 helices (the N-lobe and the C-lobe) connected by a central loop that is partly structured into an α-helix running parallel to the membrane plane (Figure 2). Most of the polypeptide is clearly visible in the electron density and allowed unambiguous building of the model with the exception of residues 195 to 197 in the 3rd intracellular loop, residues 251 to 256 in the 4th extracellular loop and the two C-terminal residues.
Figure 2. The structure of LmrP in complex with Hoechst 33342.
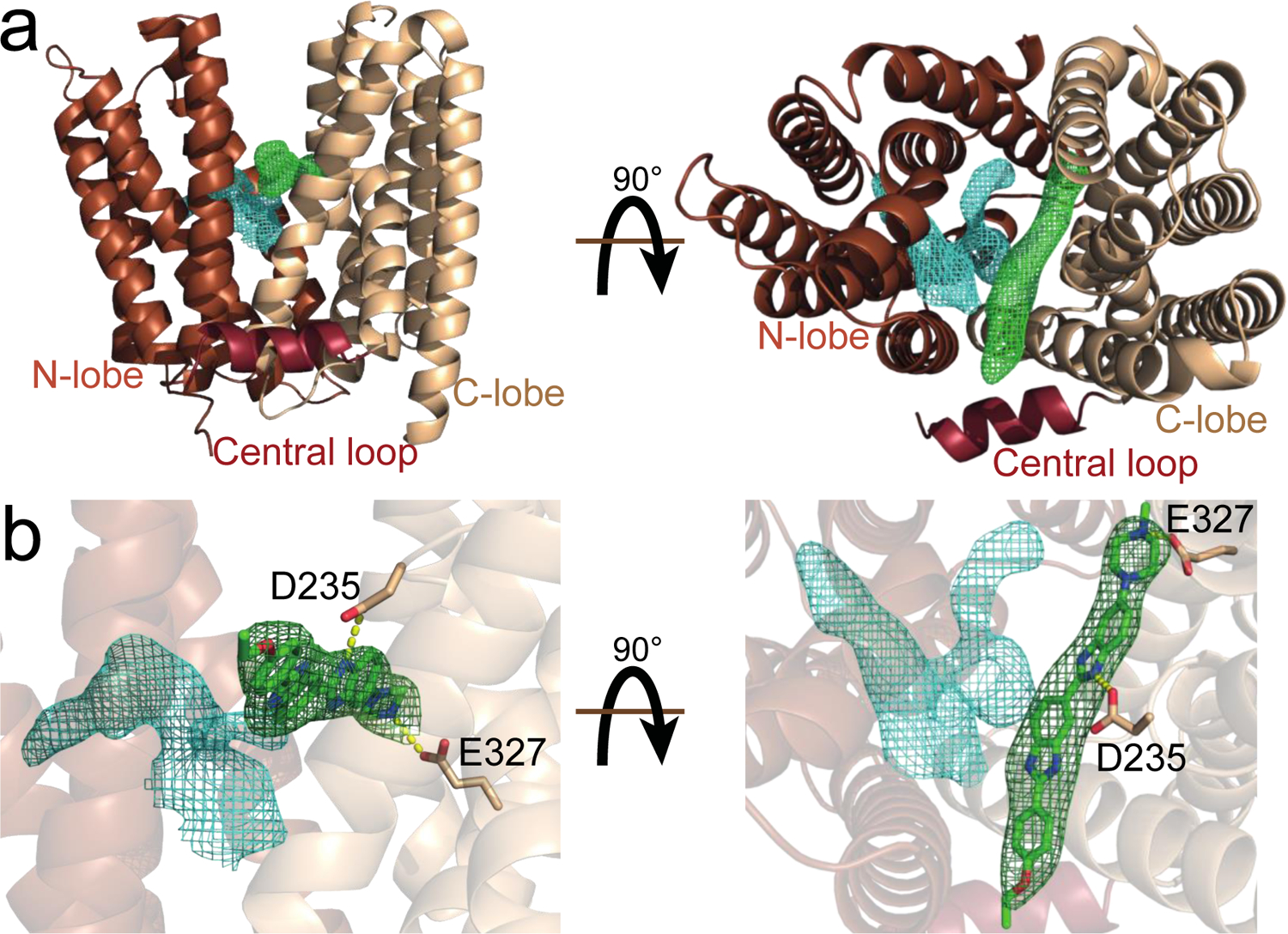
a) Cartoon representation of LmrP with N-lobe, central loop and C-lobe coloured in brown, dark red and light brown respectively. Meshes represent Fo-Fc maps at 2 σ after molecular replacement and refinement without modelled ligand. b) Hoechst 33342 (green) can be modelled (with the resultant 2Fo-Fc map following refinement shown at 1 σ in green) and forms polar interactions with D235 and E327, located in the C-lobe of LmrP.
The Hoechst 33342-bound structure adopts an outward-open state, consistent with the predominant state predicted by previous DEER studies18 (Figure 2a), and similar to the previously determined outward-open apo structure of MdfA22 (root mean square deviation of 3.0 Å across 316 residues when aligned through secondary-structure matching)23,24. The highly conserved “motif A” between TM2 and TM3 is well defined and shows how the highly conserved Asp68 in TM2 interacts with the bottom of TM11 in order to stabilise the inward-closed/outward-open state. The motif includes residues Arg72 and Asp128, which form a polar network of key residues that appear to precisely coordinate to position Asp68 towards TM11.
Ligand Binding
LmrP crystals were obtained after incubating the protein with 500 µM Hoechst 33342 and the ligand is clearly visible in the electron density, lying inside the protein, between the N- and C-lobes and parallel to the membrane plane (Figure 2b). It can be unequivocally modelled with the piperazine group buried near the centre of TM5, TM8 and TM10, while the ethoxyphenol group is sandwiched between TM2 and TM11 on the other side of the crevice, and with the ethyl moiety protruding out of the TM bundle.
The substrate makes two well defined polar interactions with negative side chains, Asp235 and Glu327, both of which have previously been identified as important residues for binding25–28. Glu327 in TM10 interacts with the distal nitrogen of the piperazine group while Asp235 coordinates the nitrogen of the central benzimidazole group. As all LmrP substrates are lipophilic with at least one positive charge at neutral pH, it is possible that the negative charges provided by the carboxylic acid groups on Asp235 and/or Glu327 could stabilise the positive charge(s) of the substrate to accommodate substrate binding. Remarkably, the substrate does not appear to make other interactions with the protein and, in particular, we do not observe hydrophobic interactions with aromatic or aliphatic sidechains (as typically suggested for other multidrug transporters). Thus, as the ligand seems to interact only with the C-lobe of LmrP it does not appear to be stricto senso coordinated.
The LmrP binding cavity holds an embedded lipid
Further inspection of the electron density revealed the presence of another molecule inside the transporter that cannot be accounted for by a part of the protein (Figure 2). The extra density shows a bidentate morphology composed of two elongated parts connected at one end. The density is clearly different from those observed for detergent molecules, and the branched architecture is strongly suggestive of a phospholipid, where one acyl chain would be inserted between TM1 and TM4, and the other would be located between the N-lobe (TM1 and TM5) and the substrate. While no clear signal is observed for a headgroup, the phosphatidic acid moiety common to many phospholipids can be readily modelled into this density (Figure 3a). Interestingly, the density located between the N- and C-lobes also neighbours the substrate, suggesting that it could provide a hydrophobic counterpart that would partially fill the binding pocket and may thus actively participate in ligand binding/coordination. In this way, the embedded active site lipid might substitute for the striking lack of hydrophobic side chains in the LmrP binding pocket.
Figure 3. A lipid within the binding cavity of LmrP stabilises the observed structure.
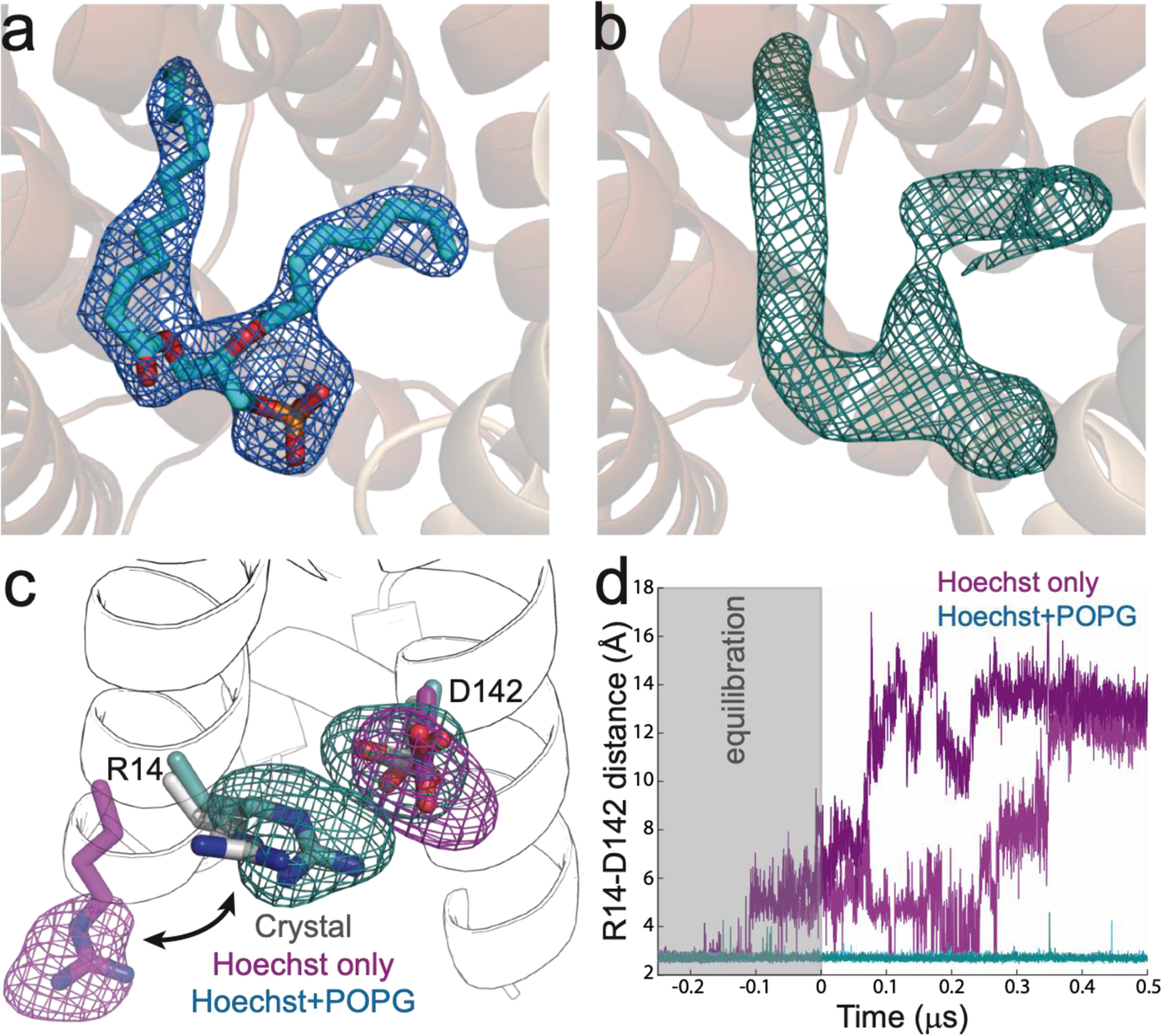
a) Phosphatidic acid, shown as cyan sticks, can be modelled into the unaccounted density observed in the binding cavity of LmrP, shown as a blue mesh of the 2Fo-Fc map at 1 σ. b) Across microsecond simulations of LmrP bound to Hoechst 33342 and with POPG within the binding pocket, the 3D mass-density map of POPG (teal mesh) adopts a similar conformation to that of the electron density of the lipid observed in the crystal structure. c) The polar residues R14 and D142 within the binding pocket of LmrP adopt a similar position to that observed in the crystal structure during simulations in the presence of Hoechst 33342 and POPG, but re-orient in simulations without POPG, and R14 is ultimately exposed to the intracellular space. The side-chains of R14 and D142 observed in the crystal structure are shown in grey, with 3D mass-density maps superposed from the simulation with both Hoechst 33342 and POPG (teal), and with only Hoechst 33342 (magenta). The conformations of R14 and D142 at the end of each simulation are shown as a visual aid. d) In simulations without POPG present (pink and magenta) the distance between R14 and D142 increases above that observed in the crystal structure. When POPG is present (teal and cyan) the distance between R14 and D142 is consistent with that observed in the crystal structure. Two independent simulations for each simulation condition are shown.
Molecular dynamics simulations support the presence of a PG lipid in the binding pocket
The most common phospholipids in bacteria include cardiolipin, phosphatidylglycerol and phosphatidylethanolamine, the latter being a minor component in L. lactis29. As cardiolipin could not fit the observed density, and as phosphatidylglycerol (POPG) is the next most abundant lipid in L. lactis membranes29, we hypothesised that POPG is bound to the protein in our structure of outward-facing LmrP, and employed all-atom molecular dynamics simulations to assess this interpretation. Specifically, we carried out simulations with LmrP either bound to Hoechst 33342 only or bound to Hoechst 33342 and POPG simultaneously (see Methods). In the simulated trajectories, both the substrate and POPG remain stably bound to the protein on a microsecond timescale. The calculated density map for POPG is for the most part well-defined, particularly for the alkyl chain residing within the N-lobe, in between TM1 and TM4; by contrast, the end of the other alkyl chain, residing in the central cavity, is disordered (Figure 3b). It is worth noting that within this simulation the POPG headgroup is mobile, interacting with a number of different residues within the binding pocket of LmrP (Supplementary Figure 3). It is therefore likely that this mobility is responsible for the unresolved headgroup within the actual crystal structure. This mobility notwithstanding, when comparing the trajectories with and without POPG, we observe that the presence of the lipid stabilises the conformation observed in the X-ray structure (Supplementary Figure 4). Specifically, the presence of POPG appears to decrease the structural dynamics of the N-lobe region, making it more comparable to the equivalent region in the C-lobe, where Hoechst 33342 binds.
More direct evidence of the presence of POPG derives from analysis of the dynamics of the only three charged residues that point inside the binding pocket, near the putative lipid headgroup. In the crystal structure, Arg14 and Asp142 form a salt-bridge between TM1 and TM5 in the N-lobe while Lys357 lies across from them, in TM11 of the C-lobe (Figure 3c and Supplementary Figure 5). In the simulation, we observed that the Arg14-Asp142 salt-bridge is stable in the presence of POPG, but readily and reproducibly dissociates in its absence (Figures 3c and 3d). In the latter case, Arg14 changes its orientation entirely upon dissociation from Asp142, and projects into the intracellular solution rather than into the interior of the binding cavity (Figure 3c). This change appears to be driven by an uncompensated electrostatic repulsion with Lys357, which remains in the cavity throughout the simulations. Consistent with this effect, Asp142 also reorients towards Lys357 in the absence of POPG (Figure 3c and Supplementary Figure 5). These observations suggest that the binding cavity is electrostatically out-of-balance when the negative charge of POPG is omitted from the input structure. This result is further supported by MD simulations where the zwiterionic lipid POPE was modelled into the density instead of POPG (Supplementary Figure 5). In these simulations, we also clearly see that the R14-D142 bridge is unstable, a further indication that the lipid captured in the crystal is the negatively charged PG. It is worth noting that an acidic residue at position 142 has previously been identified as required for transport of Hoechst 33342, while mutation of Lys357 has also been shown to reduce the transport of Hoechst 33342 and ethidium bromide25,30. Our simulation data strongly indicates that the configuration of these side chains in the crystal structure reflects the presence of POPG.
Native mass spectrometry indicates the presence of an embedded PG lipid
In order to experimentally confirm the presence and nature of the bound lipid, we performed native mass spectrometry on LmrP. As removing detergent micelles in the gas phase typically requires very high activation energy that disrupts most non covalent interactions, we first reconstituted LmrP in MSP-based lipid nanodiscs composed of 80% DOPE and 20% DOPG before analysing the sample in the mass spectrometer (see Methods). This permitted the identification of peaks corresponding to the exact mass of LmrP at lower activation energies, when compared to DDM-solubilised protein. Interestingly we could also resolve several additional peaks corresponding to one or several lipid adducts (Figure 4a and Supplementary Figure 6). Analyses of the high-resolution spectra clearly differentiate the two lipids species (Supplementary Figure 7). Upon increasing the activation energies most of these adduct peaks disappeared, with the exception of the LmrP+DOPG (single DOPG) peak, which remained present even at the highest energy tested (230V) suggesting a high affinity binding site for this lipid. In our structure, asparagine 116 is located inside the protein interior in close proximity to the lipid density whilst not directly contributing to Hoechst 33342 co-ordination. Mutation of N116 to the bulkier residue tyrosine is predicted to create a steric clash with the embedded lipid density (while remaining about 6 Å away from Hoechst, Figure 4b). Therefore we used an N116Y point mutant to confirm that the tightly bound PG corresponds to the embedded lipid seen in the X-ray structure. Native MS analysis of the nanodisc-reconstituted N116Y mutant showed a complete delipidation at higher energies (Figure 4a). Specifically we observe that the N116Y+DOPG peak entirely disappeared above 200V, suggesting that the “high” affinity PG binding site is either absent or strongly perturbed in the mutant. Considering the location of N116 near the ordered arm of the lipid in the N-lobe of LmrP, this indicates that the high affinity site is in the protein interior and thus that a PG molecule is embedded in the binding pocket of LmrP.
Figure 4. Native mass spectra of LmrP and the N116Y mutant reconstituted in nanodiscs made of DOPE (80%) and DOPG (20%).
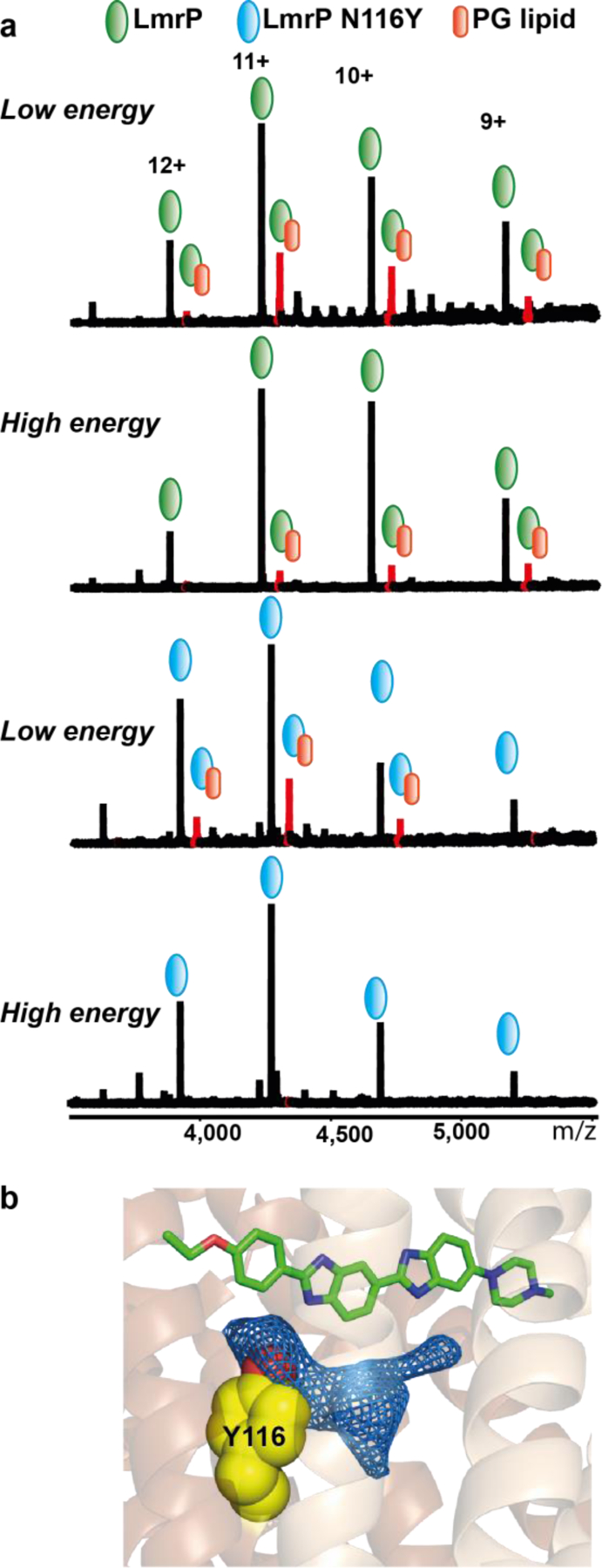
a) At low activation energy (160V) we observe multiple lipids bound to LmrP, including a peak corresponding to a single DOPG bound (in red). At high energy (200V) this peak is still present. In the case of the N116Y mutant, while we observe a bound DOPG at low energy, it disappears at high activation energy. b) Residue 116 is positioned inside the protein, and mutation to tyrosine (as depicted by the placement of a common tyrosine rotamer, yellow spheres) is expected to perturb the binding of the interior lipid (which is shown by the corresponding 2Fo-Fc map at 1 σ).
Functional importance of the embedded lipid
To assess whether the embedded lipid plays a functional role, we tested the functional effect of the N116Y mutation which inhibits binding of PG, as demonstrated by our MS experiment. We also tested another two point mutants that were predicted to sterically affect lipid binding without directly influencing substrate co-ordination: S52Y and T56Y. When comparing Hoechst 33342 transport using inside-out vesicles, we observed that T56Y and N116Y had greatly reduced transport activity compared to wild type LmrP, more consistent with the functionally disabled D68N mutant (Figure 5a), while the S52Y mutant displayed an intermediate behaviour. These observations indicate that the embedded lipid is important for LmrP activity, presumably by perturbing structural integrity of the transporter, inhibiting conformational cycling during transport, or influencing its substrate binding.
Figure 5. The S52Y, T56Y and N116Y mutations alter substrate specificity without directly interacting with Hoechst 33342.
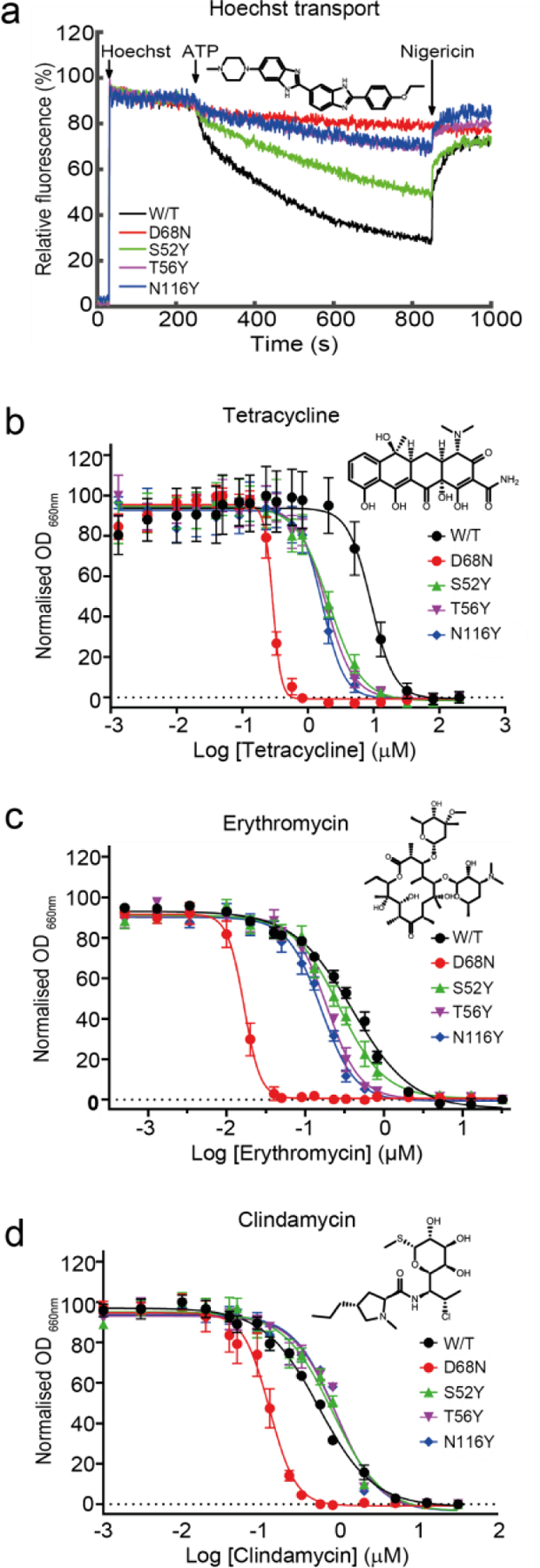
a) Transport of Hoechst 33342 is significantly reduced in the T56Y (purple) and N116Y (blue) mutants compared to wild-type LmrP (black). b) Cell survival assay demonstrating S52Y (green), T56Y (purple) and N116Y (blue) LmrP mutants cannot grow in as high a concentration of tetracycline as wild-type LmrP (black). c) Cell survival assay demonstrating S52Y (green), T56Y (purple) and N116Y (blue) LmrP mutants cannot grow in as high a concentration of erythromycin as wild-type LmrP (black). d) Cell survival assay showing that S52Y (green), T56Y (purple) and N116Y (blue) LmrP mutants are able to grow in equivalent concentrations of clindamycin to wild-type LmrP (black). The non-functional LmrP mutant D68N (red) is included as a negative control in panels. Chemical structures of Hoechst 33342, tetracycline, erythromycin and clindamycin are presented alongside the accompanying data. Error bars in panels b, c and d show standard-error mean (n=6).
We then investigated whether the loss of the embedded lipid had similar detrimental effects on the transport of other substrates. Because no direct transport assays were available, we monitored in vivo LmrP efflux activity by measuring cell growth in the presence of different antibiotics. Using three structurally diverse antibiotics (clindamycin, erythromycin and tetracycline), the IC50 of each was determined for L. lactis cells expressing wild-type LmrP, the transport deficient D68N mutant and each of the S52Y, T56Y or N116Y mutants. In the presence of tetracycline we observed an approximately 5-fold decrease in IC50 for cells expressing the N116Y or T56Y mutants and a greater than 4-fold decrease in IC50 for cells expressing the S52Y mutant in comparison to wild-type LmrP (Figure 5b). Similarly, in the presence of erythromycin, cells expressing the T56Y and N116Y mutants present a more than 2-fold decrease in IC50 in comparison to cells expressing wild-type LmrP, with the S52Y mutant also displaying a reduced IC50, but not to the same extent (Figure 5c). However with clindamycin we observed a slight increase in IC50 for all of the tyrosine mutant strains over wild-type LmrP (Figure 5d). This remarkable difference in survival showed that the tyrosine mutants did not perturb LmrP integrity or transport function per se, but rather altered activity in a substrate specific manner. These observations could imply that different substrates take different mechanistic routes, or more likely, that the mutations alter substrate specificity of the transporter. Based on our combined data, we hypothesise that the embedded lipid in the substrate binding site of LmrP contributes to the binding of some substrates and the polyspecificity of substrate recruitment.
Conclusion
The structure of LmrP bound to Hoechst 33342 confirmed substrate coordination through the side chains of amino acids previously demonstrated as required for transport, but also revealed the presence of a lipid within the substrate binding pocket. The embedded lipid is in direct interaction with the bound Hoechst 33342, and through molecular dynamics studies we find that a negatively charged lipid such as PG is required to stabilise the protein in the substrate-bound conformation. The nature of the embedded lipid was experimentally confirmed through native MS, while functional studies show that perturbing the positioning of this lipid alters transport activity, and does so in a substrate-specific manner. We therefore propose that the presence of a lipid within the substrate-binding pocket could form a key component to LmrP mediated multidrug transport. We hypothesise that compared to protein side chains, the embedded lipid may have greater conformational plasticity, thus providing a malleable hydrophobic environment to accommodate structurally diverse substrates. Structural confirmation of LmrP bound to different substrates and further research into how widely this mechanism is used and how such a lipid enters into the binding site may provide deeper insight into the nature of multidrug transport.
Methods
Cloning and expression of LmrP
The lmrp gene fragment was cloned into the pHLP5–3C expression plasmid as previously described14,26,29. Mutants were then introduced into the lmrp gene plasmid using Q5 site-directed mutagenesis (NEB). These plasmids were transformed into L. lactis strain NZ9000 through electroporation, which was used for protein expression. Cells were grown at 30 °C in M17 medium supplemented with 0.5% (w/v) glucose and 5 μg/ mL chloramphenicol until the OD660nm reached 0.8. Overexpression of LmrP mutants was then induced by addition of 1:1000 dilution of the supernatant of the nisin-producing L. lactis strain NZ970031,32. After 2 h of further incubation, cells were harvested by centrifugation at 5,000 g.
Preparation of inside-out membrane vesicles
Cells were thawed and supplemented with lysozyme to a final concentration of 5 mg/ mL before incubation for 1 hour at 30 °C, with 10 mM MgSO4 and 10 μg/ mL DNase I added for the last 10 minutes. Cells were then broken with six passes at ~15,000 psi using a high-pressure homogeniser. Cell debris and unbroken cells were subsequently removed through centrifugation three times at 17,000 g, 4 °C, each for 15 minutes. Membrane vesicles were then isolated by ultracentrifugation at 125,000 g for 1 hr at 4 °C before resuspension in buffer A (100 mM HEPES, pH7, 300 mM NaCl and 20% (v/v) glycerol) (2 mL per L of cultured cells). Vesicles were then frozen in liquid nitrogen and stored at −80 °C for further use.
Purification of LmrP
Membrane vesicles were thawed and solubilised with an equal volume of 2.4% (w/v) n-dodecyl-β-D-maltoside (βDDM; Inalco) in water for 1 hr at 4 °C. Insoluble fragments were removed through centrifugation at 100,000 g for 1 hr. Centrifugation supernatant was supplemented with 10 mM imidazole, added to Ni-NTA resin (Qiagen) and incubated under agitation for 2 hrs at 4 °C, before washing with 10 CV of buffer B (50 mM HEPES, pH7, 150 mM NaCl, 10% (v/v) glycerol and 0.05% βDDM) containing 20 mM imidazole and elution with 3 CV of buffer B containing 250 mM imidazole at 4 °C. Imidazole was removed with PD10 desalting columns (GE healthcare) and buffer B. LmrP was then further purified through size-exclusion chromatography (SDX 200 10/300 GL increase; GE lifescience) in buffer C (20 mM HEPES pH7, 100 mM NaCl, 10% (v/v) glycerol, 0.02% βDDM) and concentrated as desired with through the use of spin-concentrators (VWR).
Labelling of DEER samples
Following purification of LmrP, the protein concentration was determined (based upon the 280 nm UV absorbance) and a 30-fold molar excess of MTSSL (Enzo Life Sciences) was added from a 100 μM stock solution in DMF. The labelling reaction was allowed to proceed at room temperature for 2 h, followed by another addition of MTSSL, a further 2 h incubation at room temperature and incubation on ice overnight. Unbound MTSSL was then removed through size exclusion chromatography (SDX 200 10/300 GL increase; GE lifescience) in TMA buffer (50 mM Tris-MES-Acetate, 100 mM NaCl, 10% (v /v) glycerol and 0.05% (w /v) β-DDM at pH8).
DEER experiments
The protein in detergent micelles was concentrated using a 50K MWCO concentrator to a spin label concentration of 100–150 µM (determined by the integration of the CW spectrum) after which glycerol was added to a final concentration of 23% (v/v). Ligands were added to a final concentration of 1 mM. DEER measurements were performed on a Bruker 580 pulsed ESR spectrometer operating at Q-band (33.4 GHz) using a standard four pulse protocol33–35. Data was collected with the samples at 83 K with 23% (v/v) glycerol as cryoprotectant. Analysis of the DEER data to determine the distance distributions was carried in DeerAnalysis2011 or DeerAnalysis201336. The data was fit with Tikhonov regularisation and L-curve determination of the optimal regularisation parameter. Optimal background correction was established by statistical analysis of the fits. The acquisition time was optimised for each spin pair. For a few samples, we observed evidence of partial protein aggregation35. This was manifested in the raw DEER decays by a deviation of the baseline from a stretched exponential and the lack of an oscillation in the echo decay even at longer collection times. Aggregation results in a component at the tail end of the distance distribution. The artifactual nature of these peaks (marked with an asterisk) could thus be demonstrated by varying the measured duration of the echo intensity oscillation.
Lipidation and crystallisation of LmrP
For crystallisation experiments LmrP∆199–202 was purified as detailed for LmrP above, but with the α-isomer of DDM (Avanti) used in buffer B and buffer C, and with the His-tag removed and the protein lipidated prior to SEC purification. To achieve this, after buffer exchange the protein was concentrated to 4 mg/ mL and incubated with E. coli polar lipids (Avanti), solubilised at 15 mg/mL in buffer C with 1% αDDM, at a 50:1 lipd:LmrP molar ratio. Simultaneously, LmrP was also incubated with 6His-MBP-3C protease37 (at a 10:1 ratio of LmrP:3C, as determined by mass) at 4 °C overnight, with uncleaved LmrP, free His-tag and 3C protease subsequently removed through reverse-phase IMAC.
Immediately prior to crystallisation trials LmrP was concentrated to 4 mg/ mL, supplemented with 0.5 mM Hoechst 33342 and 0.8% n-Octyl-β-D-Glucopyranoside, (OG, Anatrace) and centrifuged 3 times at 16,000 g, 4 °C. Vapour diffusion crystallisation trials were established using 0.2 μL sitting drops, with the best diffracting crystals grown at 20 °C in 80 mM SPG pH 7, 20% (w/v) PEG 1500 (Molecular dimensions, PACT Premier Eco crystallisation screen), and cryo-protected with 15% (w/v) sucrose.
Selenomethionine labelling
Selenomethionine labelling of LmrP was performed in vivo using a previously established protocol38. Briefly, L. lactis NZ9000 containing the lmrp-pHLP5–3C plasmid were grown at 30 °C in M17 medium supplemented with 0.5% glucose and 5 μg/mL chloramphenicol until the OD660nm reached 0.8. Cells were isolated through centrifugation at 6,000 g, 15 mins, 30 °C, followed by washing to remove excess M17 medium and centrifugation once more at 6,000 g, 15 mins, 30 °C. Cells were then resuspended in selenoMethionine Medium base (Molecular dimensions) supplemented with 5 μg/ mL chloramphenicol, 160 μg/ mL L-selenomethionine (Acros Organics) and selenoMethionine nutrient mix (Molecular dimensions, 100 mL per litre of selenoMethionine medium). The culture was incubated statically at 30 °C for 1 hour before LmrP expression was induced through the addition of 1:1000 dilution of the supernatant of the nisin-producing L. lactis strain NZ9700. After a further 2 hours static incubation at 30 °C, cells were harvested through centrifugation for 15 mins at 6,000 g, and resuspended in buffer A. Labelled LmrP was purified and crystallised using the same methods outlined above, with selenomethionine incorporation confirmed through mass spectroscopy. Highest resolution crystals grew in 160 mM sodium actetate trihydrate, 0.8 mM Bis-Tris propane and 16% (w/v) PEG 3350 (Molecular dimensions, PACT Premier Eco crystallisation screen).
Data Collection, structure solution and refinement
For selenomethionine anomalous data collection, three datasets were collected on the same crystal at Proxima-1 (Soleil synchrotron), using a wavelength of 0.97925 Å (corresponding to the Se peak) at 100 K. Each dataset was processed with XDS (through XDSME)39,40 to a maximum resolution of 3.8 Å, before being scaled together with XSCALE and run through XDSME/XDSCONV39,40 to generate SHELX input files. Selenium sites were identified using SHELXC and SHELXD41 before phase calculation with SHARP42. Phases obtained from this Se-SAD experiment were used to build a model of LmrP, with selenomethionine positions used to guide sequence assignment. This model was then used for molecular replacement into the highest resolution native dataset, using molrep43.
The highest resolution native data was collected at I03 (Diamond Light Source), at 100 K, using four datasets processed with XDS/XDSME39,40 and scaled together with XSCALE to a maximum resolution of 2.9 Å. Anisotropic correction was then applied to this collective dataset through use of STARANISO44. Ellipsoidal resolution limits were defined at 2.76 Å in the a* orientation, 4.33 Å in the b* orientation and 2.90 Å in the c* orientation, as determined using the default CC1/2 and (I/sigI) thresholds.
For both datasets, the protein model was built with COOT45, and refined with BUSTER46. Model validation was performed with MolProbity47. Figures of all structures were prepared using Pymol version 2.2.048.
Molecular Dynamics
All MD simulations were carried out with NAMD version 2.949, using the CHARMM36 force field50,51 with an NBFIX correction of protein-sodium interactions52. Temperature and pressure were maintained constant at 298 K and 1 atm, respectively. An integration time-step of 2 fs was used. Electrostatic interactions were calculated using PME with a real-space cut-off of 12 Å; van-der-Waals interactions were also cut-off at 12 Å, with a switching function turned on at 10 Å. Periodic boundary conditions were used.
Simulations were based on the crystal structure of outward-facing LmrP and performed on 3 different systems, LmrP bound to Hoechst 33342, LmrP bound to Hoechst and POPG as well as LmrP bound to Hoechst and POPE. In both cases the protein-ligand complexes had been embedded into a pre-equilibrated POPC-lipid bilayer using GRIFFIN53, after a 500-step energy minimization with CHARMM54. The surrounding solution contained 100 mM NaCl plus counterions to neutralise the total charge of the simulation systems. The resulting systems comprise approx. 76,300 atoms in an orthorhombic box of approx. 90 by 90 by 91 Å.
The initial coordinates for POPG/POPE were those in the fragment resolved in the structure, with the addition of the glycerol/ethanolamine headgroup and the alkyl chains extended. The initial coordinates for Hoechst were those in the crystal structure. The protein structure comprised residues 1–187, 204–246 and 257–403. DOWSER55 was used to add water molecules within the solvent-excluded surface. Among the Glu/Asp sidechains in the protein, only Glu166 and Glu388 were represented as protonated, as they form hydrogen-bonds (as donors) with nearby backbone carbonyl groups in the experimental structure. Asp235 and Glu327, by contrast, were represented as deprotonated/charged as they coordinate positively-charged groups in Hoechst. All other acidic sidechains in the structure are also in close proximity to Lys or Arg sidechains and were therefore represented as charged. Default protonation states were also used for His.
Two independent simulations were carried out for each simulation system, each comprising an equilibration phase and a production phase. The equilibration phase consisted of a sequence of simulations during which the dynamics of the protein-ligand complex is restricted, primarily through internal-conformation restraints, to a degree that is gradually diminished over approx. 250 ns. In the production phase, the structural dynamics of the protein-ligand complex was entirely unrestricted. The production simulations for LmrP bound to Hoechst were 1-microsecond long, while those of lipid-bound LmrP were 500 ns long. VMD56 was used to analyse the simulated trajectories. Mass-density maps were calculated with the VolMap tool, using a 0.5 Å-resolution grid and standard atomic radii.
Hoechst 33342 forcefield
Among all possible protonation states of Hoechst 33342, the state considered in this study is protonated at the methyl-substituted N atom in the piperazine group, as well as at the adjacent benzimidazole ring, i.e. the two interaction sites for Asp235 and Glu327 in LmrP. These two sites are also the most prone to protonate in solution, and in that order; the solution pKa values of these sites have been measured to be 7.9 and 5.7, respectively (versus 3.5 for the second benzimidazole ring)57. Interaction with Asp235 and Glu327 will further shift these pKa values upwards, and thus the doubly-protonated state is the most probable when Hoechst is bound to LmrP. Therefore, the total charge of Hoechst in our simulations is +2. Forcefield parameters for Hoechst were initially derived from CGenFF (https://cgenff.paramchem.org58–60). Partial atomic charges for aliphatic and aromatic C and H atoms were preserved, as were most of the bonded terms. Adjustments in the partial charges for polar groups (N, O and associated H) were made by comparing Hoechst-water interaction energies calculated with the CHARMM energy function and with quantum-mechanical (QM) calculations, for different sites in Hoechst. For each interaction site considered, alternative water orientations were also studied, reflecting the location of the lone pairs in Hoechst. For each interaction site and orientation considered, the minimum-energy configuration was first identified (for both energy functions) through an iterative process whereby the Hoechst-water distance was varied gradually. The energy values at the minima were then contrasted. As a result of this optimisation, CHARMM energies are within 0.2 kcal/mol of the QM result for all interaction sites considered, except for the terminal ethoxy oxygen in Hoechst. Optimisation at this site required, in addition, a slight increase in the van der Waals parameter Rmin (by 0.12 Å), relative to the value initially assigned by CGenFF. All QM calculations were carried out using Gaussian0961 at the HF/6–31G(d) level. Initial coordinates for Hoechst were those in the crystal structure reported here. This geometry was optimised prior to the interaction-energy calculations. The bonded terms in the resulting forcefield were evaluated through 100-ns long simulations of Hoechst. Adjustments were made in the angles and dihedrals involving the two nitrogen atoms in the piperazine ring, relative to the CGenFF values, to stabilise the chair geometry of this group. To evaluate the ligand-solvent interactions, another 100-ns simulation was then carried out with Hoechst and 2984 TIP3P molecules, 8 sodium ions and 10 chloride ions, in cubic simulation box of 46 Å in length. As shown in Supplementary Figure 8, interactions with the protonated groups in Hoechst order and orient water molecules in the first hydration shell. The geometries of the each of the constituent groups closely resembles that in the experimental structure. The Hoechst 33342 forcefield used in this study is freely available upon request to the authors.
MSP1E3D1 production and purification
Membrane scaffold protein (MSP1D1E3) was expressed and purified as previously described62, with the following modifications. Briefly, E. coli BL21(DE3) (New England Biolabs) cells containing the MSP1D1E3 gene in pET-28a(+) were plated on LB-agar plates supplemented with kanamycin (30 μg/ mL). A single colony was used to inoculate 30 mL of LB supplemented with 30 μg/ mL of kanamycin. A dense overnight culture of 30 mL was used to inoculate a secondary culture in 1 L Terrific broth supplemented with 30 μg/ mL kanamycin. Cultures were grown at 37 °C with shaking to an OD600 of ~2.2–2.5, and expression of MSP1D1E3 was induced by addition of 1 mM IPTG. Cultures were further grown for 4 h at 37 °C, and cells were harvested by centrifugation. Cell pellets were resuspended in 30 mL of lysis buffer (20 mM sodium phosphate, 1% Triton X-100, pH 7.4), with one-third of a Complete EDTA-free protease-inhibitor-cocktail tablet (Pierce), 10 μg/ mL Benzonase I, and were lysed by 2 passes at 15,000 psi in a high-pressure homogenizer (Emulsiflex). The lysate was centrifuged at 30,000 g for 30 min, and the supernatant was mixed with 3 mL of Ni–NTA resin equilibrated with lysis buffer. The slurry was transferred to a gravity-flow column, and the flow-through was discarded. The resin was washed with four bed volumes of buffer M1 (40 mM Tris-HCl and 0.3 M NaCl, pH 8.0) containing 1% Triton X-100, four bed volumes of buffer M1 containing 50 mM sodium cholate, four bed volumes of buffer M1 containing 20 mM imidazole and four bed volumes of buffer M1 containing 50 mM imidazole. The bound protein was eluted stepwise with buffer M1 containing 300 mM imidazole. The eluted MSP1D1E3 was buffer-exchanged into MSP buffer (50 mM Tris-HCl, 0.1 M NaCl and 0.5 mM EDTA, pH 7.5) on a PD10 column (Biorad). Concentration was determined by absorbance at 280 nm (extinction coefficient = 29,910 M−1 cm−1). The protein was concentrated to ~15 mg/ mL on a 10 K MWCO concentrator (Amicon). The purity was assessed by SDS–PAGE and Coomassie staining. The 6-his tag was removed by overnight incubation at 4 °C with TEV protease (80 µg of TEV to cleave 1 mg of MSP). Cleaved MSP was collected by reverse IMAC, and concentrated again to ~15 mg/ mL.
Lipid preparation for nanodiscs
Lipids DOPE and DOPG dissolved in chloroform (Avanti Polar Lipids) were combined in an 8:2 ratio to reach a final quantity of 100 mg, dried under nitrogen flow and desiccated overnight under vacuum. The lipid films were hydrated with MSP buffer to reach a final concentration of 40 mg/ mL. β-DDM was added to the mixture to a final concentration of 7.5% (w/v). The lipids were further homogenized by 4 cycles of freeze-thawing, divided into 100 µL aliquots and stored at −80 °C until the day of use. The choice of the chain length was motivated by the low Tm temperature of dioleoyl chains, which facilitates the reconstitution of the membrane proteins at 4 °C.
Reconstitution of LmrP into nanodiscs
For reconstitution into nanodiscs, 800 µg of LmrP or LmrP N116Y in β-DDM micelles was mixed with the DOPE:DOPG lipid mixture, MSP1D1E3 and β-DDM in the following molar ratios: lipid:MSP1D1E3, 60:1; MSP1D1E3:LmrP, 8:1; and β-DDM:lipid, 3:1. Samples were incubated at room temperature for 30 min then incubated overnight at 4 °C with agitation. In the morning, Biobeads SM-2 (Bio-Rad) were added to the mixture (700 mg per mL of sample) and incubated for 2 h at 4 °C, then 1 h at RT. Full nanodiscs were separated from empty ones on Ni-NTA high affinity beads (500 µL of beads for 1 mg of reconstituted LmrP), eluted with nanodisc purification buffer (sodium phosphate 50 mM, 100 mM NaCl, 10% glycerol) supplemented with 300mM imidazole. The full discs were further purified by size-exclusion chromatography on a Superdex200 column (GE) using nanodisc purification buffer, then were concentrated to 100 µL final volume with Amicon Ultra-50K centrifugal filter units at a speed not exceeding 2,000 g. Full nanodiscs were characterized with SDS–PAGE to assess reconstitution.
Native mass spectrometry measurements
For mass spectrometry experiments, purified nanodiscs were exchanged into 0.3 M ethylene diamine diacetate buffer (Sigma Aldrich) using micro bio-spin 6 columns (Bio-Rad). Nanodisc samples were loaded into homemade nanoelectrospray needles coated in gold. Mass spectrometry was performed using a Q-Exactive HF mass spectrometer with UHMR modifications (Thermo Fisher Scientific). Samples were ionised by nano-ESI in positive ion mode with a 1.5 kV capillary voltage and 150 °C capillary temperature. Key parameters included 12,500 resolution, mass range 1500–12000 m/z, no extended trapping, trapping gas pressure 6, ion transfer optimization and detector optimization on high m/z. Nitrogen was used as the trapping/collision gas. In-source trapping energy ranged from 150V to 230V.
Hoechst transport assay
Mutant and wild-type LmrP genes were transformed into L. lactis strain NZ9000 ΔLmrA ΔLmrCD63 through electroporation as described above. LmrP and mutant variants were expressed and membranes isolated as detailed above. Membrane vesicles were resuspended in buffer A (100 mM HEPES pH7, 300 mM NaCl, 20% glycerol) to a concentration of 0.2 g/ mL and assessed for Hoechst transport activity using a previously determined technique14,15. Briefly, 50 μL of membrane solution was added to 1.5 mL of transport buffer (50 mM HEPES KOH pH7.4, 2 mM MgCl2, 300 mM KCl) at 25 °C and placed in a stirring cuvette within a fluorimeter exciting at 355 nm and monitoring emission at 457 nm. After 30 seconds 0.75 μL of 0.1 mM Hoechst 33342 in water was added and fluorescence monitored for a further 220 seconds. LmrP was then energised by a proton gradient generated using fof1-ATPase within the membrane vesicles upon addition of 12 μL of ATP solution (250 mM MgSO4, 250 mM ATP-Na2, 50 mM HEPES pH 7). Fluorescence was then monitored for a further 10 minutes before addition of 1.5 μL of nigericin at 0.75 mg/ mL in H2O to disrupt the proton gradient. All measurements were repeated in triplicate using independent transformation colonies.
Cell survival assay
Mutant and wild-type LmrP genes were transformed into L. lactis strain NZ9000 ΔLmrA ΔLmrCD63 through electroporation as described above. Individual colonies were grown overnight and used to inoculate 12 mL cultures of M17 (supplemented with 0.5% glucose, 5 μg/ mL chloramphenicol), which were then incubated statically at 30 °C until an OD660nm of around 0.7 was reached, when expression was induced through the addition of a 1:1000 dilution of the supernatant of the nisin-producing L. lactis strain NZ900031,32. After 2 hours of expression, cells were diluted with M17 (supplemented with 0.5% glucose, 5 μg/mL chloramphenicol and 1:1000 dilution of the supernatant of the nisin-producing L. lactis strain NZ900031,32) to an OD660 of 0.2. These dilute solutions were then aliquoted into 96-well plates containing a dilution series of antibiotic within M17 (supplemented with 0.5% glucose, 5 μg/ mL chloramphenicol and 1:1000 dilution of the supernatant of the nisin-producing L. lactis strain NZ9000). All growths were allowed to proceed at 30 °C, without shaking, for 16 hours before measuring the OD660. Six independent transformation colonies were used for each mutant/antibiotic combination.
Supplementary Material
Acknowledgements
We thank Prof Ruysschaert and Garcia-Pino for helpful discussions.
We thank Dr James Ault of the Biomolecular Mass Spectrometry Facility for his support & assistance in this work and the BBSRC (BB/M012573/1) for funding.
This work was supported by the Fonds de la Recherche Scientifique F.R.S.-F.N.R.S. (grants F.4523.12, T.0057.15F, J0044.17F and T.0105.19). V.D. was a fellow of the FRIA, C.M. is Postdoctoral Researcher of the F.R.S.-F.N.R.S. C.G. is supported as a Senior Research Associate of the F.R.S.-F.N.R.S.
Footnotes
Competing Financial Interests Statement
The authors declare no competing financial interests.
References
- 1.Poole K Efflux-mediated multiresistance in Gram-negative bacteria. Clin. Microbiol. Infect 10, 12–26 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Piddock LJV Clinically relevant chromosomally encoded multidrug resistance efflux pumps in bacteria. Clin. Microbiol. Rev 19, 382–402 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fromm MF Importance of P-glycoprotein at blood-tissue barriers. Trends Pharmacol. Sci 25, 423–429 (2004). [DOI] [PubMed] [Google Scholar]
- 4.Aller SG et al. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 323, 1718–1722 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Du D et al. Multidrug efflux pumps: structure, function and regulation. Nat. Rev. Microbiol 16, 523–539 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Murakami S, Nakashima R, Yamashita E, Matsumoto T & Yamaguchi A Crystal structures of a multidrug transporter reveal a functionally rotating mechanism. Nature 443, 173–179 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Nakashima R, Sakurai K, Yamasaki S, Nishino K & Yamaguchi A Structures of the multidrug exporter AcrB reveal a proximal multisite drug-binding pocket. Nature 480, 565–569 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Eicher T et al. Transport of drugs by the multidrug transporter AcrB involves an access and a deep binding pocket that are separated by a switch-loop. Proc. Natl. Acad. Sci 109, 5687–5692 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamaguchi A, Nakashima R & Sakurai K Structural basis of RND-type multidrug exporters. Front. Microbiol 6, 1–19 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zwama M & Yamaguchi A Molecular mechanisms of AcrB-mediated multidrug export. Res. Microbiol 169, 372–383 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Johnson ZL & Chen J Structural basis of substrate recognition by the multidrug resistance protein MRP1. Cell 168, 1075–1085.e9 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Alam A, Kowal J, Broude E, Roninson I & Locher KP Structural insight into substrate and inhibitor discrimination by human P-glycoprotein. Science 363, 753–756 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heng J et al. Substrate-bound structure of the E. coli multidrug resistance transporter MdfA. Cell Res 25, 1060–1073 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Putman M, Van Veen HW, Poolman B & Konings WN Restrictive use of detergents in the functional reconstitution of the secondary multidrug transporter LmrP. Biochemistry 38, 1002–1008 (1999). [DOI] [PubMed] [Google Scholar]
- 15.Putman M, Koole LA, Van Veen HW & Konings WN The secondary multidrug transporter LmrP contains multiple drug interaction sites. Biochemistry 38, 13900–13905 (1999). [DOI] [PubMed] [Google Scholar]
- 16.Putman M, van Veen HW, Degener JE & Konings WN The lactococcal secondary multidrug transporter LmrP confers resistance to lincosamides , macrolides , streptogramins and tetracyclines. Microbiology 147, 2873–2880 (2001). [DOI] [PubMed] [Google Scholar]
- 17.Schaedler TA, Tong Z & Van Veen HW The multidrug transporter LmrP protein mediates selective calcium efflux. J. Biol. Chem 287, 27682–27690 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Masureel M et al. Protonation drives the conformational switch in the multidrug transporter LmrP. Nat. Chem. Biol 10, 149–155 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bolhuis H et al. The lactococcal lmrP gene encodes a proton motive force-dependent drug transporter. J. Biol. Chem 270, 26092–26098 (1995). [DOI] [PubMed] [Google Scholar]
- 20.Roth A & Govaerts C LmrP from Lactoccoccus lactis: a tractable model to understand secondary multidrug transport in MFS. Res. Microbiol 169, 468–477 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Goldschmidt L, Cooper DR, Derewenda ZS & Eisenberg D Toward rational protein crystallization: A web server for the design of crystallizable protein variants. Protein Sci 16, 1569–1576 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nagarathinam K et al. Outward open conformation of a Major Facilitator Superfamily multidrug/H+ antiporter provides insights into switching mechanism. Nat. Commun 9, 4005 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krissinel E & Henrick K Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. Sect. D Biol. Crystallogr 60, 2256–2268 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Winn MD et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. Sect. D Biol. Crystallogr 67, 235–242 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mazurkiewicz P, Konings WN & Poelarends GJ Acidic residues in the lactococcal multidrug efflux pump LmrP play critical roles in transport of lipophilic cationic compounds. J. Biol. Chem 277, 26081–26088 (2002). [DOI] [PubMed] [Google Scholar]
- 26.Mazurkiewicz P, Driessen AJM & Konings WN Energetics of wild-type and mutant multidrug resistance secondary transporter LmrP of Lactococcus lactis. Biochim. Biophys. Acta - Bioenerg 1658, 252–261 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Mazurkiewicz P, Poelarends GJ, Driessen AJM & Konings WN Facilitated drug influx by an energy-uncoupled secondary multidrug transporter. J. Biol. Chem 279, 103–108 (2004). [DOI] [PubMed] [Google Scholar]
- 28.Schaedler TA & Van Veen HW A flexible cation binding site in the multidrug major facilitator superfamily transporter LmrP is associated with variable proton coupling. FASEB J 24, 3653–3661 (2010). [DOI] [PubMed] [Google Scholar]
- 29.Martens C et al. Lipids modulate the conformational dynamics of a secondary multidrug transporter. Nat. Struct. Mol. Biol 23, 744–751 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang W & van Veen HW Basic residues R260 and K357 affect the conformational dynamics of the major facilitator superfamily multidrug transporter LmrP. PLoS One 7, e38715 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuipers OP, Beerthuyzen MM, Siezen RJ & de Vos WM Characterization of the nisin gene cluster nisABTCIPR of Lactococcus lactis. Requirement of expression of the nisA and nisI genes for development of immunity. Eur. J. Biochem 216, 281–291 (1993). [DOI] [PubMed] [Google Scholar]
- 32.Mierau I & Kleerebezem M 10 Years of the nisin-controlled gene expression system (NICE) in Lactococcus lactis. Appl. Microbiol. Biotechnol 68, 705–717 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Pannier M, Veit S, Godt A, Jeschke G & Spiess HW Dead-time free measurement of dipole-dipole interactions between electron spins. J. Magn. Reson 142, 331–340 (2000). [DOI] [PubMed] [Google Scholar]
- 34.Jeschke G & Polyhach Y Distance measurements on spin-labelled biomacromolecules by pulsed electron paramagnetic resonance. Phys. Chem. Chem. Phys 9, 1895–1910 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Jeschke G DEER distance measurements on proteins. Annu. Rev. Phys. Chem 63, 419–446 (2012). [DOI] [PubMed] [Google Scholar]
- 36.Jeschke G et al. Deer Analysis 2006 - a comprehensive software package for analyzing pulsed ELDOR Data. Appl. Magn. Reson 30, 473–498 (2006). [Google Scholar]
- 37.Alexandrov A, Dutta K & Pascal SM MBP fusion protein with a viral protease cleavage site: One-step cleavage/ purification of insoluble proteins. Biotechniques 30, 1194–1198 (2001). [DOI] [PubMed] [Google Scholar]
- 38.Berntsson RPA et al. Selenomethionine incorporation in proteins expressed in Lactococcus lactis. Protein Sci 18, 1121–1127 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kabsch W Xds. Acta Crystallogr. Sect. D Struct. Biol 66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Legrand P XDSME: XDS Made Easier (2017). GitHub repository https://github.com/legrandp/xdsme (2017). doi: 10.5281/zenodo.837885 [DOI]
- 41.Sheldrick GM Experimental phasing with SHELXC/D/E: Combining chain tracing with density modification. Acta Crystallogr. Sect. D Biol. Crystallogr 66, 479–485 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bricogne G, Vonrhein C, Flensburg C, Schiltz M & Paciorek W Generation, representation and flow of phase information in structure determination: Recent developments in and around SHARP 2.0. Acta Crystallogr. Sect. D Biol. Crystallogr 59, 2023–2030 (2003). [DOI] [PubMed] [Google Scholar]
- 43.Vagin A & Teplyakov A Molecular replacement with MOLREP. Acta Crystallogr. Sect. D Biol. Crystallogr 66, 22–25 (2010). [DOI] [PubMed] [Google Scholar]
- 44.Tickle IJ et al. STARANISO Cambridge, United Kingdom: Global Phasing Ltd. http://staraniso.globalphasing.org/cgi-bin/staraniso.cgi (2018). [Google Scholar]
- 45.Emsley P, Lohkamp B, Scott WG & Cowtan K Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bricogne G et al. BUSTER version 2.10.3 Cambridge, United Kingdom: Global Phasing Ltd. (2016). [Google Scholar]
- 47.Chen VB et al. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schrödinger L The PyMOL Molecular Graphics System, version 2.2.0 (2018).
- 49.Phillips JC et al. Scalable molecular dynamics with NAMD. J. Comput. Chem 26, 1781–1802 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Best RB et al. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ1 and χ2 dihedral angles. J. Chem. Theory Comput 8, 3257–3273 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Klauda JB et al. Update of the CHARMM all-atom additive force field for lipids: Validation on six lipid types. J. Phys. Chem. B 114, 7830–7843 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marinelli F et al. Sodium recognition by the Na+/Ca2+ exchanger in the outward-facing conformation. Proc. Natl. Acad. Sci 111, E5354–E5362 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Staritzbichler R, Anselmi C, Forrest LR & Faraldo-Gómez JD GRIFFIN: A versatile methodology for optimization of protein-lipid interfaces for membrane protein simulations. J. Chem. Theory Comput 7, 1167–1176 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brooks BR et al. CHARMM: The biomolecular simulation program. J. Comput. Chem 30, 1545–1614 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang L & Hermans J Hydrophilicity of cavities in proteins. Proteins Struct. Funct. Genet 24, 433–438 (1996). [DOI] [PubMed] [Google Scholar]
- 56.Humphrey W, Dalke A & Schulten K VMD: Visual molecular dynamics. J. Mol. Graph 14, 33–38 (1996). [DOI] [PubMed] [Google Scholar]
- 57.Ladinig M et al. Protonation equilibria of Hoechst 33258 in aqueous solution. Helv. Chim. Acta 88, 53–67 (2005). [Google Scholar]
- 58.Vanommeslaeghe K et al. CHARMM General Force Field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem 31, 671–690 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vanommeslaeghe K & MacKerell AD Automation of the CHARMM general force field (CGenFF) I: Bond perception and atom typing. J. Chem. Inf. Model 52, 3144–3154 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vanommeslaeghe K, Raman EP & MacKerell AD Automation of the CHARMM General Force Field (CGenFF) II: Assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model 52, 3155–3168 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Frisch MJ et al. Gaussian 09, revision a.02 Gaussian, Inc., Wallingford CT: (2009). doi: 10.1021/ja0031220 [DOI] [Google Scholar]
- 62.Boldog T, Li M & Hazelbauer GL Using nanodiscs to create water-soluble transmembrane chemoreceptors inserted in lipid bilayers. Methods Enzymol 423, 317–335 (2007). [DOI] [PubMed] [Google Scholar]
- 63.Venter H, Velamakanni S, Balakrishnan L & van Veen HW On the energy-dependence of Hoechst 33342 transport by the ABC transporter LmrA. Biochem. Pharmacol 75, 866–874 (2008). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.