

Abstract
2',7'-dichlorofluorescein (DCF) and derivatives are commonly used as fluorescent indicators of a broad spectrum of reactive oxygen species (ROS) generation in cell-based assays. However, there are numerous challenges inherent to the utilization of DCF probes for intracellular microscopic analysis, including photostability and probe efflux. Plate spectroscopy is comparatively simple and scalable compared to microscopy or flow cytometry-based acquisition, however is often subject to artefacts, including those introduced by thermal gradients and normalization methods. In this protocol we demonstrate a simple and sensitive plate spectrometry-based protocol utilizing the probes H2DCFDA and sulforhodamine B. The rapid sulforhodamine B assay (SRB) for cellular protein allows for a stable endpoint measurement of total cell population while also preserving morphology, can be combined or run in parallel with any other assay for normalization of readout to cell mass, and complemented by microscopic scoring of cell number and nuclear count. The oxidative stress and normalisation methods may enhance fields of research investigating cell differentiation, stress, or toxicity..
Graphical abstract:

Graphical overview for quantification of ROS generation and cellular protein
Keywords: Dichlorofluorescein, Sulforhodamine, DCF, SRB, Oxidative stress, Reactive oxygen species, Normalization assay
Background
Microplate assays offer a significant advantage over microscopy-based assays with regard to throughput, cost of analysis equipment, optimization and analysis time. The detection of reactive oxygen intermediates (ROS) generated by drugs in activated inflammatory cell types can be facilitated by fluorescent probes such as 2',7'-dichlorodihydrofluorescein diacetate (H2DCFDA) and dihydroethidium (DHE) in microscopy, plate spectroscopy and flow cytometry formats. Following deacetylation by intracellular esterases, H2DCF is oxidized by hydroxyl, carbonate, nitrogen or thiyl radicals to the green fluorescent DCF (Figure 1). As such DCF derivative probes are not only sensitive indicators of intracellular and drug-induced ROS generation but also prone to artefactual signal, including self-oxidation from H2O2 generated during oxidation, and photo-oxidation by an excitation source ( Wojtala et al., 2014 ; Paige Davis Volk and Moreland, 2014). Although DCF probes do not result in formation of nucleic acid binding species in comparison to DHE, DCF is not retained intracellularly for extended periods of time, limiting its window of detecting ROS for microscopy or flow cytometry. We have found that ROS generation can be more sensitively detected by a comparatively simple experimental analysis mode of plate spectroscopy.
Figure 1. Mechanism of H2DCFDA ROS probe.

Cell permeable H2DCFDA is cleaved by intracellular esterases to non-fluorescent H2DCF, which is oxidized by various ROS to green fluorescent DCF which can be detected by fluorescence plate spectroscopy.
Potentially overlooked artefactual distortions to signal in microplate format include uneven seeding, thermal gradient effects formed during heating, evaporation and background signal from small molecules. In this protocol, even seeding is addressed by a pre-incubation at ambient temperature ( Lundholt et al., 2003 ). Fluorescent background signal contributed by small molecules or other reagents can be deducted with cell-free controls. While microplate edge wells are often replaced with water to reduce loss of volume even in humidified incubators, enclosure of microplates in a simple humidifying chamber can overcome edge well evaporation ( Walzl et al., 2012 ). Thermal edge effects due to uneven heating can be addressed by use of metal heating blocks that align with base of microtitre plates and pre-heated reaction solutions ( Shellman et al., 2004 ), or room temperature incubation where possible.
Cell-based plate spectroscopy assays such as the DCF assay typically require normalization to cell population per well. Microplate assay signal to population normalizations are often performed with the bicinchronic acid (BCA) assay. However the BCA assay is compromised by low concentrations of H2O2, and practical scalability is challenged due to the lack of endpoint, or stop reaction method (Baker, 1991). Nuclear DNA stains are often employed for normalization of cell populations when scored microscopically, but overestimate populations in plate spectroscopy format where there is high DNA condensation with cell death, particularly relevant to small molecule or radiation stress experiments. The sulforhodamine B (SRB) assay provides a stable endpoint measurement of the cell population by quantifying fixed cellular protein whilst preserving morphology and avoiding drug interactions ( Skehan et al., 1990 ). SRB is an anionic aminoxanthene protein dye that under acidic conditions binds basic amino acid residues ( Han et al., 2012 ). Numerous SRB assays have been presented involving typically hour long cold acidic protein precipitation by trichloroacetic acid (TCA), SRB staining and solubilization with numerous wash steps. However the fixation reaction by cold acid precipitation ( Hayashi et al., 1999 ) and SRB staining has been reported to reach completion within minutes ( Skehan et al., 1990 ). In this protocol the DCF assay is normalized to cellular protein by a TCA-SRB assay that combines fixation and staining steps (Figure 2).
Figure 2. Rapid TCA-SRB assay schematic.

Cell cultures are simultaneously fixed and stained by cold TCA-SRB. In acidic conditions SRB binds basic amino acid residues. After a wash step, SRB is solubilized in basic conditions and released into well contents, reflecting content of cellular protein per well.
The protocol is applicable to any adherent cell culture in microplate well format, and can be executed by any basic cell culture laboratory equipped with a fluorescent spectroscopy plate reader with green and red fluorescent channels (excitation 494/emission 522 and excitation 565/emission 586) (Figure 3). The rapid SRB assay can be used to normalize any other non-destructive cell-based assay readout. All reagents required are easily commercially available at very low cost per assay plate.
Figure 3. Protocol schematic.

Cells are loaded with DCF-based probe, incubated for desired treatment period (e.g., 1 h or 24 h) before plate spectroscopy readout of DCF as an indication of ROS generation. To normalize DCF signal to cell population per well, a simultaneous acidic protein precipitation stage and cellular protein dye stage is performed before solubilization and spectroscopic readout of SRB. The TCA-SRB assay can be utilized to obtain relative cell populations for growth inhibition, cytotoxicity or to normalize other cell-based assay readouts.
Materials and Reagents
15 or 50 ml centrifuge tubes
96-well microplates (ideally black-walled, clear bottom)
DMEM/F-12, no phenol red (Thermo Fisher Scientific, Gibco, catalog number: 21041025), store at 2-8 °C
PBS Tablets (Thermo Fisher Scientific, Gibco, catalog number: 18912014), store at room temperature (RT)
H2DCFDA (Thermo Fisher Scientific, Life Technologies, catalog number: D399), store powder at -20 °C
Sulforhodamine B sodium salt (SRB) (Sigma-Aldrich, catalog number: S1402), store powder at RT
Trizma base (Sigma-Aldrich, catalog number: T1503), store powder at RT
Trichloroacetic acid (TCA) (Sigma-Aldrich, catalog number: T4885-1KG), store powder at RT
Acetic acid (Sigma-Aldrich, catalog number: A6283), store liquid at RT
Extracellular matrix solution (e.g., thermally cross-linked ECM with cold 10 µg/ml Collagen I for fibroblasts, or 1% Engelbreth-Holm-Swarm (EHS) sarcoma from Matrigel or Geltrex for iPSC, neural stem cells, incubated at 37 °C for 30 min)
30% wt. Hydrogen peroxide
100% DMSO
MilliQ grade water
10 mM H2DCFDA in DMSO (see Recipes)
10% w/v TCA in water (see Recipes)
0.004% w/v SRB in 10% w/v TCA (TCA-SRB) (see Recipes)
10 mM Trizma in water (see Recipes)
Equipment
Assumes availability of typical cell culture equipment (CO2 incubator, centrifuge, biosafety cabinet, water bath, fridge and/or cold room, etc.)
BMG LABTECH POLARstar Omega plate reader or fluorescence plate spectrometer with similar fluorescence capacity in fluorescein channel and 494/522 nm and 565/586 nm
Multichannel pipette (~10 µl and ~100 µl range) and sterile reagent reservoirs
Plastic humidifier box
Corrosives waste container
Software
Microsoft Excel or other spreadsheet processing software
Procedure
-
Cell seeding
Coat 96 well plate plates with extracellular matrix solution if required for desired cell type.
Dissociate and count cells with cell counting device.
Seed at appropriate density in 100 µl in phenol red-free complete media overnight (typically 20,000-30,000 cells/cm2).
Incubate for up to 0.5 h at room temperature (~24 °C) to prevent uneven seeding.
Plate plate(s) within humidifier chamber in CO2 incubator.
-
Dye loading and incubation
Prepare 20 µM H2DCFDA in pre-warmed base medium (DMEM/F12, no phenol red) on day of experiment.
Empty plate into waste container (inversion and flicking can be utilized if cells are strongly adherent, otherwise carefully remove media with a multichannel, for suspension cells centrifuge prior to this).
Add 100 μl 20 µM H2DCFDA to each well using a multichannel pipette.
Return plate to incubator for 30 min.
Empty plate into a waste container.
Add phenol-red free media and treatment including hydrogen peroxide as a positive control (e.g., 90 µl media, 10 µl compound).
Return plate to a humidified chamber in incubator for desired treatment period (e.g., 1 h or 24 h incubation).
-
Acquisition
Acquire with fluorescence plate spectroscopy with appropriate filter set closest to excitation 494 nm/emission 522 nm.
Empty plate and add 50 µl TCA-SRB (0.004% w/v SRB in 10% w/v TCA) (2-8 °C) for 15 min in a fridge or cold room.
Empty plate into an appropriate corrosives waste container and wash with 200 µl 1% acetic acid.
Empty place and replace with 100 µl 10 mM Trizma base solution, incubate at ambient temperature for 5 min, and gently manually agitate plate for 5 s to redistribute dye following incubation.
Acquire by fluorescence plate spectroscopy with appropriate filter closest to excitation 565 nm/emission 586 nm.
Data analysis
Export data to spreadsheet format.
Deduct the average of cell-free controls from DCF and SRB data (at least duplicate).
Calculate percentage of SRB signal relative to untreated controls.
Calculate percentage of DCF signal relative to untreated controls.
Calculate ratio of DCF:SRB percentage per well to normalize ROS generation readout to cellular protein per well.
Statistical analyses include independent t-tests, 1-way or 2-way ANOVA with post-hoc multiple comparisons tests for multiple groups (Figure 4).
Figure 4. Example of relative ROS generation.
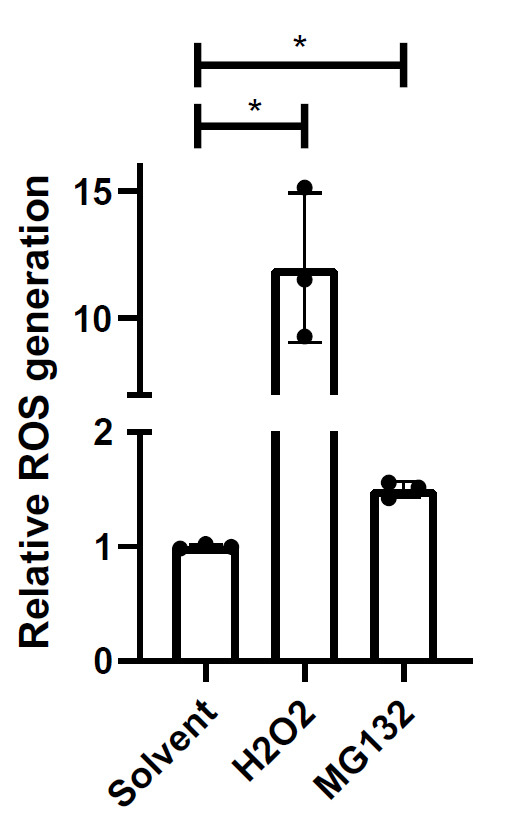
iPSC-derived astrocytes were treated with solvent (0.2% DMSO), 100 µM H2O2, or proteasomal inhibitor 0.1 µM MG132 for 24 h. MG132-induced ROS was unable to be detected under treatment conditions by intracellular DCF microscopy assay. Mean of independent replicates is presented with SD error bars (n = 3). Significant differences identified by one-way ANOVA followed by a post-hoc Holm-Sidak test. *p < 0.05.
Notes
Include at least 2 technical duplicates, empty well control, and positive control well.
As a general rule each plate requires ~12 ml of 20 µM H2DCFDA, 6 ml of 0.004% SRB + 10% TCA, 22 ml of 1% acetic acid and 12 ml of 10 mM Trizma base.
The concentration of H2O2 for a positive control should be determined by a cytotoxicity assay that does not result in total cell death (ideally > 50%), we have found 100 µM to be suitable for fibroblasts and human iPSC-induced astrocyte cultures but would vary with cell type and duration of analysis.
A parallel plate with cell-free controls is essential if there is substantial fluorescence contribution in the DCF channel by test substance added. Otherwise a single column of cell-free wells can be utilized for convenience.
The period of time to allow for cell adherence post-seeding can be confirmed by microscopy to avoid 0.5 h.
The humidifier chamber advised allows for usage of all wells without concern of evaporation for up to several days and simply involves stacking microplates on a plastic container with a platform of elevation (such as an empty pipette tip box) above a volume of water ( Walzl et al., 2012 ). Parafilm which also allows carbon dioxide and oxygen exchange is a cost-effective means of reducing evaporation if no humidifier chamber is used. Replacing edge wells with a water reservoir is a common technique but is not necessarily effective for longer incubation periods and results in a loss of ~37.5% of assayable well space.
Thermal gradients can be avoided by usage of metal blocks that fit beneath 96-well plates, however this can be cumbersome for scalability. Microplates can be loaded with H2DCFDA at ambient temperature instead of prewarmed media for large scale handling. The overnight incubation mitigates the duration of uneven heating that occurs during transition from plate from ambient temperature conditions to incubator.
Cold TCA can be replaced by 4% paraformaldehyde (PFA) in PBS which is more routinely available to laboratories performing immunocytochemistry, however PFA fixation and SRB staining cannot be combined as SRB only binds to basic protein amino acid residues effectively under mild acidic conditions ( Skehan et al., 1990 ).
For batch processing of plates, the 15 min TCA-SRB step can be replaced with an overnight duration period instead to reduce inter-well variability.
Prior publications describing the SRB protocol typically involve 3-5 wash steps with acetic acid. We have found minimal difference in outcome by staining in low volume followed by single high-volume wash.
Loading of H2DCFDA can be replaced by incubation with low concentrations of H2DCFDA (≤ 1 µM) without a wash step however cytotoxicity of probe dependent on cell type must be considered.
This protocol is intended for adherent cells; for more effective fixation of loosely adherent cells or suspension cells up to 50% w/v TCA may be utilized ( Kim et al., 1996 ).
Although more laborious, in plate spectrometry format the SRB assay is less susceptible to artefactual increase in signal per cell under apoptotic conditions than nucleic acid groove/intercalating probes. Nucleic acid binding dyes such as Hoechst 33342 result in overestimation of viable cell population (Figure 5).
For normalization to cell number or confluency, microscopic scoring of individual cells can be faciliated by a SRB cellular mask prior to, or restained after solublisation, along with nuclear scoring by non-overlapping dyes such as Hoescht 33342, DAPI or SYTOX Green. The cellular mask and total cellular protein data provided by SRB allow for correlation of total cellular protein, cellular population and nuclei which may be applicable toxicity and genotoxicity studies, and provide alternative normalization methods where applicable (Figure 6). Efficient microscopic data acquisition and analysis requires use of automated microscopy platforms and software where available.
Figure 5. Example of artefactual nuclear dye plate spectroscopy scoring of apoptotic cell populations.

iPSC-derived astrocytes were treated with solvent (0.2% DMSO) or 100 µM H2O2 for 24 h. Mean of technical duplicates is presented with SD error bars. Representative nuclear staining with Hoechst 33342 (10 µM) of control (a) in comparison to DNA-condensed apoptotic bodies that provide higher signal per cell leading to overestimation of viable cell population (b) (scale bar = 30 µm).
Figure 6. Example SRB cytoplasmic and nuclear mask.

iPSC-derived astrocytes were fixed and stained as described by TCA-SRB (a), before staining with 10 µM LCS1 dye in H2O (b). Overlay with phase contrast image (c) (scale bar = 100 µm).
Recipes
-
10 mM H2DCFDA
27.6 mg per 1 ml DMSO
Store at -20 °C and use within 3 months of preparation
Each 1 ml is sufficient for ~450 x 96-well microplate assays
-
10% TCA
Dissolve 10 g TCA per 100 ml MilliQ grade water
-
0.004% w/v SRB in 10% TCA
Dissolve 4 mg SRB per 100 ml 10% TCA
-
10 mM Trizma base (121.14 g/mol)
Dissolve 121.14 mg Trizma per 100 ml of MilliQ grade water
Acknowledgments
This research was funded by philanthropic donations and a grant from the Illawarra Health and Medical Research Institute (IHMRI). L.O. is supported by a National Health and Medical Research Council (NHMRC) of Australia Boosting Dementia Research Leadership Fellowship (APP1135720). The H2DCFDA assay is based on a method in a previously listed oxidative stress-related publication ( Han et al., 2009 ). The SRB protocol is based on findings of a previously established cytotoxicity protocol ( Skehan et al., 1990 ). We thank Dr Reece Gately for helpful discussions.
Competing interests
The authors declare they have no competing interests.
Citation
Readers should cite both the Bio-protocol article and the original research article where this protocol was used.
References
- 1. Baker W. L.(1991). Potential interference of hydrogen peroxide in the 2,2'-bicinchoninic acid protein assay. Anal Biochem 192(1): 212-214. [DOI] [PubMed] [Google Scholar]
- 2. Han F., Fan L., Wang X. and Li W.(2012). Sulforhodamine B restaining as a whole-cell label allows visualizing one more fluorochrome and its application in assaying protein nucleocytoplasmic distribution. Cytometry A 81(6): 532-540. [DOI] [PubMed] [Google Scholar]
- 3. Han Y. H., Moon H. J., You B. R. and Park W. H.(2009). The effect of MG132, a proteasome inhibitor on HeLa cells in relation to cell growth, reactive oxygen species and GSH. Oncol Rep 22(1): 215-221. [PubMed] [Google Scholar]
- 4. Hayashi K., Yonemura S., Matsui T. and Tsukita S.(1999). Immunofluorescence detection of ezrin/radixin/moesin(ERM) proteins with their carboxyl-terminal threonine phosphorylated in cultured cells and tissues. J Cell Sci 8): 1149-1158. [DOI] [PubMed] [Google Scholar]
- 5. Kim H. M., Han S. B., Kim M. S., Kang J. S., Oh G. T. and Hong D. H.(1996). Efficient fixation procedure of human leukemia cells in sulforhodamine B cytotoxicity assay. J Pharmacol Toxicol Methods 36(3): 163-169. [DOI] [PubMed] [Google Scholar]
- 6. Lundholt B. K., Scudder K. M. and Pagliaro L.(2003). A simple technique for reducing edge effect in cell-based assays. J Biomol Screen 8(5): 566-570. [DOI] [PubMed] [Google Scholar]
- 7. A. Paige Davis Volk and Moreland J. G.(2014). Chapter Thirteen: ROS-Containing Endosomal Compartments: Implications for Signaling. In: P. Michael Conn(Ed.). Methods in Enzymology. Academic Press. [DOI] [PubMed] [Google Scholar]
- 8. Shellman Y. G., Ribble D., Yi M., Pacheco T., Hensley M., Finch D., Kreith F., Mahajan R. L. and Norris D. A.(2004). Fast response temperature measurement and highly reproducible heating methods for 96-well plates. Biotechniques 36(6): 968–972., 974-966. [DOI] [PubMed] [Google Scholar]
- 9. Skehan P., Storeng R., Scudiero D., Monks A., McMahon J., Vistica D., Warren J. T., Bokesch H., Kenney S. and Boyd M. R.(1990). New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst 82(13): 1107-1112. [DOI] [PubMed] [Google Scholar]
- 10. Walzl A., Kramer N., Mazza G., Rosner M., Falkenhagen D., Hengstschläger M., D. r Schwanzer-Pfeiffer and Dolznig H.(2012). A simple and cost efficient method to avoid unequal evaporation in cellular screening assays, which restores cellular metabolic activity. Int J Appl Sci Tech 2. [Google Scholar]
- 11. Wojtala A., Bonora M., Malinska D., Pinton P., Duszynski J. and Wieckowski M. R.(2014). Methods to monitor ROS production by fluorescence microscopy and fluorometry. Methods Enzymol 542: 243-262. [DOI] [PubMed] [Google Scholar]