

SUMMARY
Experience-dependent refinement of neuronal connections is critically important for brain development and learning. Here, we show that ion-flow-independent NMDA receptor (NMDAR) signaling is required for the long-term dendritic spine growth that is a vital component of brain circuit plasticity. We find that inhibition of p38 mitogen-activated protein kinase (p38 MAPK), which is downstream of non-ionotropic NMDAR signaling in long-term depression (LTD) and spine shrinkage, blocks long-term potentiation (LTP)-induced spine growth but not LTP. We hypothesize that non-ionotropic NMDAR signaling drives the cytoskeletal changes that support bidirectional spine structural plasticity. Indeed, we find that key signaling components downstream of non-ionotropic NMDAR function in LTD-induced spine shrinkage are also necessary for LTP-induced spine growth. Furthermore, NMDAR conformational signaling with coincident Ca2+ influx is sufficient to drive CaMKII-dependent long-term spine growth, even when Ca2+ is artificially driven through voltage-gated Ca2+ channels. Our results support a model in which non-ionotropic NMDAR signaling gates the bidirectional spine structural changes vital for brain plasticity.
Graphical Abstract
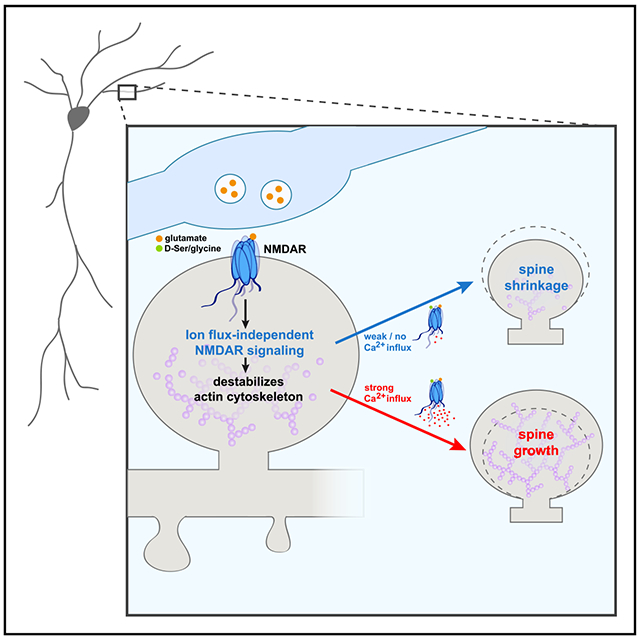
In Brief
Structural plasticity of dendritic spines is a critical step in the remodeling of brain circuits during learning. Stein et al. demonstrate a vital role for ion-flux-independent NMDAR signaling in plasticity-associated dendritic spine growth, supporting a model in which non-ionotropic NMDAR signaling primes the spine actin cytoskeleton for bidirectional structural plasticity.
INTRODUCTION
Dynamic alternations in neuronal connectivity are critical for the experience-dependent modification of brain circuits throughout development and during learning. In particular, the bidirectional structural plasticity of dendritic spines is a vital process in the refinement of synaptic circuits in the mammalian cortex (e.g., Hayashi-Takagi et al., 2015; Lai et al., 2018). Increases in synaptic strength, as through the induction of long-term potentiation (LTP), are associated with spine enlargement and new spine formation (Matsuzaki et al., 2004; Nishiyama and Yasuda, 2015), whereas decreases in synaptic strength, as through the induction of long-term depression (LTD), are associated with spine shrinkage or loss (Stein and Zito, 2019; Zhou et al., 2004). Notably, activation of the NMDA-type glutamate receptor (NMDAR) is required for both the spine growth associated with LTP and the spine shrinkage associated with LTD.
Recent studies have demonstrated that LTD (Carter and Jahr, 2016; Nabavi et al., 2013; Wong and Gray, 2018) and dendritic spine shrinkage (Birnbaum et al., 2015; Stein et al., 2015; Thomazeau et al., 2020) can occur independent of ion flux through the NMDAR. These findings have supported a model in which glutamate binding leads to conformational changes in the NMDAR that drive spine shrinkage and synaptic weakening. Indeed, imaging studies using fluorescence resonance energy transfer (FRET) reporters have shown that NMDA or glutamate binding triggers conformational changes in the NMDAR intracellular domains and changes its interaction with calcium/calmodulin-dependent protein kinase II (CaMKII) and protein phosphatase 1 (PP1) (Aow et al., 2015; Dore et al., 2015; Ferreira et al., 2017). p38 mitogen-activated protein kinase (MAPK) has been identified as a key component of the molecular pathway downstream of the NMDAR conformational signaling (Birnbaum et al., 2015; Nabavi et al., 2013; Stein et al., 2015), and a recent study further identified neuronal nitric oxide synthase (nNOS), nNOS-NOS1AP interactions, MAPK-activated protein kinase 2 (MK2), and cofilin as part of this signaling pathway (Stein et al., 2020).
Here, we made the surprising discovery that ion-flow-independent NMDAR signaling is required for long-term spine growth associated with synaptic strengthening. We show that p38 MAPK, an essential signaling component for LTD and spine shrinkage, is required for spine growth, but not for LTP, suggesting that non-ionotropic NMDAR signaling is a vital component of bidirectional spine structural plasticity. Indeed, we further show that key components of ion-flux-independent NMDAR signaling, including interactions among NOS1AP and nNOS, MK2, nNOS, and CaMKII activity are all required for LTP-induced spine growth. Importantly, we also demonstrate that, when combined with non-ionotropic NMDAR signaling, long-term spine growth can be driven by Ca2+ influx through voltage-gated Ca2+ channels. Our findings support a model in which non-ionotropic NMDAR signaling leads to disruption of the spine F-actin network, which drives spine shrinkage unless coincident Ca2+-influx converts F-actin remodeling to instead promote spine growth.
RESULTS
p38 MAPK activity is required for LTP-induced spine growth, but not for LTP-induced synaptic strengthening
Glutamate binding to NMDARs initiates a signaling cascade that drives synaptic weakening (LTD) and the shrinkage of dendritic spines (sLTD), even when ion flow is blocked pharmacologically (Carter and Jahr, 2016; Nabavi et al., 2013; Stein et al., 2015; Wong and Gray, 2018). Furthermore, in the absence of ion flux, patterns of glutamatergic stimulation that would normally drive LTP and spine growth, instead drive LTD and spine shrinkage (Nabavi et al., 2013; Stein et al., 2020; Stein and Zito, 2019). Because the non-ionotropic NMDAR signaling pathway should also be activated with glutamate binding when the ion flow through the receptor is not blocked, we wondered whether it has a role in bidirectional synaptic plasticity. We hypothesized non-ionotropic NMDAR signaling could function as part of a built-in synaptic regulatory mechanism to fine tune structural rearrangement and prevent spine overgrowth during LTP induction.
Intriguingly, inhibition of p38 MAPK activity by SB203580 (Cuenda et al., 1995; Davies et al., 2000) did not lead to excessive high-frequency uncaging (HFU)-induced spine growth but, instead, blocked persistent spine enlargement after HFU stimulation (Figures 1A-1C; vehicle [veh]: 220% ± 18%; SB203580 [SB]: 117% ± 11%). Because p38 MAPK activation previously had only been implicated in LTD and not LTP (Zhu et al., 2002), we next examined whether expression of HFU-induced single-spine LTP was normal during inhibition of p38 MAPK activity. HFU stimulation under vehicle conditions successfully induced LTP of the target spine, which lasted for at least 25 min after stimulation but did not increase the uncaging-evoked excitatory postsynaptic current (uEPSC) amplitude of the unstimulated neighboring spine (Figures 1D and 1F; target: 153% ± 11%; neighbor: 93% ± 7%). Unlike spine enlargement, functional LTP induction was not blocked by SB203580 and was indistinguishable from LTP during vehicle conditions (Figures 1E and 1F; target: 157% ± 6%; neighbor: 98% ± 9%). SB203580 did not alter baseline spine volume (Figure S1A). These results support a specific role for p38 MAPK signaling in structural, but not functional, LTP.
Figure 1. p38 MAPK activity is required for LTP-induced spine growth, but not for synaptic strengthening.

(A) Images of dendrites from DIV14-18 EGFP-expressing CA1 neurons from rat organotypic slices before and after HFU stimulation (yellow crosses) at individual dendritic spines (yellow arrowheads) with and without p38 MAPK inhibitor SB203580 (SB, 2 μM).
(B and C) Inhibition of p38 MAPK with SB (black-filled circles/bar; 11 spines/11 cells) reduced HFU-induced spine growth compared with vehicle (red-filled circles/bar; 11 spines/11 cells). Volume of unstimulated neighbors (open circles/bars) was unaffected.
(D and E) Top, average traces of uEPSCs from a target spine and an unstimulated neighbor on CA1 neurons from mouse organotypic slices before (gray) and 25 min after HFU stimulation during vehicle conditions (target, red; neighbor, dark gray) or in the presence of SB (target, black; neighbor, dark gray). Bottom, HFU-induced increases in uEPSC amplitude in vehicle (red; 8 spines/8 cells) were unaffected by p38 MAPK inhibition with SB (black; 9 spines/9 cells). uEPSC amplitudes of unstimulated neighboring spines (gray) were unaffected.
(F) HFU induced a long-lasting uEPSC amplitude increase (red filled bar) compared with baseline, which is unaffected by p38 MAPK inhibition (black-filled bar).
Two-way ANOVA with Tukey’s test used in (C) and (F) and two-way repeated-measures (RMs) ANOVA with Dunnett’s test to baseline used in (B), (D), and (E). Data are represented as means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
Non-ionotropic NMDAR signaling pathway is required for LTP-induced spine growth
The requirement of p38 MAPK for spine growth, but not for LTP, and the previously implicated role of p38 MAPK in non-ionotropic signaling during LTD and spine shrinkage led us to the unexpected hypothesis that non-ionotropic NMDAR signaling might be required for bidirectional spine structural plasticity. We have previously shown that non-ionotropic NMDAR-dependent spine shrinkage is dependent on the interaction between NOS1AP and nNOS, which during NMDAR-mediated excitoxicity is required for p38 MAPK activation (Li et al., 2013), and activation of NOS, p38 MAPK, the p38 MAPK substrate MK2, and cofilin (Stein et al., 2020). We, therefore, set out to test whether these molecules are also required for LTP-induced spine growth.
We tested whether NOS1AP binding to nNOS, MK2 activity, and nNOS activity, similar to p38 MAPK activation, are also required for persistent structural LTP. Indeed, we found that inhibition of NOS1AP binding to nNOS using L-TAT-GESV (Li et al., 2013) impaired persistent HFU-induced structural LTP (Figures 2A-2C; 110% ± 13%) compared with the inactive L-TAT-GASA control peptide (Figures 2A-2C; 173% ± 17%). Spine size of the unstimulated neighboring spines was not changed (Figures 2A-2C; L-TAT-GASA: 101% ± 4%; L-TAT-GESV: 107% ± 6%). In addition, HFU-induced spine enlargement was significantly reduced in the presence of MK2 inhibitor III (Anderson et al., 2007; Fiore et al., 2016) compared to vehicle conditions (Figures 2D-2F; veh: 241% ± 34%; MK2 inhibitor III: 152% ± 24%), and no change in the size of the unstimulated neighboring spines was observed (Figures 2D-2F; veh: 109% ± 6%; MK2 inhibitor III: 109% ± 7%). Furthermore, in the presence of the NOS inhibitor, L-NNA (Pigott et al., 2013; Reif and McCreedy, 1995), HFU-induced spine enlargement was significantly reduced compared with vehicle conditions (Figures 2G-2I; veh: 201% ± 28%; L-NNA: 136% ± 14%), and no change in the size of the unstimulated neighboring spines was observed (Figures 2G-2I; veh: 105% ± 5%; L-NNA: 110% ± 6%). L-TAT-GESV, MK2 inhibitor III, and L-NNA did not alter baseline spine volume (Figures S1B-S1D). Our findings strongly support a role for the non-ionotropic NMDAR signaling pathway in LTP-induced spine growth.
Figure 2. Non-ionotropic NMDAR signaling pathway is required for LTP-induced spine growth.

(A, D, and G) Images of dendrites from EGFP-expressing CA1 neurons from rat (A and D) and mice (G) organotypic slices at DIV14-18 before and after HFU stimulation (yellow crosses) of individual spines (yellow arrowheads) in the presence of L-TAT-GESV (1 μM) and the L-TAT-GASA control peptide (1 μM), during vehicle conditions and in the presence of MK2 inhibitor III (10 μM) or the NOS inhibitor, L-NNA (100 μM).
(B and C) Disruption of the NOS1AP/nNOS interaction with L-TAT-GESV (black-filled circles/bar; 9 spines/9 cells), but not application of the inactive L-TAT-GASA control peptide (red-filled circles/bar; 9 spines/9 cells) inhibited persistent spine growth after LTP induction. Volume of the unstimulated neighbors (open circles/bars) was unchanged.
(E and F) Inhibition of MK2 activity (black-filled circles/bar; 11 spines/11 cells) prevented HFU-induced persistent spine enlargement (red-filled circles/bar; 12 spines/12 cells). Volume of the unstimulated neighbors did not change (open circles/bars).
(H and I) Inhibition of NOS activity (black-filled circles/bar; 11 spines/11 cells) prevented HFU-induced persistent spine enlargement (red-filled circles/bar; 12 spines/12 cells). Volume of the unstimulated neighbors did not change (open circles/bars).
Two-way ANOVA with Tukey’s test used in (C), (F), and (I) and two-way RMs ANOVA with Dunnett’s test to baseline used in B, E, and H. Data are represented as means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
Non-ionotropic NMDAR signaling, in combination with VGCC-mediated Ca2+ influx, is sufficient to drive LTP-induced spine growth
Our results support a model in which non-ionotropic NMDAR signaling leads to the p38 MAPK- and cofilin-dependent disruption of the F-actin network, which, in the absence of strong Ca2+-influx, drives spine shrinkage but, during LTP, provides new actin filament nucleation and branching points for subsequent spine enlargement by Ca2+-dependent actin modifying and stabilizing proteins. Based on this model, we hypothesized that it should be possible to drive spine growth through a combination of non-ionotropic NMDAR signaling with NMDAR-independent Ca2+-influx (Figure 3A).
Figure 3. Non-ionotropic NMDAR signaling with Ca2+-influx through voltage-gated calcium channels is sufficient to drive LTP-induced spine growth.

(A) Left: proposed model in which activity-induced spine growth requires both non-ionotropic and ionotropic NMDAR signaling. Middle: schematic of experiment to test proposed model. Non-ionotropic NMDAR signaling is activated with glutamate, whereas calcium influx through the NMDAR is blocked with L-689. Instead, calcium influx is driven through VGCCs with the stronger HFU+ conditions in the presence of Bay K to favor opening of VGCCs. Right: control experiments block non-ionotropic NMDAR signaling by blocking glutamate binding to the NMDAR with CPP.
(B) Images of dendrites from CA1 neurons of acute slices from P16-20 GFP-M mice before and after HFU+ stimulation (yellow crosses) of individual spines (yellow arrowheads) in the presence of L-689 (10 μM) and Bay K (10 μM) or in combination with CPP (50 μM).
(C and D) HFU+ stimulation drives spine growth in the presence of Bay K, even when ion flow through the NMDAR is blocked with L-689 (red-filled circles/bar; 9 spines/9 cells), but not when non-ionotropic NMDAR signaling is blocked with CPP (black-filled circles/bar; 10 spines/10 cells). Volume of the unstimulated neighbors (open circles/bars) was unchanged.
(E) Images of dendrites under experimental conditions in (B)–(D), except blocking the influx of calcium through VGCCs by removing Bay K and adding NBQX (50 μM).
(F and G) HFU+ stimulation in the presence of L-689 and NBQX (blue-filled circles/bar; 7 spines/7 cells) led to spine shrinkage, which was blocked when non-ionotropic NMDAR signaling was inhibited with the presence of CPP (black-filled circles/bar; 6 spines/6 cells). Volume of the unstimulated neighbors (open circles/bars) did not change.
Two-way ANOVA with Tukey’s test used in (D) and (G) and two-way RMs ANOVA with Dunnett’s test to baseline used in C and F. Data are represented as means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
See also Figure S2.
To drive non-ionotropic NMDAR signaling together with voltage-gated calcium channel (VGCC)-mediated Ca2+-influx, we used the co-agonist binding site NMDAR antagonist, L-689,560 (Leeson et al., 1992), which blocks all current through the NMDAR (Stein et al., 2020; Wong and Gray, 2018), along with the L-type Ca2+ channel agonist Bay K 8644 (Hess et al., 1984). Furthermore, to ensure sufficient L-type VGCC-mediated Ca2+ influx into the stimulated spine during glutamate uncaging, we increased extracellular Ca2+, and we increased the intensity and frequency of our high-frequency uncaging protocol (HFU+; see STAR methods) to facilitate stronger AMPA receptor (AMPAR)-dependent depolarization of the target spine. Remarkably, HFU+ stimulation in the presence of L-689 and Bay K induced long-term spine growth (Figures 3B-3D; L-689 + Bay K: 147% ± 12%). Importantly, adding the glutamate binding site NMDAR antagonist, CPP (Davies et al., 1986; Lehmann et al., 1987) to inhibit non-ionotropic NMDAR signaling, blocked HFU+-induced long-term spine growth (Figures 3B-3D; L-689 + Bay K + CPP: 102% ± 7%) without affecting the magnitude of VGCC-mediated Ca2+-influx (Figure S2). Size of unstimulated neighboring spines (Figures 3B-3D; L-689 + Bay K: 104% ± 6%; L-689 + CPP + Bay K: 89% ± 3%) and baseline spine volume (Figure S1E) were unaffected by Bay K, excluding acute and activity-independent effects of Bay K alone on spine morphology.
To confirm that our modified HFU+ stimulation paradigm still drives non-ionotropic NMDAR-signaling-induced spine shrinkage in acute hippocampal slices from mice, we replaced Bay K with NBQX (Sheardown et al., 1990) to block uncaging-induced, AMPAR-driven spine depolarization and VGCC activation and tested whether the same stimulation, now in the absence of Ca2+ influx, drives dendritic spine shrinkage. Indeed, HFU+ stimulation in the presence of L-689 and NBQX resulted in non-ionotropic, NMDAR-dependent spine shrinkage (Figures 3E-3G; L-689 + NBQX: 55% ± 9%), which was inhibited if glutamate binding, and thus conformational non-ionotropic NMDAR signaling, was blocked with CPP (Figures 3E-3G; L-689 + CPP + NBQX: 94% ± 8%). Spine size of unstimulated neighbors (Figures 3E-3G; L-689 + NBQX: 92% ± 3%; L-689 + CPP + NBQX: 89% ± 10%) and baseline spine volume (Figure S1F) were unaffected.
Our results confirm the generalizability of ion-flux-independent NMDAR signaling in driving dendritic spine shrinkage across slice preparations and species, which is further confirmed in studies of ion-flux-independent LTD, which showed consistent results across species, slice preparations, and dissociated rat cultures (Aow et al., 2015; Nabavi et al., 2013; Wong and Gray, 2018). These results strongly support our model that non-ionotropic NMDAR signaling primes the actin cytoskeleton for bidirectional structural plasticity.
CaMKII activity is required for long-term spine growth, independent of calcium source
CaMKII is a Ca2+-dependent kinase that has been extensively studied in LTP induction (Bayer and Schulman, 2019) and is required for the long-term spine growth associated with LTP (Matsuzaki et al., 2004; Murakoshi et al., 2011) (Figure S3). Notably, disruption of the interaction of CaMKII with the C terminus of the NMDAR interferes with LTP (Barria and Malinow, 2005; Halt et al., 2012; Sanhueza et al., 2011) and long-term spine stabilization (Hill and Zito, 2013), suggesting that the local calcium microdomains produced by the calcium flow through the NMDAR may be critical in activating CaMKII during LTP-induced structural and functional plasticity.
We tested whether Ca2+/CaMKII-dependent signaling is also required for long-term spine growth driven by NMDAR-independent Ca2+ influx through VGCCs using acute hippocampal slices from mice. We found that when Ca2+ is supplied through VGCCs and not through the NMDAR under our HFU+ stimulation conditions, long-term spine growth (Figures 4A-4C; L-689 + Bay K + TAT-SCR: 173% ± 17%) was also blocked in the presence of KN-62 (Hidaka and Yokokura, 1996; Tokumitsu et al., 1990) (Figures 4A-4C; L-689 + Bay K + KN-62: 106% ± 11%) or TAT-CN21 (Figures 4A-C; L689 + Bay K + TAT-CN21: 102% ± 15%), a CaMKII-specific peptide inhibitor (Vest et al., 2007). The size of the unstimulated neighboring spines (Figure 4A-C; L-689 + Bay K + TAT-SCR: 87% ± 3%; L-689 + Bay K + KN-62: 97% ± 10%; L-689 + Bay K + TAT-CN21: 100% ± 7%) and baseline spine volume (Figure S1G) were not affected. Our results demonstrate the requirement of CaMKII for LTP-induced long-term spine growth, regardless of calcium source.
Figure 4. CaMKII activity is required for LTP-induced spine growth, independent of calcium source.

(A) Images of dendrites from CA1 neurons of acute slices from P16-20 GFP-M mice before and after HFU+ stimulation (yellow crosses) of individual spines (yellow arrowheads) in the presence of L-689 (10 μM) and Bay K (10 μM) or in combination with TAT-SCR (5 μM), TAT-CN21 (5 μM), or KN-62 (10 μM).
(B and C) HFU+-induced spine growth in the presence of Bay K and L-689 is blocked by TAT-CN21 peptide (black-filled circles/bar; 6 spines/ 6 cells) or KN-62 (gray-filled circles/bars; 6 spines/6 cells), but not in the presence of control TAT-SCR peptide (red-filled circles/bar; 6 spines/ 6 cells). Volume of the unstimulated neighbors (open circles/bars) was unchanged. Two-way ANOVA with Tukey’s test used in (C) and two-way RMs ANOVA with Dunnett’s test to baseline used in B. Data are represented as means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
(D) Proposed model. Plasticity-inducing glutamatergic stimulation activates non-ionotropic NMDAR signaling, driving cofilin-dependent severing of the actin cytoskeleton, which, in the absence of strong Ca2+ influx, leads to spine shrinkage. On the other hand, during the strong Ca2+ influx associated with LTP induction, severed actin filaments serve as new starting points for actin filament nucleation and branching by the Ca2+- and CaMKII-dependent actin-modifying proteins to expand the F-actin cytoskeleton and drive spine growth.
DISCUSSION
Non-ionotropic NMDAR signaling drives bidirectional spine structural plasticity
Ion-flow-independent NMDAR signaling has been implicated by many independent studies in spine shrinkage and synaptic weakening (Aow et al., 2015; Birnbaum et al., 2015; Carter and Jahr, 2016; Nabavi et al., 2013; Stein et al., 2015, 2020; Thomazeau et al., 2020; Wong and Gray, 2018). Here, we made the unexpected discovery that this non-ionotropic NMDAR signaling pathway is also required for spine growth during synaptic strengthening. It may appear contradictory that the same signaling pathway could support both spine shrinkage and spine growth; however, it is notable that cofilin activation, which has been implicated in spine shrinkage during LTD (Hayama et al., 2013; Stein et al., 2020; Zhou et al., 2004), is also important for long-term spine growth during LTP (Bosch et al., 2014). We propose a model for spine structural plasticity (Figure 4D) in which non-ionotropic NMDAR signaling leads to the p38 MAPK- and cofilin-dependent destabilization of the F-actin network, which, in the absence of Ca2+ influx, drives spine shrinkage, but with Ca2+ influx, instead provides new actin filament nucleation and branching points for subsequent spine enlargement by Ca2+-dependent actin modifying and stabilizing proteins. Indeed, the Ca2+- and CaMKII-dependent activation of small Rho GTPases has been shown to drive LIMK-dependent phosphorylation and inactivation of cofilin and to promote Arp2/3-mediated actin branching activity (Bosch et al., 2014; Murakoshi et al., 2011; Nakahata and Yasuda, 2018). Our model highlights ion-flux-independent NMDAR signaling as a vital component for the bidirectional structural plasticity of dendritic spines.
Role of p38 MAPK in LTP-induced spine growth but not synaptic strengthening
Our studies support an unexpected role for p38 MAPK, a classical LTD molecule implicated in non-ionotropic NMDAR signaling during both LTD and spine shrinkage (Nabavi et al., 2013; Stein et al., 2015) in spine growth during LTP. Notably, we found that inhibition of p38 MAPK blocked spine growth, but not synaptic strengthening, during LTP, at least not in the short term. Our results are in line with previous studies reporting no role for p38 MAPK in tetanus- or pairing-induced NMDAR-dependent LTP (Zhu et al., 2002). In addition, MK2, a p38 MAPK substrate, identified as part of the non-ionotropic NMDAR signaling pathway in spine shrinkage (Stein et al., 2020), is required for spine growth but not for LTP (Privitera et al., 2019), and NOS signaling, presumably upstream of p38 MAPK in non-ionotropic NMDAR signaling (Stein et al., 2020), is required for both spine growth and LTP (Lu etal., 1999; O’Dell et al., 1994). Because inhibition of p38 MAPK and MK2 lead to a pharmacological dissociation of spine growth and LTP, which are normally tightly linked (Matsuzaki et al., 2004), our results suggest that these molecules are downstream of a branch point in signaling that drives synaptic strengthening and spine growth during LTP.
Role of CaMKII in bidirectional spine structural plasticity
During LTP-induced spine growth, Ca2+ influx through the NMDAR leads to the activation of CaMKII, which then activates small Rho GTPases, whose concerted activity drives spine enlargement via the actin regulatory proteins LIMK and Arp2/3, promoting actin polymerization and branching and spine growth (Bosch et al., 2014; Hedrick et al., 2016; Kim et al., 2013; Lee et al., 2009; Murakoshi et al., 2011; Nakahata and Yasuda, 2018; Okamoto et al., 2004; Saneyoshi et al., 2019). Our results, surprisingly, show that the calcium influx that drives LTP-induced spine growth does not need to enter the spine through the NMDAR; instead, it can be supplied by VGCCs, as long as non-ionotropic NMDAR signaling is intact. Although it has been shown that specific local signaling microdomains are important for independently driving LTP and LTD (Zhang et al., 2018), our results suggest that local calcium microdomains produced by ion flows through the NMDAR are not critical for driving LTP-induced spine growth.
CaMKII has been extensively studied in LTP induction (Bayer and Schulman, 2019) and LTP-induced spine growth (Nishiyama and Yasuda, 2015); however, lately CaMKII also has been implicated in LTD (Coultrap et al., 2014; Woolfrey et al., 2018) and spine shrinkage driven by non-ionotropic NMDAR signaling (Stein et al., 2020). A key question is whether and how CaMKII carries out multiple roles in bidirectional spine structural plasticity. It is possible that the kinase mediates ion-flux-dependent and -independent signaling pathways sequentially during spine structural plasticity. Such a situation has been shown for VGCCs, in which an ion flux before conformational signaling drives nuclear transcription (Li et al., 2016). Another possibility is that CaMKII acts simultaneously in the two pathways through different populations, perhaps one Ca2+/CaM bound and the other not. Indeed, synaptic activity has been proposed to activate different populations of CaMKII (Pi et al., 2010; Yasuda et al., 2003), which could then have different substrate specificities, as shown for Ca2+-bound versus autonomous CaMKII activity during LTP and LTD (Coultrap et al., 2014; Woolfrey et al., 2018).
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Karen Zito (kzito@ucdavis.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
This study did not generate any unique datasets or code.
Experimental model and subject details
C57BL/6J or GFP-M (Feng et al., 2000) mice and Sprague-Dawley rats of both sexes were used for the preparation of acute hippocampal slices at postnatal day 16-20 (P16-20) and organotypic hippocampal slice cultures at P6-8. All experimental protocols were approved by the University of California Davis Institutional Animal Care and Use Committee.
METHOD DETAILS
Preparation and transfection of organotypic hippocampal slice cultures
Organotypic hippocampal slices were prepared from P6-P8 Sprague-Dawley rats or C57BL/6J mice of both sexes, as described (Stoppini et al., 1991). Cultures were transfected 1-2 d (EGFP) or 2 d (GCaMP6f) before imaging via biolistic gene transfer (180 psi), as previously described (Woods and Zito, 2008). 10-15 μg of EGFP-n1 or 5 μg GCaMP6f (gift from Lin Tian and Karel Svoboda; [Chen et al., 2013]) and 10 μg pCAG-CyRFP1 (Laviv et al., 2016) were coated onto 6-8 mg of 1.6 μm gold beads.
Preparation of acute hippocampal slices
Acute hippocampal slices were prepared from P16-P20 GFP-M mice (Feng et al., 2000) of both sexes in C57BL/6J background. Coronal 400 μm slices were cut (Leica VT100S vibratome) in cold choline chloride dissection solution containing (in mM): 110 choline chloride, 2.5 KCl, 25 NaHCO3, 0.5 CaCl2, 7 MgCl2, 1.3 NaH2PO4, 11.6 sodium ascorbate, 3.1 sodium pyruvate, and 25 glucose, saturated with 95% O2/5% CO2. Slices were recovered first at 30°C for 45 min and then at room temperature for an additional 45 min, in oxygenated artificial cerebrospinal fluid (ACSF) containing (in mM): 127 NaCl, 25 NaHCO3, 1.25 NaH2PO4, 2.5 KCl, 25 glucose, 2 CaCl2, and 1 MgCl2, before imaging experiments were initiated.
Time-lapse two-photon imaging
Transfected CA1 pyramidal neurons [14-18 days in vitro (DIV)] at depths of 10-50 μm were imaged using a custom two-photon microscope (Woods et al., 2011) controlled with ScanImage (Pologruto et al., 2003). Image stacks (512 × 512 pixels; 0.02 μm per pixel) with 1-μm z-steps were collected. For each neuron, one segment of secondary or tertiary basal dendrite was imaged at 5 min intervals at 29-30°C in recirculating artificial cerebral spinal fluid (ACSF; in mM: 127 NaCl, 25 NaHCO3, 1.2 NaH2PO4, 2.5 KCl, 25 d-glucose, aerated with 95%O2/5%CO2, ~310 mOsm, pH 7.2) with 1 μM TTX, 0 mM Mg2+, and 2 mM Ca2+, unless otherwise stated.
10 μM L-689,560 (L-689, 15 mM stock in DMSO), 100 μM 7CK (100 mM stock in H2O), 2 μM SB203580 (4 mM stock in DMSO), 100 μM NG-Nitro-L-arginine (L-NNA, 200 mM stock in 0.25 N HCL), 10 μM Bay-K (10 mM stock in DMSO), 50 μM (RS)-CPP (50 mM stock in H2O), 50 μM NBQX (10 mM stock in H2O); 10 μM MK2 inhibitor III (20 mM stock in DMSO); and 5 μM TAT-CN21 (5 mM stock in H2O; (Vest et al., 2007)) and 5 μM TAT-SCR (5 mM stock in H2O) were included, as indicated. Slices were pre-incubated for at least 30 min with the drug or vehicle control before glutamate uncaging. Peptides were obtained from GenicBio: L-TAT-GESV: NH2-GRKKRRQRRRYAGQWGESV-COOH, L-TAT-GASA: NH2-GRKKRRQRRRYAGQWGASA-COOH. Slices were pre-incubated with 1 μM (2 mM stock in H2O) peptide for at least 60 min before stimulation.
Photolysis of MNI-caged glutamate with HFU and HFU+ stimulation
High-frequency uncaging (HFU) consisted of 60 pulses (720 nm; 2 ms duration, ~7-10 mW at the sample; adjusted to evoke an average response of ~10 pA at the soma) at 2 Hz, delivered in ACSF containing (in mM): 2 Ca2+, 0 Mg2+, 2.5 MNI-glutamate, and 0.001 TTX. The beam was parked at a point ~0.5-1 μm from the spine at the position farthest from the dendrite. HFU+ stimulation was designed to increase Ca2+-influx through voltage-gated calcium channels. HFU+ consisted of 60 pulses (720 nm; 8 ms duration, ~7 mW at the sample) at 6 Hz, delivered in ACSF containing (in mM): 10 Ca2+, 0 Mg2+, 5 MNI-glutamate, and 0.001 TTX. Healthy and stimulus responsive cells were selected based on a test HFU stimulus onto a test spine on a test dendrite before the application of pharmacological reagents. Only cells on which the test dendritic spine displayed transient growth in response to HFU were used for experiments. Spines targeted for HFU stimulation during experimental data collection were on a different dendrite than the test spine, and were well-isolated and of an average size and that did not fluctuate during baseline imaging. All remaining isolated spines within the region of interest were analyzed as unstimulated neighbors.
Electrophysiology
Whole-cell recordings (Vhold = −65 mV; series resistance 20-40 MΩ) were obtained from visually identified CA1 pyramidal neurons in slice culture (14-18 DIV, depths of 10-50 μm) at 25°C in ACSF containing in mM: 2 CaCl2, 1 MgCl2, 0.001 TTX, 2.5 MNI-glutamate. 2 μM SB203580 was included, as indicated. Recording pipettes (~5-7 MΩ) were filled with cesium-based internal solution (in mM: 135 Cs-methanesulfonate, 10 HEPES, 10 Na2 phosphocreatine, 4 MgCl2, 4 Na2-ATP, 0.4 Na-GTP, 3 Na L-ascorbate, 0.2 Alexa 488, and ~300 mOsm, ~pH 7.25). For each cell, baseline uncaging-evoked excitatory postsynaptic currents (uEPSCs) were recorded from two spines (2-12 μm apart) on secondary or tertiary basal branches (50-120 μm from the soma). The high-frequency glutamate uncaging stimulus (720 nm, 1 ms duration, 8-10 mW at the sample) was then applied to one spine, during which the cell was depolarized to 0 mV. Following the HFU stimulus, uEPSCs were recorded from both the target and neighboring spine at 5 min intervals for 25 min.
Calcium imaging
CA1 pyramidal neurons (13-18 DIV) in slice culture co-expressing GCaMP6 and CyRFP1 were imaged in line-scan mode (500 Hz) to assess if they were healthy and responsive using a test stimulation of a single glutamate uncaging pulse at a dendritic spine. Using a different dendritic segment than the test spine, responsive CA1 neurons were then imaged in frame-scan mode (64 pixels per line, 7.8 Hz) before and after glutamate uncaging (720 nm, 60 pulses, 8 ms duration at 6 Hz, ~7 mW at sample) adjacent to the spine head at 27°C in ACSF containing the following (in mM): 10 Ca2+, 0 Mg2+, 5 MNI-glutamate, and 0.001 TTX. Neurons with high baseline GCaMP6f and neurons that exhibited large calcium transients extending across the dendritic shaft were excluded.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data analysis was performed blind to the experimental condition. Cells for each condition were obtained from at least three independent preparations of either hippocampal acute slices or slice cultures, and only one cell per slice was imaged for any experimental condition.
Quantification of data from imaging experiments
For spine structural plasticity experiments, stimulated spine volume was estimated from background-subtracted green fluorescence using the integrated pixel intensity of a boxed region surrounding the spine head, as previously described (Woods et al., 2011). Previous studies comparing the results from this method to those obtained from electron microscopy have shown that the use of integrated fluorescence intensity to be a valid method for estimating spine volume (Holtmaat et al., 2005). For calcium imaging experiments, Ca2+ transient peak amplitude (ΔF/F0) was measured from background-subtracted green fluorescence in the spine as the ratio of fluorescence during HFU (of specified windows after HFU) to basal fluorescence (2.4 s window before uncaging). All images are maximum projections of three-dimensional (3D) image stacks after applying a median filter (3 × 3) to the raw image data.
Quantification of data from electrophysiology experiments
uEPSC amplitudes from individual spines were quantified as the average of 5-6 test pulses at 0.1 Hz) from a 2 ms window centered on the maximum current amplitude within 50 ms following uncaging pulse delivery relative to the baseline.
Statistical analysis
All data are represented as mean ± standard error of the mean (SEM). All statistics were calculated across cells. Statistical significance was set at p < 0.05 (ANOVA). Data in the bar graphs of Figures 1, 2, 3, 4, and S3 were analyzed using two-way ANOVA with Tukey’s multiple comparison test. Data in the line graphs of Figures 1, 2, 3, 4, and S3 were analyzed using two-way repeated-measure ANOVA with Dunnett’s post hoc test for comparison of HFU stimulation at each time point to baseline. Data in Figures S1C and S1E were analyzed using two-way repeated-measure ANOVA with Bonferroni’s multiple comparison test and for Figure S1F using one-way ANOVA with Tukey’s multiple comparison test. Data were analyzed for Figures S2A-S2F using unpaired t test and for Figure S2G using one-way ANOVA with Tukey’s multiple comparison test. All p and n values are presented in the figure legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| MNI-glutamate | Tocris | Cat #: 1490 |
| TTX citrate | Tocris | Cat #: 1069 |
| L-689,560 | Tocris | Cat #: 0742 |
| 7-Chlorokynurenic acid (7CK) | Tocris | Cat #: 0237 |
| SB203580 | Tocris | Cat #: 1202 |
| L-NNA | Tocris | Cat #: 0664 |
| (S)-(−)-Bay K 8644 | Tocris | Cat #: 1546 |
| (RS)-CPP | Tocris | Cat #: 0173 |
| NBQX | Tocris | Cat #: 1044 |
| KN-62 | Tocris | Cat #: 1277 |
| MK2 inhibitor III | Cayman Chemical | Cat #: 15943 |
| L-TAT-GESV: NH2-GRKKRRQRRRYAGQWGESV-COOH | GenicBio | N/A |
| L-TAT-GASA: NH2-GRKKRRQRRRYAGQWGASA-COOH | GenicBio | N/A |
| TAT-CN21 (tatCN21) | Ulli Bayer (Vest et al., 2007) | N/A |
| TAT-SCR (tatSCR) | Ulli Bayer (Vest et al., 2007) | N/A |
| Experimental models: organisms/strains | ||
| C57BL/6J | Jackson Laboratory | Stock No: 000664 |
| Thy1-GFP line M | Jackson Laboratory | Stock No: 007788 |
| Sprague Dawley rats | Envigo | N/A |
| Recombinant DNA | ||
| EGFP | pEGFP-N1 | Clontech |
| pCAG-CyRFP1 | Laviv et al., 2016 | Addgene #84356 |
| GCaMP6f | Chen et al., 2013 | Addgene #40755 |
Highlights.
p38 MAPK activity is vital for LTP-induced spine growth not synaptic strengthening
Non-ionotropic NMDAR signaling is required for LTP-induced spine growth
Non-ionotropic NMDAR signaling with NMDAR-independent Ca2+ can drive spine growth
CaMKII activity is required for spine growth, independent of calcium source
ACKNOWLEDGMENTS
This work was supported by the NIH (R01 NS062736, T32 MH112507), an ARCS scholar award (D.K.P.), and a grant from the Howard Hughes Medical Institute through the Gilliam Fellowships for Advanced Study (N.C.). We thank Ulli Bayer for tatCN21; Julie Culp, Jennifer Jahncke, and Lorenzo Tom for support with experiments and analysis; and Ulli Bayer, Juan Flores, and Samuel Petshow for critical reading of the manuscript.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108664.
REFERENCES
- Anderson DR, Meyers MJ, Vernier WF, Mahoney MW, Kurumbail RG, Caspers N, Poda GI, Schindler JF, Reitz DB, and Mourey RJ (2007). Pyrrolopyridine inhibitors of mitogen-activated protein kinase-activated protein kinase 2 (MK-2). J. Med. Chem 50, 2647–2654. [DOI] [PubMed] [Google Scholar]
- Aow J, Dore K, and Malinow R (2015). Conformational signaling required for synaptic plasticity by the NMDA receptor complex. Proc. Natl. Acad. Sci. USA 112, 14711–14716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barria A, and Malinow R (2005). NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron 48, 289–301. [DOI] [PubMed] [Google Scholar]
- Bayer KU, and Schulman H (2019). CaM Kinase: Still Inspiring at 40. Neuron 103, 380–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnbaum JH, Bali J, Rajendran L, Nitsch RM, and Tackenberg C (2015). Calcium flux-independent NMDA receptor activity is required for Aβ oligomer-induced synaptic loss. Cell Death Dis. 6, e1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch M, Castro J, Saneyoshi T, Matsuno H, Sur M, and Hayashi Y (2014). Structural and molecular remodeling of dendritic spine substructures during long-term potentiation. Neuron 82, 444–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter BC, and Jahr CE (2016). Postsynaptic, not presynaptic NMDA receptors are required for spike-timing-dependent LTD induction. Nat. Neurosci 19, 1218–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, et al. (2013). Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coultrap SJ, Freund RK, O’Leary H, Sanderson JL, Roche KW, Dell’Acqua ML, and Bayer KU (2014). Autonomous CaMKII mediates both LTP and LTD using a mechanism for differential substrate site selection. Cell Rep. 6, 431–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuenda A, Rouse J, Doza YN, Meier R, Cohen P, Gallagher TF, Young PR, and Lee JC (1995). SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett. 364, 229–233. [DOI] [PubMed] [Google Scholar]
- Davies J, Evans RH, Herrling PL, Jones AW, Olverman HJ, Pook P, and Watkins JC (1986). CPP, a new potent and selective NMDA antagonist. Depression of central neuron responses, affinity for [3H]D-AP5 binding sites on brain membranes and anticonvulsant activity. Brain Res. 382, 169–173. [DOI] [PubMed] [Google Scholar]
- Davies SP, Reddy H, Caivano M, and Cohen P (2000). Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J 351, 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dore K, Aow J, and Malinow R (2015). Agonist binding to the NMDA receptor drives movement of its cytoplasmic domain without ion flow. Proc. Natl. Acad. Sci. USA 112, 14705–14710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, and Sanes JR (2000). Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron 28, 41–51. [DOI] [PubMed] [Google Scholar]
- Ferreira JS, Papouin T, Ladépêche L, Yao A, Langlais VC, Bouchet D, Dulong J, Mothet JP, Sacchi S, Pollegioni L, et al. (2017). Co-agonists differentially tune GluN2B-NMDA receptor trafficking at hippocampal synapses. eLife 6, e25492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiore M, Forli S, and Manetti F (2016). Targeting mitogen-activated protein kinase-activated protein kinase 2 (MAPKAPK2, MK2): medicinal chemistry efforts to lead small molecule inhibitors to clinical trials. J. Med. Chem 59,3609–3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halt AR, Dallapiazza RF, Zhou Y, Stein IS, Qian H, Juntti S, Wojcik S, Brose N, Silva AJ, and Hell JW (2012). CaMKII binding to GluN2B is critical during memory consolidation. EMBO J. 31, 1203–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayama T, Noguchi J, Watanabe S, Takahashi N, Hayashi-Takagi A, Ellis-Davies GC, Matsuzaki M, and Kasai H (2013). GABA promotes the competitive selection of dendritic spines by controlling local Ca2+ signaling. Nat. Neurosci 16, 1409–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi-Takagi A, Yagishita S, Nakamura M, Shirai F, Wu YI, Loshbaugh AL, Kuhlman B, Hahn KM, and Kasai H (2015). Labelling and optical erasure of synaptic memory traces in the motor cortex. Nature 525, 333–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrick NG, Harward SC, Hall CE, Murakoshi H, McNamara JO, and Yasuda R (2016). Rho GTPase complementation underlies BDNF-dependent homo- and heterosynaptic plasticity. Nature 538, 104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess P, Lansman JB, and Tsien RW (1984). Different modes of Ca channel gating behaviour favoured by dihydropyridine Ca agonists and antagonists. Nature 311, 538–544. [DOI] [PubMed] [Google Scholar]
- Hidaka H, and Yokokura H (1996). Molecular and cellular pharmacology of a calcium/calmodulin-dependent protein kinase II (CaM kinase II) inhibitor, KN-62, and proposal of CaM kinase phosphorylation cascades. Adv. Pharmacol 36, 193–219. [DOI] [PubMed] [Google Scholar]
- Hill TC, and Zito K (2013). LTP-induced long-term stabilization of individual nascent dendritic spines. J. Neurosci 33, 678–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtmaat AJ, Trachtenberg JT, Wilbrecht L, Shepherd GM, Zhang X, Knott GW, and Svoboda K (2005).Transient and persistent dendritic spines in the neocortex in vivo. Neuron 45, 279–291. [DOI] [PubMed] [Google Scholar]
- Kim IH, Racz B, Wang H, Burianek L, Weinberg R, Yasuda R, Wetsel WC, and Soderling SH (2013). Disruption of Arp2/3 results in asymmetric structural plasticity of dendritic spines and progressive synaptic and behavioral abnormalities. J. Neurosci 33, 6081–6092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai CSW, Adler A, and Gan WB (2018). Fear extinction reverses dendritic spine formation induced by fear conditioning in the mouse auditory cortex. Proc. Natl. Acad. Sci. USA 115, 9306–9311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laviv T, Kim BB, Chu J, Lam AJ, Lin MZ, and Yasuda R (2016). Simultaneous dual-color fluorescence lifetime imaging with novel red-shifted fluorescent proteins. Nat. Methods 13, 989–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Escobedo-Lozoya Y, Szatmari EM, and Yasuda R (2009). Activation of CaMKII in single dendritic spines during long-term potentiation. Nature 458, 299–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeson PD, Carling RW, Moore KW, Moseley AM, Smith JD, Stevenson G, Chan T, Baker R, Foster AC, Grimwood S, et al. (1992). 4-Amido-2-carboxytetrahydroquinolines: structure-activity relationships for antagonism at the glycine site of the NMDA receptor. J. Med. Chem 35, 1954–1968. [DOI] [PubMed] [Google Scholar]
- Lehmann J, Schneider J, McPherson S, Murphy DE, Bernard P, Tsai C, Bennett DA, Pastor G, Steel DJ, Boehm C, et al. (1987). CPP, a selective N-methyl-D-aspartate (NMDA)-type receptor antagonist: characterization in vitro and in vivo. J. Pharmacol. Exp. Ther 240, 737–746. [PubMed] [Google Scholar]
- Li LL, Ginet V, Liu X, Vergun O, Tuittila M, Mathieu M, Bonny C, Puyal J, Truttmann AC, and Courtney MJ (2013).The nNOS-p38MAPK pathway is mediated by NOS1AP during neuronal death. J. Neurosci 33, 8185–8201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Tadross MR, and Tsien RW (2016). Sequential ionic and conformational signaling by calcium channels drives neuronal gene expression. Science 351, 863–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YF, Kandel ER, and Hawkins RD (1999). Nitric oxide signaling contributes to late-phase LTP and CREB phosphorylation in the hippocampus. J. Neurosci 19, 10250–10261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, and Kasai H (2004). Structural basis of long-term potentiation in single dendritic spines. Nature 429, 761–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakoshi H, Wang H, and Yasuda R (2011). Local, persistent activation of Rho GTPases during plasticity of single dendritic spines. Nature 472, 100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabavi S, Kessels HW, Alfonso S, Aow J, Fox R, and Malinow R (2013). Metabotropic NMDA receptor function is required for NMDA receptor-dependent long-term depression. Proc. Natl. Acad. Sci. USA 110, 4027–4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahata Y, and Yasuda R (2018). Plasticity of spine structure: local signaling, translation and cytoskeletal reorganization. Front. Synaptic Neurosci 10, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama J, and Yasuda R (2015). Biochemical computation for spine structural plasticity. Neuron 87, 63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Dell TJ, Huang PL, Dawson TM, Dinerman JL, Snyder SH, Kandel ER, and Fishman MC (1994). Endothelial NOS and the blockade of LTP by NOS inhibitors in mice lacking neuronal NOS. Science 265, 542–546. [DOI] [PubMed] [Google Scholar]
- Okamoto K, Nagai T, Miyawaki A, and Hayashi Y (2004). Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat. Neurosci 7, 1104–1112. [DOI] [PubMed] [Google Scholar]
- Pi HJ, Otmakhov N, Lemelin D, De Koninck P, and Lisman J (2010). Autonomous CaMKII can promote either long-term potentiation or long-term depression, depending on the state of T305/T306 phosphorylation. J. Neurosci 30, 8704–8709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pigott B, Bartus K, and Garthwaite J (2013). On the selectivity of neuronal NOS inhibitors. Br. J. Pharmacol 168, 1255–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pologruto TA, Sabatini BL, and Svoboda K (2003). ScanImage: flexible software for operating laser scanning microscopes. Biomed. Eng. Online 2, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Privitera L, Hogg EL, Gaestel M, Wall MJ, and Corrêa SAL (2019). The MK2 cascade regulates mGluR-dependent synaptic plasticity and reversal learning. Neuropharmacology 155, 121–130. [DOI] [PubMed] [Google Scholar]
- Reif DW, and McCreedy SA (1995). N-nitro-L-arginine and N-monomethyl-L-arginine exhibit a different pattern of inactivation toward the three nitric oxide synthases. Arch. Biochem. Biophys 320, 170–176. [DOI] [PubMed] [Google Scholar]
- Saneyoshi T, Matsuno H, Suzuki A, Murakoshi H, Hedrick NG, Agnello E, O’Connell R, Stratton MM, Yasuda R, and Hayashi Y (2019). Reciprocal activation within a kinase-effector complex underlying persistence of structural LTP. Neuron 102, 1199–1210.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanhueza M, Fernandez-Villalobos G, Stein IS, Kasumova G, Zhang P, Bayer KU, Otmakhov N, Hell JW, and Lisman J (2011). Role of the CaMKII/NMDA receptor complex in the maintenance of synaptic strength. J. Neurosci 31, 9170–9178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheardown MJ, Nielsen EO, Hansen AJ, Jacobsen P, and Honoré T (1990). 2,3-Dihydroxy-6-nitro-7-sulfamoyl-benzo(F)quinoxaline: a neuroprotectant for cerebral ischemia. Science 247, 571–574. [DOI] [PubMed] [Google Scholar]
- Stein IS, and Zito K (2019). Dendritic spine elimination: molecular mechanisms and implications. Neuroscientist 25, 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein IS, Gray JA, and Zito K (2015). Non-ionotropic NMDA receptor signaling drives activity-induced dendritic spine shrinkage. J. Neurosci 35, 12303–12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein IS, Park DK, Flores JC, Jahncke JN, and Zito K (2020). Molecular mechanisms of non-ionotropic NMDA receptor signaling in dendritic spine shrinkage. J. Neurosci 40, 3741–3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, and Muller D (1991). A simple method for organotypic cultures of nervous tissue. J. Neurosci. Methods 37, 173–182. [DOI] [PubMed] [Google Scholar]
- Thomazeau A, Bosch M, Essayan-Perez S, Barnes SA, De Jesus-Cortes H, and Bear MF (2020). Dissociation of functional and structural plasticity of dendritic spines during NMDAR and mGluR-dependent long-term synaptic depression in wild-type and fragile X model mice. Mol. Psychiatry, Published online July 1, 2020. 10.1038/s41380-020-0821-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokumitsu H, Chijiwa T, Hagiwara M, Mizutani A, Terasawa M, and Hidaka H (1990). KN-62, 1-[N,O-bis(5-isoquinolinesulfonyl)-N-methyl-L-tyrosyl]-4-phenylpiperazi ne, a specific inhibitor of Ca2+/calmodulin-dependent protein kinase II. J. Biol. Chem 265, 4315–320. [PubMed] [Google Scholar]
- Vest RS, Davies KD, O’Leary H, Port JD, and Bayer KU (2007). Dual mechanism of a natural CaMKII inhibitor. Mol. Biol. Cell 18, 5024–5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong JM, and Gray JA (2018). Long-term depression is independent of GluN2 subunit composition. J. Neurosci 38, 4462–4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods G, and Zito K (2008). Preparation of gene gun bullets and biolistic transfection of neurons in slice culture. J. Vis. Exp (12), 675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods GF, Oh WC, Boudewyn LC, Mikula SK, and Zito K (2011). Loss of PSD-95 enrichment is not a prerequisite for spine retraction. J. Neurosci 31, 12129–12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolfrey KM, O’Leary H, Goodell DJ, Robertson HR, Horne EA, Coultrap SJ, Dell’Acqua ML, and Bayer KU (2018). CaMKII regulates the depalmitoylation and synaptic removal of the scaffold protein AKAP79/150 to mediate structural long-term depression. J. Biol. Chem 293, 1551–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda R, Sabatini BL, and Svoboda K (2003). Plasticity of calcium channels in dendritic spines. Nat. Neurosci 6, 948–955. [DOI] [PubMed] [Google Scholar]
- Zhang L, Zhang P, Wang G, Zhang H, Zhang Y, Yu Y, Zhang M, Xiao J, Crespo P, Hell JW, et al. (2018). Ras and Rap signal bidirectional synaptic plasticity via distinct subcellular microdomains. Neuron 98, 783–800.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Homma KJ, and Poo MM (2004). Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron 44, 749–757. [DOI] [PubMed] [Google Scholar]
- Zhu JJ, Qin Y, Zhao M, Van Aelst L, and Malinow R (2002). Ras and Rap control AMPA receptor trafficking during synaptic plasticity. Cell 110, 443–455. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.