

ABSTRACT
CRISPR type III systems, which are abundantly found in archaea, recognize and degrade RNA in their specific response to invading nucleic acids. Therefore, these systems can be harnessed for gene knockdown technologies even in hyperthermophilic archaea to study essential genes. We show here the broader usability of this posttranscriptional silencing technology by expanding the application to further essential genes and systematically analysing and comparing silencing thresholds and escape mutants. Synthetic guide RNAs expressed from miniCRISPR cassettes were used to silence genes involved in cell division (cdvA), transcription (rpo8), and RNA metabolism (smAP2) of the two crenarchaeal model organisms Saccharolobus solfataricus and Sulfolobus acidocaldarius. Results were systematically analysed together with those obtained from earlier experiments of cell wall biogenesis (slaB) and translation (aif5A). Comparison of over 100 individual transformants revealed gene-specific silencing maxima ranging between 40 and 75%, which induced specific knockdown phenotypes leading to growth retardation. Exceedance of this threshold by strong miniCRISPR constructs was not tolerated and led to specific mutation of the silencing miniCRISPR array and phenotypical reversion of cultures. In two thirds of sequenced reverted cultures, the targeting spacers were found to be precisely excised from the miniCRISPR array, indicating a still hypothetical, but highly active recombination system acting on the dynamics of CRISPR spacer arrays. Our results indicate that CRISPR type III – based silencing is a broadly applicable tool to study in vivo functions of essential genes in Sulfolobales which underlies a specific mechanism to avoid malignant silencing overdose.
KEYWORDS: CRISPR type III, knockdown, essential genes, archaea, hyperthermophilic, Sulfolobales
Introduction
Proteins that execute fundamental processes are often indispensable for the life of the organism. Genome-wide gene disruption studies and computational approaches have identified many of these essential proteins from across the tree of life [1], with 300 to 500 genes being assigned as essential in prokaryotic models [2–5]. Functional prediction studies revealed that a large proportion of these essential proteins are intimately involved in information processing and storage [6]. However, in archaea, many remain poorly characterized, as exemplified by a recent gene-disruption library of the hyperthermophilic archaeon Sulfolobus islandicus where the functions of 17% of essential genes could not be categorized based on sequence [3]. Often, neither homology-based nor in vitro approaches do suffice to fully characterize the physiological role of a protein. Additionally, essential proteins were shown to have significantly more protein interaction partners than non-essential ones [7], which suggests multiple sites of action and a broader spectrum of complexity. This emphasizes the need for new tools that will make it possible to carry out comprehensive screens to explore the native role of essential genes in the context of living archaeal cells. However, such approaches are not trivial given that, by definition, a certain dose of the essential protein must be available for the organism to grow and survive. In eukaryotes, heterozygous diploids have proven useful in the study of phenotypes caused by the loss of one essential allele [8]. Similarly, heterologous expression of a (conditionally) defective gene version has successfully been applied in prokaryotes [9]. However, these studies often involve laborious modification of the essential gene on the host chromosome or require the addition of inducing or repressing agents that might cause side effects masking the phenotype.
Gene silencing tools represent a simple and convenient alternative for studying the physiological role of essential genes. Silencing tools generally employ small RNAs that guide an effector protein to complementary sites on the target mRNA or DNA, where the effector cleaves the RNA or blocks effective transcription. Because the endogenous locus remains intact, in both cases, gene expression tends to be reduced but is not abolished, allowing survival. Besides the well-established RNAi (RNA interference) pathway that has been widely used to silence genes in virtually every eukaryotic model [10], new fruitful knockdown tools have recently emerged from the prokaryotic virus-defence system CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) [11–18]. CRISPR systems are widespread in bacteria and archaea and function to protect the host cell from viral infection [19,20]. Small CRISPR RNAs (crRNAs) are transcribed from spacers in genomic CRISPR loci and incorporated into specific CRISPR effector complexes which further bind and degrade complementary virus DNA or RNA. CRISPR systems are subdivided into six types, each accommodating effector proteins specifically recognizing either DNA or RNA, respectively [21]. For essential gene studies in eukaryotes and bacteria, mainly nuclease-inactive derivatives of DNA-targeting CRISPR type II and V proteins (i.e. dCas9 and ddCas12a) have been employed that, when expressed in trans, bind to the respective promoter or coding region to block transcription [22–26]. Similarly, a mutated variant of the DNA-targeting type I protein complex (CASCADE) deficient in Cas3 nuclease activity, was successfully harnessed to silence essential genes in a halophilic archaeon [18]. In contrast to the above-mentioned DNA-targeting proteins, CRISPR type III systems are ribonucleolytic protein complexes that can directly target and degrade RNA upon crRNA binding [27,28]. By heterologously expressing synthetic crRNAs matching a host mRNA at specific protospacers, we and others have repurposed the endogenous CRISPR type III pathway in hyperthermophilic archaea for gene silencing in the closely related strains Saccharolobus solfataricus and Sulfolobus islandicus (cf. Fig. 1) [29–34]. Recently, we used this technology to silence the gene encoding an essential translation initiation factor, aIF5A [32], and the S-layer anchor, SlaB [31] in S. solfataricus, enabling a phenotypic analysis of the depletion cultures. Silencing of slaB caused severe growth defects and perturbed cell division, limiting our ability to obtain stable lines with a silence efficiency of greater than 75% [31]. This indicated that, under the applied growth conditions, SlaB might execute an essential function in S. solfataricus, even though it was shown to be non-essential in S. islandicus [3].
Figure 1.
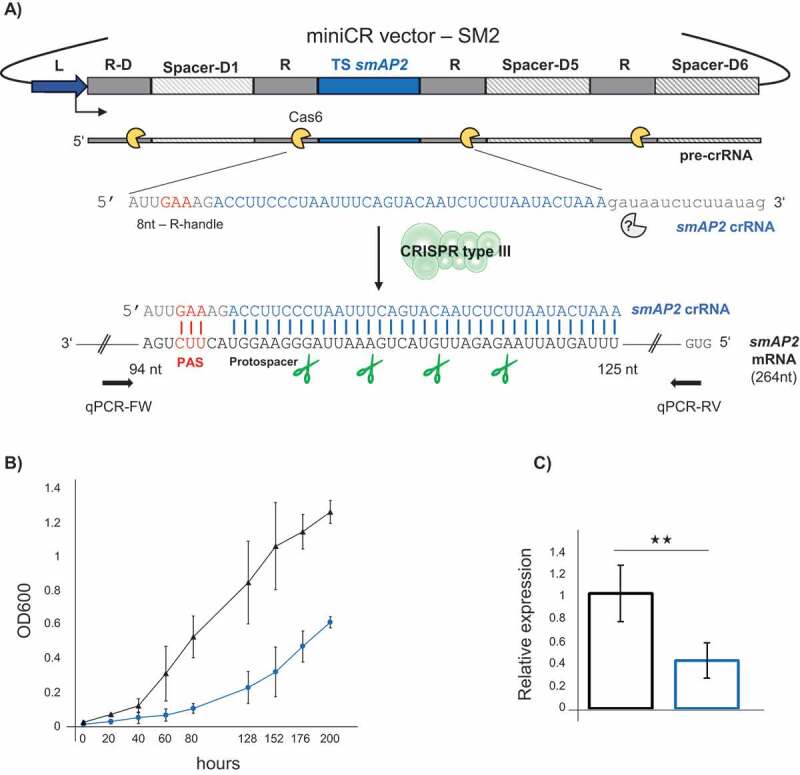
CRISPR type III – mediated silencing of the smAP2. A) Schematic representation of the miniCRISPR (section of native CRISPR locus D of S. solfataricus P2) vector used for gene silencing: Dark blue: leader; dark grey: repeats; light grey: native spacer-D1, spacer-D5 and spacer-D6; blue: synthetic smAP2 targeting spacer (37 bp). A pre-crRNA is first transcribed from the leader and subsequently cleaved by Cas6 (yellow) resulting in a mature crRNA consisting of an 8 nt – repeat-derived 5ʹ handle (grey), a targeting spacer (blue) and a 3ʹ repeat remnant of variable size, trimmed by an unknown exonuclease (grey). The crRNA incorporates into the type III system and binds to the complementary protospacer on the smAP2 mRNA; the 5ʹ-handle must match a 3ʹ PAS (protospacer adjacent sequence) in at least the three nucleotides (red). The type III degrades the mRNA at indicated positions (green scissors) leading to silencing of the smAP2 gene. Arrows indicate primer sites of RT-qPCR primers used for quantification of smAP2 mRNA. B) Growth profiles (OD600) of control (black, triangle) and smAP2 silenced cultures (blue, circles). Error bars, mean ± SD (n = 3). C) Expression of the smAP2 mRNA relative to the 16S rRNA measured by RT-qPCR on reverse transcribed RNA extracts of control (black, left bar) and smAP2 silenced cultures (blue, right bar), harvested at OD600 = 0.2 each. Error bars, mean ± SD (n ≥ 3); Significant differences to Ctrl are indicated by asterisks (two-tailed t test, n ≥ 3, p ≤ 0.01)
In this study, we demonstrate the CRISPR type III-based technology to efficiently deplete two further essential genes, encoding the Sm-like protein SmAP2 and the CdvA cell division protein. By comparing the silencing efficiencies, phenotypes, and escape mutants obtained from all four silencing experiments, we identify common traits and boundaries when silencing an essential gene in S. solfataricus. Finally, we investigate the applicability of the CRISPR type III tool in another Sulfolobales species by silencing the essential gene encoding the RNA polymerase (RNAP) subunit Rpo8 in the more distantly related strain S. acidocaldarius.
Materials and methods
Culturing and transformation
The uracil-auxotrophic derivate M18 of S. solfataricus P1 (DSM 1616) was cultured in Brock T/S/U medium supplemented with 0.1% tryptone (T) (Roth) (wt/vol), (+) D-Sucrose (S) (Serva) (wt/vol) and uracil (U) (Sigma Aldrich) at a final conc. of 0.0125 mg/ml under shaking conditions [30]. The uracil-auxotrophic strain MW001 [35] of S. acidocaldarius strain (DSM 639) cultivated in the same medium under same conditions, with the exception that tryptone was exchanged with N-Z-amine AS (NZ) (Fluka Analytics) and added to same concentrations. Liquid incubation of cells transfected with viral – based pDEST – vectors (see below) were incubated under the same conditions in Brock T/S medium lacking uracil. Liquid cultures of plasmid pIZ-transformants (see below) were cultivated in Brock NZ/S medium under the same conditions.
Electroporation of S. solfataricus was performed as described before [31]. Aliquots of the electroporation suspensions of pDEST – vector transfectants were used in inverse plaque assays directly after electroporation [29]. Plaques were further purified and inoculated in liquid medium as described [29], followed by analysis of cultures. Remaining electroporation solutions were directly inoculated into liquid culture and growth was monitored as intrinsic control and backup, but these cultures were not further analysed.
Electroporation suspensions of pIZ-transformants were directly inoculated into liquid medium without plating beforehand (primary cultures) and analysed [31], or plated on NZ/S gellan gum plates as described in ref [31] (colony). Single colonies were recovered and transferred to liquid medium and analysed. Unless stated otherwise, we did not further differentiate between pIZ or pDEST-vector – transformants in this study, as the vector backbone did not influence the phenotypic outcome. Electroporation of pIZ-plasmid-based vectors into S. acidocaldarius ∆cas3 cells (see below) was performed as described in ref [35] and electroporation suspensions were directly plated on NZ/S gellan gum plates. Colonies were recovered after 6 days of incubation and transferred to liquid medium (see above) and analysed.
Construction of miniCR vectors and cloning
MiniCR targeting aif5A and slaB were already designed in previous studies [31,32]. CA (cdvA targeting), SM2 (smAP2 targeting) and rpo8 miniCRs (rpo8 targeting) were constructed by overlap-PCR as described before [29] using anchor primers MoE-FW/RV [29] in combination with the respective miniCR construction primers listed in Table S2. Chromosomal locations of protospacers on cdvA, smAP2 and rpo8 targeted by the respective miniCR spacers are listed in Table S3. For CA-1, CA-2, CA-3 and CA-123 miniCR-constructs, as well as SB-23, SB-123, SB-3x6 miniCR-constructs, miniCR arrays were cloned onto both, pDEST and pIZ vectors, respectively [31]. All others were transferred to pIZ (rpo8 miniCR) or pDEST vectors only.
Cas3 gene disruption in S. acidocaldarius MW001
The procedure to disrupt Saci cas3 (saci_1872/SACI_RS09010) gene followed the method that uses pyrEF as marker gene and the uracil-auxotrophic Saci strain MW001 as parental strain [35]. The upstream and downstream flanking gene of saci_1872 were PCR-amplified (primers listed in Table S2). The PCR products for each construct were fused by overlap extension PCR and inserted into the NcoI and BamHI sites of pSVA406. The plasmid was methylated in E. coli EsaBC41 strain [36] and then transformed into electrocompetent MW001 cells. Single-crossover integrants were selected on uracil-deficient Brock plates [35]. Single-crossover integrants (detected by PCR, primers listed in Table S2) were then plated on 5-fluoroorotic acid containing plates for counter-selection against PyrEF triggering excision of pSVA406-∆cas3. Positive mutants (detected by PCR, primers listed in Table S2) were transferred to Brock liquid NZ/S medium and incubated as described above.
Growth profiles, culture PCR and sampling
Growth of all liquid cultures was monitored at OD (600 nm) (Beckam Coulter, DU 800 Spectrophotometer). Growth profiles depicted in the manuscript were all performed with transfectants/transformants freshly generated for this study. Aliquots were taken at regular intervals to verify the integrity of the miniCRs on the vectors by culture PCR. For culture PCR (Phusion Polymerase, ThermoFisher, Scientific), 2 µl of the culture were applied as template in a reaction using 406-FW and 406-RV primers (Table S2) hybridizing to the vector backbones in regions of ~ 100 bp up- and downstream of the miniCR array (Table S2). Additionally, vector-specific backbone primers (pIZ: ORF-904 FW/RV; pDEST: D291 FW/RV, Table S2), were used in separate PCR reactions on same cultures to verify the vector backbone. The entire volume of a PCR reaction was loaded on 1% agarose for gel-electrophoresis. The gel bands were gel purified (Monarch, PCR & DNA Gel Cleanup Kit, NEB) and sent for sequencing using the above-mentioned FW and RV primers in independent reactions, respectively. Sequencing was carried out by Eurofins Genomics (Germany). 5 ml of cultures showing an OD600 between 0.1 and 0.2 were harvested for RNA preparations.
RNA and cDNA preparation
For S. solfataricus cultures, RNA was isolated as described before [30]. Pellets of S. acidocaldarius cultures were resuspended directly in three volumes ice-cold TRIzolTM LS (Thermo Fisher Scientific) and the RNA was isolated according to manufacturer’s protocol. For all RNA extracts, DNAse digestion was performed as in ref [31] and complete removal of DNA was verified by PCR. Integrity of purified RNA was checked by gel electrophoresis. One microgram of RNA was reverse transcribed using ProtoScript II Reverse transcriptase (NEB) according to the manufacturer`s protocol and cDNA was further purified (Monarch, PCR & DNA Cleanup Kit, NEB). Concentrations of all RNA and cDNA were measured by Qubit using RNA BR, and ssDNA Assay Kit for cDNA, respectively (ThermoFisher Scientific).
Quantitative RT-qPCR
RT-qPCR on cDNA sampled from S. solfataricus was executed using GoTaq qPCR Master Mix (Promega) in an Eppendorf Mastercycler epgradient S relplex2 (Eppendorf) as described in ref [31] and relative expression of the individual target gene was calculated relative to either gapN-3 (SSOP1_3283, encoding glyceraldehyde-3-phosphate dehydrogenase) or 16S rRNA in same cDNA preparations as described in ref [37]. Primers used are listed in Table S2 (q3194 FW/RV for gapN-3). Linearized vectors carrying the target gene or reference gene (RS0486-FW/RV_flank primers for cdv operon plus flanking genes; SM2_FW/RV_Seq primers for smAP2 gene), respectively, served as standards which were also used to calculate transcript copies for quantitative analysis.
RT-qPCR on S. acidocaldarius cDNA was carried out using Fast SYBR Green master mix (Thermo Fisher Scientific) on QuantStudio™ 6 Flex Real-Time PCR System (Thermo Fisher Scientific). For analysis, values for the negative controls were subtracted, followed by normalization to the corresponding value of 16S rRNA. Genomic DNA isolated from Saci MW001 strain was used to obtain a standard curve. Efficiencies for all RT-qPCR ranged between 98 and 94%.
Immuno-detection
Cell lysates of miniCR-rpo8-PS and miniCR-SM2 harbouring cells were resolved on 12% Tris-tricine SDS gels and blotted onto nitrocellulose membranes as described in ref [32]. Immuno-detections of Rpo8 and the transcription factor TBP were carried out using polyclonal rabbit antiserum raised against recombinant Saci_Rpo8 and Saci_TBP in combination with donkey anti-rabbit IgG Dylight680 (Bethyl Laboratories). The blots were visualized using Typhoon FLA 9500 biomolecular imager (GE Healthcare) and analysed using ImageQuant TL Software (GE Healthcare). Immunodetection of SmAP2 and SmAP1 were performed using Anit-Sm-AP2 and Anti-Sm-AP1 antibodies followed by incubation with the secondary antibody Anti-rabbit IgG linked to horseradish peroxidase (Cell Signaling Technology). The blot was developed as described in ref [32].
Microscopy
For light microscopy of fresh samples, 5 µl of growing cdvA-silenced (transformed with miniCR-CA-2) and control cultures (transformed with miniCR-Ctrl) were transferred to microscope slides (Marienfeld superior) and immediately (within 2 min after sampling) recorded using a Jenoptik microscope camera (ProgRes MF Cool CCD 1.4 M.P) attached to a Nikon ECLIPSE Ni-E. For DAPI staining (Sigma), cells of same cultures were fixed at 4°C overnight in 70% ice-cold ethanol, subsequently washed and resuspended in buffer A (20 mM TRIS-Acetate buffer pH 5.5) and incubated according to the manufacturer's protocol and imaged.
Results
Depletion of the SmAP2
Sm-like proteins are major players in RNA metabolism across the tree of life. The halophilic archaeon Haloferax volcanii encodes one Lsm (‘like-Sm’) protein and disruption of the corresponding gene resulted in upregulation of genes involved in motility [38]. In contrast, members of the Sulfolobales contain three Lsm paralogs. One of these, SmAP2, appears to be essential, as knockout attempts failed repeatedly (U. Bläsi, unpublished data). Even though the physiological function of the SmAP2 is still elusive, 53 mRNA substrates were recently identified that all carried a specific motif that is tightly bound by SmAP2 in vitro [39,40]. To aid the characterization of the in vivo role of SmAP2, we sought to study the phenotype of the SmAP2-depletion in S. solfataricus P1. For the gene-silencing approach (as it was in principle used for all silenced genes), a 37bp protospacer sequence of the smAP2 gene was chosen that served as a target site in the silencing assay (CDS: SSOP1_RS00995, genome coordinates: complement: 174,756–174,792) (Fig. 1A). The smAP2 protospacer was selected based on the presence of a 3ʹ flanking protospacer adjacent sequence (PAS) which binds the repeat-derived 5ʹ handle of the crRNA in three positions (5ʹ GAA) within three base pairs of the start of the protospacer (Fig. 1A, red). Such PAS-handle binding was previously shown to suppress all CRISPR-mediated DNA interference in S. solfataricus [30,41], as well as the type III-mediated synthesis of secondary messengers (cyclic oligoadenylates) that induce non-specific RNA shredding by activating CARF-domain nucleases [42]. Thus, PAS binding in our experimental setup permits degradation of the target mRNA only. Further, a targeting spacer (TS) sequence was designed to be complementary to the protospacer and inserted between repeats of the miniCRISPR locus located on the virus-based expression vector pDEST – MJ [30]. Constructs carrying these miniCR sequences were then used to transfect S. solfataricus P1 cells. As a control, a pDEST vector (Ctrl) carrying the same miniCR array devoid of the smAP2 targeting spacer [31], was transfected independently. These transfections generated plaques on plates (see Materials and Methods). For each, three were picked and grown up in liquid medium. The growth of silenced cultures was significantly delayed and retarded in comparison to Ctrl cultures (Fig. 1B). An RT-qPCR analysis of reverse-transcribed RNA extracted from transferred cultures harvested at OD600 = 0.2 showed a 60% reduction in smAP2 RNA levels relative to the 16S rRNA in knockdown cultures compared to control cultures (Fig. 1C, Fig. S1, upper panel). This was accompanied by a reduction in the levels of the corresponding SmAP2 protein as detected in individual replicates by Western Blot (Fig. S1, lower panel). These data showed that the essential gene smAP2 could be efficiently silenced using the CRISPR type III knockdown technology, leading to a dramatic reduction in the growth of the culture. Transcriptomic studies that allow a closer inspection of effects on the mRNA pools are under way.
Silencing of an essential operon: the cell division initiation machinery CdvABC
As a second test of our technology, we silenced the cell division machinery by targeting CdvA. In members of the Sulfolobales, cell division is a tightly regulated process initiated concomitantly with the expression of three genes from one chromosomal locus [43], cdvA, cdvB and cdvC (cell division), late in G2 phase. These genes encode an archaea-specific protein CdvA, an ESCRT-III homologue, CdvB, and a homologue of eukaryotic Vps4, CdvC [44–46], all of which are thought to be involved in cytokinesis [44,45]. To explore the function of these proteins in S. solfataricus, we identified the conserved cdv gene cluster [43] with the cdvA gene (SSOP1_RS04680) lying upstream of cdvB (SSOP1_RS04675), followed by cdvC (SSOP1_RS04670) (Fig. 2A). A closer inspection of this gene cluster and transcription profiles (I. Zink, unpublished data) revealed potential regulatory regions for transcription (TATA-Box) and translation (ribosomal binding sites) upstream of the CDS of cdvA, but potentially also upstream of cdvB (closer investigation of regulatory elements provided in Supplemental Result), as was previously suggested for Sulfolobus acidocaldarius [43]. In S. acidocaldarius, independent as well as co-transcription with cdvBC has been reported for cdvA [43,45]. By targeting the locus, we, therefore, hoped to carry out a functional analysis of CdvA and to use this as a model to shed light on the impact of CRISPR silencing on polycistronic transcripts.
Figure 2.
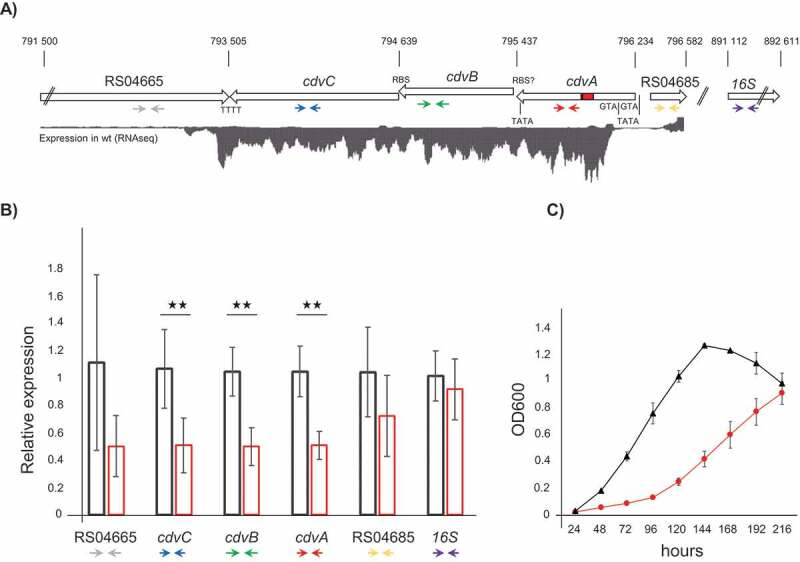
Silencing of the cdvABC locus. A) Schematic representation of the S. solfataricus P1 chromosomal regions 791 500–796 582, and 891,112–892,611, containing the genes SSOP1_RS04665, cdvC (SSOP1_RS04670), cdvB (SSOP1_RS04675) cdvA (SSOP1_RS04680), SSOP1_RS04685 and the 16S rRNA gene (SSOP1_RS05165), respectively. Red rectangle in cdvA = protospacer targeted by miniCR-CA-2; TATA boxes, RBS and ATG are indicated to scale. Expression levels of the first chromosomal region are represented by a read coverage histogram obtained from wild type S. solfataricus P1 RNA-Seq data. Coloured arrows below genes represent binding sites of primers used in RT-qPCR (see B). B) Relative expression of each gene (using primer pairs indicated above) relative to the gapN-3 mRNA measured by RT-qPCR on reverse transcribed RNA extracts of control (black left bar) and cdvA-targeted cultures (red right bar), harvested at OD600 = 0.2 each. Error bars, mean ± SD (n ≥ 3); Significant differences to Ctrl are indicated by asterisks (two-tailed t test, n ≥ 3, p ≤ 0.009). C) Growth profiles (OD600) of control (black triangle) and cdvABC – silenced cultures (red circles). Error bars, mean ± SD (n = 3)
We challenged S. solfataricus cells with a single-spacer-miniCR (miniCR-CA-2, cf. Table 1, cf. Fig. 3) targeting the mRNA at a protospacer located within the highly transcribed region of the cdvA gene, downstream of the second ATG (Fig. 2A indicated in red, Table S3). The growth of silenced cultures was significantly delayed when compared to control cultures (Fig. 2C). RT-qPCR analysis using primers binding cdvA (Fig. 2A, red arrows, Table S2) revealed a 50% reduction of the cdvA RNA compared to control cultures measured relative to the housekeeping genes gapN-3 and 16S rRNA (Fig. 2A, purple arrows) in all samples (Fig. 2B). To check for downstream effects of cdvA silencing, we also quantified the expression of cdvB and cdvC, as well as the upstream and downstream flanking genes, SSOP1_RS04665 and SSOP1_RS04685 (Fig. 2A) encoding the Topoisomerase 1 and a hypothetical protein, respectively (primers used indicated by coloured arrows in Fig. 2A, Table S2). While neither the neighbouring genes (for which gene expression of RS04665 fluctuated strongly in all cultures, see Fig. S3) nor the 16S rRNA showed significant downregulation upon cdvA silencing, cdvB and cdvC were clearly affected as they were silenced to an almost identical degree as cdvA (Fig. 2B, blue and green arrows). These observations suggest that the cdvABC locus is expressed as a polycistronic mRNA, which in our case is probably degraded by exonucleases upon type III-mediated cleavage within the cdvA open reading frame (ORF). It also suggests that the type III system can be used to efficiently silence operons when targeting the first gene. Interestingly, a microscopic analysis of freshly harvested (untreated) cdvABC-depletion cultures revealed heterogenous cell sizes and very large cells, when compared to images of control wild type Saccharolobus cultures (Fig. S2A). Staining of fixed cells from the same cultures suggested that the enlarged cells have an increased DNA content, (Fig. S2B), as has been reported for cells that fail in division [44,47–49].
Table 1.
Overview of 102 essential gene silencing experiments
| Target gene | Cultures# | Stable kd * | Stable miniCR | Maximum kd (%)§ | Revertants& | Strong miniCR$ |
|---|---|---|---|---|---|---|
| slaB+ | 52 | 30 | SB-2, SB-3, SB-3x2, SB-3x6 | 75 | 22 | SB-123, SB-23, SB-3x6 (colony) |
| aif5A+ | 11 | 6 | aIF5A-II | 40 | 5 | aIF5A-I |
| cdvABC | 23 | 10 | CA-2 | 50 | 13 | CA-1, CA-3, CA-123 |
| smAP2 | 16 | 13 | SM2 | 60 | 3 | SM2 (colony) |
#Cultures: Number of independently transformed cultures carrying a miniCRISPR vector; *Stable Kd: Number of cultures with measurable and stable knockdown of the essential gene; §Maxmimum kd: maximum knockdown observed in stably silenced cultures expressed as % mRNA lost compared to Ctrl; &Revertants: Number of cultures reverted to wild type owing to strong miniCRs; $Strong miniCR: miniCR conveying strong silencing leading to their instability; + Data pooled from published studies (ref. [31,32]) and from freshly transformed cultures. Colony = constructs changed after recovery from a single colony
Figure 3.
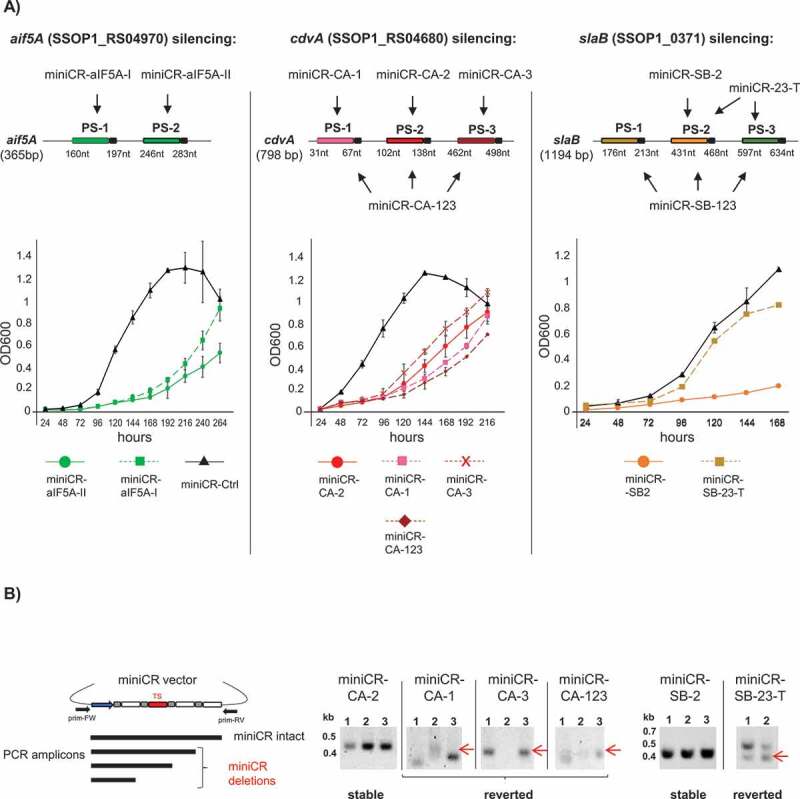
Stably silenced and reverting cultures in aif5A, cdvA and slaB silencing experiments. A) Upper panel: Schematic representation of mRNAs of each gene and corresponding silencing miniCR constructs where arrows point to protospacers regions targeted by each construct (cf. Table 1). The positions of protospacers (PS) including PAS (coloured boxes with black tail) on respective mRNAs are given with respect to the gene length. Lower panel: Growth profiles (OD600) of cells transformed with miniCR constructs leading to stably silenced cultures (coloured continuous lines)* and strong miniCR construct leading to reverted cultures (coloured discontinuous lines). Black lines (triangle) represent control cultures transformed with miniCR-Ctrl, devoid of a targeting spacer. Error bars, mean ± SD (n = 3). MiniCR-SB-23-T: culture transformed with miniCR-23 and transferred to fresh medium once. *Growth profiles of miniCR-SB-2/miniCR-aIF5A-II of another sample set have been published [31,32], but fresh transformants grown in parallel with the depicted revertant cultures are presented here. B) Left panel: Schematic representation of PCR products generated by culture PCR on stably silenced/reverted cultures using primers (prim-FW, prim-RV) binding up and downstream of the miniCR cassette, respectively. Amplicons of different lengths reflect the integrity of the miniCR array. Right panel: Agarose gels depicting culture PCR amplicons of miniCR cassettes in respective transformants (same cultures as in A sampled at OD600 = 0.2 were used as templates). Band heights emerging from deletions in miniCR arrays are indicated by red arrows and often appear as multiple or fuzzy bands in reverted cultures. Position of DNA ladder is indicated (kb). Representative agarose gels of culture PCRs on miniCR-SB-123 and miniCR-aIF5A-I carrying revertant cultures can be found in refs. [31] and [32], respectively
Phenotypes of maximally silenced genes and reverting cultures
In addition to cdvABC and smAP2 genes, our team recently silenced two additional genes of pivotal function: the gene encoding the translation factor aIF5A [32] and slaB coding for the S-layer anchor [31]. Data of these published studies as well as fresh cultures transformed with the slaB/aif5A-targeting miniCR constructs were added to the quantitative analysis. Apart from smAP2 carrying only one suitable protospacer, all other genes have been targeted at different protospacers independently or simultaneously from different miniCR constructs (Table 1, Fig. 3A). Having analysed at least three biological replicates per targeting miniCR construct transformed in these four silencing experiments, a total of 102 cultures (representing 15 different miniCR constructs) were screened for common features and patterns of essential gene silencing (Table 1). Among the 102 cultures, 59 were stably silenced with specific miniCR constructs (‘stable miniCRs’) enabling them to be used for phenotypic analysis. 43 of the 102 cultures however, were transformed with miniCR constructs that proved unstable, leading to a reversion to the wild type phenotype (Table 1, Fig. 3A, B). Both sample sets are analysed below, referred to either as ‘stably silenced’ or “reverted” cultures, respectively. For all four essential genes, stable silencing was achieved by expressing a single crRNA species targeting the respective mRNA at a specific protospacer: PS-2 for cdvABC and aif5A; PS-2 and PS-3 for slaB and PS-1 for smAP2 (the constructs are listed in Table 1 and illustrated in Figs. 3A, and 1A for smAP2). Across all 59 stably silenced cultures, the highest levels of silencing were seen for slaB, where we were able to achieve a 75% knockdown (i.e. 25% residual mRNA) [31] (Table 1). Maximum levels of silencing for cdvABC, aif5A and smAP2 ranged between 40 and 60% (Table 1, and as shown earlier in ref [32]). The strongest silencing levels were always accompanied by strong growth retardation, with cultures exhibiting a prolonged lag phase and a lower endpoint OD600 (Figs. 1A, 2C, 3A continuous lines). This decreased cell fitness was also reflected in the inability of stably silenced cultures to grow on plates as single colonies that still harboured an intact miniCR (see below and ref [31]). Only plaques (when using a virus vector), or direct liquid inoculation (when silencing from a plasmid) could be used to generate stably silenced cultures of essential genes in S. solfataricus. During incubation, stably silenced and control cultures were periodically screened by light microscopy to check cell morphology. Morphological alterations were only observed following the silencing of cdvABC (cf. Fig. S2) or slaB [31], and thus were not a general consequence of essential gene silencing or the accompanying decrease in the growth rate and/or fitness.
As observed in a small subset of slaB and aif5A silenced cultures [31,32], cultures transformed with ‘strong’ miniCRs (Table 1, Fig. 3A) either targeting a single mRNA at multiple sites, or at a certain protospacer (Fig. 3A shaded PS), were consistently seen reverting to wild type. This was readily apparent in the growth profiles of replicate cultures of all targets. Thus, although growth was retarded within the first 5 days of incubation, growth rates recovered significantly thereafter (Fig. 3A, aIF5A-I, CA-3 discontinuous lines, see ref. [31] for SB-123), upon longer incubation (CA-1, −123, Fig. S4 discontinuous lines), or upon transfer to fresh medium (SB23-T discontinuous line). PCR tests on revertants (sampled at OD600 = 0.1–0.2) using vector-specific primers spanning the miniCR often revealed a reduced miniCR, apparent as fuzzy smaller amplification products (Fig. 3B, CA-1, CA-3, CA-123, see ref. [32] for aIF5A-I) or double bands (Fig. 3B, SB-23-T see ref. [31] for SB-123) on the gel. These alterations of the miniCRs were further analysed by sequencing and identified as deletions spanning the targeting spacers (see below) which functioned to inactivate silencing. Notably, such alterations of the miniCR locus were never observed in the 59 stably silenced cultures sampled in exponentially growing cells incubated in liquid medium (Fig. 3B cf. aIF5A-II, CA-2, SB-2). However, when stably silenced cultures were plated directly after electroporation, single colonies again appeared with altered targeting spacers (identified by sequencing of the colony PCR product), as observed for miniCR-SM2 and miniCR-SB3x6 (Table 1, Fig. 4B, C). These observations indicate that an essential gene can only be silenced to a critical threshold level – below which levels of the essential protein are not sufficient for cell survival.
Figure 4.
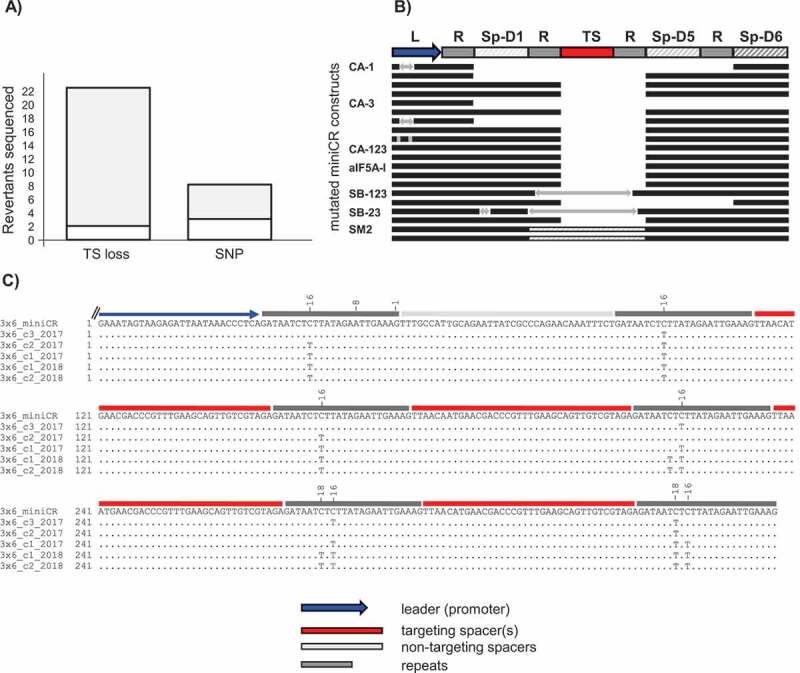
Analysis of mutations of sequenced miniCR array in reverted cultures. A) Stacked bar plot representing the absolute number of sequenced miniCR recovered from reverted cultures exhibiting deletions (TS loss) or single nucleotide polymorphisms (SNP) of/in the targeting spacer, respectively. Grey area in TS loss = spacer losses at repeat junctions, white area in TS loss = imprecise spacer deletions; Grey area in SNP = SNPs in repeat regions -16/-18 (cf. C), white area in SNP = SNPs found elsewhere (cf. Table S1). B) Schematic alignment of miniCR arrays exhibiting spacer losses at repeat junctions. An intact miniCR is schematically represented as reference; arrows represent insertions; dashed lines in SM2 represent a doubling event of Sp-D5. C) Sequences of miniCR-SB3x6 retrieved from five colonies of individual transformants aligned to the intact miniCR-SB3x6 array (3x6_miniCR); four spacer shown, as only these regions were affected
Unstable spacers are predominantly cut out at CRISPR repeats
In order to investigate the nature of revertant mutations in more detail, we sequenced the miniCR vectors from 30 out of the 43 revertants. This did not include some cultures carrying the ‘strong’ slaB-targeting constructs miniCR-SB-123 (10 cultures) and miniCR-SB-23 (three cultures), since material was no longer available for these cultures. However, these cultures had been screened by PCR before-hand and showed a shortened miniCR (see above, Fig. 3B and ref [31]). The remaining 30 consisted of 13 revertants that emerged from cdvABC-targeting, 5 from aif5A-targeting, 9 from slaB-targeting and 3 from smAP2-targeting experiments, respectively (see Table S1). Within the 30 sequenced constructs, 22 were verified as being devoid of the targeting spacer, and eight constructs carried mutations in the targeting spacer or adjacent repeat regions, respectively (Fig. 4A, Table S1). In 20 of the 22 constructs that had lost the targeting spacer in the miniCR cassette (TS loss, Fig. 4A) deletions occurred precisely at repeat boarders (whilst retaining the last nt of the repeat), where one repeat unit plus the targeting spacer(s) and sometimes additionally backbone Spacer D1 or Spacer D2 were excised (Fig. 4A TS loss grey, Fig. 4B). Interestingly, five cultures that exhibited losses at repeat regions concomitantly showed insertions of various lengths either within the leader, within the backbone Sp-D1 miniCR spacer, or within the targeting spacer region (Fig. 4B, arrows). However, when examining these insertions using BLAST, we were unable to find any significant match to the database, identifying them as potential remnants of imprecise recombination. SmAP2 revertants only emerged when recovering colonies from smAP2-silenced cultures (see above). These revertants had undergone a duplication event of the backbone spacer Sp-D5, which was inserted again precisely between repeats (Fig. 4B, dashed panels). Investigations of the eight constructs that exhibited a mutated targeting spacer revealed three that carried SNPs (single nucleotide polymorphisms) all along the TS (Fig. 4A SNP white, Table S1) and five that exhibited specific mutations within the repeats (Fig. 4A SNP grey). These specific mutations appeared to have been introduced upon plating of cells transformed with the slaB-silencing miniCR-SB3x6 plasmid vector (Fig. 4A SNP grey, Fig. 4C), a construct that conferred strong and stable silencing of slaB to 75% in liquid incubations as a result of six crRNAs, that all target one protospacer [31]. The revertants of the independently transformed and plated miniCR-SB3x6 carrying cultures acquired SNPs in the defined position of −16 and/or −18 with respect to the downstream spacer. In both positions, C was exchanged with a T, mutating the sequence 5ʹ CTC into 5ʹ TTT. This inhibited the silencing effect, since no growth retardation or silencing was detected when these revertants were grown up in liquid medium (data not shown). Furthermore, we were unable to identify other mutations in these miniCRs. Note that when aliquots of SB3x6-transformed cells retrieved from the same electroporation experiments were grown up in liquid culture without plating, these point mutations were not observed [31], indicating that they accumulated during colony growth.
Application of the type III silencing system in a related strain: silencing of rpo8 in S. acidocaldarius
In order to investigate whether the CRISPR type III-mediated silencing technology could be applied to other Saccharolobus/Sulfolobus species besides S. solfataricus and S. islandicus, we decided to test the technology in the well-studied and more distantly related S. acidocaldarius. This strain carries a CRISPR type III-D and CRISPR type I-D system as well as four CRISPR spacer-repeat arrays (Fig. 5A and ref. [21]). The two CRISPR arrays 11 and 5 (Fig. 5A and ref. [50]) carry repeats that would generate crRNAs with the same 5ʹ handle as developed for our miniCR locus. Therefore, we expected that crRNAs expressed from the backbone miniCR would incorporate into the native type III complex of S. acidocaldarius and that we could apply the commonly used PAS sequence for protection of any unspecific DNA/RNA shredding of the complex. However, in order to avoid any unforeseen cross-reactions with the type I-D CRISPR system, we deleted the gene encoding the nuclease Cas3 to make it catalytically inactive (c.f. Fig. 5A, Fig. S5A). S. acidocaldarius ∆cas3 did not show any substantial growth rate phenotype in comparison to Saci MW001 (Fig. S5B). As a target gene for silencing, we selected the rpo8 gene (saci_0661/SACI_RS03150), which encodes a subunit of the archaeal RNAP with unknown function [51]. Rpo8 likely plays a crucial role since knockout attempts in S. acidocaldarius repeatedly failed (Fouqueau and Werner, unpublished data), and it is shared between all classes of eukaryotic RNA polymerases including RNAPI, II, III, IV and V. In archaea, Rpo8 is conserved in the TACK and Asgard superphyla [52]. For the knockdown of rpo8, we selected two protospacers on the rpo8 gene (Fig. 5B, Table S3), each preceded by a PAS matching the 8 nt 5ʹ-handle in at least four positions. We designed two single-spacer miniCRs, individually targeting either of the protospacers, as well as one multiplex miniCR-rpo8-PS12, expressing both crRNA species for simultaneous targeting, which were cloned into pIZ plasmids. Whereas expression of the single miniCR targeting at either PS1 or PS2, respectively, did not cause any significant changes in the growth profile when compared to the control construct (miniCR without TS), immunodetection of Rpo8 by Western blot showed that the protein production was moderately reduced (Fig. S6). By contrast, transformation of multiplex miniCR-rpo8-PS12 resulted in mild growth retardation and significant depletion of the rpo8 mRNA down to 30% residual mRNA (Fig. 5C, D). Furthermore, western blot analyses verified a marked reduction in Rpo8 protein levels (Fig. 5E). As the growth defect was only mild, the 70% knockdown does probably not define the maximum silencing level of this mRNA. However, our results demonstrate that the miniCR technology can be applied to study genes in S. acidocaldarius by harnessing its native type III-D endonuclease.
Figure 5.
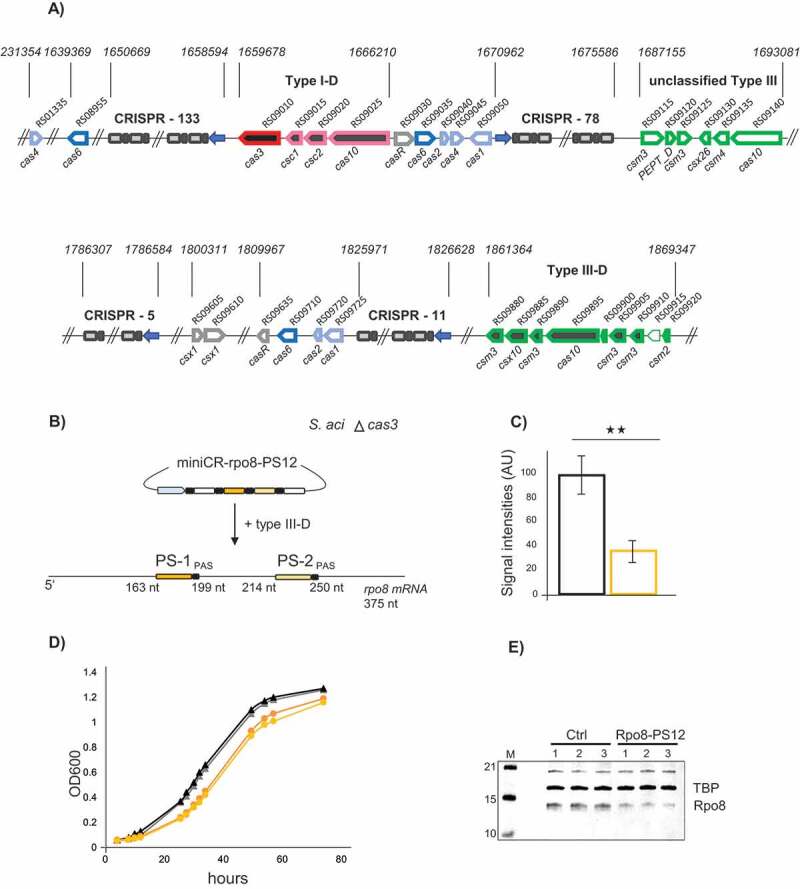
Silencing of rpo8 in S. acidocaldarius using the native type III-D system. A) Schematic representation of the CRISPR-Cas loci in S. acidocaldarius including all characterized adaptation cassettes (light blue), cas6 processing genes (blue), csx1 and casR accessory genes (grey), type I and type III effector complexes (pink, green) as well as CRISPR arrays (spacer number indicated); the unclassified type III cluster is of unknown function [21]; pseudogenes without annotation are indicated by a light border; the gene cas3 (red) was deleted in the mutant strain used in the rpo8 silencing experiment. B) Schematic representation of the silencing experiment using miniCR-rpo8-PS12 targeting the rpo8 mRNA at two protospacers (PS1, PS2); PS positions in respect to the gene length are indicated. C) Relative expression of the rpo8 mRNA relative to the 16S rRNA measured by RT-qPCR on reverse transcribed RNA extracts of control (black left bar) and rpo8 silenced transformants (orange right bar), harvested at OD600 = 0.3 each. Error bars, mean ± SD (n ≥ 3); Significant differences to Ctrl are indicated by asterisks (two-tailed t test, n ≥ 3, p ≤ 0.01). D) Growth profiles (OD600) of control (black triangles) and rpo8 silenced cultures (orange circles); two representative replicates grown up from separate colonies are shown. E) Western blot analyses using Anti-Rpo8 detecting Rpo8 (lower band) and Anti-TBP binding the transcription factor TBP (loading control, upper band). Three biological replicates of control (left) and rpo8- silenced cultures (harvested at OD600 = 0.3) are presented
Discussion
In this study, we showed that essential genes can efficiently be silenced by employing an endogenous CRISPR type III system and synthetic crRNAs expressed from a miniCR vector in hyperthermophilic archaea of the order Sulfolobales. The silencing approach is simple: synthetic crRNAs heterologously expressed from a miniCR vector, incorporate into a native CRISPR type III endonuclease and guide it to complementary loci on the mRNA of the essential gene, which is subsequently cleaved by the Cas7 backbone of the endogenous complex [53]. While we have recently used this technology for targeted knockdown of slaB, encoding the S-layer anchor and aif5A encoding an essential translation factor [31,32], we here present the successful depletion of the essential SmAP2 protein and the polycistronic mRNA of the operon encoding the cell division initiation machinery CdvABC. By pooling data of 102 independent silencing experiments of these four genes and carrying out a detailed analysis, we consistently found two outcomes: either cells were stably silenced up to a gene-specific maximum level (between 40 and 75%), which caused distinct phenotypes, or cells reverted to the wild type phenotype owing to acquired mutations in the miniCR locus of particular constructs that potentially conferred too strong gene silencing.
Successful silencing of essential genes
When comparing the successful silencing experiments, i.e. stably silenced cultures, we made several interesting observations: First, all of the four genes could be efficiently silenced by one crRNA species targeting the mRNA at one position. Second, in contrast to the almost 100% knockdown efficiency measured for a non-essential gene [29], the maximal silencing level we could achieve here was 75%, but varied between gene candidates (from 40 to 75%). Third, all maximally silenced cultures exhibited a loss of cell fitness as seen by strong growth retardation and the inability to grow as stably silenced single colonies on plates. When comparing these observations to other essential gene silencing studies in prokaryotes, we find several parallels. For instance, when essential gene RNAse P was silenced in Haloferax volcanii up to a maximum of 78% by employing a recombinant CRISPR type I system, which blocks transcription, cultures suffered from an attenuated growth profile with a prolonged lag phase [18]. Similar trends were reported for silencing assays using an antisense RNA approach in E. coli, where 65% silencing of ftsZ, encoding the central cell division protein was achieved which led to 50% reduction of the growth rate [54]. Furthermore, heterologous expression of the catalytically inactive CRISPR-dCas9 enzyme, was used to inhibit transcription of essential genes in other bacterial genera, such as Lactobacillus [24], Staphylococcus [55] or Mycobacterium [22], which again all showed strong growth retardation and a reduction in viability upon maximum silencing. Thus, the phenotypes observed in our survey of a hyperthermophilic archaeon are similar to findings in other mesophilic organisms where different silencing approaches were used to target essential genes. Notably, although there was a correlation between increasing silencing levels and decline in growth when targeting the same mRNA using multiplex miniCRs [31], we did not observe such similar correlations among the different gene candidates, as slaB depleted cultures show a similar growth profile compared to the ‘more mildly silenced’ smAP2 and aif5A cultures, respectively. Thus, the required protein dosage for proper growth differs amongst essential genes, as expected, and as previously observed in E. coli [54].
Reversion of phenotypes through specific deletion of targeting spacers
Strong miniCR constructs targeting the mRNA at multiple sites or at certain, probably more efficient, protospacer was not tolerated and led to the above-mentioned second outcome, that is abrogation of silencing [31] and reversion of the cultures to the wild type phenotype. The intolerance of cells to stronger silencing reflects a certain level of the essential protein necessary for cell survival. Reverted cultures escaped silencing by either deleting or mutating parts of the miniCR cassette, while leaving the vector backbone intact. Perhaps these mutations were promoted or accelerated by a potentially higher mutation rate of the expression vectors. Intriguingly however, these alterations predominantly happened in one of two specific ways: either the targeting spacers plus one repeat were excised precisely at the repeat junction, or SNPs exchanged a C to a T in the repeat in positions −16 or/and −18 upstream of the targeting spacer. The latter is likely to interfere with processing of the pre-crRNA by the Cas6 enzyme to inhibit the formation of a mature and functional targeting crRNA [50,56]. Alternatively, as these specific point mutations changed the sequence 5ʹ CTCTTAT to a consecutive T-stretch (5ʹ TTTTTAT), these mutations could potentially interfere with proper transcription of the miniCR array, by introducing a terminator sequence.
Interestingly, spacer excision at repeat junctions from the host CRISPR arrays was previously observed in bacteria and archaea harbouring different CRISPR interference systems [57–62]. For instance, when Lactobacillus gasseri, was challenged with an antibiotic-resistance carrying plasmid recognized by a host spacer, cells tended to delete the targeting spacer at repeat regions from the host CRISPR array, rather than mutating the protospacer on the plasmid in order to survive. Notably, as DNA targeting of the L. grasseri CRISPR type II Cas9 system relies on the recognition of a four-nucleotide PAM (protospacer adjacent motif) preceding the protospacer, a single point mutation in the PAM would have potentially sufficed to abrogate targeting of the plasmid [60]. Also in other Sulfolobales carrying type I and type III CRISPR systems, deletion of host spacers at repeat regions was observed under selective pressure or in order to maintain a beneficial plasmid or virus [57,62].
Under environmental conditions, native CRISPR arrays are known to be hot spots of rearrangements and recombination events that are driven by the constant integration of new spacers from most recent invaders in order to maintain immunity [63,64]. Thus, deletions of spacer-repeat units might occur frequently, to regulate CRISPR array size and spacer content, by efficiently eradicating obsolete or even malignant spacers, such as chromosome-targeting spacers [57,62,65,66]. This poses the question as to whether the precise spacer deletion represents a specific mechanism acting on the dynamics of the CRISPR spacer arrays. Given the prevalence of specific CRISPR repeat-binding proteins [67,68] and two active CRISPR-adaptation complexes that cleave at repeat borders to integrate new spacers into the CRISPR array in S. solfataricus [69], it is tempting to speculate whether such proteins might be involved in deletion processes. Indeed, the spacer-acquisition hallmark integrase Cas1 was shown to mediate transesterification reactions of branched DNA substrates in reverse in S. solfataricus in vitro, which would suggest that it could catalyse steps of spacer disintegration [70]. Furthermore, as CRISPR-Cas DNA repair are closely intertwined [71,72], their proteins might cooperate in such a process. Although repeats might be too short to trigger efficient homologous recombination in our study (24 bp), microhomology-based repair pathways, which were recently shown to be active in Sulfolobus [73], could also be induced by Cas protein-induced DNA breaks.
Notably, we have never observed any mutations or deletions in the type III genomic loci in any reverter. This is in contrast to findings in the halophilic archaeon Haloferax volcanii, where, besides spacer loss, survival by deletion of cas genes encoding the type I-B effector complex was frequently observed [18,61]. These deletions were attributed to recombination events between upstream and downstream homologous CRISPR arrays enclosing the type I-B operon [61]. As type III operons in S. solfataricus are located further away from CRISPR islands (and are not bordered by homologous CRISPR arrays), deletions of those loci might not as easily be triggered as in H. volcanii and therefore might constitute rather rare events.
Specificity of protospacer targeting
Our study shows that type III silencing specifically decreases the transcript levels of the target genes or genes that are co-transcribed. Because of the latter, the type III technology can be used as a tool for targeted knockdown of whole operons. In our analysis, we do not find neighbouring genes or housekeeping genes (located elsewhere in the genome) to be significantly differently regulated in silenced cultures compared to control cultures, which indicates that the type III targeting does not affect transcript levels other than that of the targeted genes. However, a tendential downregulation of SSOP1_RS04665 encoded upstream of the cdvABC locus and transcribed from the opposite strand, can be observed upon cdvABC silencing (cf. 2B). We believe that this owes to the transcriptional regulation of the gene itself, as the high standard deviations of transcript numbers in control as well as silenced cultures indicate that this gene might be generally stochastically transcribed. An alternative explanation could also be that SSOP1_RS04665 underlies a specific posttranscriptional regulation possibly depending on structure formation in the 3ʹ end, as has been observed in Sulfolobales [74]. This could probably be somehow impaired by the reduced transcript of the silenced cdvC, as it is indicative by the RNAseq chromatogram (c.f. Fig. 2A), that the cdvC transcript traverses the terminator and ‘reaches into’ the reading frame of SSOP1_RS04665.
In slaB depletion experiments, the independent expression of two different crRNA species targeting at two different protospacers of the slaB mRNA caused an identical phenotype. Furthermore, the expression of slaB from a close relative, S. islandicus, rescued the silencing phenotypes generated using different miniCRs [31]. This indicates that silencing via the type III technology is specific and makes it clear that the phenotypes observed in our studies are a direct effect of the depleted gene, rather than the result of off-target effects, i.e. non-specific mRNA cleavage. Investigations of the type III-B-mediated RNA interference in the close relative S. islandicus revealed a seed motif located within the 3ʹ spacer part of the crRNA that was recently shown to function as ‘capture motif’ aiding the initial recognition and proper binding of mRNA targets via the Cmr1 subunit of type III-B complexes [75,76]. These studies further suggest that a few mismatches between the crRNA capture sequence and the 5ʹ end of the target mRNA are likely to be sufficient to decrease the initial binding affinity and hence, cleavage efficiency of the target mRNA [75]. This emphasizes the target specificity of type III complexes, which potentially reduces the risk of off-target cleavage events. However, not all type III complexes carry a Cmr1 subunit [77]. Thus, target recognition and subsequent cleavage requirements might differ among different type III types [77] and are probably further influenced by cell dynamics. Therefore, off-target effects in the context of type III-mediated gene silencing in S. solfataricus remain to be further investigated.
Conclusion
Based on our combined results, we conclude that silencing via the type III system is a very powerful tool to carry out a functional analysis of essential genes. Besides identifying key roles for the S-layer in cell division and virus infection, our analysis demonstrates strong phenotypes also for aif5A, cdvA and smAP2 knockdown. In addition, it shows that the targeting of cdvA silences the cdvABC operon to abrogate regulated cell division, leading to the formation of enlarged cells in silenced cultures, a phenotype that will be explored in detail in future studies. Furthermore, we showed that the system could be successfully applied to deplete the essential Rpo8 RNAP subunit in a more distant Sulfolobus species, which demonstrates a broader application of type III silencing. As in other gene knockdown systems, an overdose of silencing leads to the selection of mutant variants, which can be easily identified by monitoring the genotype and phenotype, and which reveal new details regarding the mechanisms underpinning CRISPR array evolution and the specificity of the type III system. Since we have observed that the strength of the silencing depends on the choice of guide RNA it would be very helpful to be able to predict how strong the silencing of a chosen guide RNA will be. But this is currently impossible due to the complexity of RNA stability and folding particularly under high temperature as in these hyperthermophilic organisms. Therefore, it will remain necessary to use different sets of guide RNAs in order to find the best silencing level that leads to stable gene knockdowns.
Supplementary Material
Acknowledgments
The authors thank Thomas Pribasnig, Kevin Pfeifer and Erika Wimmer for lab assistance. Also, special thanks go to Prof. Silvia Bulgheresi, Melina Kerou and Philipp Weber for fruitful discussions. This work was financed through a DOC-fellowship of the Austrian Academy of Sciences (OEAW) and a PhD completion grant of the University of Vienna to IZ as well as an ERC - Adv grant TACKLE (No. 695192) and FWF grant (P29399) to CS.
Funding Statement
This work was supported by the ERC under Grant [695192]; and FWF under Grant [P29399]; and by the Austrian Academy of Sciences (OEAW) under a [DOC fellowship]; and by University of Vienna under a [PhD completion grant].
Data availability
The genome sequences are published and available under GenBank assembly accession GCA_900079115.1 (S. solfataricus P1) and GCA_000012285.1 (S. acidocaldarius DSM 639). The raw sequencing data of culture PCRs on miniCR vectors and raw quantitative PCR data are available upon request.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Luo H, Lin Y, Gao F, et al. DEG 10, an update of the database of essential genes that includes both protein-coding genes and noncoding genomic elements. Nucleic Acids Res. 2014;42:D574–D580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Glass JI, Assad-Garcia N, Alperovich N, et al. Essential genes of a minimal bacterium. Proc Natl Acad Sci U S A. 2006;103:425–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Zhang C, Phillips APR, Wipfler RL, et al. The essential genome of the crenarchaeal model sulfolobus islandicus. Nat Commun. 2018;9:4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sarmiento F, Mrázek J, Whitman WB.. Genome-scale analysis of gene function in the hydrogenotrophic methanogenic archaeon methanococcus maripaludis. Proc Natl Acad Sci U S A. 2013;110:4726–4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Goodall ECA, Robinson A, Johnston IG, et al. The essential genome of Escherichia coli K-12. MBio. 2018;9. DOI: 10.1128/mBio.02096-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bartha I, Di Iulio J, Venter JC, et al. Human gene essentiality. Nat Rev Genet. 2018;19:51–62. [DOI] [PubMed] [Google Scholar]
- [7].Jeong H, Mason SP, Barabási A-L, et al. Lethality and centrality in protein networks. Nature. 2001;411:41–42. [DOI] [PubMed] [Google Scholar]
- [8].Giaever G, Shoemaker DD, Jones TW, et al. Genomic profiling of drug sensitivities via induced haploinsufficiency. Nat Genet. 1999;21:278–283. [DOI] [PubMed] [Google Scholar]
- [9].Hupert-Kocurek K, Sage JM, Makowska-Grzyska M, et al. Genetic method to analyze essential genes of Escherichia coli. Appl Environ Microbiol. 2007;73:7075–7082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Mohr S, Bakal C, Perrimon N.. Genomic screening with RNAi: results and challenges. Annu Rev Biochem. 2010;79:37–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Qi LS, Larson MH, Gilbert LA, et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bikard D, Jiang W, Samai P, et al. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res. 2013;41:7429–7437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kim SK, Kim H, Ahn W-C, et al. Efficient transcriptional gene repression by type V-A CRISPR-Cpf1 from Eubacterium eligens. ACS Synth Biol. 2017;6:1273–1282. [DOI] [PubMed] [Google Scholar]
- [14].Luo ML, Mullis AS, Leenay RT, et al. Repurposing endogenous type I CRISPR-Cas systems for programmable gene repression. Nucleic Acids Res. 2015;43:674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Fernandes LGV, Guaman LP, Vasconcellos SA, et al. Gene silencing based on RNA-guided catalytically inactive Cas9 (dCas9): a new tool for genetic engineering in Leptospira. Sci Rep. 2019;9:1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Rath D, Amlinger L, Hoekzema M, et al. Efficient programmable gene silencing by Cascade. Nucleic Acids Res. 2015;43:237–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Abudayyeh OO, Gootenberg JS, Essletzbichler P, et al. RNA targeting with CRISPR–Cas13. Nature. 2017;550:280–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Stachler A-E, Marchfelder A. Gene repression in haloarchaea using the CRISPR (Clustered regularly interspaced short palindromic repeats)-Cas I-B system. J Biol Chem. 2016;291:15226–15242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Barrangou R, Fremaux C, Deveau H, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–1712. [DOI] [PubMed] [Google Scholar]
- [20].Makarova KS, Grishin NV, Shabalina SA, et al. A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol Direct. 2006;1:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Makarova KS, Wolf YI, Iranzo J, et al. Evolutionary classification of CRISPR–Cas systems: a burst of class 2 and derived variants. Nat Rev Microbiol. 2020;18:67–83. Nature Research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Choudhary E, Thakur P, Pareek M, et al. Gene silencing by CRISPR interference in mycobacteria. Nat Commun. 2015;6:6267. [DOI] [PubMed] [Google Scholar]
- [23].Gilbert LA, Larson MH, Morsut L, et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154:442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Myrbråten IS, Wiull K, Salehian Z, et al. CRISPR interference for rapid knockdown of essential cell cycle genes in Lactobacillus plantarum. mSphere. 2019;4. DOI: 10.1128/mSphere.00007-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wensing L, Sharma J, Uthayakumar D, et al. A CRISPR interference platform for efficient genetic repression in Candida albicans. mSphere. 2019;4. DOI: 10.1128/mSphere.00002-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zheng Y, Shen W, Zhang J, et al. CRISPR interference-based specific and efficient gene inactivation in the brain. Nat Neurosci. 2018;21:447–454. [DOI] [PubMed] [Google Scholar]
- [27].Hale CR, Zhao P, Olson S, et al. RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell. 2009;139:945–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zhang J, Rouillon C, Kerou M, et al. Structure and mechanism of the CMR complex for CRISPR-mediated antiviral immunity. Mol Cell. 2012;45:303–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zebec Z, Zink IA, Kerou M, et al. Efficient CRISPR-mediated post-transcriptional gene silencing in a hyperthermophilic archaeon using multiplexed crRNA expression. G3: Genes | Genomes | Genetics. 2016;6:3161–3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zebec Z, Manica A, Zhang J, et al. CRISPR-mediated targeted mRNA degradation in the archaeon Sulfolobus solfataricus. Nucleic Acids Res. 2014;42:5280–5288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zink IA, Pfeifer K, Wimmer E, et al. CRISPR-mediated gene silencing reveals involvement of the archaeal S-layer in cell division and virus infection. Nat Commun. 2019;10:4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Bassani F, Zink IA, Pribasnig T, et al. Indications for a moonlighting function of translation factor aIF5A in the crenarchaeum Sulfolobus solfataricus. RNA Biol. 2019;16:675–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Peng W, Feng M, Feng X, et al. An archaeal CRISPR type III-B system exhibiting distinctive RNA targeting features and mediating dual RNA and DNA interference. Nucleic Acids Res. 2015;43:406–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Han W, Feng X, She Q. Reverse gyrase functions in genome integrity maintenance by protecting DNA breaks in vivo. Int J Mol Sci. 2017;18:1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wagner M, van Wolferen M, Wagner A, et al. Versatile genetic tool box for the crenarchaeote sulfolobus acidocaldarius. Front Microbiol. 2012;3:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Grogan DW. Cytosine methylation by the SuaI restriction-modification system: implications for genetic fidelity in a hyperthermophilic archaeon. J Bacteriol. 2003;185:4657–4661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Maier L-K, Benz J, Fischer S, et al. Deletion of the Sm1 encoding motif in the lsm gene results in distinct changes in the transcriptome and enhanced swarming activity of Haloferax cells. Biochimie. 2015;117:129–137. [DOI] [PubMed] [Google Scholar]
- [39].Märtens B, Bezerra GA, Kreuter MJ, et al. The heptameric SmAP1 and SmAP2 proteins of the crenarchaeon sulfolobus solfataricus bind to common and distinct RNA targets. Life (Basel). 2015;5:1264–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Märtens B, Sharma K, Urlaub H, et al. The SmAP2 RNA binding motif in the 3′UTR affects mRNA stability in the crenarchaeum Sulfolobus solfataricus. Nucleic Acids Res. 2017;45:8957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Manica A, Zebec Z, Steinkellner J, et al. Unexpectedly broad target recognition of the CRISPR-mediated virus defence system in the archaeon Sulfolobus solfataricus. Nucleic Acids Res. 2013;41:10509–10517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Rouillon C, Athukoralage JS, Graham S, et al. Control of cyclic oligoadenylate synthesis in a type III CRISPR system. Elife. 2018;7. DOI: 10.7554/eLife.36734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lindås A-C, Karlsson EA, Lindgren MT, et al. A unique cell division machinery in the Archaea. Proc Natl Acad Sci U S A. 2008;105:18942–18946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Samson RY, Obita T, Freund SM, et al. A role for the ESCRT system in cell division in Archaea. Science. 2008;322:1710–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Samson RY, Obita T, Hodgson B, et al. Molecular and structural basis of ESCRT-III recruitment to membranes during archaeal cell division. Mol Cell. 2011;41:186–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Risa GT, Hurtig F, Bray S, et al. Proteasome-mediated protein degradation resets the cell division cycle and triggers ESCRT-III-mediated cytokinesis in an archaeon. Science. 2020;369(6504):eaaz2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Long SW, Faguy DM. Anucleate and titan cell phenotypes caused by insertional inactivation of the structural maintenance of chromosomes (smc) gene in the archaeon Methanococcus voltae. Mol Microbiol. 2004;52:1567–1577. [DOI] [PubMed] [Google Scholar]
- [48].Yang N, Driessen AJM. Deletion of cdvB paralogous genes of Sulfolobus acidocaldarius impairs cell division. Extremophiles. 2014;18:331–339. [DOI] [PubMed] [Google Scholar]
- [49].Liu J, Gao R, Li C, et al. Functional assignment of multiple ESCRT-III homologs in cell division and budding in Sulfolobus islandicus. Mol Microbiol. 2017;105:540–553. [DOI] [PubMed] [Google Scholar]
- [50].Lillestøl RK, Shah SA, Brügger K, et al. CRISPR families of the crenarchaeal genus Sulfolobus: bidirectional transcription and dynamic properties. Mol Microbiol. 2009;72:259–272. [DOI] [PubMed] [Google Scholar]
- [51].Briand J-F, Navarro F, Rematier P, et al. Partners of Rpb8p, a small subunit shared by yeast RNA polymerases I, II, and III. Mol Cell Biol. 2001;21:6056–6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Fouqueau T, Blombach F, Cackett G, et al. The cutting edge of archaeal transcription. Emerg Top Life Sci. 2018;2:517–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Osawa T, Inanaga H, Sato C, et al. Crystal structure of the CRISPR-cas RNA silencing Cmr complex bound to a target analog. Molcel. 2015;58:1–14. [DOI] [PubMed] [Google Scholar]
- [54].Goh S, Boberek JM, Nakashima N, et al. Concurrent growth rate and transcript analyses reveal essential gene stringency in Escherichia coli. PLoS One. 2009;4:e6061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Zhao C, Shu X, Sun B. Construction of a gene knockdown system based on catalytically inactive (“Dead”) Cas9 (dCas9) in staphylococcus aureus. Appl Environ Microbiol. 2017;83. DOI: 10.1128/AEM.00291-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Sokolowski RD, Graham S, White MF. Cas6 specificity and CRISPR RNA loading in a complex CRISPR-Cas system. Nucleic Acids Res. 2014;42(10):6532–6541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Gudbergsdottir S, Deng L, Chen Z, et al. Dynamic properties of the sulfolobus CRISPR/Cas and CRISPR/Cmr systems when challenged with vector-borne viral and plasmid genes and protospacers. Mol Microbiol. 2011;79:35–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Cañez C, Selle K, Goh YJ, et al. Outcomes and characterization of chromosomal self-targeting by native CRISPR-Cas systems in streptococcus thermophilus. FEMS Microbiol Lett. 2019;366. DOI: 10.1093/femsle/fnz105 [DOI] [PubMed] [Google Scholar]
- [59].Jiang W, Maniv I, Arain F, et al. Dealing with the evolutionary downside of CRISPR immunity: bacteria and beneficial plasmids. PLoS Genet. 2013;9:e1003844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Stout EA, Sanozky-Dawes R, Goh YJ, et al. Deletion-based escape of CRISPR-Cas9 targeting in lactobacillus gasseri. Microbiology. 2018;164:1098–1111. [DOI] [PubMed] [Google Scholar]
- [61].Fischer S, Maier LK, Stoll B, et al. An archaeal immune system can detect multiple protospacer adjacent motifs (PAMs) to target invader DNA. J Biol Chem. 2012;287:33351–33365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Erdmann S, Le Moine Bauer S, Garrett RA. Inter-viral conflicts that exploit host CRISPR immune systems of Sulfolobus. Mol Microbiol. 2014;91:900–917. [DOI] [PubMed] [Google Scholar]
- [63].Erdmann S, Garrett RA. Selective and hyperactive uptake of foreign DNA by adaptive immune systems of an archaeon via two distinct mechanisms. Mol Microbiol. 2012;85:1044–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Nuñez JK, Lee ASY, Engelman A, et al. Integrase-mediated spacer acquisition during CRISPR-Cas adaptive immunity. Nature. 2015;519:193–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Wimmer F, Beisel CL. CRISPR-cas systems and the paradox of self-targeting spacers. Front Microbiol. 2020;10:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Lillestøl RK, Redder P, Garrett RA, et al. A putative viral defence mechanism in archaeal cells. Archaea. 2006;2:59–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Deng L, Kenchappa CS, Peng X, et al. Modulation of CRISPR locus transcription by the repeat-binding protein Cbp1 in sulfolobus. Nucleic Acids Res. 2012;40:2470–2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Kenchappa CS, Heidarsson PO, Kragelund BB, et al. Solution properties of the archaeal CRISPR DNA repeat-binding homeodomain protein Cbp2. Nucleic Acids Res. 2013;41:3424–3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Rollie C, Graham S, Rouillon C, et al. Prespacer processing and specific integration in a Type I-A CRISPR system. Nucleic Acids Res. 2018;46:1007–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Rollie C, Schneider S, Brinkmann AS, et al. Intrinsic sequence specificity of the Cas1 integrase directs new spacer acquisition. Elife. 2015;4. DOI: 10.7554/eLife.08716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Cubbon A, Ivancic-Bace I, Bolt EL. CRISPR-Cas immunity, DNA repair and genome stability. Biosci Rep. 2018;38. DOI: 10.1042/BSR20180457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Bernheim A, Bikard D, Touchon M, et al. A matter of background: DNA repair pathways as a possible cause for the sparse distribution of CRISPR-Cas systems in bacteria. Philos Trans R Soc B Biol Sci. 2019;374:20180088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Zhang C, Whitaker RJ. Microhomology-mediated high-throughput gene inactivation strategy for the hyperthermophilic crenarchaeon Sulfolobus islandicus. Appl Environ Microbiol. 2018;84:e02167-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Andersson AF, Lundgren M, Eriksson S, et al. Global analysis of mRNA stability in the archaeon Sulfolobus. Genome Biol. 2006;7:R99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Pan S, Li Q, Deng L, et al. A seed motif for target RNA capture enables efficient immune defence by a type III-B CRISPR-Cas system. RNA Biol. 2019;16(9):1166–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Li Y, Zhang Y, Lin J, et al. Cmr1 enables efficient RNA and DNA interference of a III-B CRISPR-Cas system by binding to target RNA and crRNA. Nucleic Acids Res. 2017;45:11305–11314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Tamulaitis G, Venclovas Č, Siksnys V. Type III CRISPR-cas immunity: major differences brushed Aside1. Tamulaitis, G., Venclovas, Č. & Siksnys, V. Type III CRISPR-cas immunity: major differences brushed aside. Trends Microbiol. 2017;25:49–61. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The genome sequences are published and available under GenBank assembly accession GCA_900079115.1 (S. solfataricus P1) and GCA_000012285.1 (S. acidocaldarius DSM 639). The raw sequencing data of culture PCRs on miniCR vectors and raw quantitative PCR data are available upon request.