

Abstract
Few other elements play a more central role in biology than hydrogen. The interactions, bonding and movement of hydrogen atoms are central to biological catalysis, structure and function. Yet owing to the elusive nature of a single hydrogen atom few experimental and computational techniques can precisely determine its location. This is exemplified in short hydrogen bonds (SHBs) where the location of the hydrogen is indicative of the underlying strength of the bonds, which can vary from 1–5 kcal/mol in canonical hydrogen bonds, to an almost covalent nature in single-well hydrogen bonds. Owing to the often-times inferred position of hydrogen, the role of SHBs in biology has remained highly contested and debated. This has also led to discrepancies in computational, biochemical and structural studies of proteins thought to utilize SHBs in performing chemistry and stabilizing interactions. Here we discuss in detail two distinct examples, namely the conserved catalytic triad and the photoreceptor, photoactive yellow protein, where studies of these SHB-containing systems have permitted contextualization of the role these unique hydrogen bonds play in biology.
Keywords: hydrogen bond, catalytic triad, photoactive yellow protein, low-barrier hydrogen bond, short ionic hydrogen bond
Graphical Abstract
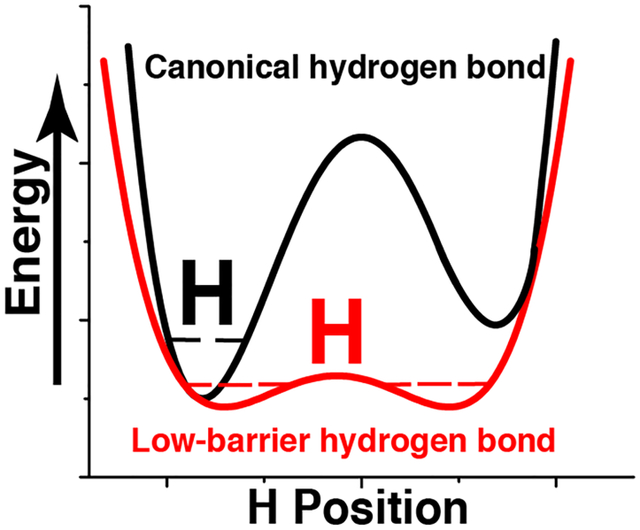
The position of a hydrogen in a short hydrogen bond (SHB) is indicative of the bond strength. Yet, few techniques can locate a hydrogen atom in either canonical, or unusually strong, low barrier hydrogen bonds. This has led to discrepancies among simulations and biochemical studies. In this review we discuss two examples of SHB-containing proteins to gain insights into the energetic role of these bonds.
1. Short Hydrogen Bonds in Biology
Hydrogen bond strength in proteins is dictated through a myriad of well understood processes, such as heavy-atom distances, geometry and electrostatics.[1] Yet, it is the less well understood properties of hydrogen bonds that perhaps provide the largest energetic contributions in biology. An enigma persists as the often-times elusive location of the hydrogen atom in short hydrogen bonds (SHBs) indicates the underlying free-energy associated with this interaction. As the ΔpKa between the donor and acceptor atoms decreases and the distance between them approaches the sum of their van der Waals radii, the overall covalent character of the hydrogen bond increases, and the position of the hydrogen atom can no longer be inferred geometrically. Several scenarios, which depend on where the hydrogen atom resides, lead to drastically distinct energetic consequences (Figure 1). In short ionic hydrogen bonds (SIHBs) the hydrogen atom remains bound to the heavy-atom donor regardless of the proximity of heavy-atoms, and energetically this interaction is on the order of normal hydrogen bonds. As the energetic barrier difference becomes less through matching of pKa values, two low energetic barrier wells of equal magnitude dictate that hydrogen atoms in low barrier hydrogen bonds (LBHBs) are found at the center (average position in a population) of the two heavy-atoms. In an LBHB the hydrogen atom can move freely between heavy-atoms while leading to ~15 kcal/mol of free-energy in certain cases.[2] Another scenario exists in what are known as single-well hydrogen bonds (SWHBs) where the approach of heavy-atoms is so close that the barrier existing between the two wells is lost entirely, and the hydrogen atom shows characteristics of being bound to both the heavy-atoms simultaneously. The energetic consequence of the latter interaction is therefore on the order of a covalent bond. Although these are discrete descriptions of various SHBs, it is likely that there is a continuum of states and free-energy existing among the two SHB extremes of a SIHB and a SWHB[3]
Figure 1. Schematic of typical potential energy profiles for different types of hydrogen bonds.
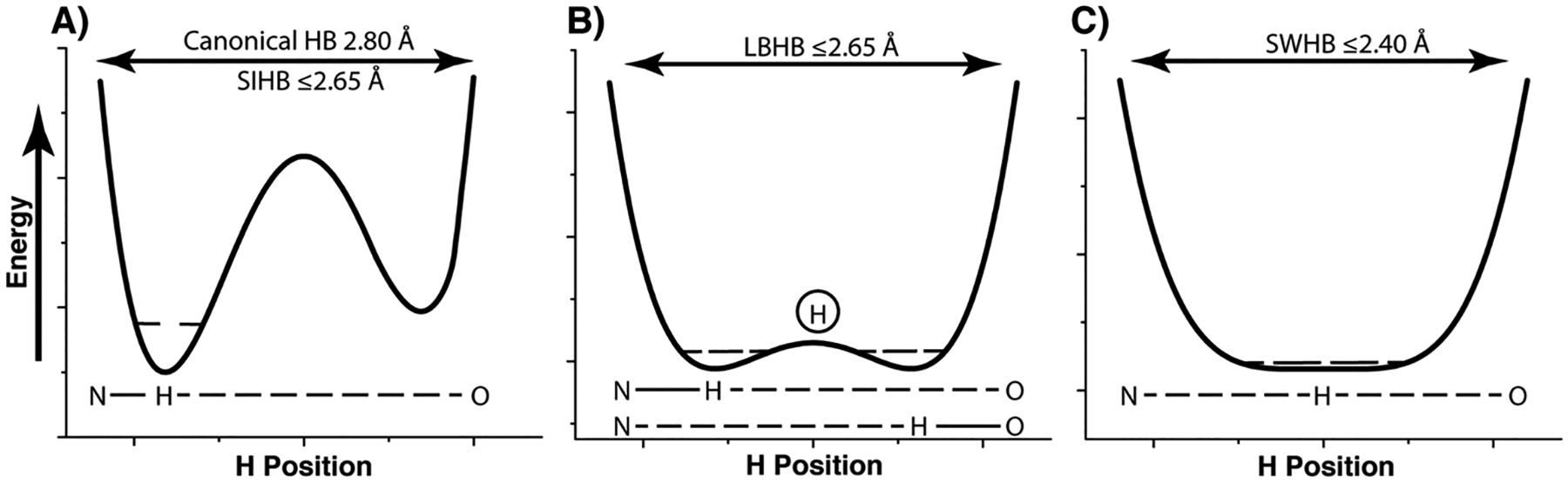
A) In a canonical hydrogen bond, an asymmetric double well potential exists, where the hydrogen remains attached to the donor heavy atom. In a short-ionic hydrogen bond (SIHB), the asymmetric double well persists even though the distance between the atoms can be less than the van der Waals radii. B) In a low barrier hydrogen bond (LBHB) matching pKa values of the donor and acceptor heavy atoms means that the transfer of the hydrogen to either heavy atoms occurs with an equal probability and the average position of the hydrogen atom is in the center, as seen in structural studies. C) In single-well hydrogen bond (SWHB) the energetic barrier for transfer of the hydrogen atom is lost completely and it is simultaneously bound to both the heavy atoms. The dashed lines within the wells represent the zero-point energies.
Although LBHBs in general were first proposed well more than 20 years ago,[4] the unusual energetic consequences of SHBs in biological catalysis were first treatised in two seminal papers in 1994.[2a, 5] At that time, it was postulated that SHBs are responsible, or perhaps the missing link, in the search for the energetic requirements needed to stabilize enzyme transition states or other intermediates. The large family of serine proteases containing the conserved catalytic triad were the first ones to be discussed and debated, and since then SHBs have been invoked in a diverse array of enzymes and reaction archetypes beyond the catalytic triad-containing trypsin, chymotrypsin, subtilisin, elastase, caspase, alpha-lytic protease and acetyltransferases.[6] Structural studies on several aspartic proteinases, such as HIV protease[7] and endothiapepsin[8] have revealed the unique functional role of SHBs in this catalytic archetype as well. Catalytically relevant SHBs have also been identified, or proposed to play roles in beta lactamase,[9] triosephosphate isomerase,[10] methylthioadenosine nucleosidase,[11] ketosteroid isomerase,[12] pyruvate oxidase,[13] transketolase,[13] aspartate aminotransferase,[14] and more recently a nucleotidyltransferase that shares structural homology with a mammalian DNA polymerase.[15] The list of enzymes proposed or demonstrated to use SHBs to perform chemistry is ever expanding.
Beyond their role in catalysis, interactions involving hydrogen atoms are the key contributors to the driving force of ligand and drug binding to target biomacromolecules, in addition to stabilization of biomolecular structure.[6a] Therefore, SHBs do not function exclusively in enzyme catalysis and have also been proposed to play a key role in processes, such as in conferring “exquisite specificity” in a phosphate binding protein, [16] carbohydrate recognition in concanavalin A,[17] chromophore tuning in green fluorescent protein,[18] as well as allosteric communication.[13] Additionally, a vast amount of biochemical, structural and theoretical studies exist on structural SHBs in photoactive yellow protein, which will be discussed in detail.
As soon as the role of SHBs in biological catalysis was first put forth, the debate regarding their relevance and energetic contributions began in earnest. Various counter arguments have been proposed to account for unusual biological energetics of SHBs, such as not being anything other than electrostatics. In fact, Arieh Warshel offered an alternative explanation to the initial series of LBHB papers by suggesting that an LBHB is an anti-catalytic versus a canonical hydrogen bond.[19] Yet beyond the debate established through theory and molecular simulations, several experimental examples now exist where the loss of a SHB through mutation has little effect on the underlying energetics, thus further confounding the original narrative on the extreme free-energy contributions of these types of interactions.[15–16, 20]
Even though their classification has been challenged by several studies, LBHBs have been routinely observed to occur in certain simple compounds, yet they were not discovered in proteins until much later. In biological systems LBHBs have been commonly inferred by far downfield nuclear magnetic resonance (NMR) chemical shift values, isotope effects on the NMR chemical shifts as well as unique H/D fractionation factors and infrared stretching frequencies. However, none of these methods allow a direct measurement of the distance between the donor and acceptor atoms, nor the location of the hydrogen atom between them.[21] Ultra-high-resolution X-ray crystallography (<1.0 Å resolution) and neutron diffraction are bridging this gap, making the direct visualization of hydrogen-atom positions possible. Moreover, details of SHBs and LBHBs have been revealed by computational investigations of biomolecular systems where the positions of hydrogen atoms in the ground states are explicitly available. Yet, despite having been proposed, debated, and tested for over 20 years now, few examples exist where the energetic role of SHBs has been unequivocally corroborated with structural, biochemical and molecular simulations studies. Some examples do exist, of which here we will discuss the role of catalytic and structural SHBs where the placement of the hydrogen atom, and the accompanying debate, has been firmly established.
2. Short hydrogen bonds in catalytic triads
The involvement of an LBHB in biological catalysis was first hypothesized by Frey et al and Cleland et al. in chymotrypsin, as well as in several other enzymes in a relatively short span of time. [2a, 5b, c] Chymotrypsin is a classical serine protease that utilizes the canonical serine, histidine, aspartic acid catalytic triad for cleaving the peptide bond (Figure 2A). The efficiency of the catalytic triad is achieved through a hydrogen bond between the aspartic acid and the Nδ of the histidine; it is thus this hydrogen bond that has been the subject of the LBHB debate in this family of enzymes. The Asp-His hydrogen bonding interaction increases the basicity of the histidine Nε while facilitating deprotonation of the serine hydroxyl and generation of the reactive nucleophile. The resulting nucleophile attacks the scissile peptide bond and generates a negatively charged covalent intermediate that is stabilized through hydrogen bonding interactions in the oxyanion hole (Figure 2A). This first tetrahedral intermediate collapses resulting in a covalent acyl-enzyme intermediate and releasing of the C-terminal fragment. This is followed by the attack of a water molecule, forming a second tetrahedral intermediate, which is resolved by the release of the N-terminal fragment and regeneration of the free nucleophile. The common architecture of the catalytic triad is found in a variety of other proteases like trypsin,[22] subtilisin,[23] elastase,[24] α-lytic protease,[25] and NS3 from Dengue virus type II,[26] yet a diversity of nucleophiles abound, such as in cysteine proteases[27] and more recently in aminoglycoside acetyltransferases.[6b, c]
Figure 2. Low barrier hydrogen bonds in catalytic triads.
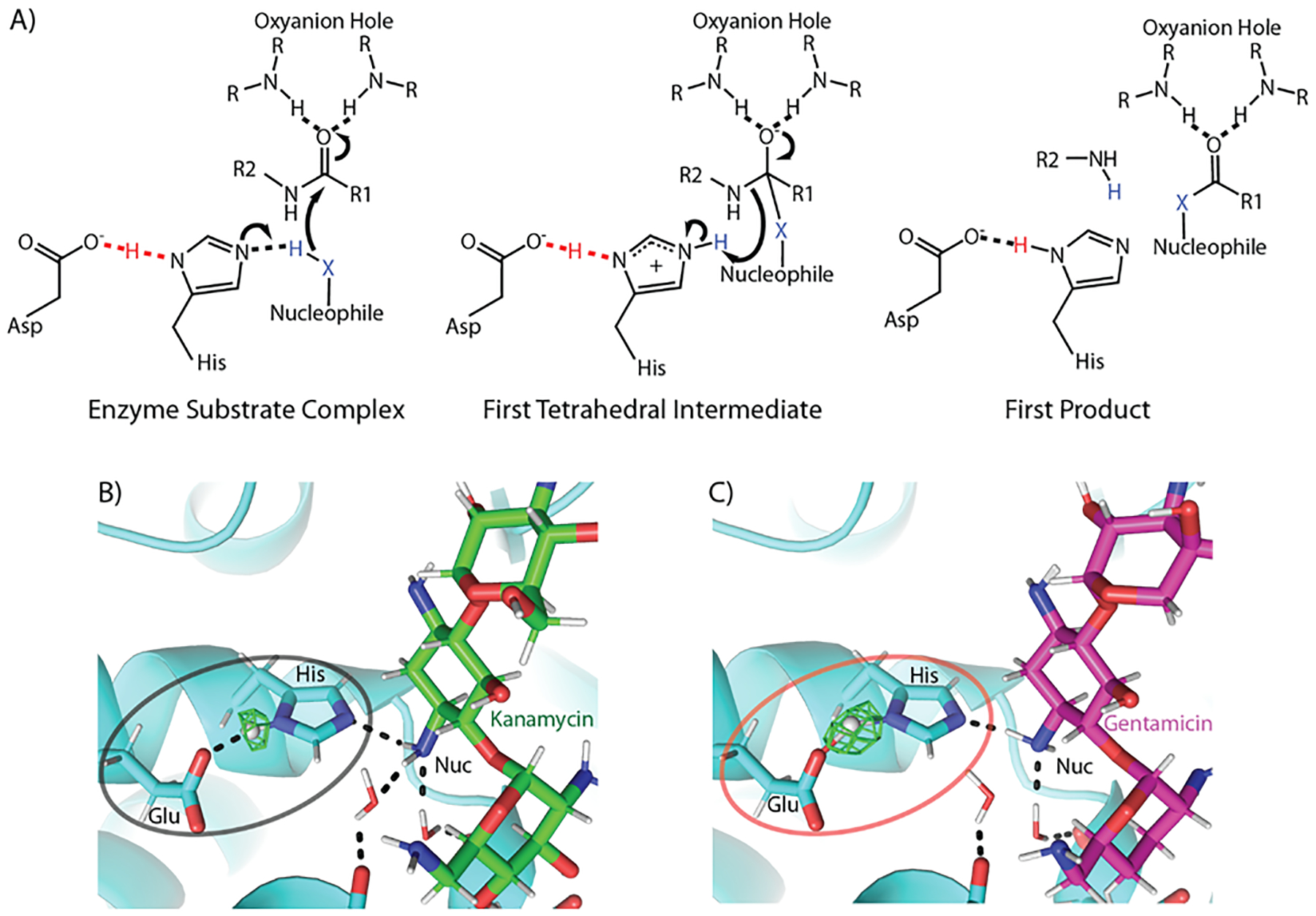
A) Three states of a general catalytic triad mechanism are shown. In the starting enzyme substrate complex, a hydrogen bond exists between the aspartic acid and the histidine Nδ, an interaction found in some instances to be an LBHB, increases the basicity of the histidine Nε (left). This permits the abstraction of a proton from the nucleophile, which subsequently attacks the peptide bond and generates the first tetrahedral intermediate (center). The first intermediate collapses resulting in the first two products of the reaction (right). R groups indicate continuation of the protein chain. B) The local chemical environment of the non-canonical (Glu-His-antibiotic amine) catalytic triad found in an aminoglycoside acetyltransferase dictates the type of hydrogen bond when bound to B) kanamycin and C) gentamicin. The Fo-Fc nuclear omit density for the hydrogen atom involved in the hydrogen bond between the catalytic residues is shown in green. For the least catalytically preferred substrate (kanamycin), a canonical hydrogen bond is found, whereas for one of the best turned over antibiotics (gentamicin) a low barrier hydrogen bond is found.
The initial hallmark of an LBHB in the serine protease family, an unusually low field proton NMR signal observed in chymotrypsin,[28], was also found in trypsin[29] and α-lytic protease.[30] Based on the results obtained by measuring the chemical shifts in model small molecule compounds, Frey et al. postulated that LBHBs can be formed in a protein active site if the difference in the pKa values of the heavy atoms is close to zero, geometry is favorable and other H-bonding possibilities are not available.[2a] According to their proposed mechanism, an LBHB forms between the histidine and the aspartic acid in the protonated state of the catalytic triad as a means to stabilize the transition-state complex while reducing the activation energy and increasing the rate of catalysis through an energetic effect on the order of 12–20 kcal/mol. Similar to the observations by Frey et al, Cleland and Kreevoy also hypothesized that formation of an LBHB is only permissible in the transition-state complex, where a conventional hydrogen bond converts into an LBHB due to the favorable conditions provided by the microenvironment of the active site.[5c]
The underlying LBHB hypothesis has been the topic of a series of theoretical and experimental studies aimed at investigating its existence and controversial energetic contribution to catalysis. The first rebuttal of the hypothesis was proposed by Warshel et al., less than a year after the first set of papers on the topic, on the grounds that purely electrostatic factors are responsible for the stabilization of hydrogen bonds in “condensed” phase systems, such as proteins. Thus the catalytic effect of an LBHB can also be considered largely electrostatic.[19] It was also maintained that the environment of an enzyme active site leads to polarization of the hydrogen bond to attain a large solvation energy, thus favoring an asymmetrical hydrogen bond configuration over the symmetrical one of an LBHB (Figure 1) and resulting in LBHBs having an anti-catalytic effect. This is caused by the polar microenvironment of active site causing the solvation of the hydrogen bond to achieve electrostatic stabilization of the ionic transition state.[31] As has been demonstrated in the case of SN2-type reactions, solvation energy for an asymmetric configuration of the hydrogen bond, where the hydrogen atom is localized to one of the heavy atoms, is much larger compared to that of a delocalized charge system of an LBHB.[32] Thus in an enzyme active site, an asymmetric non-LBHB configuration would be preferred. Additionally, it was advocated that enzymes promote a pre-organized polar microenvironment in the active site, which allows for transition state stabilization and facilitates catalysis, instead of the process being driven by LBHB formation. Theoretical studies on several other enzyme systems, like trypsin,[33] subtilisin,[34] carbonic anhydrase,[35] triose phosphate isomerase,[36] staphylococcal nuclease[37] have also reported that electrostatic factors suffice for transition state stabilization and the resultant catalytic effects and no other explanations need be invoked.
The observations made by Warshel were subsequently refuted by Frey and Cleland. It was argued that the microenvironment found in proteins cannot be assigned as a liquid solvent like state and the idea that all hydrogen bonds in a condensed state are weak, with an energy of ~5 kcal/mol, was considered incorrect (Response to [19]). Countering Warshel’s argument it was also maintained that the pKa values of the groups engaged in hydrogen bonding need not match under aqueous conditions, rather only in the microenvironment of an enzyme’s active site. Cleland and Kreevoy also argued that most chemists prefer to reserve the term “electrostatic” for first-order coulombic interactions, therefore, all effects cannot be included in electrostatics and that by this criterion hydrogen bonds are not simply electrostatic (Response to [19]). Further experimental studies were carried out by Cassidy et al., where they studied the NMR properties of chymotrypsin in complex with trifluoromethyl ketones (TFKs), which are chemical mimetics of the serine protease transition state[38]. Based on these studies, they postulated that LBHB formation is induced upon substrate binding and leads to steric compression between the catalytic histidine and aspartate. LBHB formation stabilizes the tetrahedral intermediate by relieving the steric strain between these residues. This mechanism aids in increasing the basicity of the histidine while making the proton abstraction from the serine more effective. The difference in free-energy for formation of an LBHB in the chymotrypsin-TFK complex and the conventional hydrogen bond in the case of chymotrypsin-imidazolium ion was found to be 7 kcal/mol, in agreement with the observations made by Frey et al.[2a] Based on their results, Cassidy et al. concluded that LBHBs are a significant, but not the sole factor contributing to catalysis.[38] Additional interactions between the substrate and the active site residues were proposed to also aid in catalysis by facilitating the proper orientation of the reactive groups.
Schutz and Warshel continued to further question the LBHB hypothesis based on results from theoretical study of chymotrypsin in complex with TFK, using a protein dipoles Langevin dipoles (PDLD/S-LRA) model for calculating electrostatic energies and pKa values, and an empirical valence bond (EVB) method based calculation for examining the proton transfer profile.[39] Their observations supported their previous hypothesis that enzymes function by providing a pre-organization effect and that the LBHB proposal corresponds to an anti-catalytic effect, since it implies that the ionic state is destabilized in the enzyme. Based on the results of their calculations, they demonstrated that experimental LBHB definitions are equally consistent with SIHBs. Additionally, a positive free-energy profile was obtained from both the PDLD/S-LRA and EVB calculations, in the absence or presence of TFK, countering the existence of an LBHB. The authors maintained that the sole criterion to conclude the existence of LBHBs should not be the distance between the donor and acceptor moieties or the strength of hydrogen bond, or NMR chemical shifts or fractionation factors. The pKa values of the donor and acceptor moieties are crucial in determining the energy barrier required to move the hydrogen atom between the two moieties, thus free-energy profile of proton transfer needs to be evaluated. It was also suggested that another important factor to consider is the energetics of the different ionization states of the system. The relative contribution of the relevant resonance structures needs to be considered while designating an LBHB to be catalytically important.[39]
More recently, Ishida adopted two complementary approaches to study the downfield proton chemical shift observed in trypsin; an ab initio QM/MM framework to calculate the NMR chemical shifts of the histidine imidazole ring, and QM/MM calculations combined with molecular dynamics free-energy perturbation (MD-FEP) simulations to calculate free-energy profile for proton transfer.[40] A largely downfield shift of 8.4 ppm was observed for the imidazole hydrogen atom, which the author attributed to be arising from an electrostatic interaction between the histidine and negatively charged aspartate. The free-energy profile along the proton-transfer coordinate implied that the proton resides with the histidine, instead of being shared with the aspartate. In spite of the canonical hydrogen bond energy profile and the absence of the steric compression induced by substrate binding, the calculated chemical shifts were in agreement with the experimentally determined values. The downfield shift was thus concluded to result from the strong electrostatic interaction between the protonated histidine and ionized aspartate, which de-shields the electronic environment of the hydrogen bonding proton. This analysis concluded that these observations can be explained based on the concept of electrostatic stabilization alone, without invoking a covalent-like bonding character nor the symmetrical double well potential of an LBHB.[40]
In addition to the theoretical studies refuting the existence of LBHB in serine proteases, there have been some reports of experimental studies contradicting the LBHB hypothesis as well. X-ray crystallography studies of α-lytic protease bound to the peptidyl boronic acid inhibitor revealed that the hydrogen bond in the catalytic triad is a normal ionic hydrogen bond and not an LBHB.[41] In both the structures, the hydrogen bond between the two catalytic residues was too long to be considered as an LBHB (2.755 ± 0.005 Å and 2.734 ± 0.005 Å). Additionally, the electron density indicated the hydrogen atom to be bound to the histidine, leading the authors to conclude it to be a SIHB. Based on their observations, the authors also maintained that an LBHB is not necessary for efficient catalysis by serine proteases. The authors favored the ideas put forward by Warshel et al.,[19, 39] that enzymes are able to catalyze reactions effectively because of a pre-organized microenvironment consisting of several strong ionic HBs in and around the active site. This network of bonds aids in the optimal positioning and stabilization of the intermediates and drives the catalysis, rather than an exceptionally strong LBHB being the sole contributor.
In addition to several ultra-high-resolution X-ray crystal structures (<1.0 Å resolution), neutron crystallographic studies, in which explicit determination of hydrogen-atom positions is possible, should have put an end to the debate of LBHBs in the catalytic triad. Yet, these studies have only added complexities to this story. Similar to α-lytic protease, a SIHB was also observed in case of porcine pancreatic elastase complexed with a peptidic inhibitor to mimic the tetrahedral transition state.[24] The hydrogen bond length between the catalytic histidine and aspartic acid residues was determined to be 2.62 Å. However, the nuclear density maps showed a doubly protonated histidine, indicating the existence of a SIHB rather than an LBHB. Sub-Angstrom crystallographic studies of subtilisin revealed the presence of an LBHB, indicated by the distance between the donor and acceptor atoms (2.62 Å) as well as a hydrogen atom located equidistant between the two heavy atoms.[42] More recently, further direct evidence for LBHB mediated catalytic enhancement was reported in an acetyltransferase, which facilitates catalysis using a novel variation of the catalytic triad.[6b] This previously unidentified non-canonical catalytic triad consists of a histidine and a glutamic acid, while the amine of the substrate that gets acetylated serves as the nucleophile (Figure 2B). Based on X-ray and neutron diffraction studies of a catalytically competent ternary complex, an LBHB was unambiguously identified in the active site, with the proton residing equidistant between the glutamic acid and the histidine. Several other related acetyltransferases, utilize the same catalytic residues and thus likely employ an LBHB as a common catalytic mechanism as well.[43] Yet the earlier studies on the acetyltransferase demonstrated that LBHB formation afforded only a 30-fold increase in the catalytic turnover, a far cry from the enhancement that would be provided by tens of kcals/mol of energy. Interestingly, further studies of the acetyltransferase complexed with other substrates revealed that different types of SHBs exist in the same active site and were employed to catalytically and kinetically distinguish between chemically similar substrates.[6c] Indeed, an LBHB is formed in the case of the most preferred substrate, whereas a conventional hydrogen bond is found in the case of the least preferred one[6c] (Figure 2B). This study provided the first ever experimental demonstration of how the electrostatic modulation of a SHB is linked to catalytic selection, but this hasn’t been tested by theoretical studies thus far. Moreover, the catalytic enhancement is far below what should be achieved through the proposed energetic consequences of an LBHB.
3. Short hydrogen bonds in photoactive yellow protein
Photoactive yellow protein (PYP) is a blue light photoreceptor where the thermal reaction cycle entails conformational changes associated with a photon-induced proton transfer process.[44] The para-coumaric acid (pCA) chromophore of PYP undergoes a trans-to-cis isomerization upon absorption of a photon, concomitantly triggering changes in the hydrogen-bonding network with the protein.[45] PYP was one of the first proteins with crystallographic evidence of a functionally important SHB[46] (Figure 3). The SHBs between the pCA and Glu46 and Tyr42 have been observed to be important for the photo-cycling and stability of PYP,[45a, 47] while another residue located adjacent to the chromophore pocket (Arg52) has been proposed to act as a counterion to the negative charge on the pCA molecule[46, 48] (Figure 3). There has been quite some contention regarding the protonation state of Arg52 and the location of the two hydrogen atoms involved in SHBs. Based on neutron and X-ray crystallographic structures, the position of hydrogen atoms in the SHBs of PYP were explicitly determined by Yamaguchi et al.[49] These studies demonstrated the SHB between pCA and Glu46 to be an LBHB, whereas the interaction with Tyr42 was found to be a SIHB (Figure 3).[49] In addition to unambiguously identifying the LBHB in PYP, it was also determined that Arg52 was unexpectedly deprotonated. In order to explain the occurrence of a neutral Arg52 it was postulated that the strong bond strength afforded by an LBHB energetically compensates for the presence of the negative charge of an anionic pCA located in the PYP core. The direct determination of proton positions in the SHBs of PYP has led to a series of contradictory experimental and computational studies of the SHBs as well as the protonation state of Arg52.
Figure 3. The chromophore binding site in photoactive yellow protein (PYP).
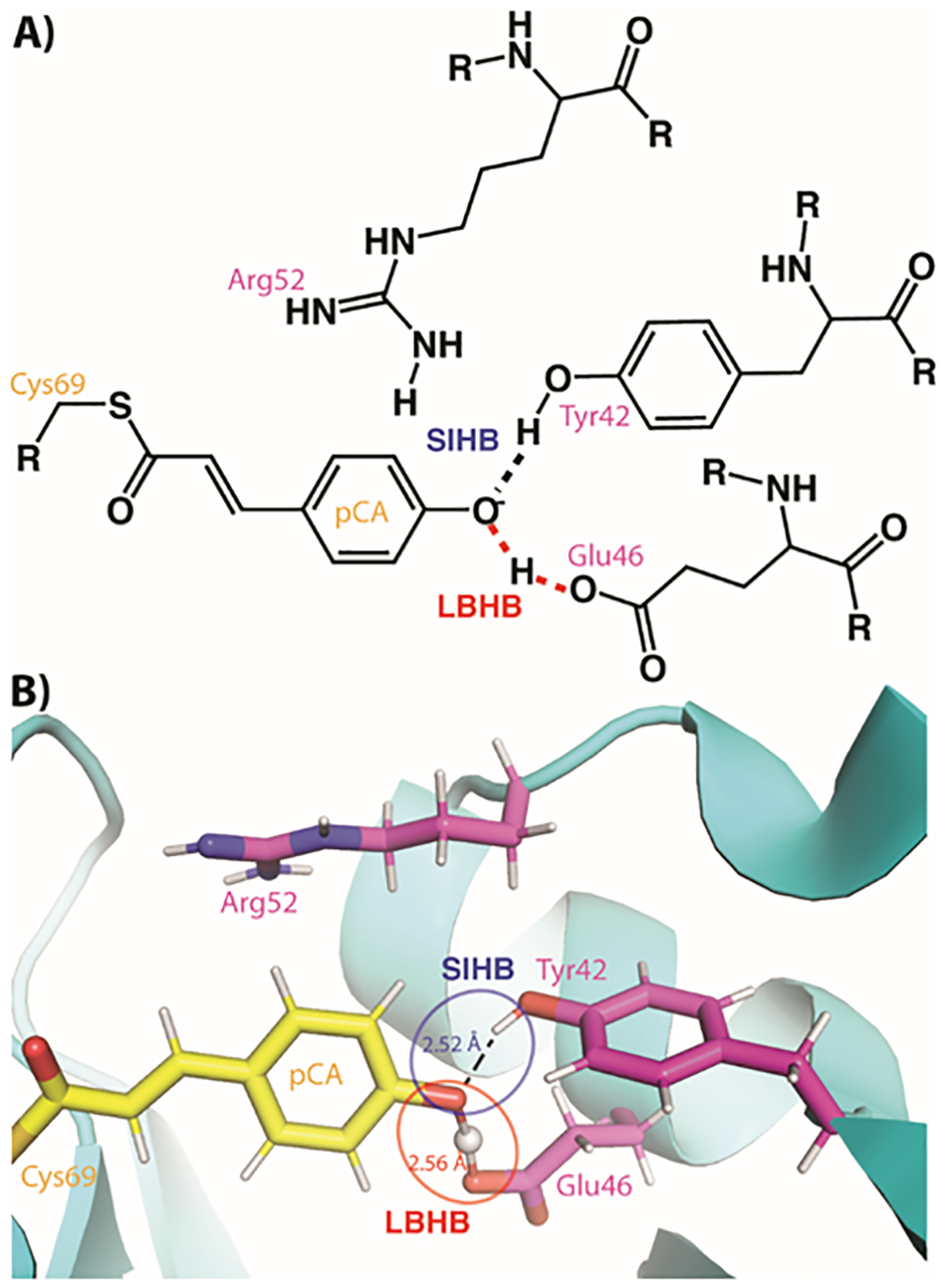
A) A schematic of the PYP chromophore (pCA) binding site with the two types of SHBs indicated. R groups indicate continuation of the protein chain. B) The neutron crystal structure of the PYP chromophore active site with the two types of SHBs indicated.
Based on the provocative nature of the LBHB and neutral protonation state of Arg52, a large number of molecular simulations have been carried out on the Yamaguchi PYP structure. Using a large-scale QM/MM approach, Saito et al.[50] determined that the bond lengths of the SHB between pCA, Tyr42 and Glu46 were supportive of the neutron crystal structure, yet the hydrogen atom involved in the Glu46 SHB was found to be a SIHB, rather than an LBHB (Figure 1). Thus, it was concluded that an LBHB is not required to stabilize the anionic chromophore, and that some stabilization is achieved by the SHB between Tyr42 and the chromophore. Additionally, the short bond length between Glu46 and pCA were said to be explained by an electrostatic interaction, rather than an LBHB, mimicking the thoughts of Warshel on the nature of LBHBs in general. In another study by Graen et al.,[51] QM/MM calculations were performed to determine the extent of delocalization of the Glu46 bound hydrogen atom under vacuum, in crystal and in solution conditions. The calculations in this study strongly supported the presence of a protonated Arg52 and an LBHB was found to exist only under vacuum, where the authors attributed it to a shift in the potential minimum due to an anisotropic protein environment in vacuum.
The neutral Arg52 found in the neutron structure was also questioned by other studies, given that the intrinsic pKa value was determined to be 13.8 and that the pH of the crystal structure state was 9.[52] pKa calculations were performed to determine the protonation states of all titratable sites of PYP and it was determined that Arg52 is in a protonated form.[50] Independent QM/MM analyses also supported the occurrence of a protonated Arg52, as models with a deprotonated Arg52 had a larger RMSD (0.35 Å) than the protonated (0.14 Å) one. The existence of a protonated Arg52 in PYP was also argued for in a study where differences between the pKa values of Arg52 in crystal and in solution were calculated.[51] An upward shift of two pKa units was reported in the crystal, as compared to the solution, thus making it unlikely for a deprotonated Arg52 to be present. Furthermore, re-examination of the crystallographic data also indicated that density maps were insufficient to conclude a fully deprotonated Arg52.
In addition to theoretical studies of the SHBs, NMR experiments have also challenged the positions of the protons in the PYP crystallographic models. Typically, shorter donor-acceptor distances of an LBHB translate to a downfield NMR chemical shift for the shared hydrogen atom. Consequently, the chemical shift of a hydrogen atom in an LBHB is observed to be in the range of 17–19 ppm, whereas a value of 20–22 ppm has been reported in the case of single-well hydrogen bonds.[53] Yet in a study by Sigala et al., two far-downfield peaks at 13.7 and 15.2 ppm in 1H-NMR spectrum were reported to correspond to the protons bound to Tyr42 and Glu46 respectively,[54] countering the presence of an LBHB. A neutral Arg52 was also refuted in other NMR experiments that showed there were four protons in the head group, indicating a protonated guanidinium in solution as well.[55]
In the midst of the multiple reports challenging the Yamaguchi structure,[49] there have been several reports in agreement with the crystallographic proton positions. Hirano and Sato reasoned that the protein electrostatic environment influences the spectral properties of pCA and consequently, the electronic properties of the LBHB and Arg52. They used the Own N-layered Integrated molecular Orbital and Molecular mechanics (ONIOM) method, to test the existence of an LBHB in PYP.[56] ONIOM uses hybrid quantum and classical calculation to allow modeling of the multiple electronic states and charge transfer associated with protein function and the nature of hydrogen bonds. Two models were used; one based on the neutron structure, with an LBHB and a deprotonated Arg52 (called the deprotonated model), and a second model that consisted of a protonated Arg52, a neutral Glu46 with the proton residing on the carboxyl group and a deprotonated and negatively charged chromophore. They calculated the potential energy curve for proton migration between Glu46 and pCA in both models and it was observed that the deprotonation of Arg52 is crucial for the existence of an LBHB.
Kanematsu et al. concluded that in order to be able to accurately obtain molecular geometries and chemical shifts from computational studies, fluctuations in hydrogen bond due to the nuclear quantum effects also need to be included in the model. An extended ONIOM with multicomponent quantum mechanics (MC-QM) method with explicit inclusion of nuclear quantum effects was used to study the changes in PYP geometry and isotope shifts due to the surrounding protein environment as well as quantum and thermal effects[57]. According to their results, the elongation of the O-H bond of Glu46 can be explained by the presence of a neutral Arg52. In another study by Kanematsu et al., they performed vibrational analysis of the Glu46 SHB using both equilibrium structure as well as the PYP crystal structure and the results supported the occurrence of an LBHB in the deuterated PYP crystal structure[58]. The equilibrium structure had an O-O distance of 2.47 Å, giving rise to a single-well shape potential energy curve where the experimentally determined crystal structure had an O-O distance of 2.56 Å and a double-well shape with a low energy barrier. The vibrationally averaged O-H bond length for the crystal structure was found to be in agreement with the corresponding crystal structure values, whereas the values for the equilibrium structure were underestimated. This suggests that the elongation of the O-O distance from the equilibrium structure to the experimentally determined structure occurs through quantum mechanical or thermal fluctuation.
An interesting case for the Yamaguchi neutron diffraction studies was made by Nadal-Ferret et al., who argued that the conflicting reports in the literature can be explained by taking into account different states of the PYP structure used, crystalline versus the solution state.[59] QM/MM simulations with density functional theory (DFT) based treatment of quantum region were carried out to compute the potential energy profile for the transfer of a hydrogen atom from Glu46 to pCA for both neutral and protonated Arg52. The energy profile along the proton-transfer coordinate was found to be very anharmonic. Delocalization of the Glu46 bound hydrogen atom was observed to occur only when Arg52 was deprotonated. Also, the chemical shift of the ground vibrational state of deuterated PYP was calculated to be 18.5 ppm, a value indicative of an LBHB. Contradicting results were obtained in the case of a solvated system, namely, a protonated Arg52 and a conventional hydrogen bond between Glu46 and the chromophore. It was concluded that the solvation of the system leads to protonation of Arg52, and the resulting thermal agitation in solution weakens the LBHB causing it to revert to a normal hydrogen bond.
Recent further experimental support for the Yamaguchi structure came in the form of neutron crystallographic studies of the E46Q mutant of PYP.[60] Overall, the structure was found to be very similar to the original. However, the Glu46 LBHB was found to be lengthened in the E46Q mutant to 2.85 ± 0.04 Å. In the E46Q mutant, the Glu46 deuterium atom was experimentally found to be covalently bound to Gln46, with a bond length of 1.02 Å, thus implying the conversion of LBHB to a canonical hydrogen bond. Additionally, inspection of the nuclear density maps revealed that the E46Q mutant showed more prominent nuclear density for the deuterium atoms of the guanidinium group than the wild type PYP. This led the authors to suggest Arg52 was more highly protonated in E46Q compared to the neutral Arg52 wild type PYP. These results conclusively demonstrated that the SHB vicinal to the chromophore influences the pKa of Arg52 in crystalline PYP.
4. Conclusions
It has been stated that the most conclusive way to categorize a SHB is through determination of the hydrogen atom position through neutron diffraction, likely in addition to ultra-high-resolution X-ray crystallography.[59] Indeed, at the outset we intended to demonstrate the position that the direct determination of hydrogen atom location is the gold-standard for the study of SHBs and the identification of SIHBs, LBHBs or SWHBs. The fact that we even failed to convince ourselves of this is only representative of the contentious nature of the role of SHBs in biology, coupled to the lack of consistency among structure, biochemistry and simulations. While we maintain that hydrogen atom location is critical to accurately define the type of SHB involved, relying solely on a single experimental or theoretical technique can lead to contradicting observations and interpretations due to the inherent limitations and errors associated with the individual techniques, thus confounding the SHB narrative. We presented in detail the two SHB-containing archetypal proteins that above and beyond have been studied in a level of detail where one would expect a consistent story to arise, yet that evidently is not the case thus far. The inconsistency in tying together theoretical and experimental studies seems to be the only consistency in the study of SHBs; so what if anything can be done to fully delineate the true thermodynamic effect of SHBs in biology and where is the disconnect among the different methodologies?
Classical computational methods are fast, but their accuracy depends on the quality of underlying force-fields. It has been shown that hydrogen bonds depend on force-field terms.[61] A large-scale study of classical force-field (with fixed atomic charges) based simulations showing the successes and failures for hydrogen bonding patterns in different protein and enzyme systems remains missing. With the emergence of polarizable force-fields,[62] classical methods may offer future opportunities for investigating the nature of hydrogen bonds in proteins. More commonly, the quantum methods (and QM/MM) have been used for investigating proteins and enzymes[63] particularly involving biochemical reactions with proton or hydride transfer.[64]
Hydrogen atoms are light particles and their quantum behavior (including tunneling) adds another layer of complexity for computational studies. Even standard QM methods have been unable to fully explain the observed experimental observations, therefore, computational methods that include the quantum behavior of the hydrogen nuclei have been developed.[65] These methods include nuclear wavefunction methods[66] and vibrational analysis, and have been successfully able to explain hydrogen bonding and catalytic rates associated with enzymes that involve hydride transfer.[64, 67] Moreover, proteins and enzymes do not stay fixed in a single conformation, and conformational sampling drives the catalytic cycle of enzymes.[68] Now it is widely acknowledged that the location of hydrogen between heavy atoms can be subtly controlled by interconversion of protein conformations between different populations.[69] Unlike most experimental techniques that inherently collect information representing ensembles, computational methods need to explicitly include means to include conformational sampling at different scales.
An enzyme’s active site environment is also crucial in the formation and the strength of bonds that drive catalysis. In the case of LBHBs, the distance at which the pKa values of the donor and acceptor atoms match depends on the local environment.[6c] This challenges the use of a threshold distance or the downfield chemical shift of the proton as the sole criterion for defining LBHBs, since both ionic and covalent hydrogen bonds can exist at shorter than conventional distances. Thus, a multitude of complementary experimental approaches need to be adopted in order to definitively ascertain the existence of LBHBs. Crystallographic data, H/D fractionation factors, chemical shifts, isotope effects on NMR chemical shifts and infrared stretching frequencies, energetics of hydrogen bond, proton transfer profiles are all critical but not mutually exclusive pieces of information required to be able to irrefutably demonstrate the existence of an LBHB. Perhaps the most rational argument regarding reconciliation of the discrepancies among structure, biochemistry (NMR) and simulations was also made in studies on PYP, where it was explicitly demonstrated and discussed that the state of matter affects the results.[59] Indeed, the microenvironment of the crystal is vastly different from that in the solution environment used for biochemical studies, yet it is often the coordinates from crystal structures that are used for molecular simulations in a pseudo-hydrated environment. The last point we will suggest, though, is that perhaps the theory underlying the energetic contribution of LBHBs in biological systems needs to be further refined to better match what is observed in simulations and biochemical experiments, therefore we would like to say that the debate over the energetic role of SHBs in biology is still in its infancy.
Acknowledgements
PKA acknowledges a grant from the National Institute of General Medical Sciences of the National Institutes of Health under award number GM105978. This work was supported by the American Lebanese Syrian Associated Charities (ALSAC) and St. Jude Children’s Research Hospital. The authors would like to thank Norman Zhaowen for assistance with graphic design.
Biographies

Prashasti Kumar received her B.Sc. in Biochemistry from the University of Delhi and her M.Sc. in Plant Biotechnology in 2011. She joined the University of Tennessee for her doctoral studies where she received a Ph.D. in 2018. She is presently pursuing postdoctoral work studying chemical modulators of protein degradation at the Icahn School of Medicine at Mt. Sinai.

Pratul Agarwal is the Assistant Vice President of Research for Cyber-Infrastructure, and the Director of High-Performance Computing Center at Oklahoma State University. He is also Associate Professor in Department of Physiological Sciences. He has over 15 years of experience in high performance computing and scientific research. He previously held positions at University of Tennessee, Knoxville and Oak Ridge National Laboratory.

Matthew Cuneo graduated from the University of North Carolina at Charlotte with a B.Sc. in Biology and a B.A. in Chemistry in 2002, and a Ph.D. in Biochemistry from Duke University in 2007. After training at the National Institutes of Health he joined Oak Ridge National Laboratory. In 2018 he joined St. Jude Children’s Research Hospital where he works on understanding human diseases at a molecular level.
References
- [1].Herschlag D, Pinney MM, Biochemistry 2018, 57, 3338–3352. [DOI] [PubMed] [Google Scholar]
- [2].a) Frey PA, Whitt SA, Tobin JB, Science 1994, 264, 1927–1930; [DOI] [PubMed] [Google Scholar]; b) Cleland WW, Frey PA, Gerlt JA, Journal of Biological Chemistry 1998, 273, 25529–25532. [DOI] [PubMed] [Google Scholar]
- [3].Cleland WW, Arch Biochem Biophys 2000, 382, 1–5. [DOI] [PubMed] [Google Scholar]
- [4].a) Kreevoy MM, Liang TM, Journal of the American Chemical Society 1980, 102, 3315–3322; [Google Scholar]; b) Cleland WW, Biochemistry 1992, 31, 317–319; [DOI] [PubMed] [Google Scholar]; c) Gerlt JA, Kreevoy MM, Cleland W, Frey PA, Chem Biol 1997, 4, 259–267. [DOI] [PubMed] [Google Scholar]
- [5].a) Gerlt JA, Gassman PG, Journal of the American Chemical Society 1993, 115, 11552–11568; [Google Scholar]; b) Gerlt JA, Gassman PG, Biochemistry 1993, 32, 11943–11952; [DOI] [PubMed] [Google Scholar]; c) Cleland WW, Kreevoy MM, Science 1994, 264, 1887–1890. [DOI] [PubMed] [Google Scholar]
- [6].a) Hosur MV, Chitra R, Hegde S, Choudhury RR, Das A, Hosur RV, Crystallography Reviews 2013, 19, 3–50; [Google Scholar]; b) Kumar P, Serpersu EH, Cuneo MJ, Sci Adv 2018, 4, eaas8667; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kumar P, Agarwal PK, Waddell MB, Mittag T, Serpersu EH, Cuneo MJ, Angew Chem Int Ed Engl 2019, 58, 16260–16266. [DOI] [PubMed] [Google Scholar]
- [7].a) Weber IT, Waltman MJ, Mustyakimov M, Blakeley MP, Keen DA, Ghosh AK, Langan P, Kovalevsky AY, J Med Chem 2013, 56, 5631–5635; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gerlits O, Wymore T, Das A, Shen CH, Parks JM, Smith JC, Weiss KL, Keen DA, Blakeley MP, Louis JM, Langan P, Weber IT, Kovalevsky A, Angew Chem Int Ed Engl 2016, 55, 4924–4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Coates L, Tuan HF, Tomanicek S, Kovalevsky A, Mustyakimov M, Erskine P, Cooper J, J Am Chem Soc 2008, 130, 7235–7237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Nichols DA, Hargis JC, Sanishvili R, Jaishankar P, Defrees K, Smith EW, Wang KK, Prati F, Renslo AR, Woodcock HL, Chen Y, J Am Chem Soc 2015, 137, 8086–8095; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Pemberton OA, Noor RE, Kumar MVV, Sanishvili R, Kemp MT, Kearns FL, Woodcock HL, Gelis I, Chen Y, Proc Natl Acad Sci U S A 2020, 117, 5818–5825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Harris TK, Abeygunawardana C, Mildvan AS, Biochemistry 1997, 36, 14661–14675. [DOI] [PubMed] [Google Scholar]
- [11].a) Lee JE, Smith GD, Horvatin C, Huang DJ, Cornell KA, Riscoe MK, Howell PL, J Mol Biol 2005, 352, 559–574; [DOI] [PubMed] [Google Scholar]; b) Banco MT, Mishra V, Ostermann A, Schrader TE, Evans GB, Kovalevsky A, Ronning DR, Proc Natl Acad Sci U S A 2016, 113, 13756–13761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zhao Q, Abeygunawardana C, Gittis AG, Mildvan AS, Biochemistry 1997, 36, 14616–14626. [DOI] [PubMed] [Google Scholar]
- [13].Dai S, Funk LM, von Pappenheim FR, Sautner V, Paulikat M, Schroder B, Uranga J, Mata RA, Tittmann K, Nature 2019, 573, 609–613. [DOI] [PubMed] [Google Scholar]
- [14].Dajnowicz S, Johnston RC, Parks JM, Blakeley MP, Keen DA, Weiss KL, Gerlits O, Kovalevsky A, Mueser TC, Nat Commun 2017, 8, 955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Selvaraj B, Kocaman S, Trifas M, Serpersu EH, Cuneo MJ, ACS Catalysis 2020, 10, 3548–3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].a) Wang Z, Luecke H, Yao N, Quiocho FA, Nature structural biology 1997, 4, 519–522; [DOI] [PubMed] [Google Scholar]; b) Elias M, Wellner A, Goldin-Azulay K, Chabriere E, Vorholt JA, Erb TJ, Tawfik DS, Nature 2012, 491, 134–137; [DOI] [PubMed] [Google Scholar]; c Luecke H, Quiocho FA, Nature 1990, 347, 402–406. [DOI] [PubMed] [Google Scholar]
- [17].Gerlits OO, Coates L, Woods RJ, Kovalevsky A, Biochemistry 2017, 56, 4747–4750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].a) Oltrogge LM, Boxer SG, ACS Cent Sci 2015, 1, 148–156; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nadal-Ferret M, Gelabert R, Moreno M, Lluch JM, Phys Chem Chem Phys 2015, 17, 30876–30888. [DOI] [PubMed] [Google Scholar]
- [19].Warshel A, Papazyan A, Kollman PA, Science 1995, 269, 102–106. [DOI] [PubMed] [Google Scholar]
- [20].a) Usher KC, Remington SJ, Martin DP, Drueckhammer DG, Biochemistry 1994, 33, 7753–7759; [DOI] [PubMed] [Google Scholar]; b) Stratton JR, Pelton JG, Kirsch JF, Biochemistry 2001, 40, 10411–10416; [DOI] [PubMed] [Google Scholar]; c) Jang DS, Choi G, Cha HJ, Shin S, Hong BH, Lee HJ, Lee HC, Choi KY, Mol Cells 2015, 38, 409–415; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Schwartz B, Drueckhammer DG, Journal of the American Chemical Society 1995, 117, 11902–11905. [Google Scholar]
- [21].Hibbert F, Emsley J, in Advances in Physical Organic Chemistry , Vol. 26, 1990, pp. 255–379. [Google Scholar]
- [22].Schiebel J, Gaspari R, Wulsdorf T, Ngo K, Sohn C, Schrader TE, Cavalli A, Ostermann A, Heine A, Klebe G, Nat Commun 2018, 9, 3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kidd RD, Sears P, Huang DH, Witte K, Wong CH, Farber GK, Protein Sci 1999, 8, 410–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Tamada T, Kinoshita T, Kurihara K, Adachi M, Ohhara T, Imai K, Kuroki R, Tada T, J Am Chem Soc 2009, 131, 11033–11040. [DOI] [PubMed] [Google Scholar]
- [25].Molina PA, Jensen JH, The Journal of Physical Chemistry B 2003, 107, 6226–6233. [Google Scholar]
- [26].Agback P, Agback T, Sci Rep 2018, 8, 10078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ganesan R, Mittl PR, Jelakovic S, Grutter MG, J Mol Biol 2006, 359, 1378–1388. [DOI] [PubMed] [Google Scholar]
- [28].Robillard G, Shulman RG, J Mol Biol 1972, 71, 507–511. [DOI] [PubMed] [Google Scholar]
- [29].Porubcan MA, Neves DE, Rausch SK, Markley JL, Biochemistry 1978, 17, 4640–4647. [DOI] [PubMed] [Google Scholar]
- [30].Bachovchin WW, Proc Natl Acad Sci U S A 1985, 82, 7948–7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Warshel A, Aqvist J, Creighton S, Proc Natl Acad Sci U S A 1989, 86, 5820–5824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].a) Chandrasekhar J, Jorgensen WL, Journal of the American Chemical Society 1985, 107, 2974–2975; [Google Scholar]; b) Hwang JK, King G, Creighton S, Warshel A, Journal of the American Chemical Society 1988, 110, 5297–5311. [Google Scholar]
- [33].a) Warshel A, Russell S, Journal of the American Chemical Society 1986, 108, 6569–6579; [Google Scholar]; b) McGrath ME, Vasquez JR, Craik CS, Yang AS, Honig B, Fletterick RJ, Biochemistry 1992, 31, 3059–3064. [DOI] [PubMed] [Google Scholar]
- [34].a) Hwang JK, Warshel A, Biochemistry 1987, 26, 2669–2673; [DOI] [PubMed] [Google Scholar]; b) Rao SN, Singh UC, Bash PA, Kollman PA, Nature 1987, 328, 551–554. [DOI] [PubMed] [Google Scholar]
- [35].Åqvist J, Warshel A, Journal of Molecular Biology 1992, 224, 7–14. [DOI] [PubMed] [Google Scholar]
- [36].a) Alagona G, Desmeules P, Ghio C, Kollman PA, Journal of the American Chemical Society 1984, 106, 3623–3632; [Google Scholar]; b) Alagona G, Ghio C, Kollman PA, Journal of Molecular Biology 1986, 191, 23–27. [DOI] [PubMed] [Google Scholar]
- [37].Aaqvist J, Warshel A, Biochemistry 1989, 28, 4680–4689. [DOI] [PubMed] [Google Scholar]
- [38].Cassidy CS, Lin J, Frey PA, Biochemistry 1997, 36, 4576–4584. [DOI] [PubMed] [Google Scholar]
- [39].Schutz CN, Warshel A, Proteins: Structure, Function, and Bioinformatics 2004, 55, 711–723. [DOI] [PubMed] [Google Scholar]
- [40].Ishida T, Biochemistry 2006, 45, 5413–5420. [DOI] [PubMed] [Google Scholar]
- [41].Fuhrmann CN, Daugherty MD, Agard DA, J Am Chem Soc 2006, 128, 9086–9102. [DOI] [PubMed] [Google Scholar]
- [42].Kuhn P, Knapp M, Soltis SM, Ganshaw G, Thoene M, Bott R, Biochemistry 1998, 37, 13446–13452. [DOI] [PubMed] [Google Scholar]
- [43].Kumar P, Selvaraj B, Serpersu EH, Cuneo MJ, J Med Chem 2018, 61, 10218–10227. [DOI] [PubMed] [Google Scholar]
- [44].Sprenger WW, Hoff WD, Armitage JP, Hellingwerf KJ, J Bacteriol 1993, 175, 3096–3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].a) Xie A, Hoff WD, Kroon AR, Hellingwerf KJ, Biochemistry 1996, 35, 14671–14678; [DOI] [PubMed] [Google Scholar]; b) Imamoto Y, Kataoka M, Tokunaga F, Biochemistry 1996, 35, 14047–14053; [DOI] [PubMed] [Google Scholar]; c) Imamoto Y, Kataoka M, Photochem Photobiol 2007, 83, 40–49. [DOI] [PubMed] [Google Scholar]
- [46].Anderson S, Crosson S, Moffat K, Acta Crystallogr D Biol Crystallogr 2004, 60, 1008–1016. [DOI] [PubMed] [Google Scholar]
- [47].Imamoto Y, Mihara K, Hisatomi O, Kataoka M, Tokunaga F, Bojkova N, Yoshihara K, J Biol Chem 1997, 272, 12905–12908. [DOI] [PubMed] [Google Scholar]
- [48].Borgstahl GE, Williams DR, Getzoff ED, Biochemistry 1995, 34, 6278–6287. [DOI] [PubMed] [Google Scholar]
- [49].Yamaguchi S, Kamikubo H, Kurihara K, Kuroki R, Niimura N, Shimizu N, Yamazaki Y, Kataoka M, Proc Natl Acad Sci U S A 2009, 106, 440–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Saito K, Ishikita H, Proc Natl Acad Sci U S A 2012, 109, 167–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Graen T, Inhester L, Clemens M, Grubmuller H, Groenhof G, J Am Chem Soc 2016, 138, 16620–16631. [DOI] [PubMed] [Google Scholar]
- [52].Fitch CA, Platzer G, Okon M, Garcia-Moreno BE, McIntosh LP, Protein Sci 2015, 24, 752–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Frey PA, in Isotope effects in chemistry and biology (Eds.: Kohen A, Limbach H-H), CRC Press, Boca Raton, FL, 2006, pp. 973–993. [Google Scholar]
- [54].Sigala PA, Tsuchida MA, Herschlag D, Proc Natl Acad Sci U S A 2009, 106, 9232–9237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Yoshimura Y, Oktaviani NA, Yonezawa K, Kamikubo H, Mulder FA, Angew Chem Int Ed Engl 2017, 56, 239–242. [DOI] [PubMed] [Google Scholar]
- [56].Hirano K, Sato H, Chemical Physics 2013, 419, 163–166. [Google Scholar]
- [57].Kanematsu Y, Tachikawa M, J Chem Phys 2014, 141, 185101. [DOI] [PubMed] [Google Scholar]
- [58].Kanematsu Y, Kamikubo H, Kataoka M, Tachikawa M, Comput Struct Biotechnol J 2016, 14, 16–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Nadal-Ferret M, Gelabert R, Moreno M, Lluch JM, J Am Chem Soc 2014, 136, 3542–3552. [DOI] [PubMed] [Google Scholar]
- [60].Yonezawa K, Shimizu N, Kurihara K, Yamazaki Y, Kamikubo H, Kataoka M, Scientific Reports 2017, 7, 9361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].a) Kuhrova P, Mlynsky V, Zgarbova M, Krepl M, Bussi G, Best RB, Otyepka M, Sponer J, Banas P, J Chem Theory Comput 2019, 15, 3288–3305; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tian C, Kasavajhala K, Belfon KAA, Raguette L, Huang H, Migues AN, Bickel J, Wang Y, Pincay J, Wu Q, Simmerling C, J Chem Theory Comput 2020, 16, 528–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Inakollu VS, Geerke DP, Rowley CN, Yu H, Curr Opin Struct Biol 2020, 61, 182–190. [DOI] [PubMed] [Google Scholar]
- [63].Senn HM, Thiel W, Angew Chem Int Ed Engl 2009, 48, 1198–1229. [DOI] [PubMed] [Google Scholar]
- [64].Agarwal PK, Billeter SR, Hammes-Schiffer S, The Journal of Physical Chemistry B 2002, 106, 3283–3293. [Google Scholar]
- [65].Li X-Z, Walker B, Michaelides A, Proceedings of the National Academy of Sciences 2011, 108, 6369–6373. [Google Scholar]
- [66].Webb SP, Iordanov T, Hammes-Schiffer S, The Journal of chemical physics 2002, 117, 4106–4118. [Google Scholar]
- [67].Billeter SR, Webb SP, Iordanov T, Agarwal PK, Hammes-Schiffer S, The Journal of Chemical Physics 2001, 114, 6925–6936. [DOI] [PubMed] [Google Scholar]
- [68].Ramanathan A, Savol A, Burger V, Chennubhotla CS, Agarwal PK, Acc Chem Res 2014, 47, 149–156. [DOI] [PubMed] [Google Scholar]
- [69].Hammes-Schiffer S, Acc Chem Res 2006, 39, 93–100. [DOI] [PubMed] [Google Scholar]