

Abstract
As an effective targeted therapy for advanced hepatocellular carcinoma (HCC), sorafenib resistance has been frequently reported in recent years, with the activation of autophagy by cancer cells under drug stress being one of the crucial reasons. Sorafenib treatment could enhance autophagy in HCC cells and autophagy is also considered as an important mechanisms of drug resistance. Therefore, the inhibition of autophagy is a potential way to improve the sensitivity and eliminate drug resistance to restore their efficacy. To determine whether autophagy is involved in sorafenib resistance and investigate its role in the regulation of HepG2 cells’ (an HCC cell line) chemosensitivity to sorafenib, we simultaneously treated HepG2 with sorafenib and 3-Methyladenine (3-MA) (a common autophagy inhibitor). First, by performing cell counting kit 8 cell viability assay, Hoechst 33342 apoptosis staining, and Annexin V-fluorescein isothiocyanate/propidium iodide apoptosis kit detection, we found that both sorafenib and 3-MA effectively inhibitted the proliferative activity of HepG2 cells and induced their apoptosis to a certain extent. This effect was significantly enhanced after these two drugs were combined, which was also confirmed by the increased expression of apoptosis-related proteins. Subsequently, by using AAV-GFP-LC3 transfection methods and transmission electron microscopy, we found that both the number and activity of autophagosomes in HepG2 cells in sorafenib and 3-MA group were significantly reduced, suggesting that autophagy activity was inhibited, and this result was consistent with the expression results of autophagy-related proteins. Therefore, we conclude that 3-MA may attenuate the acquired drug resistance of sorafenib by counteracting its induction of autophagy activity, thus enhancing its sensitivity to advanced HCC therapy.
Keywords: 3-methyladenine, autophagy, drug resistance, hepatocellular carcinoma, sorafenib
Introduction
Primary liver cancer or hepatocellular carcinoma (HCC) is considered to be one of the greatest leading causes of cancer death worldwide due to its high recurrence rate and poor prognosis [1–3]. Despite development of its therapy, HCC remains one of the most difficult cancers to treat. What’s worse, HCC is often diagnosed at advanced stages and unresectable, resulting in the loss of optimal opportunity for effective treatment for many patients [4]. Moreover, traditional systemic chemotherapy does not significantly improve the survival rate [5–7]. Regardless of the treatment strategy applied, the prognosis of patients with advanced HCC remains poor [8]. Sorafenib, a multikinase inhibitor, is the first molecular targeted drug approved by Food and Drug Administration for HCC treatment and has shown some survival benefit in patients with advanced HCC. In the initial phase II study mentioned in ‘The National Comprehensive Cancer Network (NCCN) Guidelines Insights: Hepatobiliary Cancers, version 2. 2019’ [9], recovery of liver function was a marker of response to sorafenib treatment; similarly, another large, double-blinded, placebo-controlled phase III clinical study in ‘Hepatocellular carcinoma: European Society for Medical Oncology (ESMO) Clinical Practice Guidelines for diagnosis, treatment and follow-up’ [10] also showed that sorafenib could increase patient survival. However, the resistance to sorafenib has become a major bottleneck in the treatment of advanced HCC [11,12].
Autophagy is a conservative life process characterized by self-digestion and metabolism, by which cells eliminate and recycle their own damaged proteins and organelles use to cope with environmental changes and maintain homeostasis [13,14]. It is also crucial for the normal physiological function of liver and recovery of disease [15]. On the other hand, as an effector of the immune system, autophagy has been proved to be closely related to immune response by involving in innate and acquired immune processes [16]. The immune system has dual characters, which can either suppress or promote the development of tumor. Therefore, autophagy plays a complex role in the development of tumors [17]. Notably, autophagy can either promote tumor cells death at early stages or represent a protective mechanism against apoptotic cell death under starvation, as well as contribute to resistance against therapy-induced apoptosis in cancer cells [18]. At present, some progresses have been made in studies on the role of autophagy in pathogenesis and treatment of HCC, but the mechanism of autophagy in chemotherapy has not been illuminated [19]. There are three types of autophagy, including microautophagy, macroautophagy and chaperone-mediated autophagy [20]. The difference between these autophagic processes is the substrates delivered to the lysosomes. Macroautophagy is the process during which damaged organelles are first enclosed in double-membrane vesicles (also known as autophagosomes) and then fused with lysosomes to become autophagolysosomes [21]. The autophagy we talk about in general is macroautophagy. The autophagic process can be mainly divided into four steps: (a) formation of the phagophore to wrap the damaged material; (b) elongation and closure of the phagophore followed by autophagosome generation; (c) fusion of autophagosomes and lysosomes to form autolysosomes and (d) content degradation and recycling [22]. To date, over 30 types of autophagy-related proteins (ATGs) have been found to participate in the regulation of autophagy when triggered under stressful conditions, such as starvation, hypoxia and cytotoxicity. For example, autophagosome formation is controlled by the ATG12 and light chain 3 (LC3) conjugation systems. Microtubule-associated protein 1-LC3 (also known as ATG8) is cleaved by protease ATG4 into LC3-I, which is then converted into LC3-II by conjugating with phosphatidylethanolamine [23]. In this process, LC3-II is translocated from the cytoplasm to the autophagosome membrane. LC3-II is considered an important marker for autophagosomes, whereas p62 protein plays an important role in the degradation and recycling of autophagosome [24].
3-Methyladenine (3-MA), a commonly used specific autophagy inhibitor, can block the formation of autophagic vesicles by acting on phosphoinositol 3 phosphate kinase (PI3K). It has been widely used as a pharmacological tool in the autophagy studies because it can enhance chemotherapeutic effects of anticancer drugs. However, it is not clear whether these effects of 3-MA on chemotherapy efficacy act through its inhibition of autophagy. Sheng et al.’s study [25] showed that 3-MA reduced the viability of Hela cells (a human cervix carcinoma cell line) dose-dependently but did not affect the basic autophagy responses, and they concluded that 3-MA itself could induce cell death and apoptosis with no involvement from autophagy. However, Mishima et al. [26,27] had confirmed that by blocking autophagy, the sensitivity of chemotherapy drugs could be enhanced, which could be considered as a new treatment strategy for chronic myelogenous leukemia or laryngeal cancer.
In order to clarify the role of autophagy in sorafenib-acquired drug resistance to HepG2, we simultaneously treated HepG2 with sorafenib alone or in combination with 3-MA. First, the effect of autophagy regulation on HepG2 cell proliferation was detected by cell counting kit (CCK-8) cell viability assay. Subsequently, apoptosis staining and flow cytometry were used to observe the impact of regulating autophagy on HepG2 cell apoptosis. Furthermore, the formation of autophagosomes in each group was observed by transmission electron microscopy and indirect immunofluorescence staining, and apoptosis and autophagy-related proteins were detected by western blotting. Our research aims to explore the mechanism by which 3-MA enhances sorafenib sensitivity to HepG2 cells by inhibiting autophagy.
Material and methods
Antibodies and reagents
Cell viability
CCK-8 (Sigma-Aldrich, St. Louis, Missouri, USA).
Cell apoptosis
Hoechst 33342 (Sigma-Aldrich, St. Louis, Missouri, USA), Annexin V- fluorescein isothiocyanate (FITC)/propidium iodide (PI) apoptosis kit (Keygen, Nanjing, China).
Immunofluorescence
AAV-GFP-LC3(HANBIO, Shanghai, China).
Western blotting
LC3A/B antibody (Cell Signaling Technology, Beverly, Massachusetts, USA), anti-p62 antibody (Sigma-Aldrich St. Louis, Missouri, USA), Beclin-1 antibody (Abcam Trading (Shanghai) Company, China), Bcl-2 antibody (Abcam Trading (Shanghai) Company, China), Caspase-3/Cleaved-Caspase-3 antibody (Cell Signaling Technology, Beverly, Massachusetts, USA), β-Actin antibody (Cell Signaling Technology, Beverly, Massachusetts, USA), IgG Fc horseradish peroxidase (HRP) secondary antibody (Abcam Trading (Shanghai) Company, China).
Cell culture
Human hepatocarcinoma cells HepG2 (ATCC Cat# HB-8065, RRID:CVCL_0027) were inoculated in Dulbecco’s modified eagle medium high-sugar medium containing 10% fetal bovine serum, 1% penicillin/streptomycin and continuously cultured in a saturated humidity, 37 °C and 5% CO2 incubator. HepG2 cells were digested by 0.25% trypsin and subcultured, whereas the cells confluence rose up to 80–90%.
Cell grouping and viability assay by cell counting kit 8
HepG2 cells in the logarithmic growth stage were inoculated with a density of 5000 cells/pore in 96-well plates, and then incubated overnight in a saturated humidity, 37 °C and 5% CO2 incubator. Then, they were grouped by two schemes: in one scheme, sorafenib of gradient concentration (2.5, 5, 10 and 20 µM) was added, whereas the isometric medium was added as a control group. Cells were sequentially incubated for a different time period (12, 24 and 48 h). Then, the liquid in the 96-well plates were replaced with fresh medium, followed by the addition of 10 µl CCK8 for 4 h. Then, the absorbance data [optical density (OD) value] at 450 nm were collected for cell viability analysis by a microplate reader. The OD value measured in each pore is subtracted from the original OD value (blank control, without cells). IC50, also known as half maximal inhibitory concentration, refers to the concentration when a certain drug induces tumor cell apoptosis by 50% in the apoptosis experiment, and the drug concentration when the ratio of apoptotic cells to the total number of cells is equal to 50%. Cell inhibition = 1−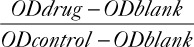 ×100%. Then, IC50 of sorafenib in different concentrations at different time points were calculated. In the other scheme, the 50% inhibitory concentration after 24 hours of Sorafenib treatment in HepG2 cells was calculated and then the cells were divided into four groups: IC5024h sorafenib, 5 mM 3-MA, IC5024h sorafenib+5 mM 3-MA were added respectively, where dimethyl sulfoxide (DMSO) was used as a control. The results was from at least triplicate independent experiments and statistical analysis of data was performed using one-way factorial analysis of variance (ANOVA) to demonstrate statistical differences between groups.
×100%. Then, IC50 of sorafenib in different concentrations at different time points were calculated. In the other scheme, the 50% inhibitory concentration after 24 hours of Sorafenib treatment in HepG2 cells was calculated and then the cells were divided into four groups: IC5024h sorafenib, 5 mM 3-MA, IC5024h sorafenib+5 mM 3-MA were added respectively, where dimethyl sulfoxide (DMSO) was used as a control. The results was from at least triplicate independent experiments and statistical analysis of data was performed using one-way factorial analysis of variance (ANOVA) to demonstrate statistical differences between groups.
Apoptosis staining by Hoechst 33342
HepG2 cells at the logarithmic growth stage were inoculated with a density of 5 × 105 cells/well in 6-well plates. In total 15 µM sorafenib dissolved in 1 µl DMSO, 5 mM 3-MA, 5 mM 3-MA combined with 15 µM sorafenib were added to the three experimental groups, whereas 1 µl DMSO was added to the control group. After being incubated for 24 h, cells were added with 10 mg/ml Hoechst 33342 and left for 15 min at room temperature. The stained nuclei were observed and photographed using a fluorescence microscope. Normal cells showed weak blue fluorescence, whereas apoptotic cells showed strong blue fluorescence. Five visual fields (200×) were selected in each group, and the total number of cells and the number of cells with strong blue fluorescence were counted. The proportion of apoptosis in each visual field was calculated using the following formula: apoptosis rate (%) = the number of cells with strong blue fluorescence/total number of cells × 100. All indicators were performed for at least triplicate independent experiments. Finally, the arithmetic mean values were obtained as the proportion of apoptosis in each group.
Flow cytometry
HepG2 cells were seeded into 6-well plates at a density of 1 × 106 cells/well and cultured for 24 h followed by treatment with: 15 µM sorafenib, 5 mM 3-MA, 15 µM sorafenib + 5 mM 3-MA or DMSO for 24 h. After washing with PBS for three times, each group of cells were digested with 0.25% trypsin without EDTA, and washed twice with pre-chilled PBS. Then, cells were centrifuged at 3000 r/min for 5 min, and the pellet was resuspended in 1 × binding buffer at a density of about 1.0 × l06 cells per mL. Of the sample solution, 100 μL was transferred to a 5 mL culture tube, and then added with 5 μL of Annexin V-FITC and 5 μL of PI. The mixed solution was gently vortexed and incubated for 15 min at room temperature (25 °C) in the dark according to the manufacturer’s instructions. A 400 μL of 1 × binding buffer was added to each sample tube for resuspension, and the samples were analyzed by flow cytometry (BD FACSCalibur) within 1 h. At least 10 000 events were recorded and represented as dot plots. The experiment was repeated for three times and statistical differences between groups were performed by one-way factorial ANOVA.
Immunofluorescence and transmission electron microscopy
HepG2 cells in 6-wells plate were transfected with AAV-GFP-LC3 for 24 h, and then washed with PBS and treated with 15 µM sorafenib, 5 mM 3-MA, 15 µM sorafenib + 5 mM 3-MA or DMSO for 6 h. Cells were fixed with 4% paraformaldehyde for 10 min at room temperature followed by washing with PBS for three times. Then, the formation of autophagosomes was observed by the fluorescence microscope and images were captured. After being treated with different drugs, HepG2 cells were collected and fixed with 0.25% glutaraldehyde overnight at 4 °C. The formation of autophagosomes was observed by transmission electron microscopy after dehydration, staining, embedding and sectioning according to the standard procedure. Images were performed at least triplicate independent experiments.
SDS-PAGE and western blotting
HepG2 cells in 6-well plates were treated with: 15 µM sorafenib, 5 mM 3-MA, 15 µM sorafenib + 5 mM 3-MA or DMSO for 24 h, and then harvested after digestion with 0.25% trypsin. The cell precipitate was washed with PBS and lysed on ice for 30 min with vortexing at 5-min intervals. After centrifugating at 12 000 rpm for 30 min at 4 °C, the supernatant was collected for detection of protein concentration. Samples were boiled for 5 min in the presence of 5 × SDS-PAGE loading buffer. Equal amounts of 50 µg protein were run on 8–12% SDS-PAGE gels and transferred onto a polyvinylidene fluoride membrane. The membrane was blocked for 2 h at room temperature in 5% W/V skim milk powder in TBS/Tween 20 (0.1%). Rabbit anti-mouse Bcl-2, Caspase-3/Cleaved-Caspase-3, Beclin-1, LC3, p62 antibodies were added to the membrane at 1:1000 dilution and incubated overnight at 4 °C. Meanwhile, β-actin was used as the internal control for protein expression. After washing with tris buffered saline tween, the membrane was probed with appropriate HRP-conjugated goat anti-rabbit IgG for 2 h at 37 °C. Finally, the membrane was visualized using the enhanced chemiluminescence purchased from Thermo Fisher Scientific Company. Densitometric analysis of protein expression was carried out using the Ultra-sensitive multifunctional imager (Amersham Imager 600, GE, USA). Then, the relative quantitative method was used to analyze the gray value of each protein. All antibodies in western blot were purchased from Cell Siginaling Technology. All indicators were performed for at least triplicate independent experiments and statistically significance comparison between multiple groups was analyzed using one-way ANOVA.
Statistics
All statistical analyses were performed by the statistical product and service solutions software (version 19.0, RRID:SCR_002865), Image J (version 1.2.4, RRID: SCR_003070) and Adobe Illustrator (version CS5, RRID: SCR_010279, and results were expressed as mean ± SEM. All dataset passed the D’Agostino and Pearson normality test before running Student’s t test. The comparison between two groups was evaluated by Student’s t test, and the comparison between multiple groups was performed by Tukey’s test using one-way ANOVA. A value of P < 0.05 was considered statistically significant.
Results
Effects of sorafenib/3-Methyladenine viability of HepG2 cells
In this study, human HCC cells HepG2 were treated with gradient concentration of sorafenib at different time points. We found that treatment with low-dose sorafenib for 12 h had not much effect on cell viability until the concentration reached 20 µM. After treatment with sorafenib for 24 h, its effect on cell viability was NS in the low-dose groups. But this was significantly decreased when the concentration reached 10 µM. In those groups treated with different concentrations of sorafenib for 48 h, cell viability was significantly decreased from 5 µM. IC50 at different time points was calculated: IC5012h (36.6 ± 2.05) µM, IC5024h (13.47 ± 1.99) µM, IC5048h (10.21 ± 1.84) µM as shown in Fig. 1a. In order to prevent the effect of high concentration of sorafenib itself, such as toxic effects, treatment time or other interference factors, rather than its pharmacological effect, 15 µM was selected as the concentration for subsequent experiments and the treatment time was 24 h.
Fig. 1.
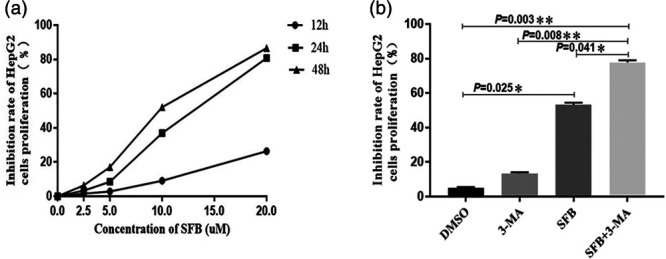
3-Methyladenine (3-MA) promoted the inhibitory effect of sorafenib on HepG2 cell proliferation. (a) HepG2 cells were treated with gradient concentrations of sorafenib (2.5, 5, 10, 20 µM) for 12, 24 and 48 h, whereas cells treated with isopyknic DMSO were used as controls. (b) Four groups of HpeG2 cells were respectively treated with 15 µM sorafenib, 5 mM 3-MA, 15 µM sorafenib + 5 mM 3-MA and DMSO for 24 h. HepG2 cell viability was detected by cell counting kit 8. The results were obtained from at least triplicate independent experiments. Values were shown as mean ± SEM (n = 5), *P value < 0.05, **P value < 0.01. DMSO, dimethyl sulfoxide.
Subsequently, cells were treated with 15 µM sorafenib, 5 mM 3-MA or 15 µM sorafenib + 5 mM 3-MA for 24 h, and cells treated with isometric DMSO were set as the control group. As shown in Fig. 1b, 3-MA had only slightly inhibit HepG2 cell proliferation (12.24 ± 4.75)%, whereas sorafenib significantly inhibited HepG2 cell proliferation (54.72 ± 9.43)% when compared with cells treated with DMSO (4.51 ± 1.76)% (P < 0.05). Interestingly, the inhibitory effect on cell proliferation was significantly increased to (79.16 ± 11.38)% (P < 0.05) after sorafenib was combined with 3-MA, which was 25% higher than the sorafenib alone group.
3-MA sensitized sorafenib-induced apoptosis of HepG2 cells
The apoptosis rate of HepG2 cells in each group was detected by the Annexin V-FITC/PI apoptosis detection kit. As shown in Fig. 2a and b, 3-MA treatment had no significant effect on the apoptosis of HepG2 cells (12.57 ± 3.91)%, whereas sorafenib enhanced the apoptosis by (23.06 ± 7.52)%. The effect on HepG2 cell apoptosis was significantly increased to (45.28 ± 10.66) % after sorafenib was combined with 3-MA, which was 22% higher than the sorafenib alone group. Therefore, our results suggested that 3-MA could increase the sensitivity of sorafenib-induced cell apoptosis, with the majority of apoptosis cells in early apoptosis.
Fig. 2.
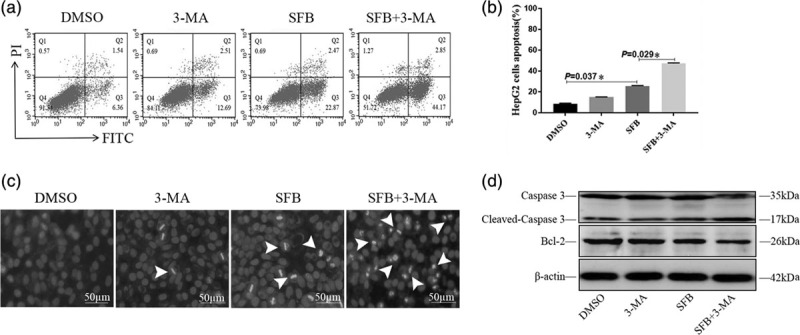
3-Methyladenine (3-MA) sensitized sorafenib-induced apoptosis of HepG2 cells. (a) Flow cytometry analysis on the apoptosis rate of HepG2 cells was detected by Annexin V-FITC/PI assay. Representative dot plots and similar results were obtained in three independent experiments. HepG2 cells in early apoptosis (FITC + /PI-) were indicated in the lower right quadrant, and late apoptosis (FITC + /PI+) were indicated in the upper right quadrant. (b) The apoptosis rate of HepG2 cells in 3-MA, sorafenib and sorafenib + 3-MA group. HepG2 cells treated with isopyknic DMSO were used as controls. (c) 3-MA sensitized sorafenib to induce apoptosis of HepG2 cells on Hoechst33342 staining. Dark blue nucleus indicates normal cells, whereas bright blue belongs to apoptotic cells. (×200) (d) Expression of apoptosis-related proteins Caspase-3/Cleaved-caspase-3 and Bcl-2 was detected by western blotting. β-actin was used as an internal control. All indicators were performed and analyzed for at least triplicate independent experiments. Values were shown as mean ± SEM (n = 5), * P value < 0.05. DMSO, dimethyl sulfoxide; FITC, fluorescein isothiocyanate; PI, propidium iodide.
Hoechst33342 staining results showed that there were more apoptotic cells in the sorafenib + 3-MA group than in the other groups, which suggested that autophagy inhibitor 3-MA could significantly enhance the sensitivity of sorafenib-induced HepG2 cell apoptosis as shown in Fig. 2c.
In order to clarify the mechanism of autophagy in sorafenib-induced apoptosis of HepG2 cells, we further analyzed the expression of apoptosis-related proteins in each group. As shown in Fig. 2d, cleaved-caspase-3 in the sorafenib treatment group was increased, whereas the apoptosis suppressor factor Bcl-2 was decreased when compared with the DMSO control group. Interestingly, these two proteins were found to be further enhanced in the sorafenib + 3-MA group.
3-MA increased the sensitivity of sorafenib in HepG2 by regulating the expression of autophagy-related proteins and the formation of autophagosomes.
HepG2 cells were first transfected with AAV-LC3-GFP for 24 h followed by treatment with DMSO, 3-MA, sorafenib, sorafenib + 3-MA. We found that high brightness dotted autophagosomes appeared around the nuclei after treatment with sorafenib, indicating that sorafenib could induce the autophagy activity of HepG2 cells, whereas this was significantly inhibited by 3-MA when combined with sorafenib in the sorafenib + 3-MA group as shown in Fig. 3a.
Fig. 3.
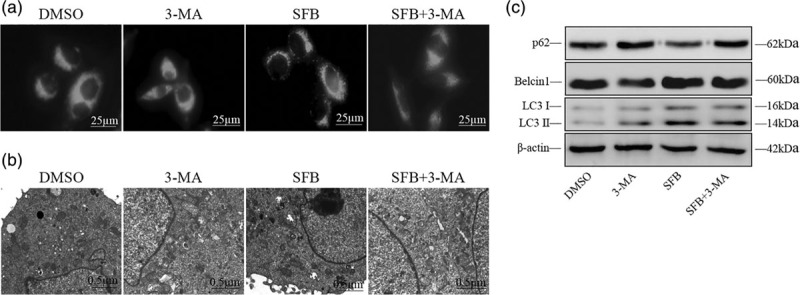
Sorafenib-induced autophagy was effectively blocked by 3-Methyladenine (3-MA). (a) Autophagy activity of HepG2 cells in different groups was observed by immunofluorescence. High brightness dots located in cytoplasm on the membrane of the sorafenib group were considered as autophagosomes (×400). (b) Transmission electron microscope was used to observe the formation of autophagosomes. The double-membrane structures which contain contents of undigested organelle were considered as autophagosome (red arrows, × 20 000). (c) Expression of autophagy-related proteins p62, Beclin-1 and LC3/II was detected by western blotting. β-actin was used as an internal control. All indicators were performed for at least triplicate independent experiments. Values were shown as mean ± SEM (n = 5).
After different treatments, HepG2 cells were processed according to the standard procedure and the formation of autophagosomes in each group was observed by transmission electron microscope. We found that there were more autophagosomes in the sorafenib group than in the DMSO group, which supported that sorafenib could enhance the autophagy activity in HepG2 cells. However, there was a significant decrease after treatment with sorafenib + 3-MA, suggesting that 3-MA successfully inhibited the formation of autophagosomes induced by sorafenib as shown in Fig. 3b.
HepG2 cells in different groups were harvested and proteins were extracted for further analysis. Western blotting was performed to detect the expression of autophagy-related proteins. We found that treatment with sorafenib significantly upregulated the expression of LC3 II (a marker protein of autophagy) as well as Beclin-1, an autophagy promoter. On the contrary, the expression of autophagy substrate p62 was decreased significantly in the sorafenib group. However, when sorafenib and 3-MA were combined, the above trends in all protein expression levels were successfully reversed as shown in Fig. 3c. These results further suggested that 3-MA could effectively inhibit the enhanced autophagy activity of HepG2 cells induced by sorafenib.
Discussion
HCC is a malignant liver tumor with high incidence. Many patients are already at advanced stages when diagnosed with HCC and, therefore, unfortunately miss the optimum opportunity for surgery and have poor response of systemic chemotherapy [28–30]. Sorafenib is the most commonly used molecular targeted drug that can be taken orally to treat advanced HCC. As a multitarget antitumor drug, it can target serine/threonine kinase and receptor lysine kinase in tumor cells or blood vessels to induce apoptosis thus inhibiting tumor growth [31,32]. Several clinical studies in the guidelines NCCN/ESMO have demonstrated that sorafenib can improve the survival rate in patients with advanced HCC [9,10]. In this study, we verified through CCK8 assay that HepG2 cell proliferation could be inhibited by sorafenib. Interestingly, this effect was significantly enhanced when sorafenib was combined with 3-MA. As an effector of cell apoptosis, Caspase-3 can be activated by upstream initiators. The activated Caspase-3 (Cleaved-Caspase-3) acts on specific downstream signaling molecules, causing a series of biological morphological changes in cells and ultimately inducing cell apoptosis [33,34]. Bcl-2 is the first protein that has been proved to reduce cell apoptosis. It maintains cell membrane permeability by adjusting the opening and closing of permeability transition holes on the mitochondrial membrane and interferes with the release of cytochrome C to block the activation of upstream caspase protease, thereby inhibiting cell apoptosis [35]. Previous studies have confirmed that the apoptosis of tumor cells is closely related to the changes of Bcl-2 and Caspase-3 [36–38]. We confirmed that the expression of Bcl-2 in HepG2 cells was significantly lower in the sorafenib treatment group compared with the control group, whereas cleaved-caspase-3 had the completely opposite trend, suggesting that sorafenib could inhibit tumor growth by promoting HepG2 cell apoptosis.
However, sorafenib resistance has been frequently reported in recent years [39]. At advanced stage of tumor, rapid proliferation of tumor cells requires abundant nutrients under hypoxia and metabolic stress, especially for solid tumors with low vascularization [40–43]. Therefore, autophagy plays an important role in tumor cell survival and recurrence after chemotherapy. Tumor cells can activate autophagy in the event of cell damage during radiation therapy and chemotherapy, eliminate the damaged proteins and organelles to store energy, and maintain cell survival against chemotherapy [17,44]. To verify this idea, we treated HepG2 cells with sorafenib and found that the formation of autophagosomes were significantly increased, whereas the expressions of autophagy initiation proteins Beclin-1 and mature marker proteins LC3 II were significantly increased by western blotting. All data above supported that sorafenib could enhance the autophagy activity of HepG2 cells, which may be one of the mechanisms for its acquired resistance to HepG2 cells.
Considering that autophagy improves the acquired resistance of HepG2 cells to sorafenib, its inhibition may be a potential treatment for eliminating resistance. 3-MA is an inhibitor of PI3K, which can specifically block the formation of autophagosome during autophagy [45–47]. In this study, by treating HepG2 cells with sorafenib + 3-MA, we found that autophagosomes were decreased and the cell proliferation inhibition level and apoptosis level were both significantly higher than in the sorafenib or 3-MA group alone, indicating that 3-MA may reduce the acquired resistance of sorafenib by inhibiting the formation of autophagosomes. Further western blotting results were consistent with these results, therefore supporting our conclusion above.
Conclusion
With the development of molecular targeted therapy, new tumor therapy modalities of biochemotherapy have received increasing attention [48]. After the positive effect of sorafenib in the treatment of HCC was verified, how to increase the efficacy of sorafenib and reduce its resistance has remained a hot topic in current researches. Given the close relationship between autophagy and tumor genesis and development, as well as its crucial role in the mechanism of drug resistance, we concluded that regulating autophagy to increase the sensitivity of antitumor drugs would be a new breakthrough in antitumor therapy in the future [49]. The limitation of this study was that we only confirmed that 3-MA could reduce the acquired resistance of sorafenib to certain extent by inhibiting the autophagy activity, and neither deeper mechanism nor an intrinsic resistance mechanism of HCC cells to sorafenib were explored, which is what we plan to investigate further in the future.
Acknowledgements
The authors would like to thank Chen Chen from the Department of Cell Biology, China Medical University, for providing language help and writing assistance. They also extend their thanks to the National Natural Science Foundation Youth Scientists Fund of China for providing great support (grant no. 81501428).
Conflicts of interest
There are no conflicts of interest.
References
- 1.Huang F, Wang BR, Wang YG. Role of autophagy in tumorigenesis, metastasis, targeted therapy and drug resistance of hepatocellular carcinoma. World J Gastroenterol. 2018; 24:4643–4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johannessen TC, Hasan-Olive MM, Zhu H, Denisova O, Grudic A, Latif MA, et al. Thioridazine inhibits autophagy and sensitizes glioblastoma cells to temozolomide. Int J Cancer. 2019; 144:1735–1745. [DOI] [PubMed] [Google Scholar]
- 3.Li HM, Ye ZH. Microenvironment of liver regeneration in liver cancer. Chin J Integr Med. 2017; 23:555–560. [DOI] [PubMed] [Google Scholar]
- 4.Harris WP, Wong KM, Saha S, Dika IE, Abou-Alfa GK. Biomarker-driven and molecular targeted therapies for hepatobiliary cancers. Semin Oncol. 2018; 45:116–123. [DOI] [PubMed] [Google Scholar]
- 5.Nishida N, Nishimura T, Kaido T, Minaga K, Yamao K, Kamata K, et al. Molecular scoring of hepatocellular carcinoma for predicting metastatic recurrence and requirements of systemic chemotherapy. Cancers (Basel). 2018; 10:E367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shen Y, Zhang ZB, Wu SD, Wu XB, Li J. Research on values of GDF-15 level in the diagnosis of primary liver cancer and evaluation of chemotherapeutic effect. Eur Rev Med Pharmacol Sci. 2018; 22:3749–3754. [DOI] [PubMed] [Google Scholar]
- 7.Ikeda M, Morizane C, Ueno M, Okusaka T, Ishii H, Furuse J. Chemotherapy for hepatocellular carcinoma: current status and future perspectives. Jpn J Clin Oncol. 2018; 48:103–114. [DOI] [PubMed] [Google Scholar]
- 8.Julie KH, Laura MK, Richard SF, Claude BS, Michael MA, Lewis RR, et al. AASLD guidelines for the treatment of hepatocellular carcinoma. Hepatology. 2018; 67:358–380. [DOI] [PubMed] [Google Scholar]
- 9.Benson AB, D’Angelica MI, Abbott DE, Abrams TA, Alberts SR, Anaya DA, et al. Guidelines insights: hepatobiliary cancers, version 2.2019. J Natl Compr Canc Netw. 2019; 17:302–310. [DOI] [PubMed] [Google Scholar]
- 10.Vogel A, Cervantes A, Chau I, Daniele B, Llovet JM, Meyer T, et al. Hepatocellular carcinoma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2018; 4:iv238–iv255. [DOI] [PubMed] [Google Scholar]
- 11.McNamara MG, Slagter AE, Nuttall C, Frizziero M, Pihlak R, Lamarca A, et al. Sorafenib as first-line therapy in patients with advanced Child-Pugh B hepatocellular carcinoma-a meta-analysis. Eur J Cancer. 2018; 105:1–9. [DOI] [PubMed] [Google Scholar]
- 12.Marisi G, Cucchetti A, Ulivi P, Canale M, Cabibbo G, Solaini L, et al. Ten years of sorafenib in hepatocellular carcinoma: are there any predictive and/or prognostic markers? World J Gastroenterol. 2018; 24:4152–4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen J, Jin R, Zhao J, Liu J, Ying H, Yan H, et al. Potential molecular, cellular and microenvironmental mechanism of sorafenib resistance in hepatocellular carcinoma. Cancer Lett. 2015; 367:1–11. [DOI] [PubMed] [Google Scholar]
- 14.Arensman MD, Eng CH. Self-digestion for lifespan extension: enhanced autophagy delays aging. Mol Cell. 2018; 71:485–486. [DOI] [PubMed] [Google Scholar]
- 15.Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal. 2014; 20:460–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang K. Autophagy and apoptosis in liver injury. Cell Cycle. 2015; 14:1631–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Katheder NS, Khezri R, O’Farrell F, Schultz SW, Jain A, Rahman MM, et al. Microenvironmental autophagy promotes tumour growth. Nature. 2017; 541:417–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Poillet-Perez L, Xie X, Zhan L, Yang Y, Sharp DW, Hu ZS, et al. Autophagy maintains tumour growth through circulating arginine. Nature. 2018; 563:569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shin JH, Park CW, Yoon G, Hong SM, Choi KY. NNMT depletion contributes to liver cancer cell survival by enhancing autophagy under nutrient starvation. Oncogenesis. 2018; 7:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kumar A, Singh UK, Chaudhary A. Targeting autophagy to overcome drug resistance in cancer therapy. Future Med Chem. 2015; 7:1535–1542. [DOI] [PubMed] [Google Scholar]
- 21.D’Arcy MS. Cell death: a review of the major forms of apoptosis, necrosis and autophagy. Cell Biol Int. 2019; 43:582–592. [DOI] [PubMed] [Google Scholar]
- 22.Camuzard O, Santucci-Darmanin S, Carle GF, Pierrefite-Carle V. Role of autophagy in osteosarcoma. J Bone Oncol. 2019; 16:100235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liao YX, Yu HY, Lv JY, Cai YR, Liu F, He ZM, He SS. Targeting autophagy is a promising therapeutic strategy to overcome chemoresistance and reduce metastasis in osteosarcoma. Int J Oncol. 2019; 55:1213–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao F, Zhai Y, Zhu J, Xiao P, Feng G. Enhancement of autophagy as a strategy for development of new DNA vaccine candidates against Japanese encephalitis. Vaccine. 2019; 37:5588–5595. [DOI] [PubMed] [Google Scholar]
- 25.Sheng Y, Sun B, Guo WT, Zhang YH, Liu X, Xing Y, Dong DL. 3-Methyladenine induces cell death and its interaction with chemotherapeutic drugs is independent of autophagy. Biochem Biophys Res Commun. 2013; 432:5–9. [DOI] [PubMed] [Google Scholar]
- 26.Mishima Y, Terui Y, Mishima Y, Taniyama A, Kuniyoshi R, Takizawa T, et al. Autophagy and autophagic cell death are next targets for elimination of the resistance to tyrosine kinase inhibitors. Cancer Sci. 2008; 99:2200–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kang R, Wang ZH, Wang BQ, Zhang CM, Gao W, Feng Y, et al. Inhibition of autophagy- potentiated chemosensitivity to cisplatin in laryngeal cancer Hep-2 cells. Am J Otolaryng. 2012; 33:678–684. [DOI] [PubMed] [Google Scholar]
- 28.Sia D, Villanueva A, Friedman SL, Llovet JM. Liver cancer cell of origin, molecular class, and effects on patient prognosis. Gastroenterology. 2017; 152:745–761. [DOI] [PubMed] [Google Scholar]
- 29.Fu J, Wang H. Precision diagnosis and treatment of liver cancer in China. Cancer Lett. 2018; 412:283–288. [DOI] [PubMed] [Google Scholar]
- 30.Ou Y, Huang J, Yang L. The prognostic significance of pretreatment serum Gamma-glutamyltranspeptidase in primary liver cancer: a meta-analysis and systematic review. Biosci Rep. 2018; 38:BSR20181058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tesori V, Piscaglia AC, Samengo D, Barba M, Bernardini C, Scatena R, et al. The multikinase inhibitor Sorafenib enhances glycolysis and synergizes with glycolysis blockade for cancer cell killing. Sci Rep. 2015; 5:9149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun T, Liu H, Ming L. Multiple roles of autophagy in the sorafenib resistance of hepatocellular carcinoma. Cell Physiol Biochem. 2017; 44:716–727. [DOI] [PubMed] [Google Scholar]
- 33.He L, Xiao D, Feng J, Yao C, Tang L. Induction of apoptosis of liver cancer cells by nanosecond pulsed electric fields (nsPEFs). Med Oncol. 2017; 34:24. [DOI] [PubMed] [Google Scholar]
- 34.Choudhary GS, Al-Harbi S, Almasan A. Caspase-3 activation is a critical determinant of genotoxic stress-induced apoptosis. Methods Mol Biol. 2015; 1219:1–9. [DOI] [PubMed] [Google Scholar]
- 35.Zhou W, Yang J, Zhang DI, Li F, Li G, Gu Y, Luo M. Role of Bcl-2/adenovirus E1B 19 kDa-interacting protein 3 in myocardial cells in diabetes. Exp Ther Med. 2015; 10:67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hassan M, Watari H, AbuAlmaaty A, Ohba Y, Sakuragi N. Apoptosis and molecular targeting therapy in cancer. Biomed Res Int. 2014; 2014:150845. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 37.Mohamed MS, Bishr MK, Almutairi FM, Ali AG. Inhibitors of apoptosis: clinical implications in cancer. Apoptosis. 2017; 22:1487–1509. [DOI] [PubMed] [Google Scholar]
- 38.Zhang X, Abdelrahman A, Vollmar B, Zechner D. The ambivalent function of YAP in apoptosis and cancer. Int J Mol Sci. 2018; 19:E3770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhu YJ, Zheng B, Wang HY, Chen L. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol Sin. 2017; 38:614–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eichner LJ, Brun SN, Herzig S, Young NP, Curtis SD, Shackelford DB, et al. Genetic analysis reveals AMPK is required to support tumor growth in murine kras-dependent lung cancer models. Cell Metab. 2019; 29:285–302.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abad E, García-Mayea Y, Mir C, Sebastian D, Zorzano A, Potesil D, et al. Common metabolic pathways implicated in resistance to chemotherapy point to a key mitochondrial role in breast cancer. Mol Cell Proteomics. 2019; 18:231–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ghosh D, Dawson MR. Microenvironment influences cancer cell mechanics from tumor growth to metastasis. Adv Exp Med Biol. 2018; 1092:69–90. [DOI] [PubMed] [Google Scholar]
- 43.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013; 19:1423–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Katheder NS, Rusten TE. Microenvironment and tumors-a nurturing relationship. Autophagy. 2017; 13:1241–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang S, Livingston MJ, Su Y, Dong Z. Reciprocal regulation of cilia and autophagy via the MTOR and proteasome pathways. Autophagy. 2015; 11:607–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bao J, Shi Y, Tao M, Liu N, Zhuang S, Yuan W. Pharmacological inhibition of autophagy by 3-MA attenuates hyperuricemic nephropathy. Clin Sci (Lond). 2018; 132:2299–2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shin D, Kim EH, Lee J, Roh JL. RITA plus 3-MA overcomes chemoresistance of head and neck cancer cells via dual inhibition of autophagy and antioxidant systems. Redox Biol. 2017; 13:219–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xiang QF, Zhan MX, Li Y, Liang H, Hu C, Huang YM, et al. Activation of MET promotes resistance to sorafenib in hepatocellular carcinoma cells via the AKT/ERK1/2-EGR1 pathway. Artif Cells Nanomed Biotechnol. 2019; 47:83–89. [DOI] [PubMed] [Google Scholar]
- 49.Méndez-Blanco C, Fondevila F, García-Palomo A, González-Gallego J, Mauriz JL. Sorafenib resistance in hepatocarcinoma: role of hypoxia-inducible factors. Exp Mol Med. 2018; 50:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]