

Abstract
A large and growing body of evidence supports functions of enzymes that regulate or effect cellular metabolism in governing the development, survival, and effector functions of immune cells—especially T cells, macrophages, and dendritic cells. Among these proteins, adenosine monophosphate-activated protein kinase (AMPK) is a conserved ATP and nutrient sensor that regulates multiple metabolic pathways to promote energy homeostasis. Although AMPK had been shown to regulate aspects of CD4+ and CD8+ T cell biology, its function in B lymphocytes has been less clear. Here, we review recent advances in our understanding of the role of AMPK in the metabolism, function, and maintenance of the B lineage.
Keywords: AMPK, B lymphocytes, humoral immunity, memory B cells, mitophagy, mTORC1, plasma cells
The B lineage, a critical branch of the adaptive immune system, gives rise to cells that confer long-lasting humoral protection against microbial diseases. This protection is mediated by the production of antibodies that recognize and potentially neutralize pathogens. B lineage cells—including plasmablasts and plasma cells derived from B cell subsets and the source of antigen-elicited antibodies—are functionally diverse, often move to pass through or reside in distinct microenvironments, and are supported by multiple metabolic pathways [1,2]. Naïve B cells, for which major residence pattern is in B cell follicles of secondary lymphoid organs, are in the G0 phase of the cell cycle with little need for rapid biosynthesis of net new proteins, lipids or metabolites. As such, they appear to be metabolically quiescent, with minimal glucose uptake and mitochondrial respiration [3]. Upon recognition of cognate antigen through the B cell receptor, however, naïve B cells are activated, undergo rapid and extensive proliferation, and can subsequently enter either extra-follicular or germinal center (GC) responses. Each of these sites potentially involves a distinct milieu, within which dividing and differentiating B cells may compete for limited nutrients and oxygen in the microenvironment [4–6]. Ultimately, B cells that survive this proliferative phase differentiate into memory B cell (MBC) subsets or terminally differentiated antibody-secreting cells, i.e. plasma cells (Figure 1A). MBCs resume quiescence in the G1 phase without cycling and depend on autophagy for their long-term persistence [7,8]. Autophagy is also critical for plasma cells, which may be short or long-lived [9]. However, unlike MBCs, plasma cells undergo substantial ER stress and need high rates of glycoprotein synthesis to meet the demands of manufacturing and secreting glycosylated antibodies at a rate of ~20 pg/cell/h [10–12]. Moreover, although there are long- as well as short-lived plasma cells (LLPC and SLPC, respectively) in secondary lymphoid organs, IgG-secreting LLPC tend to reside in specific niches in the bone marrow whereas IgA plasma cells densely populate the gut.
Figure 1. AMPK function in the B lineage and humoral immunity.
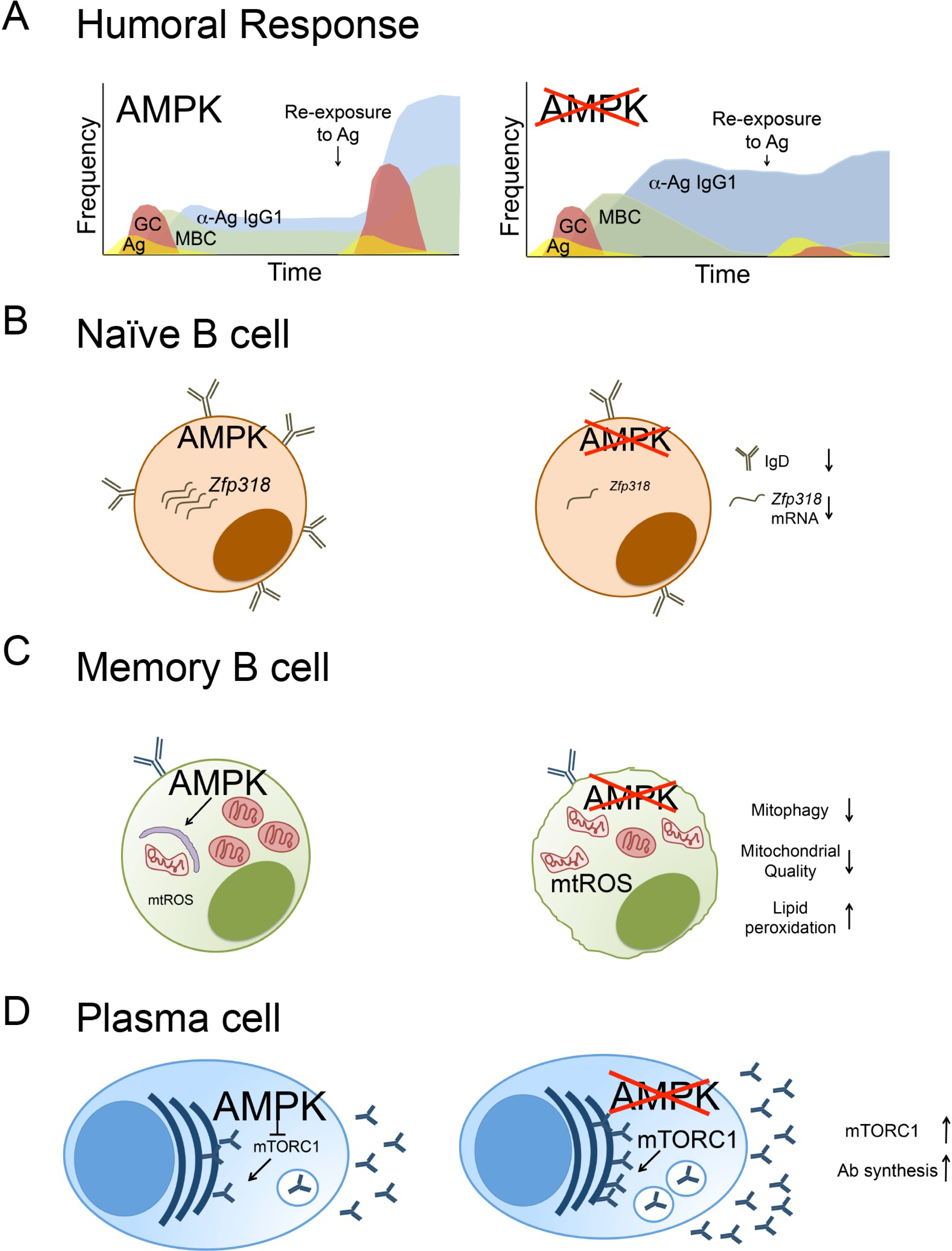
(A) Timeline of humoral responses after immunization with T-dependent antigens. In response to immunization, mice with AMPK-deficient B cells generate normal numbers of antigen-specific germinal centers but elevated levels of antigen-specific memory B cells and antigen-specific circulating antibody. Weeks after antigen exposure and the germinal center response, the memory B cell population declines but circulating antibodies remain enhanced in mice with AMPK-deficient B cells compared to wildtype mice. Consequently, re-challenge with antigen leads to ineffective recall responses in the absence of AMPK. (B) AMPK supports the expression of Zfp318 transcript and IgD expression. AMPK-deficient B cells have decreased Zfp318 mRNA, an important regulator of IgD. (C) AMPK promotes mitochondrial dynamics in memory B cells. Loss of AMPK in MBC leads to defects in mitophagy, an accumulation of nonfunctional mitochondria, and enhanced mtROS. (D) AMPK dampens antibody synthesis. Loss of AMPK leads to elevated mTORC1 signaling and antibody synthesis in plasma cells.
Accordingly, distinct subsets of cells along the B lineage must cope with changing and distinct metabolic demands, potentially in nutrient-limited microenvironments. This implies that sensors of metabolic status need to regulate multiple cellular mechanisms for B cells to adapt, survive, and function.
AMP-activated protein kinase (AMPK) is a highly conserved serine/threonine kinase that maintains energy homeostasis during times of metabolic stress by regulating multiple aspects of cellular metabolism [13]. AMPK is a heterotrimeric complex made of an α catalytic subunit and two regulatory subunits, β and γ; phosphorylation of the α catalytic subunit at the T-172 residue is critical for its activation [13–15]. Liver kinase B1 (LKB1) is a ubiquitously expressed tumor suppressor directly upstream of ~14 kinases including AMPK and has multiple roles in cellular metabolism, polarity, growth, migration, and differentiation [14]. In response to low nutrient availability, AMPK is activated by LKB1-induced T-172 phosphorylation coupled with increasing concentrations of cellular AMP or, less potently, ADP, which directly bind to the γ subunit of AMPK to inhibit dephosphorylation by an allosteric mechanism [13,16]. AMPK can also be activated independent of metabolic stress by upstream kinase Ca2+/calmodulin-dependent kinase kinase β (CaMKKβ) in T cells [17,18]. The induction of AMPK activation by CaMKKβ cells remains unexplored but we suspect that similar to in T cells, AMPK is activated independent of metabolic stress by CaMKKβ since both lineages experience an increased flux in Ca2+ ions in response to antigen. After AMPK activation, AMPK phosphorylates downstream targets that lead to ATP-generating processes such as mitochondrial biogenesis and fatty acid oxidation, and that promote autophagy. Simultaneously, AMPK inhibits ATP-consuming pathways such as protein and fatty acid synthesis through phosphorylation of protein targets in the mechanistic Target of Rapamycin (mTOR) complex 1 and of acetyl-CoA carboxylase (ACC). As summarized in [13], additional targets for regulation of metabolism—at least in other cell types—include nuclear transcription factors such as a carbohydrate-responsive element binding protein (ChREBP), a sterol regulatory element binding protein (SREBP)-1, and coactivators in the PGC1 family, which promotes mitochondrial biogenesis. B lymphocytes express the α1 isoform of the catalytic subunit, AMPKα1, which is encoded by Prkaa1 [19–21]. Although modest expression of an AMPKα2 isoform cannot be excluded, genetic ablation of Prkaa1 in B cells eliminated canonical AMPK activity as determined by the loss of phosphorylation of the established AMPK target ACC, which regulates fatty acid metabolism [20].
AMPK appears to be dispensable during B cell development [19–21]. Mice with unconditional loss of function for Prkaa1 as well as B cell specific deletion of Prkaa1 driven by Cd19-Cre or mb1-Cre have normal frequencies of B cells in the periphery [19–21]. Specifically, transitional, marginal zone, and follicular B cell populations in the spleen are normal in number in mice with Prkaa1-deficient B cells [20]. In contrast to AMPK, upstream kinase LKB1 is critical for B cell development [22,23]. Mice harboring Lkb1-deficient B cells driven by Cd19-Cre had an increase in B cell progenitors in the marrow but markedly diminished mature B cell subsets in the periphery indicating a developmental defect [22,23]. Lkb1-deficient B cells also had an approximately 5-fold increase in frequencies of cleaved caspase 3- and Annexin-V-positive cells, indicating an anti-apoptotic role for LKB1 in B cells [22,23]. These data provide evidence of an AMPK-independent role of LKB1 in early B cell development akin to findings with T cell development [24]. AMPK promotes Ighd and Zfp318 gene expression and surface IgD expression [21] and promotes IgD expression in vivo (Brookens, S. K., unpublished data) (Figure 1B). This finding suggests that AMPK may support B cell persistence in a naïve state or have an effect on alternative RNA splicing. The significance of the involvement of AMPK in IgD expression is unclear, as evidence of a major function of IgD remains elusive. However, emerging reports indicate that IgD plays a role in regulating IgM-mediated anergy and BCR signaling [25,26]. Accordingly, AMPK may have an impact on early B cell activation that has not yet been detected.
B cells can be activated through surface receptors such as the BCR, Toll-like receptors (TLRs), or CD40. As noted, AMPK is a negative regulator of mechanistic target of rapamycin complex 1 (mTORC1), a multi-subunit complex that promotes protein synthesis. mTORC1 and likely the dynamic regulation of its activity are critical for affecting outcomes of both germinal center and extra-follicular responses [27–30]. Loss of Prkaa1 in B cells led to elevated mTORC1 activity in vivo and after in vitro activation [20,31]. Similar to T cells, B cell activation can induce AMPK activity independent of metabolic stress [21]. B cells exhibited a sustained increase in pAMPKα1T172 expression despite neither an elevation of intracellular ATP nor a substantial decline in glucose or glutamine availability in the tissue culture media after anti-CD40 and IL-4 activation in vitro [21]. Though AMPK is dispensable for the expression of activation markers CD86, CD69, and MHCII, the kinetics for pAMPKα1T172 expression parallel the deceleration of biomass accumulation in B cells during activation [21]. Thus a role for AMPK in limiting excess cell growth during B cell activation, potentially through negative regulation of mTORC1 and other anabolic substrates, cannot be excluded.
AMPK may function as a metabolic switch during settings of nutrient poor conditions. Though pAMPKT172 is expressed in nutrient-replete conditions upon anti-CD40 and IL-4 activation in vitro, AMPK may manifest effects in B cells differently depending on nutrient availability [21]. In vitro activation with anti-CD40 and IL-4 reveal that not only is AMPK dispensable for glucose uptake in B cells, but in addition metabolite tracer assays found no role of AMPK in altering glucose or glutamine metabolism upon activation [21]. However, these studies were done in tissue culture conditions, which vary qualitatively and quantitatively from concentrations of metabolites in sera and tissues in vivo [32]. In other studies, the effects of AMPK were only evident in metabolically stressful environments [19,20,33]. AMPK is essential for B or T cells to survive ATP synthase inhibition through oligomycin A treatment [19]. Additionally, AMPK is critical for T cells to adapt to glucose-deficient conditions by switching to glutamine metabolism [33]. In glucose-deficient conditions, AMPK in B cells is required to induce phosphorylation of downstream target Unc51-like kinase1 (ULK1), a critical kinase for the initiation of canonical autophagy and mitophagy [20]. Furthermore, glucose withdrawal increased the expression of AMPK target pACCS79 in vitro indicating enhanced AMPK activity when glucose is limiting [20]. Nonetheless, the degree to which nutrients are limiting in various microenvironments in vivo, and thereby activate AMPK in cells in the B lineage, is yet to be definitively elucidated.
One fate for activated B cells is to enter germinal center reactions that develop 3–4 days after immunization. GCs are reported to have variegations in oxygen availability determined by using intravital hypoxia-marking dyes and HIF1α expression [4,5]. In addition to oxygen, proliferating GC B cells may also compete for extracellular glucose, fatty acids, and amino acids such as glutamine in the microenvironment to use as a carbon source, although potential questions have been raised about the relative balance of energy-generating pathways [4,6,34]. Nonetheless, unequivocal direct evidence of nutrient limitations on B cells and competition in the microenvironment of a GC is lacking at present. One might expect that GC B cells in the ostensibly nutrient-limited GC would employ AMPKα1 signaling to adapt to the metabolic stress. However, mice harboring Prkaa1-deficient B cells were able to induce GC-signature transcripts, had numbers of GC B cells comparable to wild-type controls, and exhibited affinity-matured antibodies after immunization with a T-dependent antigen [20,21]. This finding suggests that, akin to the normal B cell development, AMPK is dispensable for GC reactions, perhaps because its function is outweighed by high mTORC1 activity. It is surprising that AMPKα1 does not appear to be a significant player in regulating B cell development, activation, or the formation of GCs. The lack of detectable impact of AMPK on these B cell processes suggests that energy generation is not limiting and that AMP/ADP:ATP ratios are maintained in vivo. This hypothesis would be consistent with in vitro observations where B cells maintained AMP concentrations without a decline in AMP/ADP:ATP ratios when activated with anti-CD40 and IL-4 [21]. The tools to measure and manipulate nutrient concentrations in the microenvironment of the marrow or in secondary lymphoid structures would elucidate some of these possibilities. In contrast to AMPK, LKB1 upstream of AMPK, is critical for maintaining B-cell quiescence as Lkb1-deficient B cells experience the premature formation of GCs [22]. These findings indicate an AMPK-independent role of LKB1 in the formation of GCs.
MBCs, which can arise either from extra-follicular activated B cells or GC B cells, add to long-lasting immunity by either rapidly differentiating into plasma cells or re-entering GCs upon a later re-encounter with antigen (Figure 1A). Immunization of mice harboring B cell-specific deletion of Prkaa1 with a T-dependent antigen revealed that AMPK initially dampens the generation of B cells harboring MBC surface markers when screening at a time coincident with the peak of the GC response in vivo [20]. On the surface, this finding differs from recent evidence that the GC B cell to MBC transition is facilitated by relatively lower mTORC1 activity in GC B cells [30]. In contrast to its role in dampening the initial population numbers of MBC, AMPK supports the long-term maintenance of the MBC population: numbers of Prkaa1-deficient MBC were diminished compared to wild-type controls several weeks after the peak of the GC response [20]. The impact of AMPK on the maintenance of the antigen-specific memory B cell compartment was most evident in memory B cells that do not express CD80 or PD-L2, the subset considered most likely to function in reestablishing GCs upon antigen exposure [35]. Consequently, rechallenging mice led to a defect in numbers of secondary GCs as well as defects in recall-induced antibody production in the sera in the absence of Prkaa1 [20]. Mechanistically, AMPK was found to support mitophagy and limit mitochondrial-derived reactive oxygen species (mtROS) production as well as limit lipid peroxidation in MBCs [20]. This finding may be akin to earlier results indicating that increased oxidative stress shortened erythrocyte lifespan in mice with a universal and constitutive loss of AMPKα1 [36]. In support of a role for AMPK in mitochondrial dynamics in B cells, B lymphoblasts exhibited decreased maximal and spare respiratory capacity, which may extrapolate to its role in MBCs [20]. Collectively, these findings were most consistent with a model in which AMPK supports longevity of the MBC population by promoting mitochondrial function and turnover, limiting toxic ROS and lipid-peroxidation [20] (Figure 1C). Mitochondrial homeostasis is also associated with other long-lived cell types including CD8+ memory T cells and NK memory cells [37,38]. Supporting this model, IgM+ human CD27+ MBC have elevated mitochondrial activity and pAMPKT172 expression compared to mature naïve B cells [39]. Furthermore, metformin treatment, which indirectly activates AMPK, is associated with increased MBC and improved Ab responses to influenza vaccine in type II diabetic patients [40]. Though AMPK may support some subsets of MBC such as the IgM+ MBC, enhanced pAMPKT172 in IgD− CD27− late/exhausted human MBC was associated with senescence and the inability to respond to CpG activation [41]. Interestingly, Atg7, an autophagy-essential gene downstream of AMPK is also associated with maintaining MBC [7,8]. However, canonical autophagy was unaffected in the absence of AMPK suggesting MBC employ noncanonical AMPK-independent forms of autophagy analogous to GC B cells [20,42]. All together, it is likely that AMPK promotes MBC survival through maintaining mitochondrial homeostasis, thereby supporting the ability of the humoral immune system to mount effective responses to recurrent infections.
In addition to MBC, humoral memory in the form of durable antibodies in the serum derives from LLPC. Plasma cells confer humoral immunity by making and secreting antibodies that circulate and bind to cognate antigen. No impact on the expression of plasma cell-associated genes or numbers of SLPC or LLPC—either generated in vitro or after immunization with T-dependent antigens—was detected for AMPKα1-deficient B cells [20,21]. Other studies indicate that treatment of naïve mouse B cells or CD27+ human MBC with metformin, an indirect activator or AMPK, led to diminished plasma cell generation and antibody production [43,44]. The differences between data from pharmaceutical versus genetic approaches suggest the impact of AMPK-independent effects of metformin or compensatory alterations in signaling pathways due to persistent as opposed to transient inactivation. Interestingly, AMPK had a distinct role in supporting the longevity of MBCs but no impact on LLPCs suggesting marked differences in the metabolic programs employed by the two long-lived populations [20]. Although both are quiescent from the perspective of proliferation, the energy demands and nutrient intake by LLPC are far greater than what would be required by a resting MBC. Some divergence among papers is evident in conclusions on the role of AMPK in antibody responses. Two studies indicated that loss of Prkaa1 in B cells led to no decrease in antibody responses to T-dependent immunization strategies [19,21]. However, in our recent study using multiple genetic models and approaches, loss of AMPK in the B lineage led to elevated concentrations of circulating antigen-specific IgG1 antibodies as soon as two weeks after hapten-carrier immunization [20], a finding that may have been foreshadowed by the increase in total IgM response in mice with Prkaa1-deficient B cells [21]. The elevated antigen-specific antibody in the sera in Prkaa1-deficient animals persisted at least eight weeks post immunization suggesting that AMPK dampens antibody production in both SLPCs and LLPCs. AMPK limits antibody production specifically by modulating rates of antibody synthesis [20] (Figure 1D). Though AMPK does not give a survival advantage to LLPCs, it does dampen antibody production, likely in part through tuning down mTORC1 activity [20] (Brookens, S, unpublished observations). Effects of genetic modifications that persistently increased mTORC1 in B cells have been studied in several alternative models, yielding mixed results [29,45,46]. In one study, inactivation of the Tsc1 gene, which encodes a negative regulator of mTORC1 (tuberous sclerosis complex 1 (TSC1)), led to modestly impaired antibody responses [45]. Although TSC1 deficiency in B cells also had little impact on GC, these results did not phenocopy the loss of AMPKα1. However, differences between the two perturbations are both qualitative (both TSC1 and AMPK regulate other pathways in addition to mTORC1) and quantitative (different degrees of increase in mTORC1 activity). Analogous points apply to findings with a constitutive knock-in of a gain-of-function mutation in an upstream regulator that promotes mTORC1 activity as well as GC-focused inactivation of Tsc1 [29]. Alternatively, B cell-specific inactivation of Tsc1 with Cd19-Cre led to increased Ig synthesis [46], akin to results with induced loss of AMPKα1 [20].
LLPCs have substantial mitochondrial respiratory capacity compared to their short-lived counterparts [47]. It is unclear whether and how AMPK-deficient LLPCs maintain oxidative phosphorylation in light of evidence that AMPK is vital for maximal and spare respiratory capacity in B lymphoblasts and mitochondrial homeostasis in MBCs [20]. However, the need for AMPK to turn over accumulating mitochondria in plasma cells may be less critical since plasma cells may have lower mitochondrial mass and membrane potential than IgG1+ B cells [48].
To summarize, AMPK regulates multiple aspects of cellular metabolism. Effects of AMPK in humoral immunity and the B lineage are illustrated in Figure 1. Surprisingly, although AMPK signaling in B cells is evident during activation, AMPK has negligible impact during this period or on the formation of germinal centers. Akin to findings with memory CD8+ T cells, however, AMPK appears to be critical for the long-term maintenance of the MBC compartment [20,49] (Figure 1). Interestingly, new evidence has emerged that the rate of antibody synthesis of both SLPCs and LLPCs is fine-tuned and regulated at least in part by AMPK [20]. Distinguishing whether AMPK activity in MBC or plasma cells is due to low nutrient availability in the microenvironment is unclear. Assessing the availability of metabolites in vivo at cellular resolution in tissues where MBC and plasma cells reside will potentially shed light on how modulating the microenvironment may improve humoral responses.
ACKNOWLEDGEMENTS
The authors gratefully acknowledge helpful suggestions from the referees, and the support of NIH grants (R01 AI113292, and career development support of S. K. B., via R25 GM062459, then T32 CA009592, followed by a supplement to AI113292) and of Departmental funds from the PMI Department and Vanderbilt Institute for Infection, Inflammation, and Immunology (VI4).
Footnotes
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest to disclose.
REFERENCES
- 1.Boothby MR, Rickert RC. Metabolic regulation of the immune humoral response. Immunity. 2017;46:743–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jellusova J, Rickert RC. A brake for B cell proliferation: appropriate responses to metabolic stress are crucial to maintain B cell viability and prevent malignant outgrowth. Bioessays. 2017;39:1700079. [DOI] [PubMed] [Google Scholar]
- 3.Waters LR, Ahsan FM, Wolf DM, Shirihai O, Teitell MA. Initial B cell activation induces metabolic reprogramming and mitochondrial remodeling. iScience. 2018;5:99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jellusova J, Cato MH, Apgar JR, Ramezani-Rad P, Leung C, Chen C, et al. GSK3 is a metabolic checkpoint regulator in B cells. Nat Immunol. 2017;18:303–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cho SH, Raybuck AL, Stengel K, Wei M, Beck TC, Volanakis E, et al. Germinal Center hypoxia and regulation of antibody qualities by a hypoxia response system. Nature. 2016;537:234–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vijay R, Guthmiller JJ, Sturtz AJ, Surette FA, Rogers KJ, Sompallae RR, et al. Infection-induced plasmablasts are a nutrient sink that impaires humoral immunity to malaria. Nat Immunol. 2020;7:790–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen M, Hong MJ, Sun H, Wang L, Shi X, Gilbert BE, et al. Essential role for autophagy in the maintenance of immunological memory against influenza infection. Nat Med. 2014;5:503–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen M, Kodali S, Jang A, Kuai L, Wang J. Requirement for autophagy in the long-term persistence but not initial formation of memory B cells. J Immunol. 2015;194:2607–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pengo N, Scolari M, Oliva L, Milan E, Mainoldi F, Raimondi A, et al. Plasma cells require autophagy for sustainable immunoglobulin production. Nat Immunol. 2013;14:298–305. [DOI] [PubMed] [Google Scholar]
- 10.Gass JN, Gunn KE, Sriburi R, Brewer JW. Stressed-out B cells? Plasma-cell differentiation and the unfolded protein response. Trends Immunol. 2004;25:17–24. [DOI] [PubMed] [Google Scholar]
- 11.Arnold JN, Wormald MR, Sim RB, Rudd PM, Dwek RA. The impact of glycosylation on the biological function and structure of human immunoglobulins. Annu Rev Immunol. 2007;25:21–50. [DOI] [PubMed] [Google Scholar]
- 12.Hibi T, Dosch H. Limiting dilution analysis of the B cell compartment in human bone marrow. Eur J Immunol. 1986;16:139–145. [DOI] [PubMed] [Google Scholar]
- 13.Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. 2017;19:121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumor suppression. Nat Rev Cancer. 2009;9:563–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hawley SA, Davison M, Woods A, Davies SP, Beri RK, Carling D, et al. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J Biol Chem. 1996;271:27879–87. [DOI] [PubMed] [Google Scholar]
- 16.Oakhill JS, Steel R, Chen Z, Scott JW, Ling N, Tam S, et al. AMPK is a direct adenylate charge-regulated protein kinase. Science. 2011;332:1433–5. [DOI] [PubMed] [Google Scholar]
- 17.Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, et al. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2:9–19. [DOI] [PubMed] [Google Scholar]
- 18.Tamás P, Hawley SA, Clarke RG, Mustard KJ, Green K, Hardie DG, et al. Regulation of the energy sensor AMP-activated protein kinase by antigen receptor and Ca2+ in T lymphocytes. J Exp Med. 2006;203:1665–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mayer A, Denanglaire S, Viollet B, Leo O, Andris F. AMP-activated protein kinase regulates lymphocyte responses to metabolic stress but is largely dispensable for immune cell development and function. Eur J Immunol. 2008;38:948–956. [DOI] [PubMed] [Google Scholar]
- 20.Brookens SK, Cho SH, Basso PJ, Boothby MR. AMPKα1 in B Cells Dampens primary antibody responses yet promotes mitochondrial homeostasis and persistence of B cell memory. J Immunol. 2020;205:3011–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Waters LR, Ahsan FM, Hoeve JT, Hong JS, Kim DNH, Minasyan A, et al. Ampk regulates IgD expression by not energy stress with B cell activation. Sci Rep. 2019;9:8176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walsh NC, Waters LR, Fowler JA, Lin M, Cunningham CR, Brooks DG, et al. LKB1 inhibition of NFκB in B cells prevents T follicular helper cell differentiation and germinal center formation. EMBO Rep. 2015;16:753–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Souroulass GP, Fedoriw Y, Staudt LM, Sharpless NE. Lkb1 deletion in murine B lymphocytes promotes death and cancer. Exp Hematol. 2017;51:63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.MacIver NJ, Blagih J, Saucillo DC, Tonelli L, Griss T, Rathmell JC, et al. The liver kinase B1 is a central regulator of T cell development, activation, and metabolism. J Immunol. 2011;187:4187–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chaturvedi A, Siddiqui Z, Bayiroglu F. A GPI-linked isoform of IgD receptor regulates resting B cell activation. Nat Immunol. 2002;3:951–7. [DOI] [PubMed] [Google Scholar]
- 26.Gutzeit C, Chen K, Cerutti A. The enigmatic function of IgD: some answers at last. Eur J Immunol. 2018;48:1101–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raybuck AL, Cho SH, Li J, Rogers MC, Lee K, Williams CL, et al. B cell-intrinsic mTORC1 promotes germinal center-defining transcription factor gene expression, somatic hypermutation, and memory B cell generation in humoral immunity. J Immunol. 2018;200:2627–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gaudette BT, Jones DD, Bortnick A, Argon Y, Allman D. mTORC1 coordinates an immediate unfolded protein response-related transcriptome in activated B cells preceding antibody secretion. Nat Commun. 2020;723:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ersching J, Efeyan A, Mesin L, Jacobsen JT, Pasqual G, Grabiner BC, et al. Germinal center selection and affinity maturation require dynamic regulation of mTORC1 kinase. Immunity. 2017;46:1045–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Inoue T, Shinnakasu R, Kawai C, Ise W, Kawakami E, Sax N, et al. Exit from germinal center to become quiescent memory B cells depends on metabolic reprogramming and provision of a survival signal. J Exp Med. 2020;218:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adams WC, Chen Y, Kratchmarov R, Yen B, Nish SA, Lin WW, et al. Anabolism-associated mitochondrial stasis driving lymphocyte differentiation over self-renewal. Cell Rep. 2016;17:3142–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ackermann T, Tardito S. Cell culture medium formulation and its implications in cancer metabolism. Trends Cancer. 2019;5:329–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blagih J, Coulombe F, Vincent EE, Dupay F, Galicia-Vázquez G, Yurchenko E, et al. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity. 2015;42:41–54. [DOI] [PubMed] [Google Scholar]
- 34.Weisel FJ, Mullett SJ, Elsner RA, Menk AV, Trivedi N, Luo W, et al. Germinal center B cells selectively oxidize fatty acids for energy while conducting minimal glycolysis. Nat Immunol. 2020;21:331–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zuccarino-Catania GV, Sadanand S, Weisel FJ, Tomayko MM, Meng H, Kleinstein SH, et al. CD80 and PD-L2 define functionally distinct memory B cell subsets that are independent of antibody isotype. Nat Immunol. 2014:15:631–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang S, Dale GL, Song P, Viollet B, Zou MH. AMPKalpha1 deletion shortens erythrocyte life span in mice: role of oxidative stress. J Biol Chem. 2010;285:19976–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. 2012;36:68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O’Sullivan TE, Johnson LR, Kang HH, Sun JC. BNIP3- and BNIP3L-mediated mitophagy promotes the generation of natural killer memory. Immunity. 2015;43:331–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mensah FF, Armstrong CW, Reddy V, Bansal AS, Berkovitz S, Leandro MJ, et al. CD24 expression and B cell maturation shows a novel link with energy metabolism: potential implications for patients with myalgic encephalomyelitis/chronic fatigue syndrome. Front Immunol. 2018;9:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Diaz A, Romero M, Vazquez T, Lechner S, Blomberg BB, Frasca D. Metformin improves in vivo and in vitro B cell function in individuals with obesity and type-2 diabetes. Vaccine. 2017;35:2694–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frasca D, Diaz A, Romero M, Thaller S, Blomberg BB. Metabolic requirements of human pro-inflammatory B cells in aging and obesity. PLoS One. 2019;7:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martinez-Martin N, Maldonado P, Gasparrini F, Frederico B, Aggarwal S, Gaya M, et al. A switch from canonical to noncanonical autophagy shapes B cell responses. Science. 2017;355:641–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee SY, Moon SJ, Kim EK, Seo HB, Yang EJ, Son HJ, et al. Metformin suppresses systemic autoimmunity in Roquinsan/san mice through inhibiting B cell differentiation into plasma cells via regulation of AMPK/mTOR/STAT3. J Immunol. 2017;198:2661–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Torigoe M, Iwata S, Nakayamada S, Sakata K, Zhang M, Hajime M, et al. Metabolic reprogramming commits differentiation of human CD27+IgD+ B cells to plasmablasts or CD27−IgD− cells. J Immunol. 2017;199:425–34. [DOI] [PubMed] [Google Scholar]
- 45.Ci X, Kuraoka M, Wang H, Carico Z, Hopper K, Shin J, et al. TSC1 promotes B cell maturation but is dispensable for germinal center formation. PLoS One. 2015;10:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Benhamron S, Pattanayak SP, Berger M, Tirosh B. mTOR activation promotes plasma cell differentiation and bypasses XBP-1 for immunoglobulin secretion. Mol Cell Biol. 2015;35:153–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lam WY, Becker AM, Kennerly KM, Wong R, Curtis JD, Llufrio EM, et al. Mitochondrial pyruvate import promotes long-term survival of antibody-secreting plasma cells. Immunity. 2016;45:60–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jang KJ, Mano H, Aoki K, Hayashi T, Muto A, Nambu Y, et al. Mitochondrial function provides instructive signals for activation-induced B cell fates. Nat Commun. 2015;6:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rolf J, Zarrouk M, Finlay DK, Foretz M, Viollet B, Cantrell DA. AMPKα1: A glucose sensor that controls CD8 T-cell memory. Eur J Immunol. 2013;43:889–96. [DOI] [PMC free article] [PubMed] [Google Scholar]