

ABSTRACT
Epithelial-to-mesenchymal transition (EMT) is a hallmark of aggressive, mesenchymal-like high-grade serous ovarian carcinoma (HGSOC). The SRC kinase is a key driver of cancer-associated EMT promoting adherens junction (AJ) disassembly by phosphorylation-driven internalization and degradation of AJ proteins. Here, we show that the IGF2 mRNA-binding protein 1 (IGF2BP1) is up-regulated in mesenchymal-like HGSOC and promotes SRC activation by a previously unknown protein-ligand-induced, but RNA-independent mechanism. IGF2BP1-driven invasive growth of ovarian cancer cells essentially relies on the SRC-dependent disassembly of AJs. Concomitantly, IGF2BP1 enhances ERK2 expression in an RNA-binding dependent manner. Together this reveals a post-transcriptional mechanism of interconnected stimulation of SRC/ERK signalling in ovarian cancer cells. The IGF2BP1-SRC/ERK2 axis is targetable by the SRC-inhibitor saracatinib and MEK-inhibitor selumetinib. However, due to IGF2BP1-directed stimulation, only combinatorial treatment effectively overcomes the IGF2BP1-promoted invasive growth in 3D culture conditions as well as intraperitoneal mouse models. In conclusion, we reveal an unexpected role of IGF2BP1 in enhancing SRC/MAPK-driven invasive growth of ovarian cancer cells. This provides a rationale for the therapeutic benefit of combinatorial SRC/MEK inhibition in mesenchymal-like HGSOC.
KEYWORDS: IGF2BP1, invasion, SRC, ERK2, resistance, saracatinib, selumetinib, signalling
Introduction
EOC (epithelial ovarian carcinoma) and particularly its most common form HGSOC (high-grade serous ovarian carcinoma) have the highest death rates among female cancers and improvement of therapeutic efficacies remains dismal [1]. Besides ubiquitous mutations in the tumour suppressor TP53, the mutational landscape of HGSOC is heterogeneous with clonal alterations occurring in a subset of cells during cancer progression [2–4]. HGSOC is referred as a c-class malignancy defined by high DNA copy number variations [5]. This provides a rationale for their sensitivity towards platinum therapies and the potential use of poly(ADP-ribose) polymerase (PARP) inhibitors [1,6,7]. Although promising initially, PARP- and SRC-directed therapies suffer from occurring resistances and the lack of biomarkers required to identify patients benefitting from the respective treatments [8–11].
Unsupervised clustering of patient samples by transcriptome data identified molecular subtypes of HGSOCs with distinct characteristics, including an invasive, mesenchymal-like subtype, observed in approximately 30% of patients [1,6,12,13]. EMT, characterized by the loss of CDH1 (E-cadherin) positive cell-cell contacts termed adherens junctions (AJs), is thought to underly progression of the mesenchymal-like subtype. Recent single-cell sequencing and bulk sequencing deconvolution analyses identified cellular HGSOC subtypes on the basis of non-genetic heterogeneity, demonstrating that HGSOC tumours with a high proportion of cells with EMT signature are associated with a worse prognosis [14]. The SRC protein kinase is a crucial activator of AJ disassembly, that initiates EMT, with elevated activity in EOC patients [15,16]. SRC phosphorylates AJ components like CTNNB1 (β-Catenin), CDH1 or CDH2 (N-cadherin) resulting in their internalization and/or degradation [15,17–19]. In EOC-derived OVCAR-3 cells, SRC promotes EMT, and this activity is further pronounced by ERK2 [20]. ERK2 activates SRF (serum response factor) driving the transcription of ECM molecules and integrins [21]. This was suggested to promote tumour growth, mesenchymal properties and metastasis. Accordingly, small molecule inhibitors of SRC and ERK1/2-activating MEK, in particular saracatinib and selumetinib, are tested individually in clinical trials (ClinicalTrials.gov Identifiers: NCT01196741; NCT00610714; NCT00551070; NCT03162627) [11,22]. In EOC xenograft models, their co-application proved beneficial over single drug therapies, since MEK inhibition re-sensitized saracatinib resistant cells [8,16]. These findings provide preclinical evidence suggesting that the combination of SRC and MEK inhibitors is beneficial in the treatment of ovarian cancer and highlights the need for identifying biomarkers for selecting patients for clinical trials [8].
IGF2BP1 is an oncofetal RNA-binding protein (RBP) up-regulated in a variety of solid tumours [23]. In cancer, IGF2BP1’s main function is the partially m6A-dependent (N [6]-methyladenosine) impairment of miRNA-directed mRNA decay by binding to target transcripts via its four KH (hnRNPK homology) domains [21,24–31]. This results in elevated expression of oncogenic factors like LIN28B, HMGA2, MYC, SRF, E2F1 and ERK2 [21,24,25,32,33]. During development, the spatially restricted synthesis of ACTB is essentially controlled by the SRC-directed phosphorylation of IGF2BP1 [34]. SRC associates with IGF2BP1 at an SH3-binding VP4SS motif conserved in the SRC substrate and cell adhesion protein Paxillin (PXN) [35].
IGF2BP1 is up-regulated in EOC and associated with adverse patient outcome [25,36]. Its depletion or deletion decreases the spheroid growth, migration and invasion of ovarian cancer cells in vitro and reduces tumour growth as well as metastasis in vivo [24,25,37]. In melanoma-derived cell lines, IGF2BP1 enforces mesenchymal tumour cell properties in a LEF1-dependent manner [38]. However, if and how IGF2BP1 modulates EMT in EOC remained elusive.
Here we reveal a novel, RNA-independent mechanism of IGF2BP1-dependent SRC activation. In concert with enhancing ERK2 expression, IGF2BP1 promotes SRC/ERK2-signalling in EOC-derived cells providing a rationale for the therapeutic inhibition of SRC and MEK in mesenchymal-like HGSOC.
Results
IGF2BP1 is a pro-mesenchymal driver up-regulated in the C5 subtype of HGSOC
Consistent with previous findings, IGF2BP1 expression is associated with adverse prognosis in EOC tumours [24,25,36] (Supplementary Fig. 1A, B). Correlation analyses in three independent transcriptome datasets indicated that IGF2BP1 mRNA expression is strongly associated with the C5 subtype of HGSOC (Fig. 1A; Supplementary Fig. S1C–F). In agreement, IHC (immunohistochemistry) revealed a higher Remmele score for IGF2BP1 in C5 tumours derived from a local tumour cohort (Fig. 1B, C; Supplementary Table T1B). To identify candidate effector pathways of IGF2BP1 in EOC, the TCGA-provided transcriptome data set was separated in IGF2BP1 low (<5 cpm) and high (>5 cpm) expressing tumours. Median IGF2BP1 mRNA expression was more than 25-fold up-regulated in one-third of patients (Fig. 1D; Supplementary Table T1A). Gene set enrichment analyses (GSEA) using the fold change of gene expression identified significant up-regulation of proliferation- and EMT-associated gene sets in IGF2BP1-high vs. low tumours (Fig. 1E; Supplementary Fig. S1G and Tables T1A-3). In agreement with a pro-mesenchymal role of IGF2BP1, the protein was markedly elevated in a subset of mesenchymal-like EOC-derived cell lines (Fig. 1F, G). These were characterized by high abundance of mesenchymal markers, VIM, CDH2 and ZEB1, and low levels of epithelial markers, CDH1, KRT8 and EPCAM (Fig. 1F, G; Supplementary Table T4). Immunostaining of CTNNB1 and F-actin labelling confirmed a pronounced mesenchymal-like morphology of ES-2 cells with diminished CTNNB1-positive cell-cell contacts when compared to epithelial-like OVCAR-3 cells (Fig. 1H, I). To test if IGF2BP1 promotes a mesenchymal-like phenotype in EOC-derived cells, the protein was depleted or over-expressed in a panel of EOC-derived cells. IGF2BP1 depletion reduced 3D spheroid growth and invasion in all EOC cell lines tested (Fig. 1J, K; blue boxes). Strikingly, the forced expression of GFP-fused IGF2BP1 significantly elevated the invasive growth of OVCAR-3 and ES-2 cells (Fig. 1J, K; red boxes). In sum, this indicated that IGF2BP1 is a marker of the C5 subtype of HGSOC, promoting invasive growth in EOC-derived cell models.
Figure 1.
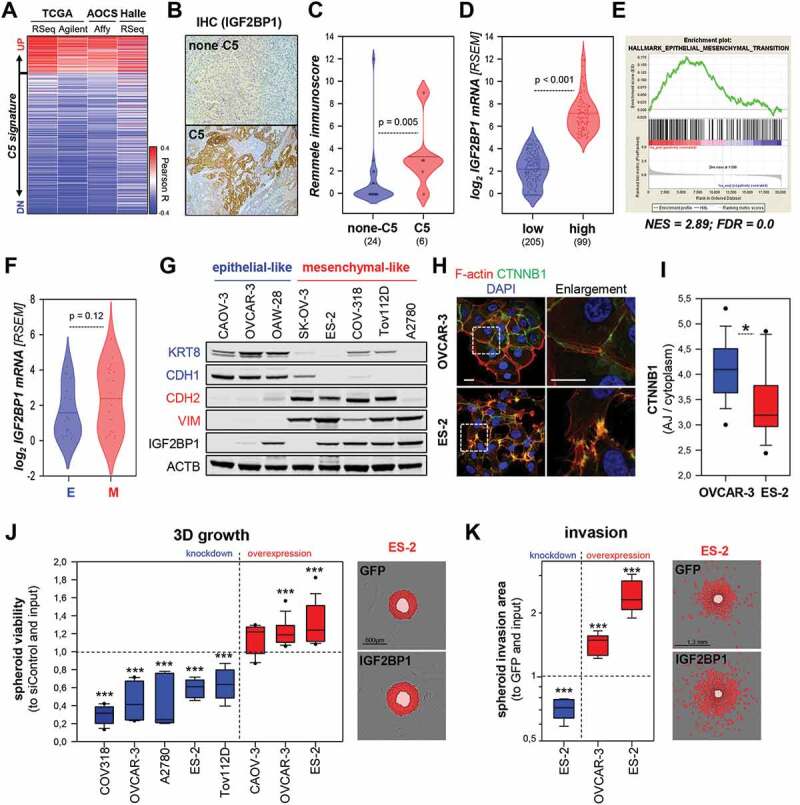
IGF2BP1 is associated to the C5 signature and promotes mesenchymal properties. (A) IGF2BP1 expression was correlated to the C5 signature using indicated data sets. Pearson correlation coefficients (R) are shown as heat map. (B, C) IHC staining of EOC samples classified as C5 or none-C5 via NGS based GSEA analyses using IGF2BP1-directed antibodies (B). IGF2BP1 staining was quantified using the Remmele immune score in none-C5 (24) and C5 (6) samples (C). (D) IGF2BP1 mRNA expression is shown by violin plots, in the TCGA-OV-RNA-Seq cohort distinguished in IGF2BP1-high (log2 RSEM ≥ 5) or -low (log2 RSEM ≤ 5). (E) GSEA plot of the HALLMARK_EMT gene set based on gene ranking by fold change expression between IGF2BP1-high vs -low samples as in (D). (F) Violin plot of IGF2BP1 mRNA expression in EOC-derived cells, classified as epithelial-like (E, blue) or mesenchymal-like (M, red) by the differential mRNA expression (Cancer Cell Line Encyclopaedia; CCLE expression data) of epithelial (CDH1, KRT8 and/or EPCAM) and mesenchymal (VIM, ZEB1 and/or CDH2) markers. (G) Western blotting of IGF2BP1 and indicated epithelial (blue) and mesenchymal (red) markers. ACTB (beta-actin) served as loading control. (H, I) Immunostaining of CTNNB1 and F-actin using Phalloidin-TRITC in indicated cell lines. Dashed boxes depict enlarged regions. Scale bar 25 µm. The ratio of CTNNB1 membrane to cytoplasmic localization was determined in 15 cells over three independent analyses. (J) 3D growth of indicated cell lines upon IGF2BP1 knockdown (blue) or over-expression (red), as determined by Cell Titre Glo relative to controls and input. Representative images of ES-2 cells expressing GFP or GFP-IGF2BP1 overlaid with growth detection masks (red) and input spheroids (light red) are shown. (K) Invasion of pre-formed Matrigel embedded spheroids of ES-2 cells upon IGF2BP1 knockdown (blue) or over-expression (red) was monitored in an Incucyte S3. Representative images overlaid with invasion detection masks (red) or input spheroids (light red) are shown. Box plots in J and K are derived from analyses of 6–9 spheroids in three independent experiments. Statistical significance was determined by Student’s T-test or Mann-Whitney rank-sum test. *, p < 0.05; ***, p < 0.001
IGF2BP1 promotes the disassembly of adherens junctions
How aberrant IGF2BP1 expression modulates cell morphology was investigated in epithelial-like OVCAR-3 and mesenchymal-like ES-2 cells. IGF2BP1 over-expression in OVCAR-3 enhanced the formation of lamellipodia and led to partial disassembly of cell colonies (Fig. 2A, arrowheads). IGF2BP1 depletion in ES-2 cells reduced spindle-like morphologies in favour of a compact, flattened shape and enhanced the formation of colonies, as previously observed [38]. This implied IGF2BP1-dependent regulation of cell-cell contacts, in particular the modulation of AJ integrity, analysed further by CDH1 and CTNNB1 immunostaining. IGF2BP1 overexpression in OVCAR-3 cells converted mature AJs to premature, zipper-like AJs (Fig. 2B). Quantification of AJ/cytoplasm fluorescence intensities confirmed decreased AJ localization of CDH1 and CTNNB1, suggesting enhanced internalization and/or decay due to IGF2BP1 overexpression. In agreement, CTNNB1 protein levels and CTNNB1-mediated transcription determined by transcription reporters are modestly reduced upon IGF2BP1 overexpression in OVCAR-3 cells (Supplementary Fig. S2A, B).
Figure 2.
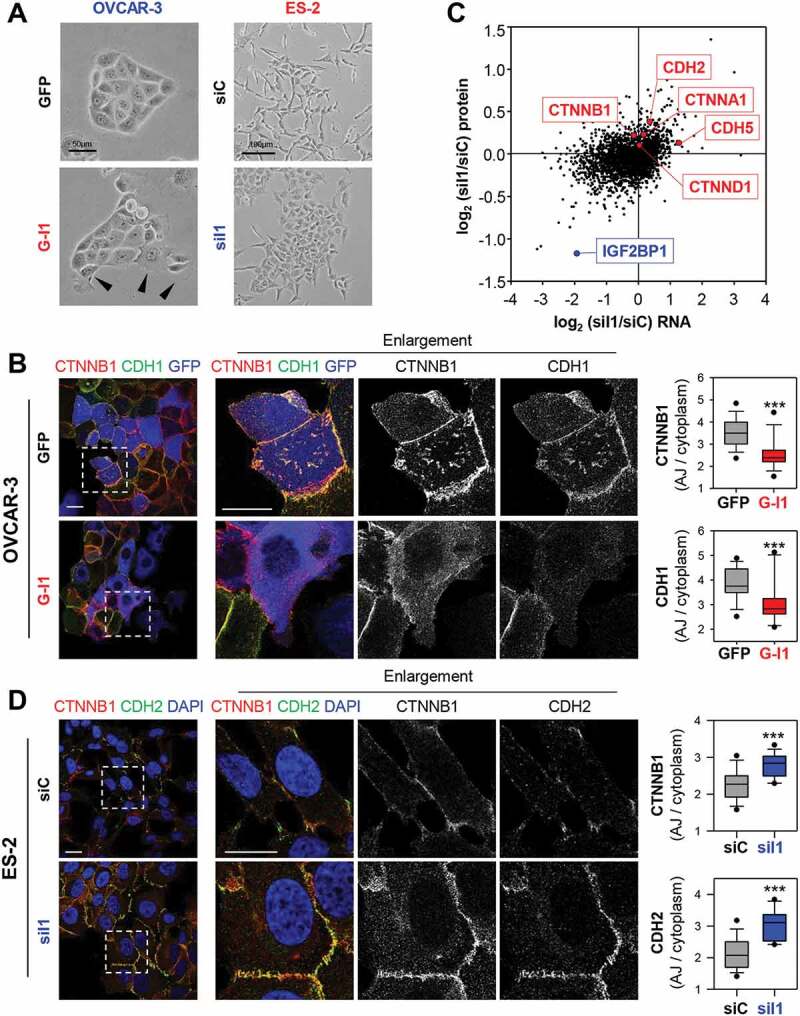
IGF2BP1 promotes AJ disassembly. (A) Phase-contrast images of OVCAR-3 and ES-2 cells upon GFP or GFP-IGF2BP1 (G-I1) over-expression and IGF2BP1 (siI1) or control depletion (siC). Arrowheads indicate colony-detaching cells. (B) Immunostaining of IGF2BP1 over-expressing or control OVCAR-3 cells with indicated antibodies. Over-expression is pseudo-coloured in blue. Dashed boxes depict enlarged region shown in middle panels. The localization of CDH2 and CTNNB1 was assessed by immunostaining. The membrane/cytoplasmic ratio of fluorescence intensities is shown by box plots (right panels). (C) Log2 fold change of protein expression (quantitative proteomics) upon IGF2BP1 depletion in ES-2 cells plotted over the respective log2 fold changes in RNA abundance (NGS). Selected AJ proteins are indicated in red, IGF2BP1 is highlighted in blue. (D) ES-2 cells with transient IGF2BP1 depletion were analysed as in (B). Scale bars 25 µm. 15 cells per condition were analysed in three independent experiments. Statistical significance was tested by student’s T-test. ***, p < 0.001
In ES-2 cells, the depletion of IGF2BP1 induced severe morphological changes reminiscent of a partial mesenchymal-to-epithelial (MET) transition (Fig. 2A, D). This was indicated by markedly increased CDH2 and CTNNB1 AJ-recruitment, although CDH1 expression was not restored (Fig. 2D). Furthermore, IGF2BP1 knockdown enforced reorganization of the intermediate as well as the microfilament system and induced the formation of premature AJs (Supplementary Fig. S2C-E). These even showed increased recruitment of the AJ-stabilizing factor CTNND1 (δ-Catenin) (Supplementary Fig. S2D; [39]). MET-like transition, indicated by enriched AJ-localization of CDH2 and CTNNB1 was conserved in a panel of EOC-derived cells and cell lines of other origin depleted for IGF2BP1 (Supplementary Fig. S2F, G). The deletion of IGF2BP1 in ES-2 cells led to drastic MET-like morphological changes culminating in the formation of mature AJs (Supplementary Fig. S3A). On the contrary, IGF2BP1 overexpression further reduced the AJ-localization of CTNNB1 and CDH2 in ES-2 and COV-318 cells (Supplementary Fig. S3B, C). This re-localization was essentially abolished by depleting exogenous IGF2BP1 in ES-2 (Supplementary Fig. S3B).
How IGF2BP1 influences the expression of AJ proteins, and was analysed further by monitoring mRNA and protein abundance by mRNA-sequencing and quantitative MS analyses upon IGF2BP1 depletion in ES-2 cells (Fig. 2C; Supplementary Table T5). This was associated with up-regulation of AJ proteins, CDH2, CDH5, CTNNA1 and CTNNB1. Together these observations supported the notion that IGF2BP1 up-regulation is a driver of AJ disassembly and EMT in EOC cells.
IGF2BP1 regulates SRC activity
IGF2BP1 is a key regulator of mRNA turnover in cancer cells, suggested to control CTNNB1 mRNA turnover in breast cancer cells [21,25,26,40]. However, with the exception of CDH5, steady-state mRNA levels or mRNA turnover of AJ components including CTNNB1 remained unchanged upon IGF2BP1 depletion (Fig. 2C; Supplementary Fig. S4A). Moreover, the activity of CDH2- and CTNNB1-3ʹUTR luciferase reporters remained unaffected by IGF2BP1 knockdown (Supplementary Fig. S4B). This largely excluded 3ʹUTR-dependent regulation by IGF2BP1, as previously reported for ACTB and MAPK4 mRNAs [34,37]. Instead, our findings implied that elevated AJ formation upon IGF2BP1 depletion involves altered turnover of AJ proteins [39,41]. In agreement, the decay of CDH2 and CTNNB1 proteins was significantly reduced by IGF2BP1 knockdown (Fig. 3A, B).
Figure 3.
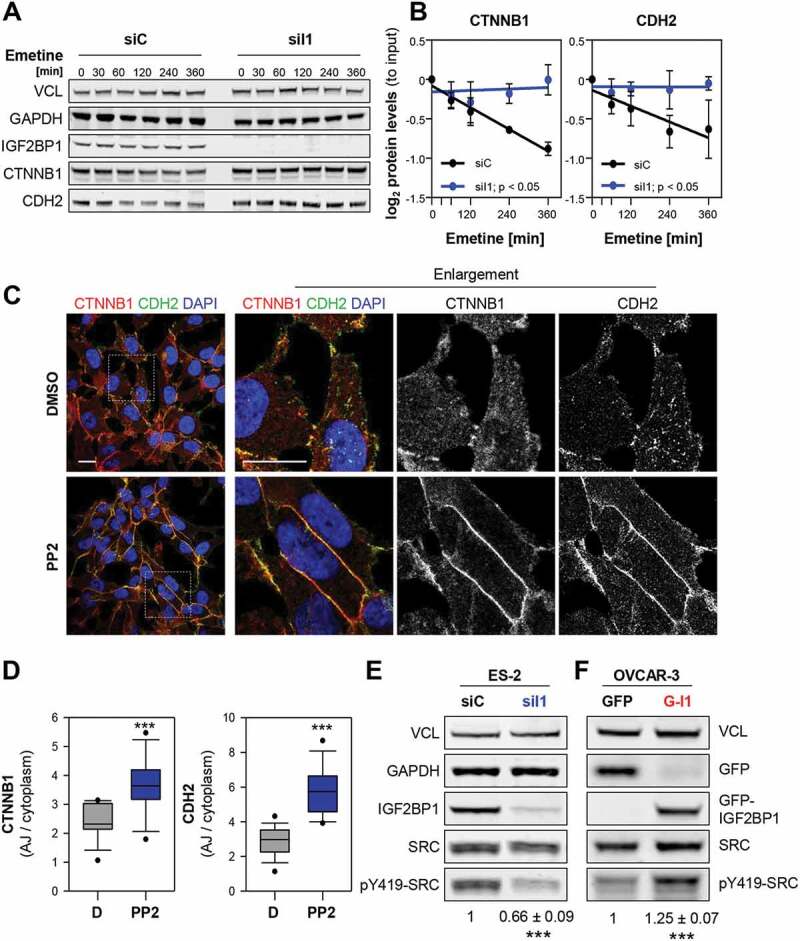
IGF2BP1 destabilizes AJs in a SRC-dependent manner. (A, B) Turnover of indicated proteins was determined by blocking protein synthesis with Emetine (100 µM) by Western blotting upon IGF2BP1 (siI1) and control (siC) knockdown. GAPDH and VCL served as loading controls to determine protein abundance relative to input levels. (C,D) Immunostainings of ES-2 cells treated with PP2 (1 µM) or DMSO for 48 h using indicated antibodies (C). Dashed box indicates enlargement. Scale bars 25 µm. The AJ/cytoplasmic ratio of CDH2 and CTNNB1 is shown by box plots (D). (E, F) Western blot analyses of indicated proteins in ES-2 cells upon IGF2BP1 depletion (E) and OVCAR-3 cells (F) with GFP or GFP-IGF2BP1 (G-I1) over-expression. SRC activity, indicated by Y419 phosphorylation, was normalized to SRC expression, as depicted under the lanes. VCL served as loading control. Error bars show SD determined in three independent experiments. Statistical significance was tested by Student’s T-test. *, p < 0.05; ***, p < 0.001
The phosphorylation of some AJ proteins, e.g. CDH2, by SRC kinase promotes AJ disassembly and subsequently protein decay [15,17–19]. Consistently, AJ localization and total abundance of CTNNB1 and CDH2 were elevated upon treatment of ES-2 cells with the SRCi PP2 (Fig. 3C,D; Supplementary Fig. S4C). In view of IGF2BP1’s association with the SH3 domain of SRC [34], it appeared tempting to speculate that IGF2BP1 influences SRC activity. Western blotting revealed that SRC activity, indicated by phosphorylation at tyrosine 419 (Y419) in the kinase domain, was substantially reduced upon IGF2BP1 depletion in all EOC-derived cell lines tested (Fig. 3E; Supplementary Fig. S4D). Conversely, increased Y419-SRC phosphorylation was observed upon the overexpression of IGF2BP1 in OVCAR-3 cells (Fig. 3F). SRC expression remained essentially unchanged by aberrant IGF2BP1 expression. In sum, these findings suggested that IGF2BP1-directed activation of SRC promotes AJ disassembly in EOC-derived cells.
IGF2BP1 promotes SRC activation via a SH3-ligand-binding mechanism
Previous studies identified IGF2BP1 as an SRC substrate and suggested association of the kinase’s SH3 domain at a VP4SS motif in IGF2BP1 [34]. This putative SRC-binding site is highly conserved among IGF2BP1 orthologues and identical to the SRC-docking site of the adhesion molecule Paxillin (Fig. 4A) [34,35,42]. SRC activity is repressed by an intramolecular interaction (closed conformation) [43]. Activation, indicated by an open conformation enabling auto-phosphorylation at pY419 in the kinase domain, is controlled by various means including ligand-binding induced mechanisms [44]. The latter were described for some SRC substrates including Paxillin and the RNA-binding proteins hnRNPK and SAM68 [42,45,46]. To test if IGF2BP1 promotes SRC activity via its VP4SS motif, increasing amounts of GFP-tagged proteins were expressed in ES-2 cells and SRC activity was monitored by Western blotting for pY419-SRC (Fig. 4B, C). This revealed dose-dependent up-regulation of pY419-SRC by wild type (WT) and RNA-binding deficient (KHmut) full length IGF2BP1 containing mutations in the GXXG loops of all four KH domains (Fig. 4A–C) [25,30]. In contrast, SRC activity remained unchanged upon deletion of the VP4SS motif in IGF2BP1 (∆SH3). Although less efficient, SRC-activity was also enhanced by over-expression of the GFP-fused VP4SS peptide (SH3motif). In sum, this unravelled that IGF2BP1 promoted SRC activity in a dose-dependent manner by a ligand-binding induced, but largely RNA-binding independent mechanism.
Figure 4.
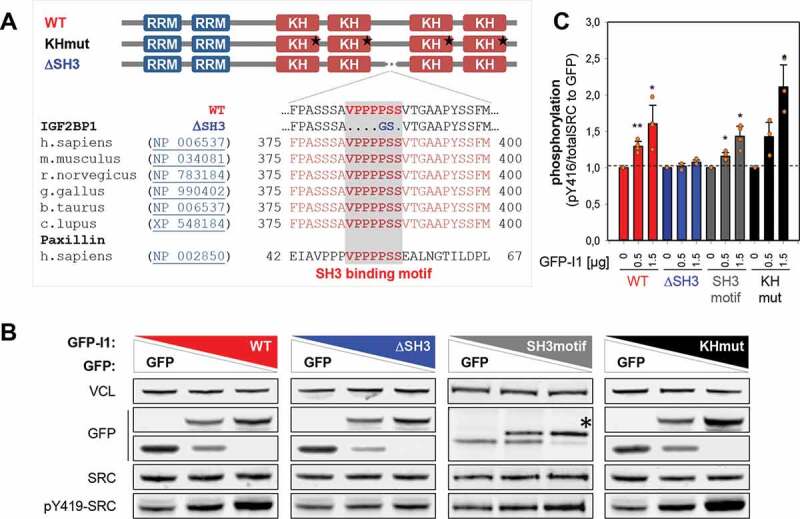
IGF2BP1 promotes SRC activation via its SH3-binding VP4SS motif. (A) Schematic representation of VP4SS conservation among IGF2BP1 orthologs and human paxillin. The domain organization of IGF2BP1 wild type and mutants is shown in the upper panel (RRM, RNA recognition motif; KH, hnRNPK homology domain). Stars depict mutations of the GXXG loop inhibiting RNA binding [30]. (B) Representative Western blot analyses of indicated proteins in ES-2 cells transiently expressing GFP alone (lane 1) a mixture of GFP and indicated GFP-fused proteins (lane 2) or only GFP-fused proteins. WT, GFP-IGF2BP1WT; ∆SH3, GFP-IGF2BP1∆SH3; SH3motif, GFP-VP4SS; KHmut, GFP-IGF2BP1KHmut. VCL served as loading control. (C) Quantification of Western blots shown in (B). Error bars indicate SD determined in three independent experiments. Statistical significance was tested by Student’s T-test. *, p < 0.05; **, p < 0.01
The IGF2BP1-SRC axis enhances EMT and invasive growth of EOC-derived cells
The physiological relevance of IGF2BP1-dependent control of SRC activity was analysed by monitoring AJ integrity using immunostaining (Fig. 5A, B). IGF2BP1 over-expression in OVCAR-3 or re-expression in IGF2BP1-deleted ES-2 cells impaired the AJ localization of CTNNB1, CDH1 or CDH2 compared to GFP controls. Similar to wild type IGF2BP1, the RNA-binding deficient (KHmut) protein substantially impaired AJ assembly. In contrast, AJ localization of the respective proteins was essentially unchanged by over- or re-expression of IGF2BP1 lacking the VP4SS motif (∆SH3).
Figure 5.
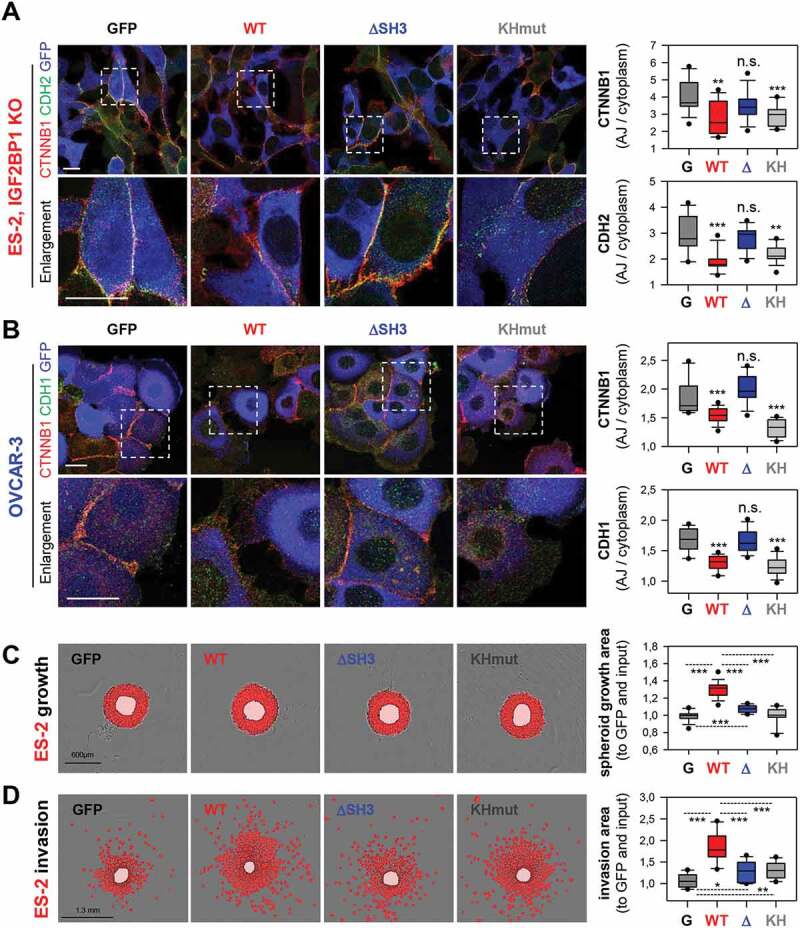
IGF2BP1 promotes AJ disassembly VP4SS-dependent. (A, B) Immunostaining of CTNNB1 and CDH2 or CDH1 upon the over-expression of GFP or indicated IGF2BP1 proteins (pseudo-coloured in blue) in IGF2BP1-deleted ES-2 (A) or parental OVCAR-3 cells (B) were performed and analysed as in Fig. H, I. Dashed boxes indicate enlargements. Scale bars 25 µm. (C,D) Spheroid growth (C) or invasion (D) of ES-2 cells stably expressing GFP or indicated IGF2BP1 proteins were performed and analysed as in Fig. 1J, K. Statistical significance was tested by Student’s T-test or Mann-Whitney rank-sum test. *, p < 0.05; **, p < 0.01; ***, p < 0.001
However, only wild type IGF2BP1 substantially promoted both, 3D spheroid growth and invasion of ES-2 cells (Fig. 5C, D). In contrast, invasive growth was only modestly elevated by ∆SH3 and KH mutant proteins. To investigate the role of IGF2BP1-dependent SRC-activation in invasive growth, the kinase was inhibited by PP2 in ES-2 cells overexpressing IGF2BP1 (Supplementary Fig. S4E). This essentially abolished IGF2BP1-directed invasion. Collectively, these findings indicated a pivotal role of IGF2BP1-directed SRC activation in AJ integrity and invasive growth of EOC-derived cells. However, full potential in promoting invasive growth requires both, the RNA-independent activation of SRC and presumably the RNA-dependent regulation of mRNA fate by IGF2BP1.
Combined SRCi/MEKi effectively impairs IGF2BP1-driven invasive growth
The SRC inhibitor saracatinib showed anti-tumour activity in HGSOC preclinical studies [47]. However, activation of the MAPK pathway is associated with resistance to saracatinib. MEK inhibition by selumetinib proved effective in re-sensitizing saracatinib resistant cells [8]. Co-activation of SRC and MAPK was reported in HGSOC patients and combined SRCi/MEKi impaired tumour growth in vivo more effectively than monotherapies [16]. We recently demonstrated that IGF2BP1 promotes ERK2 (MAPK1) and SRF expression by impairing miRNA-directed repression [21,25]. Consequently, SRF-dependent MAPK signalling was reduced upon IGF2BP1 depletion [21]. Analyses in a panel of EOC-derived cells confirmed, IGF2BP1 is a conserved enhancer of ERK2 expression and MAPK signalling (Supplementary Fig. S5A–C).
Currently, no clinically evaluated IGF2BP1 inhibitor is available. The small-molecule inhibitor BTYNB impairs tumour cell growth and IGF2BP1-RNA binding in vitro [48]. However, it appeared unlikely to inhibit the RNA-binding independent control of SRC activity by IGF2BP1. Hence, we analysed if saracatinib and/or selumetinib impair IGF2BP1-directed control of invasive growth.
To this end, we determined the EC50 concentrations for both inhibitors in the 3D growth and invasion of ES-2 cells over-expressing GFP or GFP-IGF2BP1. Irrespective of IGF2BP1 over-expression, selumetinib repressed 3D growth and saracatinib invasion at nearly equal potency (Supplementary Fig. S5D, E). Conversely, IGF2BP1 over-expression induced a 60% higher tolerance towards saracatinib under 3D growth conditions and four-fold higher invasion upon selumetinib treatment (Fig. 6A, B). In support of this, EOC-derived cell lines were sensitized to growth inhibition by saracatinib (Sa; approximated EC50 concentration of 3 µM) upon IGF2BP1 depletion and became more resistant by IGF2BP1 over-expression (Fig. 6C). The inhibition of invasion by selumetinib (Se; approximated Imax concentration of 3 µM) was elevated by IGF2BP1 depletion and decreased by its overexpression (Fig. 6D).
Figure 6.
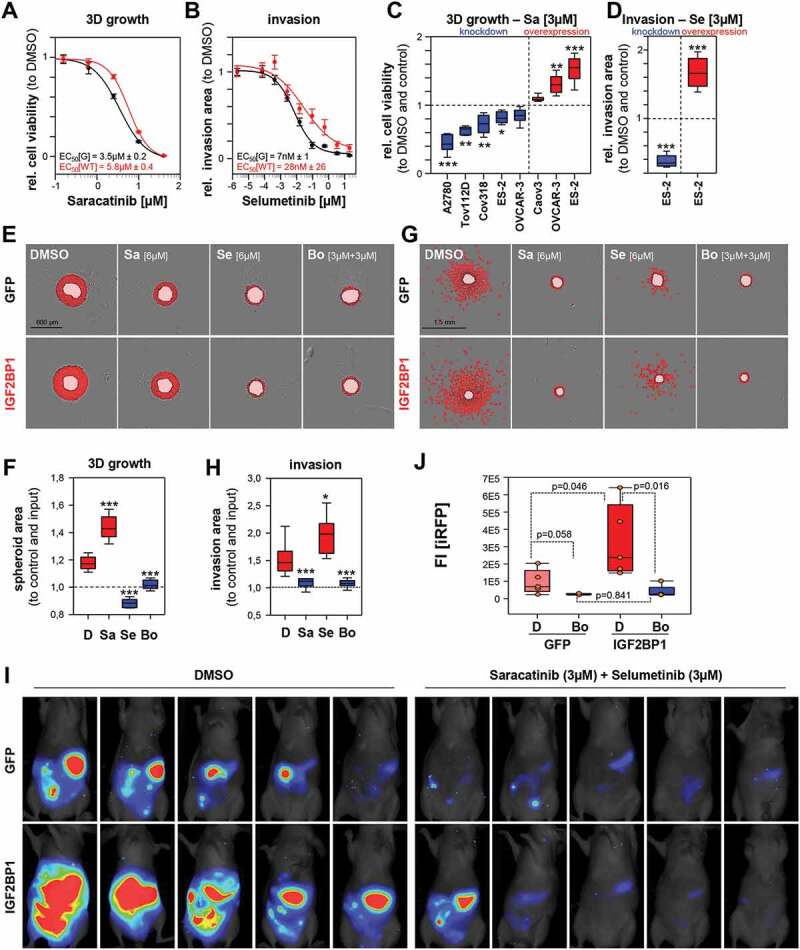
IGF2BP1-promoted invasive growth is abolished by combined SRCi/MEKi treatment. (A–H) Spheroid growth (A, C, E, F) or invasion (B, D, G, H) of GFP (controls) or IGF2BP1 over-expressing ES-2 cells was monitored and analysed as in Fig. (1J, K). All treatments at indicated concentrations were performed on pre-formed (24 h) spheroids for indicated time and concentrations. Error bars indicate SE (A, B). (I, J) iRFP-labelled GFP or IGF2BP1 over-expressing ES-2 cells pre-treated with saracatinib (3 µM) and selumetinib (3 µM) or DMSO for 48 h, were IP injected into nude mice with compounds at indicated concentrations. Tumour growth of five mice per conditions (I) was monitored and quantified (J) by infra-red imaging of the abdominal region 2 weeks post injection. Statistical significance was tested by Student’s T-test or Mann-Whitney rank-sum test. *, p < 0.05; **, p < 0.01; ***, p < 0.001
To address if both inhibitors act in a synergistic or additive manner, combinatorial treatment was performed using a 2D growth drug matrix analysis (Supplementary Fig. S6A, B). This revealed additivity, indicated by synergy scores in-between −10 and 10, determined by three independent synergy models [49]. The maximal additive concentrations for IGF2BP1-expressing were approximately 6 µM for both inhibitors. According to HSA model assumptions [49], the potential benefit of additive inhibition in 3D growth and invasion was analysed at 3 µM per drug in combined and 6 µM in mono-treatments. Western blotting confirmed the inhibition of both kinases by the respective inhibitors at these concentrations (Supplementary Fig. S6C).
However, even at 6 µM single saracatinib treatment remained substantially less efficient than selumetinib or combined treatment in 3D growth analyses (Fig. 6E, F; Supplementary Fig. S6D). The same was observed for selumetinib in 3D invasion studies (Fig. 6G, H; Supplementary Fig. S6E). Thus, only combined treatment was effective in inhibiting both, 3D growth and invasion irrespective of IGF2BP1 overexpression.
Invasive growth in the peritoneum is a major complication in the progression of ovarian cancer. If combined treatment also impairs invasive growth in vivo was analysed by intraperitoneal (IP) injection of iRFP-labelled (near-infrared red fluorescent protein) ES-2 cells expressing GFP or GFP-IGF2BP1. Cells were pre-treated with both inhibitors (3 µM each) and DMSO for 48 h and injected in the presence of both inhibitors at the same concentrations. Tumour growth was monitored by total iRFP signal in the peritoneum 2 weeks post injection of viable tumour cells (Fig. 6I,J). IGF2BP1 over-expression led to higher tumour burden in DMSO treated controls, supporting the growth- and invasion-enhancing role of IGF2BP1 observed in cellulo and previously reported [25]. Irrespective of IGF2BP1 expression, tumour growth was essentially abolished by combined treatment.
Together these observations indicated that combined treatment with selumetinib and saracatinib effectively impairs the IGF2BP1-dependent enhancement of invasive growth in EOC-derived cells in vitro and in vivo.
Discussion
Enhanced expression of IGF2BP1 is associated with a mesenchymal/proliferative-like gene signature observed in approximately 30% of serous ovarian cancers [6,12,13]. This identifies IGF2BP1 as a novel marker of the mesenchymal-like C5 subtype of HGSOCs [12]. If IGF2BP1 is also a marker for non-genetic heterogeneity associated with a cellular subtype, will need to be addressed. GSEA analyses (Fig. 1E and Supplementary Fig. 1G) and functional studies suggest a putative connection to the secretory cell signatures ‘Cell Cycle’ and ‘EMT’ [14]. Moreover, IGF2BP1 was recently shown to modulate the cell cycle in an E2F1-dependent manner [32]. In EOC-derived cells, IGF2BP1 promotes invasive growth by stimulating SRC/ERK-signalling. The protein enhances ERK2 abundance by stabilizing the respective mRNA, as previously reported [25]. In contrast, SRC-activation by IGF2BP1 is RNA-independent and solely relies on the conserved SRC/SH3-binding motif of IGF2BP1. This suggests that IGF2BP1 promotes SRC kinase activation by a ligand-binding-induced mechanism prior to unknown for IGF2BP1. This is highly important for perspective drug-design approaches currently addressing IGF2BP1’s RNA-binding activity only [48]. However, full potential of IGF2BP1 enhanced invasive growth requires both, protein-dependent activation of SRC and RNA-binding associated regulation.
The activation of SRC is crucial for the pro-invasive role of IGF2BP1 since it promotes EMT by inducing the disassembly of AJs. This is abolished by ablating SRC activity using pharmacological inhibitors. This reveals a fundamental novel role of IGF2BP1, the interconnection of deregulated gene expression and cancer cell signalling. Consistent with this dual function promoting ERK/SRC signalling, the combined inhibition of MAPK- and SRC-signalling by selumetinib and saracatinib proves substantially more effective than monotherapies. This was validated IGF2BP1-dependent for the impairment of 3D growth and invasion in cellulo and the peritoneal spread of tumour cells in experimental mouse tumour models. In support of this, previous studies report a substantial therapeutic benefit of combined inhibition of MAPK/SRC signalling in EOC tumour models [8,16]. Thus, our findings suggest IGF2BP1 as a novel marker for EOC therapy. Considering the major problem of invasive growth associated with ovarian cancer spread in the abdominal cavity, the further investigation of IGF2BP1-driven cancer progression, in particular its roles in the mesenchymal subtype of HGSOCs, appears mandatory. Along these lines, the direct targeting of IGF2BP1-dependent functions, for instance by impairing its RNA-binding by small molecules like BTYNB, needs to be evaluated.
Materials and methods
Patient samples and IHC
Human HGSOC samples, collected from patients undergoing surgery, were prepared by the pathologist and immediately frozen (liquid nitrogen). HE staining confirmed tumour the content of each sample. The study, including IHC and NGS analyses, was approved by the Ethics Committee of the Medical Faculty/Martin Luther University Halle/Wittenberg and written informed consent. IHC staining of FFPE samples was performed as previously described [33], using a Tris/EDTA buffer (pH 9) for antigen retrieval, a commercial IGF2BP1-directed antibody (MBL, Woburn, MA; Cat#. RN001M; RRID: AB_1953026; dilution 1:100), DAB Enhancer (Dako/Agilent, Santa Clara, CA; Cat# S1961) for detection and counterstaining by hemalm. IGF2BP1 staining was determined blinded by two pathologists using the immunoreactive score as described [50]: 0 – absent; 1-4 – weak; 5-8 – moderate; 9-12 – strong expression. A summary of samples, IGF2BP1 expression values and Remmele scores is provided as Supplementary Table T1B.
Animal handling and ethics approvals
Immunodeficient athymic nude mice (FOXN1nu/nu) were obtained from Charles River (Wilmington, MA). Animals were handled according to the guidelines of the Martin Luther University based on ARRIVE guidelines. Permission was granted by the County Administration Office for Animal Care Saxony-Anhalt. iRFP-labelled IGF2BP1 over-expressing or GFP control ES-2 cells (7.5 x 104 living cells determined by Trypan Blue counting) pre-treated with 3 µM saracatinib and 3 µM selumetinib or DMSO for 48 h were injected intraperitoneal (IP) into six-week-old female nude mice (five mice per condition) together with these inhibitors at the same concentration. Tumour growth of isoflurane-anaesthetized mice was monitored weekly by near-infrared imaging using a Pearl imager (LI-COR, Lincoln, NE) and quantified using the Image Studio software (LI-COR). Mice were sacrificed after 2 weeks, as IGF2BP1 over-expressing tumours reached termination criteria.
NGS and quantitative proteomics
NGS analyses of patient samples and cells were performed as previously described [25]. Total RNA-Seq data were deposited at NCBI GEO: (GSE147980) for EOC cohort or (GSE109605) for IGF2BP1 depletion in ES-2 cells. Quantitative proteomics were performed as described before [51]. In brief, TMT labelling of trypsin-digested proteins was performed according to the manufacturer’s instructions (TMT-10plex kit, Thermo Fisher, Waltham, MA). Equal volumes of all samples were mixed, concentrated in a vacuum concentrator and acidified with TFA for LC/MS/MS analysis. Samples were separated by nano-HPLC (Ultimate RSLC 3000, Thermo Fisher) using reversed phase C18 columns and 420-min gradients. The eluate was directly introduced into an Orbitrap Fusion Tribrid mass spectrometer (Thermo Fisher) equipped with nano-ESI source. Samples were analysed using a collision-induced dissociation (combined ion trap-CID/high resolution-HCD) MS/MS strategy for peptide identification and reporter ion quantification. MS/MS data were searched against the NCBI database (version 140412, taxonomy homo sapiens, 89,649 entries) using the Proteome Discoverer (version 2.0, Thermo Fisher). Quantification was performed based on the reporter ion ratios derived from high-resolution MS/MS spectra. Proteomics data are shown as Supplementary Table T5.
Public data, GSEA and databases
TCGA-OV-RNA-Seq data of the ovarian cancer cohort were obtained from the GDC portal (https://portal.gdc.cancer.gov/). NGS data of ovarian cancer cell lines were downloaded from CCLE (Cancer Cell Line Encyclopaedia) (https://portals.broadinstitute.org/ccle). Gene set enrichment analyses (GSEA; Supplementary Table T3) were performed as described before using the fold change expression of all genes between IGF2BP1 high and low expressing tumours (Supplementary Table T2) for gene ranking (18). Kaplan Meier plots for progression-free survival (A) or overall survival (B) were generated with www.kmplot.com [52] using the microarray-based cohorts for ovarian cancer including the Australian data set by Tothill et al. [12]. Synergy between saracatinib and selumetinib was tested using https://synergyfinder.fimm.fi/based on a drug matrix screen for various combinations and concentrations according to the online instructions [49].
Cell lines, culture conditions and transfections
Ovarian cancer cell lines (Supplementary Table T6) and HEK293T cells for production of lentiviral particles were cultured in DMEM (Thermo Fisher) supplemented with 10% foetal bovine serum (FBS; Biochrom/Merck, Schaffhausen, Switzerland) at 37°C and 5% CO2. The most frequently used ES-2 cell line was authenticated by Eurofins Genomics using AmpFlSTR® Identifiler® Plus PCR Amplification Kit (Thermo Fisher) for STR profiling. Mycoplasma testing was routinely performed every 2 months by PCR and DAPI staining. Reverse transfections of plasmids (Supplementary Table T8) or siRNA pools (15 nM; Supplementary Table T7) were carried out using Lipofectamine 2000 or Lipofectamine RNAiMax (Thermo Fisher), respectively, according to the manufacturer’s instructions. Lentiviral particles were produced as previously described [38].
The following inhibitors were added in indicated concentrations and time: Saracatinib (Selleckchem, Houston, TX; Cat#S1006), Selumetinib (Selleckchem; Cat#S1008) and PP2 (Sigma Aldrich, St. Louis, MO; Cat#P0042). Protein turnover was analysed by addition of Emetine (100 µM, Sigma Aldrich, Cat# E2375) and RNA decay by addition of Actinomycin D (5 µM, Sigma Aldrich, Cat# A9415) for indicated time points 72 h post siRNA transfections. Protein and mRNA abundances were analysed by Western blotting or RT-qPCR as described in the Supplementary Information. For antibodies and primers see Supplementary Table T7 and T8.
Immunostainings
Immunostainings were essentially performed as previously described [37,38]. Primary and secondary antibodies are summarized in Supplementary Table T7. Images were acquired on Leica SP5X or SP8X confocal microscopes equipped with a white light laser and HyD detectors using a 63x Oil objective and standardized settings for sequential image acquisition at the Core Facility Imaging, University of Halle. Bright field images were taken using a Nikon TE-2000-E microscope with 20x magnification and phase contrast. Representative images are shown. The localization of CTNNB1, CDH1 and CDH2 at the site of adherens junctions was quantified with respect to the cytoplasm for 14–15 cells from three independent experiments using imageJ as depicted as a box plot.
3D spheroid growth and invasion assays
For spheroid growth or invasion assays, 1 × 103 cells in 50 µL growth medium were seeded into round bottom ultra-low attachment plates (Corning, Corning, NY; Cat#7007), centrifuged for 5 min at 300 g and grown for 24 h to induce spheroid formation. For all cell lines except ES-2 and SK-OV-3, Cultrex® 10X Spheroid Formation ECM (Trevigen, Gaithersburg, MD; Cat# 3500–096-01) was added before centrifugation. For ES-2 and SK-OV-3 the addition of SFE was tested before the experiments with no improvement on 3D growth or invasion. For invasion assays, 50 µL Matrigel (Corning, Cat# 354234) was added to preformed spheroids. Growth medium (100 µL) supplemented with indicated inhibitors where stated was added before 3D growth or invasion were monitored using an Incucyte S3 (Sartorius, Göttingen, Germany) device for the indicated time. Image analyses were performed using the Incucyte software for spheroid segmentation (indicated as mask overlays in the respective figures). Growth or invasion areas excluding the spheroid body are normalized to spheroid inputs. Representative images are shown. Quantifications represent in total 6–9 spheroids from three independent experiments shown as box plots.
Antibodies, Western blotting and qRT-PCR
For Western blotting, cells were harvested by a rubber policeman to minimize degradation of transmembrane proteins. Total protein was extracted in TLB-buffer [50 mM Tris pH 7.4, 50 mM NaCl, 1% SDS, 2 mM MgCl2] followed by DNA digestion using Benzonase (Merck). Equal amounts of total protein were loaded on NuPAGE Bis-Tris (4–12%) gels (Thermo Fisher), blotted onto a nitrocellulose membrane (Amersham Bioscience) using the Mini Gel Tank Blotting system (Thermo Fisher) and analysed using the Odyssey infrared scanner (LI-COR) as previously described (26). Primary and secondary antibodies are summarized in Supplementary Table T7.
Total RNA extracted using Trizol (Thermo Fisher) served as a template for cDNA synthesis by random priming using M-MLV reverse transcription system (Promega, Madison, WI). qPCR was performed based on SYBRgreen I technology using the ORA qPCR Green ROX L Mix (HighQu, Kraichtal, Germany) on a LightCycler 480 II 384 format system (Roche, Basel, Switzerland). For all primer pairs, an annealing temperature of 60°C in a 3-step protocol was used. Relative changes of RNA abundance were determined by the ΔΔCt method using RPLP0 and GAPDH for normalization, as previously described (17). For primers see Supplementary Table T8.
Plasmids, cloning and luciferase reporter assays
Plasmid generation including the respective templates, vectors, restriction sites and oligonucleotide sequences and cloning strategies is summarized in Supplementary Table T8. All PCR (polymerase chain reaction) amplified inserts were sequenced before subcloning in the respective vectors. ES-2 cells transfected with siRNAs (24 h) were split for re-transfection of pmir-Glo luciferase plasmids (Supplementary Table T7) for an additional 48 h. Luciferase reporter analyses were performed essentially as previously described (17). Reporters containing a minimal vector-encoded 3′UTR (MCS) served as normalization controls. CTNNB1-mediated transcription was monitored using the nanoLUC reporter system (Promega, Madison, WI) 48 h after transfection into OVCAR-3 cells expressing GFP or GFP-IGF2BP1 with the following constructs: CTNNB1/TCF-LEF: pNL[NlucP/TCF/LEF-RE/Hygro]; negative control with minimal promoter: pNL3.2 [NlucP/minP]. Co-transfection of a firefly luciferase (pGL4.54[luc2/TK]) served as transfection and normalization control.
Statistical analyses
All cell culture experiments were performed in biological triplicates unless otherwise stated. Western blot quantifications are shown as mean ± SD. Whisker caps of box plots depict the 5th/95th percentile and all outliers are shown. Spheroid growth and invasion curves are shown as mean ± SE. Statistical significance was tested by Student’s t-test (two-tailed) or Mann–Whitney Rank-Sum test after data distribution was checked by Shapiro-Wilk normality test using Sigma Plot.
Supplementary Material
Funding Statement
The work was supported by Wilhelm-Roux funding to NB and DFG funding [GRK1591] to SH. Imaging, image analyses and NGS analyses were performed at the Core Facility Imaging (CFI) of the Martin Luther University Halle-Wittenberg, Germany.
Disclosure of potential conflicts of interest
The authors declare no conflicts of interest.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Bowtell DD, Bohm S, Ahmed AA, et al. Rethinking ovarian cancer II: reducing mortality from high-grade serous ovarian cancer. Nat Rev Cancer. 2015;15:668–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ahmed AA, Etemadmoghadam D, Temple J, et al. Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J Pathol. 2010;221:49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].KaramiNejadRanjbar M, Sharifzadeh S, Wietek NC, et al. A highly accurate platform for clone-specific mutation discovery enables the study of active mutational processes. Elife. 2020;9. DOI: 10.7554/eLife.55207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bley N. Finding the mutations that drive resistance. Elife. 2020;9. DOI: 10.7554/eLife.57678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ciriello G, Miller ML, Aksoy BA, et al. Emerging landscape of oncogenic signatures across human cancers. Nat Genet. 2013;45:1127–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bell D, Berchuck A, Birrer M et al. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Garsed DW, Alsop K, Fereday S, et al. Homologous recombination DNA repair pathway disruption and retinoblastoma protein loss are associated with exceptional survival in high-grade serous ovarian cancer. Clin Cancer Res. 2018;24:569–580. [DOI] [PubMed] [Google Scholar]
- [8].McGivern N, El-Helali A, Mullan P, et al. Activation of MAPK signalling results in resistance to saracatinib (AZD0530) in ovarian cancer. Oncotarget. 2018;9:4722–4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kondrashova O, Nguyen M, Shield-Artin K, et al. Secondary somatic mutations restoring RAD51C and RAD51D associated with acquired resistance to the PARP inhibitor rucaparib in high-grade ovarian carcinoma. Cancer Discov. 2017;7:984–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Mittica G, Ghisoni E, Giannone G, et al. PARP inhibitors in ovarian cancer. Recent Pat Anticancer Drug Discov. 2018;13:392–410. [DOI] [PubMed] [Google Scholar]
- [11].McNeish IA, Ledermann JA, Webber L, et al. A randomised, placebo-controlled trial of weekly paclitaxel and saracatinib (AZD0530) in platinum-resistant ovarian, fallopian tube or primary peritoneal cancer†. Ann Oncol. 2014;25:1988–1995. [DOI] [PubMed] [Google Scholar]
- [12].Tothill RW, Tinker AV, George J, et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin Cancer Res. 2008;14:5198–5208. [DOI] [PubMed] [Google Scholar]
- [13].Konecny GE, Winterhoff B, Wang C.. Gene-expression signatures in ovarian cancer: promise and challenges for patient stratification. Gynecol Oncol. 2016;141:379–385. [DOI] [PubMed] [Google Scholar]
- [14].Hu Z, Artibani M, Alsaadi A, et al. The repertoire of serous ovarian cancer non-genetic heterogeneity revealed by single-cell sequencing of normal fallopian tube epithelial cells. Cancer Cell. 2020;37:226–42 e7. [DOI] [PubMed] [Google Scholar]
- [15].Daugherty RL, Gottardi CJ. Phospho-regulation of beta-catenin adhesion and signaling functions. Physiology (Bethesda). 2007;22:303–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Simpkins F, Jang K, Yoon H, et al. Dual Src and MEK inhibition decreases ovarian cancer growth and targets tumor initiating stem-like cells. Clin Cancer Res. 2018;24:4874–4886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Roura S, Miravet S, Piedra J, et al. Regulation of E-cadherin/Catenin association by tyrosine phosphorylation. J Biol Chem. 1999;274:36734–36740. [DOI] [PubMed] [Google Scholar]
- [18].Qi J, Wang J, Romanyuk O, et al. Involvement of Src family kinases in N-cadherin phosphorylation and beta-catenin dissociation during transendothelial migration of melanoma cells. Mol Biol Cell. 2006;17:1261–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Matsuyoshi N, Hamaguchi M, Taniguchi S, et al. Cadherin-mediated cell-cell adhesion is perturbed by v-src tyrosine phosphorylation in metastatic fibroblasts. J Cell Biol. 1992;118:703–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Fang D, Chen H, Zhu JY, et al. Epithelial-mesenchymal transition of ovarian cancer cells is sustained by Rac1 through simultaneous activation of MEK1/2 and Src signaling pathways. Oncogene. 2017;36:1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Muller S, Glass M, Singh AK, et al. IGF2BP1 promotes SRF-dependent transcription in cancer in a m6A- and miRNA-dependent manner. Nucleic Acids Res. 2019;47:375–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Farley J, Brady WE, Vathipadiekal V, et al. Selumetinib in women with recurrent low-grade serous carcinoma of the ovary or peritoneum: an open-label, single-arm, phase 2 study. Lancet Oncol. 2013;14:134–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bell JL, Wachter K, Muhleck B, et al. Insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs): post-transcriptional drivers of cancer progression? Cell Mol Life Sci. 2013;70:2657–2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Busch B, Bley N, Muller S, et al. The oncogenic triangle of HMGA2, LIN28B and IGF2BP1 antagonizes tumor-suppressive actions of the let-7 family. Nucleic Acids Res. 2016;44:3845–3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Muller S, Bley N, Glass M, et al. IGF2BP1 enhances an aggressive tumor cell phenotype by impairing miRNA-directed downregulation of oncogenic factors. Nucleic Acids Res. 2018;46:6285–6303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Bi Z, Liu Y, Zhao Y, et al. A dynamic reversible RNA N(6) -methyladenosine modification: current status and perspectives. J Cell Physiol. 2019;234:7948–7956. [DOI] [PubMed] [Google Scholar]
- [27].Elcheva I, Goswami S, Noubissi FK, et al. CRD-BP protects the coding region of betaTrCP1 mRNA from miR-183-mediated degradation. Mol Cell. 2009;35:240–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Boyerinas B, Park SM, Murmann AE, et al. Let-7 modulates acquired resistance of ovarian cancer to Taxanes via IMP-1-mediated stabilization of multidrug resistance 1. Int J Cancer. 2012;130:1787–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Goswami S, Tarapore RS, Teslaa JJ, et al. MicroRNA-340-mediated degradation of microphthalmia-associated transcription factor mRNA is inhibited by the coding region determinant-binding protein. J Biol Chem. 2010;285:20532–20540. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [30].Wachter K, Kohn M, Stohr N, et al. Subcellular localization and RNP formation of IGF2BPs (IGF2 mRNA-binding proteins) is modulated by distinct RNA-binding domains. Biol Chem. 2013;394:1077–1090. [DOI] [PubMed] [Google Scholar]
- [31].Farina KL, Huttelmaier S, Musunuru K, et al. Two ZBP1 KH domains facilitate beta-actin mRNA localization, granule formation, and cytoskeletal attachment. J Cell Biol. 2003;160:77–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Müller S, Bley N, Busch B, et al. The oncofetal RNA-binding protein IGF2BP1 is a druggable, post-transcriptional super-enhancer of E2F-driven gene expression in cancer. Nucleic Acids Res. 2020. DOI: 10.1093/nar/gkaa653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Gutschner T, Hammerle M, Pazaitis N, et al. Insulin-like growth factor 2 mRNA-binding protein 1 (IGF2BP1) is an important protumorigenic factor in hepatocellular carcinoma. Hepatology. 2014;59:1900–1911. [DOI] [PubMed] [Google Scholar]
- [34].Huttelmaier S, Zenklusen D, Lederer M, et al. Spatial regulation of beta-actin translation by Src-dependent phosphorylation of ZBP1. Nature. 2005;438:512–515. [DOI] [PubMed] [Google Scholar]
- [35].Weng Z, Taylor JA, Turner CE, et al. Detection of Src homology 3-binding proteins, including paxillin, in normal and v-Src-transformed Balb/c 3T3 cells. J Biol Chem. 1993;268:14956–14963. [PubMed] [Google Scholar]
- [36].Kobel M, Weidensdorfer D, Reinke C, et al. Expression of the RNA-binding protein IMP1 correlates with poor prognosis in ovarian carcinoma. Oncogene. 2007;26:7584–7589. [DOI] [PubMed] [Google Scholar]
- [37].Stohr N, Kohn M, Lederer M, et al. IGF2BP1 promotes cell migration by regulating MK5 and PTEN signaling. Genes Dev. 2012;26:176–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Zirkel A, Lederer M, Stohr N, et al. IGF2BP1 promotes mesenchymal cell properties and migration of tumor-derived cells by enhancing the expression of LEF1 and SNAI2 (SLUG). Nucleic Acids Res. 2013;41:6618–6636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ishiyama N, Lee SH, Liu S, et al. Dynamic and static interactions between p120 catenin and E-cadherin regulate the stability of cell-cell adhesion. Cell. 2010;141:117–128. [DOI] [PubMed] [Google Scholar]
- [40].Gu W, Wells AL, Pan F, et al. Feedback regulation between zipcode binding protein 1 and beta-catenin mRNAs in breast cancer cells. Mol Cell Biol. 2008;28:4963–4974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Davis MA, Ireton RC, Reynolds AB. A core function for p120-catenin in cadherin turnover. J Cell Biol. 2003;163:525–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Schaller MD. Paxillin: a focal adhesion-associated adaptor protein. Oncogene. 2001;20:6459–6472. [DOI] [PubMed] [Google Scholar]
- [43].Yeatman TJ. A renaissance for SRC. Nat Rev Cancer. 2004;4:470–480. [DOI] [PubMed] [Google Scholar]
- [44].Yadav SS, Miller WT. Cooperative activation of Src family kinases by SH3 and SH2 ligands. Cancer Lett. 2007;257:116–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ostareck-Lederer A, Ostareck DH, Cans C, et al. c-Src-mediated phosphorylation of hnRNP K drives translational activation of specifically silenced mRNAs. Mol Cell Biol. 2002;22:4535–4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Frisone P, Pradella D, Di Matteo A, et al. SAM68: signal transduction and RNA metabolism in human cancer. Biomed Res Int. 2015;2015:528954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Simpkins F, Hevia-Paez P, Sun J, et al. Src inhibition with saracatinib reverses fulvestrant resistance in ER-positive ovarian cancer models in vitro and in vivo. Clin Cancer Res. 2012;18:5911–5923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Mahapatra L, Andruska N, Mao C, et al. A novel IMP1 inhibitor, BTYNB, targets c-Myc and inhibits melanoma and ovarian cancer cell proliferation. Transl Oncol. 2017;10:818–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ianevski A, He L, Aittokallio T, et al. SynergyFinder: a web application for analyzing drug combination dose-response matrix data. Bioinformatics. 2017;33:2413–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Remmele W, Stegner HE. Recommendation for uniform definition of an immunoreactive score (IRS) for immunohistochemical estrogen receptor detection (ER-ICA) in breast cancer tissue. Pathologe. 1987;8:138–140. [PubMed] [Google Scholar]
- [51].Ihling A, Ihling CH, Sinz A, et al. Acidosis-induced changes in proteome patterns of the prostate cancer-derived tumor cell line AT-1. J Proteome Res. 2015;14:3996–4004. [DOI] [PubMed] [Google Scholar]
- [52].Nagy A, Lanczky A, Menyhart O, et al. Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci Rep. 2018;8:9227. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.