

Abstract
Parkinson’s disease is a devastating neurodegenerative disorder affecting 2-3% of the population over 65 years of age. There is currently no disease-modifying treatment. One of the predominant pathological features of Parkinson’s disease is mitochondrial dysfunction, and much work has aimed to identify therapeutic compounds which can restore the disrupted mitochondrial physiology. However, modelling mitochondrial dysfunction in a disease-relevant model, suitable for screening large compound libraries for ameliorative effects, represents a considerable challenge. Primary patient derived cells, SHSY-5Y cells and in vivo models of Parkinson’s disease have been utilized extensively to study the contribution of mitochondrial dysfunction in Parkinson’s. Indeed many studies have utilized LUHMES cells to study Parkinson’s disease, however LUHMES cells have not been used as a compound screening model for PD-associated mitochondrial dysfunction previously, despite possessing several advantages compared to other frequently used models, such as rapid differentiation and high uniformity (e.g., in contrast to iPSC-derived neurons), and relevant physiology as human mesencephalic tissue capable of differentiating into dopaminergic-like neurons that highly express characteristic markers. After previously generating GFP+-LUHMES cells to model metabolic dysfunction, we report this protocol using GFP+-LUHMES cells for high-throughput compound screening in a restoration model of PD-associated mitochondrial dysfunction. This protocol describes the use of a robust and reproducible toxin-induced GFP+-LUHMES cell model for high throughput compound screening by assessing a range of mitochondrial and neuronal morphological parameters. We also provide detailed instructions for data and statistical analysis, including example calculations of Z’-score to assess statistical effect size across independent experiments.
Keywords: Parkinson’s disease, Mitochondria, Compound screening, LUHMES
Background
Parkinson’s disease (PD) is a neurodegenerative disorder characterised primarily by loss of dopaminergic neurons in the substantia nigra of the midbrain and the accumulation of α-synuclein in intra-neuronal inclusions. It is the second most common neurodegenerative disorder affecting 2-3% of the population over 65 years of age ( Poewe et al., 2017 ) . Patients present with resting tremors, bradykinesia and muscle rigidity, along with non-motor symptoms which can include depression, anosmia and memory problems. There is currently no disease-modifying treatment that can prevent or slow the progression of PD, presenting an urgent need for an effective therapeutic (Armstrong and Okun, 2020) .
Mitochondrial dysfunction is a key pathological hallmark of PD; many of the genetic loci associated with familial PD encode proteins that are involved in mitochondrial function or regulation (Hauser and Hastings, 2013). A variety of mitochondrial pathways are disturbed in PD pathology including ATP production, mitophagy, trafficking, biogenesis and calcium buffering ( Park et al., 2018 ) . It is therefore unsurprising that mitochondrial dysfunction is a popular target for therapeutic discovery in PD, which aims to identify compounds capable of enhancing those mitochondrial pathways disturbed in the disease state back towards normal physiological levels. This presents the challenge of finding an effective method to model mitochondrial dysfunction in vitro whereby large compound libraries can be screened efficiently within a disease-relevant model.
We have previously reported the use of primary patient fibroblasts to screen for compounds which rescue mitochondrial phenotypes seen in those fibroblasts ( Mortiboys et al., 2013 ). Furthermore, recently a patient derived iPSC neuron model using a high-throughput, semi-automatic, imaging system was described to identify compounds which ameliorate mitochondrial clearance deficits ( Yamaguchi et al., 2020 ). Both of these systems utilize patient derived cells, one potential issue when using patient derived models is the amount of material required and uniformity of sample. An alternative approach is the use of Lund human mesencephalic (LUHMES) cells. LUHMES cells are a subclone of the MESC2.10 cell line, derived from human embryonic ventral mesencephalic tissue and immortalised by integration of a v-myc retroviral factor which is tetracycline regulatable ( Lotharius et al., 2002 and 2005) . Briefly, LUHMES cells exist in a proliferative state until the addition of tetracycline, dibutyryl cyclic AMP (dCAMP) and glial cell derived neurotrophic factor (GDNF) which halts proliferation and induces uniform differentiation into a dopaminergic-like phenotype. As PD predominantly affects the dopaminergic neurons of the substantia nigra, the differentiated LUHMES cell phenotype is highly physiologically relevant as a model. Further characterisation has demonstrated that differentiated LUHMES cells resemble primary neuron cultures in many aspects: broad upregulation of neuronal markers, extensive neurite outgrowth and basic electrophysiological features ( Scholz et al., 2011 ). Unlike many other transformed neuronal cell lines, LUHMES cells in the differentiated state have c-myc switched off and no dysregulation of the cell cycle is seen. This is particularly important when considering cell cycle mechanisms involved in neuronal DNA repair and neurodegeneration modelling. LUHMES cells are candidates for use in high-throughput screening due to their ease of handling, uniformity and high purity following differentiation induction (> 99%) compared to the often capricious and phenotypically variable culturing of primary neurons ( Scholz et al., 2011 ). A comparative study also showed LUHMES cells express higher levels of neuronal markers (TUBB3, ENO2, MAP2) and have increased neurotoxicant sensitivity compared to two other commonly used neuronal models, SH-SY5Y neuroblastoma cells and human foetal neural stem cells ( Tong et al., 2017 ) .
LUHMES cells have been used extensively in high-throughput screening for the identification of neurotoxicants ( Stiegler et al., 2011 ; Krug et al., 2013 ), including one specific to mitochondrial toxicity ( Delp et al., 2019 ). However, we have only found one study to date that has exploited the high-throughput potential of LUHMES cells in PD therapeutic discovery. Höllerhage et al. used a previously developed alpha-synuclein LUHMES model to screen 1,600 FDA-approved drugs for protective effects by measuring cell viability after treatment ( Höllerhage et al., 2017 ). Previously, we reported the generation of GFP+-LUHMES and the use of them to model metabolic dysfunction in co-culture with astrocytes ( Ratcliffe et al., 2018 ).
Here we utilise the GFP+-LUHMES which we previously generated and validated as a useful model for metabolic dysfunction, to present a detailed and reproducible protocol for high-throughput screening of compounds in a toxin-induced differentiated LUHMES cell model relevant to PD. This protocol improves on previous studies by using high-content live cell imaging and analysis to assess mitochondrial parameters and neuronal morphology.
In the approach described here, differentiated LUHMES cells are treated with rotenone to model PD-associated mitochondrial dysfunction and neuronal loss in accordance with previous studies ( Krug et al., 2013 ; Dolga et al., 2014 ). LUHMES cells are grown to full confluency before the addition of differentiation factors, and then replated into 384-well plates after two days. The cells remain in differentiation media for the full time course of the experiment. The plate is treated with rotenone on the fifth day, then treated with test compounds on the sixth day and live imaged on the seventh day (Figure 1). When designing a toxin induced model for screening; there are generally two paradigms. The first being a protection model, where the potential beneficial compound is added to the cells before the toxin. Secondly, and is the case in this protocol, is a restoration model; where the toxin is added first and then the potential beneficial compounds are added afterwards. This is testing the beneficial compounds to restore a defect already present as opposed to protecting from damage occurring. The advantage of this technique lies in the relative ease and speed in generating a uniform population of disease-relevant human dopaminergic neurons that can be used for large-scale compound screening. Furthermore, high-content live cell imaging and analysis allows the assessment of compound dose-response effects on a broad range of mitochondrial and neuronal morphological parameters.
Figure 1. Timeline of the GFP-LUHMES drug screening protocol.

Materials and Reagents
Materials
384-well black plates (Greiner-Bio, catalog number: 781091)
384-well LDV source plates (Labcyte, catalog number: LP-0200)
384-well source plate seals (Fluidx, catalog number: 41-1011)
5 ml serological pipettes (Fisher Scientific, catalog number: 13-676-10H)
10 ml serological pipettes (Fisher Scientific, catalog number: 13-676-10J)
25 ml serological pipettes (Fisher Scientific, catalog number: 13-678-11)
10 μl pipette tips (Fisher Scientific, catalog number: 02-707-441)
200 μl pipette tips (Fisher Scientific, catalog number: 02-707-422)
1,000 μl pipette tips (Fisher Scientific, catalog number: 02-707-402)
15 ml Falcon tubes (Greiner-Bio, catalog number: 188271)
50 ml Falcon tubes (Greiner-Bio, catalog number: 227261)
T75 flasks (Greiner-Bio, catalog number: 658175)
Cryovials (Greiner-Bio, catalog number: 122261)
Reagents
LUHMES cells (ATCC® CRL-2927TM)
GFP-LUHMES cells (RRID: CVCL_B056, Ratcliffe et al., 2018 )
GFP-expressing lentiviral particles
Advanced DMEM/F-12 (Thermo Scientific, catalog number: 12634010)
Fibronectin (Sigma-Aldrich, catalog number: FC010)
Poly-L-Ornithine (Sigma-Aldrich, catalog number: P4957)
PBS Tablets (Thermo Scientific, catalog number: BR0014G)
Trypsin 10x (Lonza, catalog number: BE02-007E)
L-Glutamine (Lonza, catalog number: BE-17-605E)
N-2 Supplement (Gibco, catalog number: 11520536)
Pen-Strep (Lonza, catalog number: DE17-603E)
FGF-basic (Peprotec, catalog number: 100-18B)
dCAMP (Sigma-Aldrich, catalog number: D0627)
GDNF (Peprotech, catalog number: 450-10)
Tetracycline (Sigma-Aldrich, catalog number: T7660)
Hoechst (Sigma, catalog number: 94403)
TMRM (Invitrogen, catalog number: T668)
Sterile 1x PBS (see Recipes)
Sterile 1x Trypsin (see Recipes)
-
LUHMES Base Media (see Recipes)
Advanced DMEM/F-12
N-2 Supplement
Pen-Strep
L-Glutamine
-
LUHMES Proliferation Media (see Recipes)
LUHMES Base Media
FGF-basic
LUHMES Differentiation Media
Tetracycline
dCAMP
GDNF
Equipment
Mechanical pipette gun for serological pipettes
Mechanical pipettes (P20, P200, P1000)
Multichannel mechanical pipettes (P10, P200)
4 °C fridge
-20 °C freezer
-80 °C freezer
Sterile tissue culture hood
Autoclave
Centrifuge for tissue culture (MSE Harrier 15/80, catalog number: MSB080.CX1.5)
Centrifuge for source plates
CO2 Incubator (Sanyo, model: MCO-19AIC)
Echo 550 Liquid Handler (Labcyte, catalog number: Echo 550)
MultiPod Controller (Roylan Developments, catalog number: SPOD0012)
StoragePod Enclosure (Roylan Developments, catalog number: SPOD0010)
Opera Phenix High-Content Screening System (PerkinElmer)
Software
Echo Liquid Handler Software (Labcyte)
Echo Plate Reformat (Labcyte)
Harmony High-Content Imaging and Analysis Software (PerkinElmer)
Columbus Image Data Storage and Analysis System (PerkinElmer)
Excel 2016 (Microsoft)
GraphPad Prism 8.2 (GraphPad)
Procedure
Generation of GFP-LUHMES cells
GFP-LUHMES cells are generated by the transduction of proliferative LUHMES cells with GFP-expressing lentiviral particles under the control of a PGK promoter ( Ratcliffe et al., 2018 ).
Seed proliferative LUHMES cells (p3-p5) at 1.5 x 106 per T75 flask.
Transduce with GFP-expressing lentiviral particles for 24 h. MOI of 8; for transduction, a 1:100 dilution was used. Transduction efficiency was previously assessed by FACS sorting to be 98.5%. GFP-expressing lentiviral particles were a kind gift from Eva Karyka and Mimoun Azzouz at the University of Sheffield.
-
After 24 h, maintain in LUHMES Proliferation Media for 72 h before beginning experiments.
Part I: GFP-LUHMES cell Maintenance Protocol
-
Defrost and seed GFP-LUHMES cells in a sterile culture hood
Prepare LUHMES Proliferation Media and sterile 1x PBS according to recipes below.
-
Prepare a Fibronectin Poly-L-Ornithine coated T75 flask.
Prepare 10 ml of Fibronectin Poly-L-Ornithine Coating Solution per T75 flask by adding 1 μg/ml Fibronectin and 50 μg/ml Poly-L-Ornithine to sterile distilled water.
Mix the Fibronectin Poly-L-Ornithine Coating Solution and transfer 10 ml to the T75 flask using a 10 ml serological pipette.
Incubate the flask with the Fibronectin Poly-L-Ornithine Coating Solution at 37 °C for at least 3 h or overnight.
Remove the Fibronectin Poly-L-Ornithine Coating Solution from the flask using a 10 ml serological pipette.
Wash the coated surface by transferring 5 ml of sterile 1x PBS to the flask using a 10 ml serological pipette and gently tilting the flask from side to side allowing the PBS to run across the whole surface. The PBS should be left in the flask until immediately before the cells are seeded in the flask to prevent the coating from drying out. The flasks can be prepared up to 1 hour before use.
-
Defrost and seed GFP-LUHMES cells into LUHMES Proliferation Media.
Remove prepared LUHMES Proliferation Media from the fridge and allow to warm to room temperature.
Take one frozen vial of GFP-LUHMES cells from freezer storage. Generally, we recommend to revive a vial of passage 1-10 so that revived cells can undergo numerous splits before retiring at passage 15.
Add 10 ml room temperature LUHMES Proliferation Media to a 15 ml Falcon using a 10 ml serological pipette.
Transfer ~500 μl of room temperature LUHMES Proliferation Media to the GFP-LUHMES cell vial using a P1000 mechanical pipette and slowly mix by aspirating and expelling the liquid in the tip repeatedly.
When the solution in the GFP-LUHMES cell vial is fully defrosted, transfer the solution to the room temperature LUHMES Proliferation Media in the 15 ml Falcon.
Remove all 1x PBS from the coated T75 flask and add all of the cell solution from the 15 ml Falcon to the T75 flask using a 10 ml serological pipette.
Tilt the flask back and forward and then side to side 2-3 times to distribute the GFP-LUHMES cells evenly over the coated surface.
Incubate the flask in a 37 °C/5% CO2 incubator.
-
On the following day, change the GFP-LUHMES cell media to remove any cells not fully attached.
Remove all media from the T75 flask using a 10 ml serological pipette.
Add 10 ml room temperature LUHMES Proliferation Media to the T75 flask using a 10 ml serological pipette.
Incubate the flask in a 37 °C/5% CO2 incubator.
-
Split the GFP-LUHMES cells every 3-4 days or when confluency is 80-90% in a sterile culture hood
Prepare two Fibronectin Poly-L-Ornithine coated T75 flasks according to the procedure described above (Step A2).
Prepare LUHMES Proliferation Media and 1x Trypsin according to the recipes below. If pre-prepared, remove from the fridge and allow to warm to room temperature.
-
Split GFP-LUHMES cells into two flasks containing LUHMES Proliferation Media. One flask will be used for stock maintenance and one flask will be used for differentiation.
Remove all LUHMES Proliferation Media from GFP-LUHMES cell flask using a 10 ml serological pipette and transfer 5 ml into a 15 ml Falcon.
Add 5 ml room temperature 1x Trypsin to GFP-LUHMES cell flask using a 10 ml serological pipette.
Incubate GFP-LUHMES cell flask for 5 min in a 37 °C/5% CO2 incubator.
Firmly smack the side of the GFP-LUHMES cell flask 2-3 times to dislodge cells into the solution. If successful, cell clouds should be visible in the solution.
Transfer all cell solution from GFP-LUHMES cell flask into the 15 ml Falcon containing 5 ml of used LUHMES Proliferation Media.
On the bench, centrifuge the 15 ml Falcon at 400 × g for 4 min.
Remove the supernatant using a 10 ml serological pipette and discard.
Resuspend the pelleted GFP-LUHMES cells in an appropriate amount of LUHMES Proliferation Media according to desired splitting ratio. We recommend a 1:15 splitting ratio of a fully confluent flask to be ready to split again in 3 days, although this may need to be adjusted slightly depending on cell behavior.
Remove all 1x PBS from the coated T75 flasks and add 10 ml LUHMES Proliferation Media per flask using a 10 ml serological pipette.
Add an appropriate amount of re-suspended GFP-LUHMES cell solution to the LUHMES Proliferation Media in both T75 flasks.
-
Change the Proliferation Media on the GFP-LUHMES cells every 2 days (following a split)
Remove a Falcon tube of LUHMES Proliferation Media from the fridge and allow to warm to room temperature.
-
Change the media on the GFP-LUHMES cells to fresh Proliferation Media.
Remove all LUHMES Proliferation Media from GFP-LUHMES cell flask using a 10 ml serological pipette and discard.
Add 10 ml of room temperature LUHMES Proliferation Media to the GFP-LUHMES cell flask using a 10 ml serological pipette.
Incubate the flask in a 37 °C/5% CO2 incubator.
Part II: GFP-LUHMES cell Differentiation and Replating Protocol
-
Initiate differentiation of GFP-LUHMES cells (Day 1)
Wait 2-3 days following a split of the GFP-LUHMES cells to allow proliferation.
Prepare LUHMES Differentiation Media according to the recipe below. This must be made fresh before every use.
-
Change the Proliferation Media on the GFP-LUHMES cells to Differentiation Media.
Remove all LUHMES Proliferation Media from GFP-LUHMES cell flask using a 10 ml serological pipette and discard.
Add 10 ml of room temperature LUHMES Differentiation Media to the GFP-LUHMES cell flask using a 10 ml serological pipette.
Incubate the flask in a 37 °C/5% CO2 incubator.
-
Replate differentiating GFP-LUHMES into a 384-well plate (catalog number: 781091) (Day 3)
-
Prepare a Fibronectin Poly-L-Ornithine coated 384-well plate.
Prepare 15 ml of Fibronectin Poly-L-Ornithine Coating Solution per 384-well plate by adding 1 μg/ml Fibronectin and 50 μg/ml Poly-L-Ornithine to sterile distilled water.
Mix the Fibronectin Poly-L-Ornithine Coating Solution and transfer 15 ml to a suitable reservoir for multichannel pipetting (for example but not limited to an upturned pipette box lid, clean and sterile).
Transfer 50 μl of the Fibronectin Poly-L-Ornithine Coating Solution to each well of the 384-well plate except for the two outermost columns and rows using a P200 multichannel mechanical pipette.
Transfer 40 μl of sterile 1x PBS to each of the empty outermost wells to act as a firewall using a P200 multichannel mechanical pipette.
Incubate the flask with the Fibronectin Poly-L-Ornithine Coating Solution at 37 °C for at least 3 h or overnight.
Remove the Fibronectin Poly-L-Ornithine Coating Solution from the 384-well plate using a P200 multichannel mechanical pipette.
Wash the coated surface by transferring 50 μl of sterile PBS to each coated well using a P200 multichannel mechanical pipette.
Prepare LUHMES Differentiation Media according to the recipe below.
-
Lift GFP-LUHMES cells and seed 10,000 cells/well into a coated 384-well plate.
Remove all LUHMES Differentiation Media from the differentiating GFP-LUHMES cell flask using a 10 ml serological pipette and transfer 5 ml into a 15 ml Falcon.
Add 5 ml room temperature 1x Trypsin to the differentiating GFP-LUHMES cell flask using a 10 ml serological pipette.
Incubate differentiating GFP-LUHMES cell flask for 5 min in a 37 °C/5% CO2 incubator.
Firmly smack the side of the differentiating GFP-LUHMES cell flask 2-3 times to dislodge cells into the solution.
Transfer all cell solution from differentiating GFP-LUHMES cell flask into the 15 ml Falcon containing 5 ml of used LUHMES Differentiation Media.
On the bench, centrifuge the 15 ml Falcon at 400 x g for 4 min.
Remove the supernatant using a 10 ml serological pipette and discard.
Resuspend the pelleted GFP-LUHMES cells in 5 ml of LUHMES Differentiation Media, homogenising thoroughly by aspirating and expelling the GFP-LUHMES resuspension 10-15 times with a P1000 mechanical pipette.
Prepare a Haemocytometer and transfer 10 μl of GFP-LUHMES resuspension onto the counting grid.
Count the number of GFP-LUHMES cells and calculate the cells/ml in the GFP-LUHMES resuspension.
Remove quantity of GFP-LUHMES resuspension containing 4 x 106 cells and transfer to a 15 ml Falcon. This will be the plating suspension.
Top up the plating suspension to a final volume of 12 ml with LUHMES Differentiation Media using a 10 ml serological pipette.
Remove the PBS from the coated wells on the first half of the 384-well plate using a P200 multichannel mechanical pipette.
Homogenise the plating suspension thoroughly by aspirating and expelling the solution 10-15 times with a 10 ml serological pipette.
Transfer half of the plating suspension (6 ml) into a suitable reservoir for multichannel pipetting.
Transfer 30 μl of plating suspension to each coated well of the 384-well plate.
Remove the PBS from the coated wells on the second half of the 384-well plate using a P200 multichannel mechanical pipette.
Re-homogenise the plating suspension in the 15 ml Falcon thoroughly by aspirating and expelling the solution 10-15 times with a 10 ml serological pipette.
Transfer the rest of the plating suspension to the reservoir and fill the remaining coated wells with 30 μl of plating suspension per well using a P200 multichannel mechanical pipette.
Move the plate back and forth, then side to side, several times to ensure the cells are evenly distributed across the well bottoms.
-
Incubate the 384-well plate in a 37 °C/5% CO2 incubator.
Part III: GFP-LUHMES Drug Screening Protocol (timeline continued from Part II)
-
Part III: GFP-LUHMES Drug Screening Protocol (timeline continued from Part II)
-
Treat differentiated GFP-LUHMES 384-well plate with rotenone (Day 5)
Prepare LUHMES Differentiation Media according to the recipe below.
-
Prepare rotenone-treated media and vehicle-treated media in a sterile culture hood.
To avoid cell disturbance, we avoid removing media and instead top up each well with 10 μl rotenone-treated media giving a final volume of 40 μl media per well. Therefore, rotenone-treated media must be prepared at four times the desired final concentration (4 μM).
Transfer 4 ml of LUHMES Differentiation Media to a 15 ml Falcon and label “Rotenone”. Transfer 4 ml of LUHMES Differentiation Media to another 15 ml Falcon and label “Vehicle”.
Prepare 10 mM rotenone in ethanol. Rotenone powder should be weighed in a fume hood.
Add 6.4 μl 10 mM rotenone to the media in the 15 ml Falcon labeled “Rotenone” using a P20 mechanical pipette. The final rotenone concentration in the well will be 4 μM.
Add an equal volume of ethanol (6.4 μl) to the 15 ml Falcon labeled “Vehicle” using a P20 mechanical pipette.
Mix both 15 ml Falcons by slowly inverting 3-4 times.
-
Transfer treated media to the differentiated GFP-LUHMES 384-well plate. We suggest a plate map as shown in Figure 2A (i.e., “DMSO vehicle” wells are treated with vehicle-treated media, and all other wells treated with rotenone-treated media).
Transfer the contents of “Rotenone” media Falcon and “Vehicle” media Falcon into separate reservoirs for multichannel pipetting. Label the reservoirs accordingly.
Transfer 10 μl of rotenone-treated media to each well of the 384-well plate using a P10 multichannel mechanical pipette, ensuring the pipette tips are placed deep enough in the well to ensure ejected liquid mixes with the contents of the well. Leave some wells untreated for addition of vehicle-treated media to measure rotenone effect later. We suggest including vehicle-treating some wells in each DMSO only control column as explained in Step B3 below. Transfer 10 μl of vehicle-treated media to each untreated well of the 384-well plate using a P10 multichannel mechanical pipette.
Incubate the 384-well plate in a 37 °C/5% CO2 incubator.
-
Prepare compounds in DMSO and write protocols for using the Echo 550 Liquid Handler and Opera Phenix High-Content Screening System
Prepare compound library in 100% anhydrous DMSO on 384-well source plates (catalog number: LP-0200) with plate seal. Store in the StoragePod Enclosure and maintain a dry nitrogen atmosphere using the MultiPod Controller.
Write a compound transfer protocol using the Echo Liquid Handler software. The source plate should be set to “384LDV-DMSO”, the destination plate to “Griener_384PS_781096” and custom mapping should be selected.
Set the protocol to transfer an appropriate volume from each well of the source plate to the corresponding well on the destination plate to give the desired final drug concentration. We suggest splitting the destination plate into quarters and using the first column of each quarter for DMSO only treatment to act as a negative control (Figure 2). Parameter values for each compound-treated well in each quarter can then be normalised to the respective DMSO control well values in that quarter. For a full dose response curve, we recommend testing 8 concentrations for each compound at every half-log, i.e., 3 nM, 10 nM, 30 nM, 100 nM, 300 nM, 1,000 nM, 3,000 nM, 10,000 nM. We also recommend including at least one positive control compound on each plate; we used a compound previously identified in house that showed beneficial effects on mitochondrial and morphological parameters.
Write an imaging protocol (details below) for imaging the LUHMES cells on the Harmony High-Content Imaging and Analysis Software. Use the DAPI, Alexa 488 and Cy3 channels to capture the Hoechst signal, GFP signal and TMRM signal respectively. Use test images to ensure appropriate exposure time for each laser; this needs to be set by the user as it depends on the signal strength of laser power of each imaging system. Use a 40x water confocal objective, set the number of fields to 10 and set the number of stacks to 6. Select the wells to be imaged (note the outermost two columns and rows cannot be imaged by the Opera Phenix) and select the location of the fields in each well.
-
Treat differentiated GFP-LUHMES 384-well plate with compounds (Day 6)
Depressurise the StoragePod Enclosure using the Multipod Enclosure and remove the 384-well source plate.
Centrifuge the 384-well source plate at 2,500 x g for 2 min to remove any gas bubbles from the plate contents.
Calibrate and focus on the Echo 550 liquid handler before each independent plate shoot.
Survey the 384-well ldv source plate using the Echo 550 liquid handler. Ensure the volume in each well is high enough for transfer of the desired volume to the destination plate according to the compound transfer protocol. Note that the volume of each well in the 384-well source pate should not exceed 12 μl. Ensure that the water content of the contents of each well does not exceed 30% (this is inferred from the measurement of DMSO content which should not be lower than 70%; if this is the case then the contents of that well must be refreshed).
Remove the seal from the 384-well source plate and initiate the compound transfer protocol. Transfer the 384-well source plate and the destination plate into the machine when prompted.
When the protocol is complete, remove the destination plate and incubate immediately in a 37 °C/5% CO2 incubator.
Replace the seal on the 384-well source plate and depressurise the StoragePod Enclosure using the Multipod Controller. Replace the 384-well source plate in the StoragePod Enclosure. Lock and repressurise the StoragePod Enclosure using the MultiPod Controller.
-
Assay and image the GFP-LUHMES 384-well plate (Day 7)
Prepare LUHMES Differentiation Media according to the recipe below.
-
Prepare the assay media.
Prepare 10 mM Hoechst in sterile distilled water.
Prepare 80 μM TMRM in DMSO.
Transfer 4 ml of LUHMES Differentiation media into a 15 ml Falcon.
Add 20 μl of x mM Hoechst solution to the 15 ml Falcon.
Add 20 μl of x mM TMRM solution to the 15 ml Falcon.
Mix the contents of the 15 ml Falcon (assay media) by slowly inverting the Falcon 4-5 times.
-
Incubate the GFP-LUHMES 384-well plate with assay media for 1 h.
Transfer the assay media to a suitable reservoir for multichannel pipetting.
Transfer 10 μl of assay media to each well of the 384-well plate using a P10 multichannel mechanical pipette, ensuring the pipette tips are placed deep enough in the well to ensure ejected liquid mixes with the contents of the well.
Incubate the GFP-LUHMES 384-well plate in a 37 °C/5% CO2 incubator for 1 h.
-
Image the GFP-LUHMES 384-well plate using the Opera Phenix High-Content Screening System.
Set the TCO (temperature and CO2) settings on the Opera Phenix to 37 °C and 5% CO2 and set both to “On”. Open the connected CO2 gas cylinder when prompted. Allow the system to reach the desired temperature and gas level.
Open the prepared imaging protocol.
Load the GFP-LUHMES 384-well plate.
Flush the water objective lens.
Run a test and adjust the exposure time of each channel so that the intensity is significant over background levels. Adjust the height of the stacks if needed so that images are well focused across the height of the cell.
Run the imaging protocol (Figure 2).
Figure 2. Plate map and example images following assaying of a GFP-LUHMES 384-well plate drug screen.

A. Plate map showing the suggested arrangement of control wells and test compound wells. All wells are treated with rotenone except for “DMSO vehicle” wells. The colours represent the concentration in nM for each compound. B-D. Blue = DAPI, orange = TMRM, green = GFP, scale bars = 20 μm. Z stack maximum projection confocal images captured using the Opera Phenix High-Content Screening System from (B) a vehicle-treated DMSO control well, (C) a rotenone-treated DMSO control well and (D) a rotenone-treated positive control well (1,000 nM).
Data analysis
The images will be analysed in batch using the Columbus Image Data Storage and Analysis System. The GFP-signal will be used to assess neuronal morphology parameters and the TMRM-signal will be used to assess mitochondrial function and morphology.
A short video (Video 1) is available to show the analysis software and provide guidance to parts of the protocol, with detailed instructions below.
Video 1. Analysis set up using Columbus analysis software.
Once the imaging protocol is completed, export the measurement file to a convenient location. Ensure “Measurements Inc. Associated Files” is selected and note the export path.
Import the exported files to the Columbus Image Data Storage and Analysis System.
Select the Image Analysis tab and open the imported file.
-
Write an analysis protocol using the following building blocks. Add a new building block by clicking the blue plus symbol.
Input Image. This is where the images are loaded into the analysis tool. Select “Maximum Projection” for Stack Processing and select “Basic” for Flatfield Correction. Uncheck the box next to Quick Tune.
Find Nuclei. This first Find Nuclei building block is used to aid the GFP-neurite detection in the following building blocks. The Hoechst signal will be used later to more accurately count the nuclei. Set the channel to “Alexa 488” (GFP) and use Method A. Label the Output Population as “Nuclei (GFP neurites)”.
Find Neurites. Set the Channel to “Alexa 488" (GFP) and ensure Population is set to “Nuclei (GFP neurites)” and Region to “Nucleus”. Two selections will be made, shown by the tabs above the image. The first is made via the positions of the nuclei selected above, and labeled “Nuclei (GFP neurites)” (Figure 3A). The second detects any neurite structures in the image, labeled “Neurite Segments”. Click the black downward-facing arrow and adjust the values for each detection parameter so that as many neurites are selected as possible. Do not be concerned about background detection in the “Neurite Segments” selection as this will be removed in the following building block. Label the Output Population as “Neurite Segments”.
Select Population. We will now filter out background debris detected in the Find Neurites selection above. Set the Population to “Neurite Segments” and the Method to “Filter by Property”. Expand the drop down menu by clicking the black downward-facing arrow. Select “Segment Length”, “Neurite Segment Area” and “Neurite Segment Roundness” and adjust the values until most of the debris is removed from selection but the ‘real’ neurites remain selected. We used values of > 12.5, > 0.7, and > 0 respectively. Label the Output Population as “Neurite Segments Selected”.
Calculate Morphology Properties. Select Population “Neurite Segments Selected” and use Method “Standard”. Expand the drop down menu and check the box next to each parameter.
Calculate Morphology Properties (2). Select Population “Nuclei (GFP neurites)”, Region “Neurite Tree” and Method “Standard”. Expand the drop down menu and check the box next to each parameter.
Find Image Region. Select Channel “Cy3”, ROI Population to “None” and Method to “Common Threshold”. Expand the drop down menu and change the threshold to an appropriate level to select most of the signal without excessive background. Tick “Split into Objects”, expand the further drop down menu and change the minimum area to an appropriate value to remove background (we used > 25 μm2). Leave “Fill Holes” unticked. Label the Output Population “Image Region (Cy3)”.
Find Image Region (2). Repeat as in (g.) but use Channel “Alexa 488” to select the GFP-neurite image region (Figure 3B). Label the Output Population “Image Region (GFP-neurites)”.
Filter Image. We will now segment the mitochondria. Set Channel “Cy3” and Method to “Texture SER”. Expand the drop down menu and set Filter to “SER Ridge”, Scale to 1 px and Normalization to “Unnormalized”. Label the Output Image as “SER Ridge”.
Calculate Image. This step amplifies the signal to improve segmentation. Set Method to “By Formula” and enter the Formula “A*1000”. Set Channel A to “SER Ridge” and label the Output Image as “Calculated Image”.
Find Spots. Set Channel “Calculate Image” and the ROI Population to “Image Region (GFP-neurites)”. This ensures only mitochondria within the GFP-neurite selection will be segmented. Set Method “D” and label the Output Population as “Mitochondria”.
Select Population. Set Population to “Mitochondria” and Method to “Filter by Property”. Choose the parameter “Spot Area [px2]”, select “>” and enter a minimum value to remove any debris in the selection (we used 8 px2) (Figure 3C). Label the Output Population as “Mitochondria Selected”.
Calculate Intensity Properties (2). Set Channel “Cy3”, Population “Image Region (GFP-neurites)” and Region “Image Region”. Set Method to “Standard” and expand the drop down menu to tick “Mean”. This will measure the TMRM signal intensity within the GFP-neurite selection.
Calculate Morphology Properties. Set Population “Mitochondria Selected” and Method “Standard”. Expand the drop down menu and tick “Area” and “Roundness”. This will measure the morphological properties of the segmented mitochondria. Label Output Properties as “Mitochondria Morphology”.
Calculate Texture Properties. Set Channel “Cy3”, Population “Mitochondria” and use Method “SER Features”. This will measure the texture properties of the segmented mitochondria. Label Output Properties as “Mitochondria Texture”.
Find Nuclei (2). Set Channel “DAPI” and use Method “C”. Expand the drop down menu and adjust the Common Threshold appropriately. Adjust the minimum area to remove background debris (we used > 20 μm2) (Figure 3D). Label the Output Population “Nuclei (DAPI)”.
Define Results. Use Method “List of Outputs”. For each Population, expand the drop down menu and select the type of output you wish from each measured parameter. Alternatively, use “Apply to All” to select an output type for all measured parameters in that population. Click the green arrow at the top of the building block to apply changes and then save the protocol.
Select the Batch Analysis tab and select the data folder from the left-hand panel that you wish to analyse. Then, select the analysis protocol written above. Click the green arrow to start the analysis.
When the analysis finishes, the results will be shown in the corresponding data folder in the left-hand panel. Select the Browse tab. Open the corresponding data folder by clicking the plus symbol next to it, and select the “Result” file to open it. In the bottom-right-hand panel, the “Summary” tab will show the data from that analysis. Click the adjacent tab corresponding to the name of the “Result” file to see the raw data in full. Download the file using the blue “Download” link.
Open the downloaded file in Excel 2016. Organise the data so that parameter values for each compound at each concentration as well as control wells are ordered sequentially.
Input the data for each compound into GraphPad Prism 8.2 using a separate data table for each parameter. Ensure each compound is organised by column and each concentration by row. Include the data for positive control compounds.
Figure 3. Analysis protocol selections using the Columbus Image Data Storage and Analysis System.

A. “Find Neurites” selection using the Alexa 488 channel, see step 4c of the Data analysis protocol. B. “Find Image Region” selection using the Alexa 488 channel, see step 4h. C. “Select Population” using the segmented Mitochondria channel, see step 4l. The indented image shows a close up of the individual selections. D. “Find Nuclei” selection using the DAPI channel, see step 4q. All scale bars = 20 μm.
Statistical analysis
The statistical analysis will depend on the experimental setup. However, some important plate effects should be controlled to ensure robustness and reproducibility of the assay.
Create another data table and plot the nuclei number (DAPI) across the whole plate. This should remain consistent to minimise plate drift effects.
Create another data table and plot the values for DMSO vehicle control wells against DMSO rotenone control wells for each parameter (Figure 4A). A significant difference should be seen for each parameter.
-
Calculate Z’ and SW scores in order to ensure the robustness of the assay. The formulae are:
In this protocol, ‘max’ would refer to values from DMSO vehicle control wells, and ‘min’ would refer to values from DMSO rotenone control wells. Example data for Z’ scores for various parameters in this assay are given in Figure 4B. In general, a Z’ score of above 0.5 and an SW score of above 2 are acceptable for use in phenotypic screening assays.
Typically, One-Way ANOVA with Dunnet’s post-hoc test would be performed to compare single concentration drug effects against vehicle treated wells. Alternatively, if a concentration response plate map is undertaken, a Nonlinear regression (curve fit) analysis on the data, using log(agonist) vs. response (three parameters) is typically used. All analysis is carried out in GraphPad Prism 8.2.
Figure 4. Example data showing rotenone effect, Z’-scores from GFP-LUHMES 384-well plate drug screening.
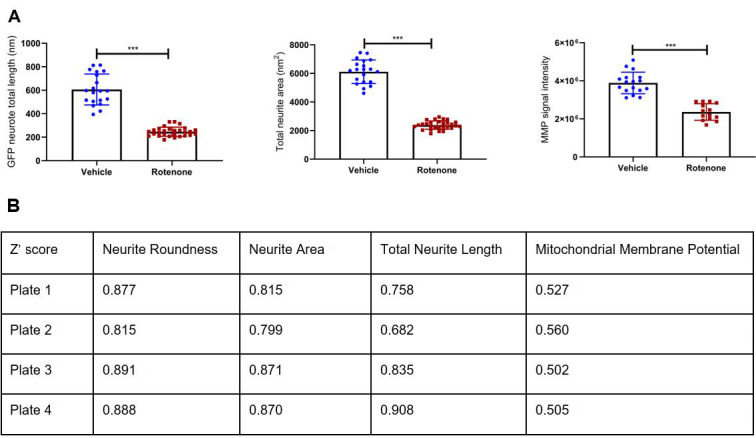
A. Rotenone effect for three example parameters is shown by comparing values for DMSO vehicle-treated wells against DMSO rotenone-treated wells. In both cases, the mean parameter value for rotenone-treated wells is significantly different to vehicle-treated wells (*** = P < 0.0001). B. Z’-scores for several parameters across four separate plates, each performed three weeks apart. The parameter ‘Neurite Roundness’ represents a measure of cell shape by comparing cell area to cell perimeter; a healthy neuron with extensive neurites would have a very low score approaching 0, compared to a spherical cell which should score up to 1.
Notes
GFP-LUHMES cells should be retired once they reach passage 15 and replaced with younger stocks.
When freezing GFP-LUHMES stocks, we recommend to split a fully confluent T75 flask, resuspend the pellet in 5 ml LUHMES Proliferation Media, add 10% DMSO and 20% FBS, then divide equally between 5 cryovials.
When counting GFP-LUHMES cells on the haemocytometer, only circular, bright spheres should be counted.
When transferring the differentiated GFP-LUHMES cells into the 384-well plate, act quickly and re-homogenise the cell suspension in the reservoir using the multichannel pipette between transfers. This is to ensure the number of cells plated per well is as consistent as possible across the plate.
The analysis software can output several additional parameters, including cell number and number of cells with neurites which can be useful parameters to assess cell viability, drift across plates and conditions as well as variability between assay days.
Recipes
-
Sterile 1x PBS
Add 5 PBS tablets to 500 ml of distilled water in a flask suitable for autoclaving
Mix until tablets dissolve and autoclave
Store at room temperature
-
Sterile 1x Trypsin
Add 500 ml of distilled water to a flask suitable for autoclaving
Autoclave the flask and transfer to a sterile culture hood
In the hood, add 45 ml of sterile distilled water to 5 ml of Trypsin 10x in a 50 ml Falcon
Mix by inverting 2-3 times
Store at 4 °C
-
LUHMES Base Media
Store at 4 °C
500 ml Advanced DMEM/F-12
5 ml N-2 Supplement
5 ml Pen-Strep
5 ml L-Glutamine
-
LUHMES Proliferation Media
Store for one week at 4 °C
50 ml LUHMES Base Media
40 ng/ml FGF-basic
-
LUHMES Differentiation Media
Prepare fresh for each use
20 ml LUHMES Base Media
1 μg/ml Tetracycline
1 mM dCAMP
2 ng/ml GDNF
Acknowledgments
This work was funded by a Parkinson’s UK Senior Fellowship award to HM (F-1301) and a Parkinson’s UK Virtual Biotech project grant to HM (I-1904). LF is supported by the Academy of Medical Sciences (SBF002\1142) and the Medical Research Council (MRC, grant 1812144). IV was supported by Consejo Nacional de Ciencia y Technologia (CONACyT), Mexico.
The generation of GFP-LUHMES cells was based on a previously established protocol from Ratcliffe et al., 2018 .
Competing interests
The authors declare no competing interests.
Citation
Readers should cite both the Bio-protocol article and the original research article where this protocol was used.
References
- 1. Armstrong M. J. and Okun M. S.(2020). Diagnosis and Treatment of Parkinson Disease: A Review. JAMA 323(6): 548-560. [DOI] [PubMed] [Google Scholar]
- 2. Delp J., Funke M., Rudolf F., Cediel A., Bennekou S. H., van der Stel W., Carta G., Jennings P., Toma C., Gardner I., van de Water B., Forsby A. and Leist M.(2019). Development of a neurotoxicity assay that is tuned to detect mitochondrial toxicants. Arch Toxicol 93(6): 1585-1608. [DOI] [PubMed] [Google Scholar]
- 3. Dolga A. M., de Andrade A., Meissner L., Knaus H. G., Höllerhage M., Christophersen P., Zischka H., Plesnila N., Hoglinger G. U. and Culmsee C.(2014). Subcellular expression and neuroprotective effects of SK channels in human dopaminergic neurons. Cell Death Dis 5: e999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hauser D. N. and Hastings T. G.(2013). Mitochondrial dysfunction and oxidative stress in Parkinson's disease and monogenic parkinsonism. Neurobiol Dis 51: 35-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Höllerhage M., Moebius C., Melms J., Chiu W.-H., Goebel J. N., Chakroun T., Koeglsperger T., Oertel W. H., Rösler T. W., Bickle M. and Höglinger G. U.(2017). Protective efficacy of phosphodiesterase-1 inhibition against alpha-synuclein toxicity revealed by compound screening in LUHMES cells. Sci Rep 7(1): 11469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Krug A. K., Balmer N. V., Matt F., Schonenberger F., Merhof D. and Leist M.(2013). Evaluation of a human neurite growth assay as specific screen for developmental neurotoxicants. Arch Toxicol 87(12): 2215-2231. [DOI] [PubMed] [Google Scholar]
- 7. Lotharius J., Barg S., Wiekop P., Lundberg C., Raymon H. K. and Brundin P.(2002). Effect of mutant alpha-synuclein on dopamine homeostasis in a new human mesencephalic cell line. J Biol Chem 277(41): 38884-38894. [DOI] [PubMed] [Google Scholar]
- 8. Lotharius J., Falsig J., van Beek J., Payne S., Dringen R., Brundin P. and Leist M.(2005). Progressive degeneration of human mesencephalic neuron-derived cells triggered by dopamine-dependent oxidative stress is dependent on the mixed-lineage kinase pathway. J Neurosci 25(27): 6329-6342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Park J. S., Davis R. L. and Sue C. M.(2018). Mitochondrial dysfunction in parkinson's disease: new mechanistic insights and therapeutic perspectives. Curr Neurol Neurosci Rep 18(5): 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mortiboys H., Aasly J. and Bandmann O.(2013). Ursocholanic acid rescues mitochondrial function in common forms of familial Parkinson's disease. Brain 10):3038-50. [DOI] [PubMed] [Google Scholar]
- 11. Yamaguchi A., Ishikawa K. I., Inoshita T., Shiba-Fukushima K., Saiki S., Hatano T., Mori A., Oji Y., Okuzumi A., Li Y., Funayama M., Imai Y., Hattori N. and Akamatsu W.(2020) Identifying Therapeutic Agents for Amelioration of Mitochondrial Clearance Disorder in Neurons of Familial Parkinson Disease. Stem Cell Reports 14(6):1060-1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Poewe W., Seppi K., Tanner C. M., Halliday G. M., Brundin P., Volkmann J., Schrag A. E. and Lang A. E.(2017). Parkinson disease. Nat Rev Dis Primers 3: 17013. [DOI] [PubMed] [Google Scholar]
- 13. Ratcliffe L. E., Vázquez Villaseñor I., Jennings L., Heath P. R., Mortiboys H., Schwartzentruber A., Karyka E., Simpson J. E., Ince P. G., Garwood C. J. and Wharton S. B.(2018). Loss of IGF1R in human astrocytes alters complex I activity and support for neurons. Neuroscience 390: 46-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Scholz D., Pöltl D., Genewsky A., Weng M., Waldmann T., Schildknecht S. and Leist M.(2011). Rapid, complete and large-scale generation of post-mitotic neurons from the human LUHMES cell line. J Neurochem 119(5): 957-971. [DOI] [PubMed] [Google Scholar]
- 15. Stiegler N. V., Krug A. K., Matt F. and Leist M.(2011). Assessment of chemical-induced impairment of human neurite outgrowth by multiparametric live cell imaging in high-density cultures. Toxicol Sci 121(1): 73-87. [DOI] [PubMed] [Google Scholar]
- 16. Tong Z. B., Hogberg H., Kuo D., Sakamuru S., Xia M., Smirnova L., Hartung T. and Gerhold D.(2017). Characterization of three human cell line models for high-throughput neuronal cytotoxicity screening. J Appl Toxicol 37(2): 167-180. [DOI] [PMC free article] [PubMed] [Google Scholar]