

Abstract
Objective:
While rare variants in the COL5A1 gene have been associated with classical Ehlers-Danlos Syndrome (EDS) and rarely with arterial dissections, recurrent variants in COL5A1 underlying a systemic arteriopathy have not been described. Monogenic forms of multifocal fibromuscular dysplasia (mFMD) have not been previously defined.
Approach and Results:
We studied four independent probands with the COL5A1 pathogenic variant c.1540G>A, p.(Gly514Ser) who presented with arterial aneurysms, dissections, tortuosity, and mFMD affecting multiple arteries. Arterial medial fibroplasia and smooth muscle cell disorganization were confirmed histologically. The COL5A1 c.1540G>A variant is predicted to be pathogenic in silico and absent in gnomAD. The c.1540G>A variant is on a shared 160.1kb haplotype with 0.4% frequency in Europeans. Further, exome sequencing data from a cohort of 264 individuals with mFMD were examined for COL5A1 variants. In this mFMD cohort, COL5A1 c.1540G>A and six additional relatively rare COL5A1 variants predicted to be deleterious in silico were identified and were associated with arterial dissections (p=0.005).
Conclusions:
COL5A1 c.1540G>A is the first recurring variant recognized to be associated with arterial dissections and mFMD. This variant presents with a phenotype reminiscent of vascular EDS. A shared haplotype among probands supports the existence of a common founder. Relatively rare COL5A1 genetic variants predicted to be deleterious by in silico analysis were identified in ~2.7% of mFMD cases, and as they were enriched in patients with arterial dissections, may act as disease modifiers. Molecular testing for COL5A1 should be considered in patients with a phenotype overlapping with vascular EDS and mFMD.
Keywords: Aneurysm, Arterial, Fibromuscular Dysplasia, Ehlers-Danlos Syndrome, Collagen
INTRODUCTION
Non-atherosclerotic and non-inflammatory dysplasia associated arterial disease (DAAD) includes a spectrum of vascular manifestations including stenoses, aneurysms, dissections, and tortuosity of medium sized muscular arteries. Multifocal fibromuscular dysplasia (mFMD) is a well-defined clinically entity, being one form of a DAAD. It is manifest histologically by medial fibroplasia1,2, and angiographically by multiple arterial stenoses with intervening mural dilations3. Arterial dissections and macroaneurysms represent a more severe DAAD phenotype.
Classical EDS (cEDS) is a rare heritable connective tissue dysplasia with an estimated population prevalence of approximately 1:20,000 or 0.005%4. It is an autosomal dominant condition characterized by a triad of skin hyperextensibility, atrophic scars and joints hypermobility, but presenting with significant inter- and intra-familial variability. Approximately 75–78% of cases of cEDS4 are caused by pathogenic variants in the COL5A1 gene while another 14% are caused by variants in COL5A2 or in COL1A1 (1%)5. There are no prior reports of a unique COL5A1 genetic variant associated with arterial dissections across multiple unrelated families. Arterial dissections have been previously reported in only five families with cEDS with different pathogenic variants,6–10 and only one of these families included more than one affected individual. In contrast, diffuse vascular dissections are frequently encountered in vascular EDS (vEDS). vEDS has a population prevalence of approximately 1:50,000, and disease-causing variants in the COL3A1 gene are identified in at least 96% of patients with vEDS11.
This study reports the first recurrent pathogenic variant occurring in independent families of the collagen, type V, alpha-1 gene (COL5A1, MIM: 120215), c.1540G>A, p.(Gly514Ser), to be associated with a DAAD, most notably arterial dissections, mFMD, and cervical artery tortuosity.. A shared haplotype amongst the four probands containing this genetic variant, supports a founder effect. All four probands fulfilled diagnostic criteria for an arteriopathy consistent with the clinically recognized phenotypic spectrum of mFMD, including arterial aneurysms, dissections and multifocal stenoses3. Only one of the probands met the 2017 diagnostic criteria for cEDS12. In a follow-up analysis of a cohort of sporadic mFMD cases (n=264) analyzed by whole exome sequencing (WES), low-frequency COL5A1 genetic variants predicted to be deleterious by in silico analysis were observed in 2.7% of mFMD cases and were associated with arterial dissections in this mFMD cohort. These findings support COL5A1 as a monogenic etiology of a DAAD phenotypically mimicking vEDS and encompassing mFMD, particularly the c.1540G>A, p.(Gly514Ser) genetic variant.
MATERIALS AND METHODS
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Study samples and clinical phenotyping
After being referred to medical genetics or cardiovascular genetics clinics and undergoing clinical testing, three probands harboring the COL5A1 c.1540G>A, p.(Gly514Ser) genetic variant and arterial lesions of DAAD were identified. One research subject had been enrolled in the Arterial Dysplasia Genetic Study at the University of Michigan as part of a mFMD cohort study in which WES was performed and identified the COL5A1 c.1540G>A variant. The probands, including one man and three women, presented clinically in the fourth and fifth decades (Table 1). In addition to clinical examinations and imaging reviews, each proband underwent documentation of family history to at least the level of first degree relatives (Table I, Online Supplement). All probands underwent vascular imaging of the head through pelvis, and aortic root z-scores were calculated13,14, and, when available, arterial specimens were subjected to histopathologic analysis.
Table 1a. Clinical and molecular findings in probands and family members with COL5A1 p.Gly524Ser substitution.
All vascular beds described refer to lesions of the arterial circulation. Hyperextensible skin was defined as extensibility >2cm on the volar surface of the forearm. F, female; M, male; nl, normal; MINOCA, myocardial infarction with nonobstructive coronary arteries; SCAD, spontaneous coronary artery dissection, b/l, bilateral; R, right-sided; CIA, common iliac artery; EIA, external iliac artery; VA, vertebral artery; AAA, abdominal aortic aneurysm; ICA, internal iliac artery; FMD, fibromuscular dysplasia
| Family | Individual | Sex | Age at last assessment | Vascular Involvement | Meets cEDS 2017 criteria | Skin phenotype | Joint phenotype | Other connective tissue features | History of hypertension | History of migraines |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Proband | M | 54 | Yes (Aneurysm, dissection, tortuosity) | Yes | Atrophic scarring, Hyperextensible skin. Skin velvety and doughy | Recurrent subluxations of L shoulder joint, b/l knee replacements; Beighton score 2/9 | Raphee of the uvula, Long face, Dolicocephaly, Down-slanted palpebral fissures, Enophthalmos. Mild scoliosis | No | Yes |
| Daughter | F | 24 | Yes Tortuosity | No | Hyperextensible skin | Subluxations knees, hips, thumbs, and jaw; Beighton score 9/9 | Raphee of the uvula, Blue sclera, thin nose and lips. Mild micrognatia | No | Yes | |
| Nephew | M | 40 | No | No | Hyperextensible skin, Velvety and doughy skin. Varicose veins | Subluxations of R thumb; Beighton score 8/9 | Bulbous uvula, Sleep apnea, GERD, Long face, Dolicocephaly, Enophthalmos, Thin nose, Mild pectus excavatum, Loss of lumbar lordosis, Piezogenic papules | No | Yes | |
| 2 | Proband* | F | 44 | Yes (Aneurysm, dissection, FMD, tortuosity) | No | Abdominal striae, Skin wound dehiscence | Joint pain, Beighton score of 5/9 | Downsloping palpebral fissures, Pectus excavatum | No | Yes |
| 3 | Proband | F | 52 | Yes (Aneurysm FMD, dissection, tortuosity) | No | Atrophic scars; required repeat suturing of skin wound dehiscence | Ankle instability with recurrent ankle sprains, Joint pain (also diagnosed with RA), Beighton score 2/9 (Thumb apposition to forearm) | Thin nose, Short uvula, Pes planus, GERD/hiatal hernia | Yes | Yes |
| 4 | Proband* | F | 64 | Yes (Aneurysm FMD, dissection, tortuosity) | No | Mildly translucent skin, Easy bruising | Beighton score 2/9, could still easily touch the floor without bending her knees at 64 years old | Pes planus, High arched palate, Dental crowding, Mild malar hypoplasia | Yes | No |
Additional molecular findings included in Proband #2 a variant (p.Leu1405Arg) in FBN1 that been observed 31 times in gnomAD, a frequency which would support this variant being benign and in Proband #4 a heterozygous variant (p.Tyr2258*) in TNXB, a gene associated with autosomal recessive inheritance of disease.
Independent of the study of the four probands harboring the COL5A1 p.(Gly514Ser) variant, unrelated adult subjects with mFMD (N=264) were enrolled from 2010–2015 in one of two participating repositories: the University of Michigan Arterial Dysplasia Genetic Study FMD cohort, or the Cleveland Clinic FMD Biorepository. Age and sex matched control subjects without vascular disease (N=284) were enrolled in the Cleveland Clinic GeneBank Biorepository. Control skin biopsies were collected from age and sex matched individuals without clinical evidence of DAAD. All participants provided informed consent and study activities received institutional IRB approval. Haplotype analysis utilized data from the Michigan Genomics Initiative (MGI) which includes >50,000 participants who gave blood samples for use in genetic studies (54.8% females, average age 53.1 years with sd=16.4 years)15.
Genotyping of Single Nucleotide Variants for Phased Haplotype Analysis of the COL5A1 p.(Gly514Ser) Variant
Genotyping of genome-wide single nucleotide markers of the probands and MGI samples was undertaken by the University of Michigan DNA Sequencing Core using the IlluminaInfinium HTS Assay Protocol, a semi-custom Infinium CoreExome-24v1.1 BeadArray with 607,778 SNP markers (Online Supplement). After quality control, 594,493 polymorphic variants remained for analysis (Online Supplement)16,17. Confirmation genotyping of the COL5A1 c.1540G>A variant and neighboring haplotype-informative markers was performed by Sanger sequencing of cloned amplicons (Table II, Online Supplement).
Ancestry Estimation and Haplotype Phasing
From the Illumina BeadArray genotypes, the four COL5A1 G514S probands were analyzed with 1000Genome EUR population (n=503), excluding variants in regions of known long-range (>2MB), or high linkage disequilibrium18 (Table III, Online Supplement). 253,097 loci with minor allele frequency (MAF) ≥0.1 were used for principal component analysis (PCA) using PLINK1.9 (hg19, v1.90b3.36) to examine the ancestry of the four probands. 166 variants in the chromosome 9:137–138M (hg19) region with minor allele frequency ≥0.01 defined by MGI samples were extracted from 1000 randomly selected MGI samples and the four probands. fastPHASE v1.4.819,20 was applied to this data set (n=1004) to identify haplotypes in the region. To validate the phasing results, LD blocks were generated from 1000 Genomes Data phase 3 with Haploview v4.2 and reviewed for haplotype boundaries and frequency (Online Supplement)21,22.
Histopathologic Analysis and Collagen Quantification in Artery and Skin Samples of COL5A1 G514S Probands
Histologic and immunohistochemistry preparations of surgical specimens from Proband #1 and Proband #3, as well as control arterial samples, were accomplished using standard techniques, including hematoxylin-and-eosin, Verhoeff-Van Gieson, and Masson’s trichrome staining. Immunostaining and quantitative analyses were standardized (Online Supplement). Dermal biopsies from Proband #3 and controls were subjected to multiphoton microscopy to detect second harmonic generation (SHG) signals from collagen and auto-fluorescent signals from elastic fibers23,24. Collagen fibril diameters were measured on transmission electron microscopy (TEM) images (Online Supplement).
Whole Exome Sequencing of a mFMD cohort and COL5A1 Genetic Variant Annotation and Analysis
DNA samples from individuals in the mFMD cohort and GeneBank control group were isolated and quantitated from blood or saliva samples (N=548). DNA samples were then prepared and sequenced by the Northwest Genomics Center (NWGC) (Online Supplement). dbNSFP version 3.5a was used to estimate SNV pathogenicity for variants including multiple scores from conservation and functional predictions25–28, along with gnomAD allele frequency ≤ 0.00129, (Online Supplement), for those variants in the mFMD cohort but not in the control group. Variants were annotated by ANNOVAR30 and through query of several resources (Table 2). Protein structure analysis was performed using an in silico approach (Online Supplement). COL5A1 genetic variants detected by exome sequencing and that were predicted to be deleterious by in silico analysis were analyzed in aggregate, for association with arterial dissection, among individuals with mFMD in this cohort. Variant frequencies were evaluated in gnomAD, LOVD, and the National Heart, Lung and Blood Institute (NHLBI) Trans-Omics for Precision Medicine (TOPMed) BRAVO server of whole genome sequences from several cohorts (N=62,784) to a median depth of 39X, with joint genotype calling across all samples.
Table 2.
COL5A1 single nucleotide variants with predicted deleterious effect by in silico analysis identified among individuals enrolled in the mFMD cohort study. All variants are located on chromosome 9, cytoband 9q34.3. Position denotes genomic location on reference genome GRCh37.
| Reference SNP ClusterID | rs147589613 | rs142114921 | rs772379819 | rs878853652 | rs147329970 | rs368305377 | rs765217611 |
|---|---|---|---|---|---|---|---|
| Genome Location | |||||||
| Position | 137591818 | 137591844 | 137623481 | 137642433 | 137648614 | 137702117 | 137709645 |
| COL5A1 Exon | 3 | 3 | 8 | 12 | 17 | 44 | 54 |
| Variant Effect | |||||||
| Base Change | c.C341A | c.C367G | c.C1304T | c.G1540A | c.C1831T | c.C3491T | c.C4198T |
| Amino Acid Change | p.(Ala114Asp) | p.(Gln123Glu) | p.(Pro435Ala) | p.(Gly514Ser) | p.(Arg611Trp) | p.(Pro1164Leu) | p.(Pro1400Ser) |
| Population Allele Frequency | |||||||
| gnomAD | 0.0005773 | 0.0002093 | 3.65E–05 | NR | 7.31E–05 | 0.0001082 | 1.11E–05 |
| ExAC | 0.0006122 | 0.0001655 | 0.0000498 | NR | 0.00009902 | 0.0001323 | 0.00001147 |
| Locus Location | |||||||
| Residue Modification | None | None | None | None | None | None | None |
| Domain | N-pro | N-pro | N-pro | Helix | Helix | Helix | Helix |
| Pathogenicity Estimation | |||||||
| Ensemble Score Prediction | |||||||
| MetaSVM | D | T | D | D | D | D | D |
| MetaLR | D | T | D | D | D | D | D |
| M.CAP | D | T | D | D | D | D | D |
| Functional Prediction | |||||||
| SIFT | D | T | D | D | D | D | T |
| Polyphen2 HDIV | P | D | D | D | D | D | D |
| Polyphen2 HVAR | P | D | D | D | D | P | D |
| LRT pred | U | U | U | U | U | U | U |
| MutationTaster | D | D | D | D | D | D | D |
| MutationAssessor | M | M | M | H | L | M | L |
| FATHMM | T | T | D | D | D | D | D |
| PROVEAN | D | N | D | D | D | D | N |
| General Prediction | |||||||
| CADD Phred Score | 34 | 24.6 | 24.2 | 32 | 35 | 26 | 22.5 |
| ACGS/ACMG Criteria | VUS | VUS | VUS | LP | VUS | VUS | VUS |
Abbreviations: gnomAD, Genome Aggregation Database; ExAC, Exome Aggregation Consortium; GERP, Genomic Evolutionary Rate Profiling; CADD, Combined Annotation Dependent Depletion; D, Deleterious; T, tolerated; P, possibly damaging; N, Neutral; U, Unknown; H, high; M, medium; L, low; NR, Not Reported; VUS, Variant of uncertain significance; LP, Likely pathogenic
RESULTS
Identification of COL5A1 c.1540G>A, p.(Gly514Ser) Variant in Independent Families
A COL5A1 c.1540G>A, p.(Gly514Ser) variant was discovered in four unrelated individuals with arterial tortuosity, aneurysms, dissections and/or mFMD (Figure 1, Table 1, Table IV, Online Supplement). The heterozygous c.1540G>A variant was confirmed in all probands by Sanger sequencing (Figure I, Online Supplement). Analysis of identity-by-descent using genome-wide SNV markers verified no close familial relationship between the four probands (PiHat<0.2).
Figure 1.

Computed tomography, magnetic resonance imaging and digital subtraction angiography demonstrating various arterial dysplasia findings in probands harboring the COL5A1 G514S: significant vertebral artery tortuosity and internal carotid artery 360° redundancy (arrow) (A), and internal carotid artery with mFMD with serial stenoses (arrows)and intervening dilation (B); celiac artery aneurysm (C), dissection (D) and mFMD with stenosis (arrows) and intervening dilation (E); dissections of a common iliac artery aneurysm (arrow) (F), and external iliac artery (arrow) (G).
The initial clinical presentations of the probands were protean, including stroke secondary to carotid artery mFMD and dissecting aneurysms, acute leg and hip pain due to a ruptured iliac artery dissecting aneurysm, myocardial infarction precipitated by spontaneous coronary artery dissection (SCAD), and a pulmonary aneurysm identified incidentally in an individual with hypertension requiring three drugs. Additional vascular lesions in each of the probands were identified using contemporary angiographic imaging3 (Figure 1, Table 1). Vertebral and/or internal carotid arteries were tortuous in all probands, celiac and iliac artery aneurysm and/or dissection was also present in three probands, while the fourth proband had multifocal stenosis of the celiac artery and iliac artery luminal irregularity (Table 1). Variable features of cEDS were noted: skin hyperextensibility >2cm was observed in Proband #1’s family; joint hypermobility affected two of four families and segregated with the COL5A1 G514S variant in the Proband #1’s family. Segregation of the variant was seen in the Proband #1 family with incomplete penetrance of arterial dissection phenotype and more complete penetrance of arterial tortuosity, although two affected relatives were age 40 years or less at the time of imaging (Table I, Online Supplement). Atrophic scars were present in three probands, including two experiencing postoperative wound dehiscences. Only proband #1 fulfilled the 2017 cEDS diagnostic criteria. Notably, vascular tissue friability was described intraoperatively proband #1. Medial fibrosis, smooth muscle cell disorganization and localized loss of elastin affected the arteries of two probands having iliac artery and carotid artery dissecting aneurysms (Figure 2).
Figure 2.

Histopathologic evidence of fibroplasia of the arterial media (arrows) in pathologic carotid and iliac artery specimens from two COL5A1 G514S variant probands as compared to the arteries of two organ donors as controls. Collagen fibers were increased and there was medial disorganization (arrow)in trichrome stained sections (A – Proband #1 iliac artery (n=1), B –Control iliac artery (n=1), C – Proband #3 internal carotid artery (n=1), D – Control carotid artery (n=1)). Medial collagen content of the arterial wall in carotid and iliac arteries was digitally quantified as the mean and standard deviation across 60 equivalent-sized subfields spanning the media (normalized medial area) (E,F). Medial collagen in probands was increased, consistent with medial fibrosis. Verhoeff-Van Gieson staining for elastin staining (inserts) demonstrated fragmentation (arrowheads) of elastin fibers (black), Overall, these findings are consistent with arterial fibroplasia. Bars =200μm.
COL5A1 p.(Gly514Ser) Variant Frequency and Interpretation
The recurrent COL5A1 c.1540G>A p.(Gly514Ser) variant (NM_000093.3) was not reported in gnomAD (accessed 6/19/2020, n=125,748) or the LOVD COL5A1 mutation database (accessed 4/16/2020). The NHLBI TOPMed database of 62,784 individuals (accessed 4/16/2020) contained two individuals with the COL5A1 c.1540G>A variant. One proband in the current series was reported in ClinVar after clinical testing (Variation ID 236997). ClinVar includes an additional report for this variant (Accession # SCV001155819),for a female individual who presented with joint pain, her echocardiogram reportedly revealed normal aortic dimensions and no other vascular imaging was pursued. The residue in question is the penultimate glycine of the first triple helical repeat within the protein’s interrupted collagenous region, and therefore predicted to be deleterious on this basis alone, as other glycine substitutions have been previously noted to underlie collagen gene mutations causing disease31,32. Algorithmic estimation of variant effect on protein structure and function unanimously predicted the substitution to be deleterious (Table 2), with a CADD phred score of 32. Bioinformatic structural analysis predicted the glycine to serine substitution at this position to create an externalized residue at this position that would otherwise be internal in the collagen fibril structure (Table VII, Online Supplement). According to the 2020 ACGS criteria, which is aligned with the most recent ACMG criteria for pathogenicity estimation, the c.1540G>A variant is classified as likely pathogenic (PS4, PM1, PM2, PP3).The predicted probability of loss-of-function intolerance in COL5A1 is 1.0, corresponding to a predicted deleterious effect of damaging variants in this gene33.
A Founder Haplotype Carries the COL5A1 p.(Gly514Ser) Pathogenic Variant
Ancestry was mapped against 1000 Genomes data, revealing probands to be of central European ancestry (Figure 3C). Using 25 variants genotyped on the Illumina BeadArray platform, one shared haplotype was identified among all four probands, being 160.1 kilobase pairs in length and containing the COL5A1 c.1540G>A, p.(Gly514Ser) variant (chromosome 9:137530346–137642577) (Figure 3). The predicted heterozygous haplotype structure was confirmed by Sanger sequencing with accurate strand placement of the COL5A1 c.1540G>A variant as in phase with the nearest haplotype-informative markers (Online Supplement). Additional confirmation was demonstrated by regional contig cloning and observing the expected restriction enzyme digestion pattern at c.1540G>A (Figure I, Online Supplement). The presumed founder haplotype had a frequency of 0.7% in MGI samples and 0.4% in 1000G data. The statistical probability of identifying a shared 0.4% frequency haplotype among our four probands is exceedingly low by a two-tailed binomial test (P=1.77×10−8).
Figure 3.
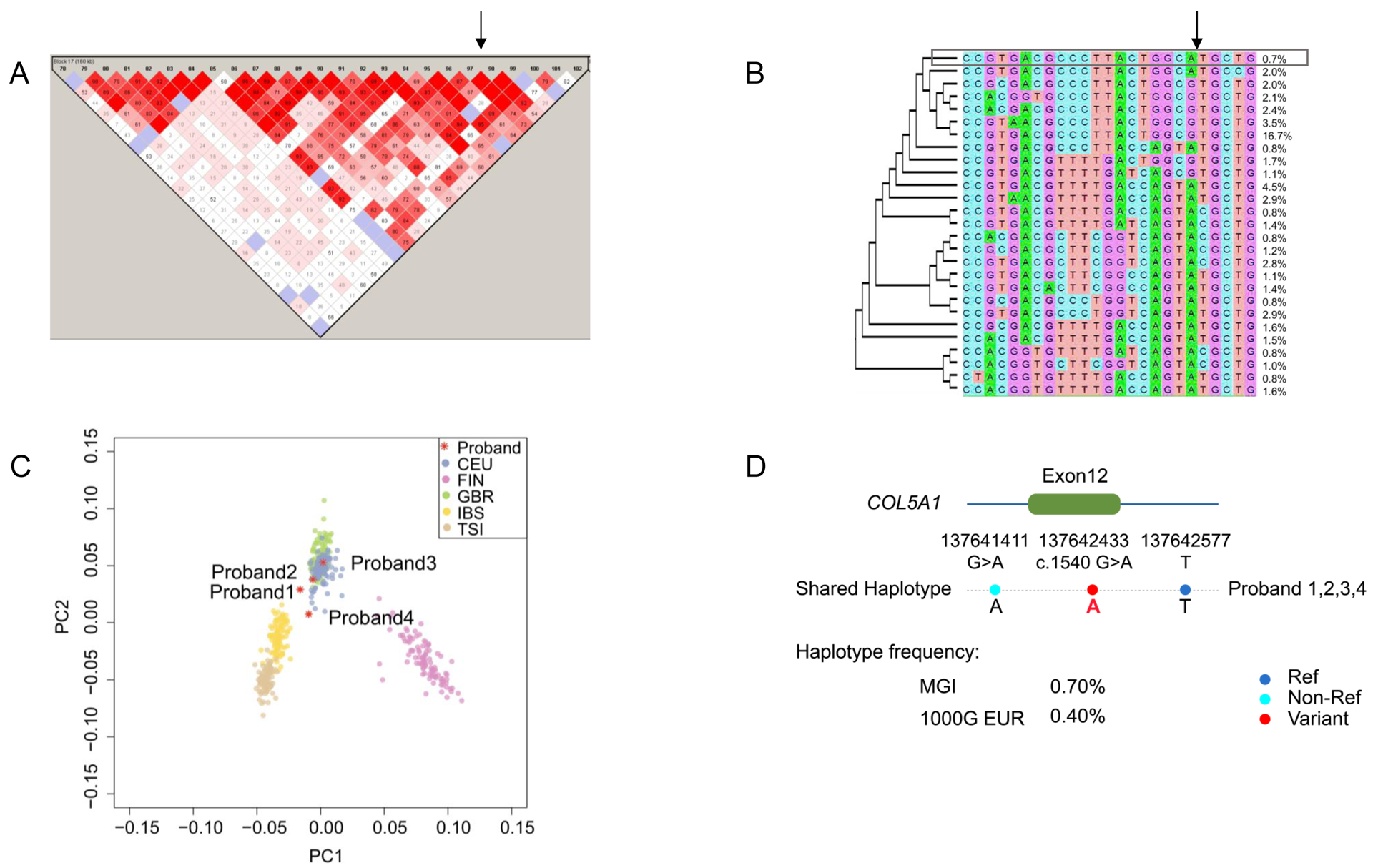
Linkage disequilibrium site estimation mapped c.1540G>A to a 160.1kbp haplotype (chromosome 9:137,496,881–137,656,942). (A) Linkage disequilibrium plot of the shared haplotype region is shown with the c.1540G>A variant location identified by the arrow. The haplotypes were defined by fastPhase with four probands and 1000 randomly selected MGI samples, and then visualized with Haploview. (B) Haplotypes in the shared 160.1kbp block containing the c.1540 genomic locus are shown with the frequencies of each haplotype, with a neighbor-joining phylogenic tree and the haplotype shared by the four probands shown in the box. (C) Principal components analysis demonstrates central European ancestry of the four probands, plotted against all European ancestry population samples in the 1000 Genomes reference, consistent with common regional origin of the probands. The number and description of each EUR subpopulation were: CEU (n=99): Utah Residents (CEPH) with Northern and Western European Ancestry; FIN (n=99): Finnish in Finland; GBR (n=91): British in England and Scotland; IBS (n=107): Iberian Population in Spain; TSI (n=107): Toscani in Italia. (D) The shared haplotype containing COL5A1 c.1540G>A, p.(Gly514Ser) is depicted.
Histopathologic Analysis
Histologic assessment was performed on arterial and skin samples obtained from COL5A1 c.1540G>A, p.(Gly514Ser) probands. Aneurysmal segments showed extensive fibrosis, and non-aneurysmal areas of the probands’ arteries exhibited multifocally and irregularly increased medial collagen, smooth muscle cell disorganization and elastin fragmentation. Fibrosis was digitally quantitated by color segmentation in one common iliac artery case from Proband #1 and one internal carotid artery case from Proband #3 in comparison to respective controls of one common iliac artery and one control common carotid artery obtained from organ donors. Medial fibrosis was higher in the media of proband versus control arteries. (Figure 2 and Table Va, Online Supplement). The medial fibrosis distribution was patchy, and the arterial walls exhibited both thinned and thickened areas within a given arterial segment (Figure 2, Figure II, Online Supplement). Mucoid extracellular matrix accumulation suggesting cystic medial degeneration was not observed. Although smooth muscle bundles were thin in areas of fibrosis, there was no loss of arterial smooth muscle nuclei (i.e. medial necrosis). These findings support that the primary process is arterial medial fibroplasia. Digital quantitation of immunohistochemical stains in the same proband samples as above documented a decrease in medial COL5A1 (Figure III, Online Supplement) and minimally increased or equivalent medial phosphorylated SMAD2 (pSMAD2), a specific marker of activated canonical TGF-β signaling, in comparison to respective control organ donor arteries (Figure IV, Online Supplement and Table Vb, Online Supplement).
Dermal analyses revealed no difference in collagen quantity of Proband #3 compared to controls (Table VI, Online Supplement), nor any change in 3-dimensional collagen architecture by SHG imaging (Figure V, Online Supplement). Dermal TEM analysis exhibited modestly larger collagen fibril diameters in Proband #3 relative to those of matched controls (Figure VI, Online Supplement). Notably, there was not excess variability in fibril diameters.
Analysis of COL5A1 Genetic Variants Identified by WES in a FMD Cohort
To follow up the findings of arterial dissection and mFMD among the COL5A1 G514S carriers, we next assessed the impact of genetic variation in the COL5A1 gene on arterial dissection in patients with mFMD by conducting an analysis for association of COL5A1 variation, predicted in silico to be deleterious but not necessarily meeting pathogenicity criteria34,35 with any dissection in a cohort of unselected mFMD patients, as the primary analysis. As a secondary analysis, all vascular phenotypes in the cohort were systematically tallied and analyzed. All COL5A1 variants identified by WES were annotated and curated. Among those with mFMD (n=264), 134 had dissections or macroaneurysms. Exome sequencing identified 33 exonic variants, with 19 (58%) uniquely in exomes of affected individuals relative to controls (n=284), consisting of 8 nonsynonymous and 11 synonymous variants. All nonsynonymous variants were of low frequency in the general population (MAF ≤ 0.001 in gnomAD non-Finish Europeans); seven were predicted to be pathogenic by in silico analysis. These 7 variants were the only identified COL5A1 variants in the cohort with gnomAD allele frequency ≤ 0.001 with a CADD phred score ≥ 20. One of the identified variants was the COL5A1 G514S variant identified in Proband #3 who had initially been included in the WES study enrollments through the routine clinical enrollment into the cohort (Table 2). Deleterious COL5A1 SNVs were heterozygous alterations, each identified in one unique individual with mFMD. Among the seven individuals with a COL5A1 variant predicted to be deleterious by in silico analysis, there were no pathogenic variants in COL3A1, COL5A2, ACTA2, FBN1, TGFBR1/2, TGFB2/3, SMAD 2/3/4/6, PRKG1, or LOX).
The seven loci included variants in the N-pro non-helical region (N=1), the interrupted collagenous region, (N=1), and the triple-helical region (N=3) (Figure VII, Online Supplement). The p.(Gly514Ser) variant was predicted to alter the COL5A1 protein structure most extensively, with several residues that would typically be internal to the protein structure becoming exposed in the presence of the p.(Gly514Ser) variant (Table VII, Online Supplement). Only one other variant, p.Pro1164Leu, was related to any structural change (Table VII, Online Supplement). Further, using criteria for clinical estimation of pathogenicity, and concordant with the in silico protein structure predictions, only the p.(Gly514Ser) variant met criteria for pathogenicity whereas the others may be clinically classified as variants of uncertain significance (VUS) (Table 2).
Of the seven variants, two variants were reported in either the literature or the LOVD database: c.341C>A and c.367C>G. Specifically, COL5A1 c.367C>G p.(Gln123Glu) was reported three times in the EDS variant database (https://eds.gene.le.ac.uk/variants.php?select_db=COL5A1&action=search_all&search_Variant%2FDNA=c.367C%3EG): once for a patient who underwent sequencing for what is listed at EDS1 (prior nomenclature for cEDS), once for an individual identified as part of a study sequencing candidate genes for keratoconus36, and once in a WES study on thoracic aortic aneurysm in one individual with a 4.6cm ascending aortic aneurysm, Beighton score of zero, and no skin abnormalities, but cystocele and rectocele repairs as well as a hysterectomy37. The c.341C>A is listed six times in ClinVar and reported all six times as a “likely benign” variant with 167 gnomAD observations. This variant was also reported in a candidate gene sequencing study for keratoconus38 and in a patient with a thoracic aortic aneurysm and a FBN1 pathogenic variant in the context of a WES study on genetic modifiers of thoracic aneurysm (https://eds.gene.le.ac.uk/variants.php?select_db=COL5A1&action=view&view=0001537%2C0000214%2C21; ID: COL5A1_00205). The other 4 variants were: c.341C>A reported six times in ClinVar as likely benign, c.367C>G reported six times in ClinVar (five times as VUS, once as likely benign (LB)), c.1304C>T noted once in ClinVar as VUS, c.1831C>T reported twice in ClinVar as VUS, c.3491C>T reported twice in ClinVar (once as LB, once as VUS) and c.4198C>T reported once in ClinVar as LB.
None of the seven subjects in the mFMD cohort had either a personal medical history or reported family history of a connective tissue disorder of any type. Among individuals with mFMD, presence of a predicted deleterious COL5A1 variant was associated with the occurrence of arterial dissections (p=0.005) in the primary analysis, and in secondary analyses of arterial bed involvement, visceral artery and lower extremity artery dissections were noted (Table 3).
Table 3: Clinical characteristics associated with the presence of a COL5A1 SNV predicted as deleterious by in silico analysis in the multifocal FMD cohort.
Values are presented as count (percentage) or mean ± standard deviation. Tests of independence were carried out using Student’s t-test for continuous variables and Fisher’s exact test for binary variables, comparing individuals in the FMD cohort with a predicted deleterious COL5A1 SNV and individuals without a deleterious COL5A1 SNV. The primary analysis was for the proportion of individuals with any dissection, provided with all other arterial bed involvement in the FMD exome-sequenced cohort. P values are provided for comparisons with all counts >3.
| No Deleterious COL5A1 SNV | Deleterious COL5A1 SNV | |||||
|---|---|---|---|---|---|---|
| Variable | Total Sample | N | % | N | % | P |
| Total N Per Group | 264 | 257 | 100.0% | 7 | 100.0% | |
| Demographics | ||||||
| Female | 257 | 250 | 97.3% | 7 | 100.0% | |
| Caucasian | 255 | 248 | 96.5% | 7 | 100.0% | |
| Age at vascular diagnosis | 50.3 ± 10.4 | 50.4 ± 10.5 | - | 48.4 ± 5.1 | - | |
| Arteriopathy Risk Factors & Comorbidities | ||||||
| Hypertension | 170 | 165 | 64.2% | 5 | 71.4% | 1 |
| Smoking history | 98 | 94 | 36.6% | 4 | 57.1% | 0.50 |
| Stroke | 45 | 43 | 16.7% | 2 | 28.6% | 0.62 |
| Family History of FMD | 14 | 14 | 5.4% | 0 | 0.0% | - |
| Ehlers Danlos Syndrome* | 3 | 3 | 1.2% | 0 | 0.0% | - |
| Marfan Syndrome | 1 | 1 | 0.4% | 0 | 0.0% | - |
| Loeys-Dietz Syndrome | 0 | 0 | 0.0% | 0 | 0.0% | - |
| Dissection | ||||||
| Proportion of Individuals With Any Dissection† | 0.35 ± 0.48 | 0.34 ± 0.47 | - | 0.86 ± 0.47 | - | 0.005 |
| Number of Affected Arteries | 0.55 ± 0.91 | 0.51 ± 0.87 | - | 2.0 ± 0.87 | - | 0.016 |
| Aorta | 4 | 4 | 1.6% | 0 | 0.0% | - |
| Cerebral | 1 | 1 | 0.4% | 0 | 0.0% | - |
| Cervical | 70 | 66 | 25.7% | 4 | 57.1% | 0.25 |
| Internal Carotid Artery | 63 | 60 | 23.3% | 3 | 42.9% | 0.41 |
| Vertebral artery | 22 | 20 | 7.8% | 2 | 28.6% | - |
| Coronary | 11 | 11 | 4.3% | 0 | 0.0% | - |
| Lower Extremity | 7 | 5 | 1.9% | 2 | 28.6% | - |
| Upper Extremity | 0 | 0 | 0.0% | 0 | 0.0% | 1 |
| Visceral | 12 | 9 | 3.5% | 3 | 42.9% | 0.006 |
| Mesenteric | 6 | 4 | 1.6% | 2 | 28.6% | - |
| Renal | 7 | 5 | 1.9% | 2 | 28.6% | - |
| Aneurysm | ||||||
| Proportion of Individuals With Any Aneurysm‡ | 0.24 ± 0.43 | 0.23 ± 0.42 | - | 0.43 ± 0.42 | - | 0.19 |
| Number of Affected Arteries | 0.41 ± 0.89 | 0.40 ± 088 | - | 0.71 ± 0.89 | - | 0.24 |
| Aorta | 6 | 6 | 2.3% | 0 | 0.0% | - |
| Cerebral | 19 | 19 | 7.4% | 0 | 0.0% | - |
| Cervical | 27 | 26 | 10.1% | 1 | 14.3% | - |
| Internal Carotid Artery | 25 | 25 | 9.7% | 0 | 0.0% | - |
| Vertebral artery | 2 | 1 | 0.4% | 1 | 14.3% | - |
| Coronary | 0 | 0 | 0.0% | 0 | 0.0% | - |
| Lower Extremity | 1 | 0 | 0.0% | 1 | 14.3% | - |
| Upper Extremity | 0 | 0 | 0.0% | 0 | 0.0% | - |
| Visceral | 29 | 27 | 10.5% | 2 | 28.6% | - |
| Mesenteric | 2 | 1 | 0.4% | 1 | 14.3% | - |
| Renal | 23 | 22 | 8.6% | 1 | 14.3% | - |
| Multifocal FMD | ||||||
| Number of Affected Arteries | 2.00 ± 1.31 | 1.98 ± 1.31 | - | 2.57 ± 1.31 | - | 0.13 |
| Aorta | 2 | 2 | 0.8% | 0 | 0.0% | - |
| Cerebral | 29 | 28 | 10.9% | 1 | 14.3% | - |
| Cervical | 200 | 196 | 76.3% | 4 | 57.1% | 0.76 |
| Internal Carotid Artery | 195 | 191 | 74.3% | 4 | 57.1% | 0.77 |
| Vertebral artery | 10 | 10 | 3.9% | 0 | 0.0% | - |
| Coronary | 6 | 6 | 2.3% | 0 | 0.0% | - |
| Lower Extremity | 37 | 34 | 13.2% | 3 | 42.9% | 0.11 |
| Upper Extremity | 4 | 4 | 1.6% | 0 | 0.0% | - |
| Visceral | 168 | 162 | 63.0% | 6 | 85.7% | 0.58 |
| Mesenteric | 20 | 18 | 7.0% | 2 | 28.6% | - |
| Renal | 161 | 156 | 60.7% | 5 | 71.4% | 0.77 |
No cases of cEDS or vEDS;
Number of individuals with any dissection was =92;
Number of individuals with any aneurysm was =63
DISCUSSION
This study reports the first recurring pathogenic COL5A1 genetic variant associated with an arteriopathy in four unrelated families, a single base change causing a glycine residue at position 514 to be substituted by a serine residue. The probands harboring this variant presented with variable DAAD manifestations, including arterial aneurysms, dissections, tortuosity, and mFMD, with mFMD identified in all three of the female probands in the series of four probands. The COL5A1 c.1540G>A, p.(Gly514Ser) variant occurs on a shared haplotype among the probands who are of central European ancestry. Phenotypic presentation in mid-life suggests reproductive fitness would be unaffected and thus additional individuals having this pathogenic variant likely exist in the population. Further implicating a role of the COL5A1 gene in DAAD and arterial dissection, we observed a higher burden of low frequency variants of unclear clinical significance in COL5A1 that were predicted to be deleterious by in silico analysis, with CADD phred score ≥ 20, in individuals with clinical diagnoses of mFMD who sustained an arterial dissection. These variants were associated with increased risk of spontaneous arterial dissections, including those affecting the coronary, carotid, celiac and iliac arteries. Whether the additional variants beyond p.(Gly514Ser) function as disease modifiers for another as of yet unknown mechanism of disease requires further study. Clinical genetic testing of COL5A1 pathogenic variants in adults with relevant phenotypic presentations appears warranted, specifically for the p.(Gly514Ser) variant defined as likely pathogenic in our study. Our findings support a DAAD with a pattern of arterial involvement overlapping vEDS and mFMD.
Pathogenic variants in COL5A1 have most often been associated with cEDS, a connective tissue disorder with skin and joint phenotypes caused by the altered fibrillar collagen protein12. Genetic variants resulting in COL5A1 haploinsufficiency (i.e., frameshift, splice site or nonsense mutations) are expected to lead to a reduction of type V collagen in the extracellular matrix. A minority of variants cause missense mutations, mostly consisting of substitution of glycine residues in the triple helix domain that when incorporated into the triple helix, perturbs proper fibril formation. More recently, null alleles of COL5A1 have been recognized as underlying a significant proportion of cEDS39,40, with TEM of skin confirming irregular collagen fibrils40. In a murine model of heterozygous Col5a1 deletion, skin and arterial tensile strengths were compromised41.
Arterial dissection is rare in cEDS. Nevertheless, familialoccurrence of dissections has been observed for asingle COL5A1pathogenic variant42. Recurrence across unrelated families of the same COL5A1 pathogenic variant with associated arterial dissection is a novel finding of the current study. In previous reports, arterial dissection in cEDS involved the iliac, celiac, or mesenteric vasculature, and death occurred as early as age nine years6,8,9,42.
Arterial dissections, with or without prior dilation, are a hallmark of vEDS, but are rarely described in other EDS subtypes43. Arterial dissections in vEDS typically involve the large and medium-sized aortic branch vessels, including the iliac, visceral, coronary, and carotid/vertebral (cerebrocervical) arteries. Carotid and vertebral artery tortuosity is a notable feature of Loeys-Dietz syndrome (LDS), but is also seen in other heritable connective tissue disorders. Aortic root dilation is a hallmark of LDS, which was suggested in the initial differential diagnosis of two of our probands by the presence of borderline elevated aortic root sizes.
FMD etiology is not well understood, being a genetically heterogeneous condition with both sporadic and familial forms having at least partially complex genetic basis44–46. Familial cases of FMD are not rare, with 7.3% of patients in a clinical registry having at least one relative with the disease, and a family history of aneurysms in 23.5% and sudden death in 19.8%47. Nonetheless, there is currently no recommended molecular diagnostic testing for FMD in affected patients or their relatives3. COL5A1 has not been comprehensively assessed in prior studies of FMD48,49. Systemic connective tissue features and atrophic scars are common in FMD patients without a molecular diagnosis of EDS or LDS; atrophic scars occurred in 17.3% of FMD cases having arterial dissections or multiple aneurysms50. Although FMD can involve any vascular bed, the renal and cervical (carotid and vertebral) arteries are most commonly affected. Involvement of the iliac arteries with FMD occurs less often. Iliac artery dissections are uncommon in FMD patients, reported in 2.1%−3.5%47,51,52. In contrast, iliac artery dissections in the present study affected three of the four probands harboring the COL5A1 G514S variant. Notably, 3 of the 4 probands with the COL5A1 p.(Gly514Ser) variant were female, and all three women had findings of mFMD, consistent with the known preponderance of mFMD affecting women. The lack of penetrance of mFMD in the one male proband is noteworthy, given the infrequency of mFMD in men, including male relatives of mFMD probands. The histologic findings, which would ideally be extended to additional samples if these become available, support arterial medial fibroplasia in both the male proband and the one female proband who had undergone surgery. The clinical significance of the additional six rare variants with CADD phred score ≥ 20 and associated with arterial dissection among individuals with mFMD, require further segregation and follow-up of clinical phenotypes.
In terms of the functional consequence of the COL5A1 G514S variant, the glycine to serine substituted residue would be expected to cause a structural disturbance in the resulting COL5A1 protein, and in silico protein modeling of the COL5A1 G514S variant showed a change at this position within the collagenous region, from a buried position internal to the protein structure to an exposed region. This type of change may not only have structural consequences for proper collagen fibril assembly, but may also open this part of the protein to other protein binding partners, with potential downstream alterations in cellular interactions with the extracellular matrix or signaling pathways, both of which are relevant potential mechanisms for tissue fibrosis53 and arterial dissections54. In the case of Collagen I, in studies of osteogenesis imperfecta in vitro, a key glycine (Gly505) substitution to a serine residue near an α2β1 integrin binding site leads to a local disturbance but not an overall structural change or effect on integrin binding55. However, if Glycine 502 (Gly502) is perturbed (Val<Ser<Ala) N-terminal to the integrin binding site, there is impaired integrin binding56. These mutant collagens can still be secreted and incorporated into a forming fibril. Based upon these examples, it is possible that the COL5A1 p.Gly514Ser could lead to a change in binding with a key interacting protein that has yet to be determined. Notably, in the case of our COL5A1 G514S variant, there was no evidence that fibrosis of the arterial media is mediated via the TGF-β signaling pathway, as tissue pSMAD2 was not altered; however this assessment in surgically resected and end-stage diseased tissue may not reflect the initial alterations involved in disease pathogenesis, and this would need to be further explored in model systems.
The anatomic arterial phenotype of the G514S probands includes aneurysm, dissection, and FMD of several arterial beds including the external iliac artery, which is an exceedingly uncommon location of aneurysms. As opposed to the internal iliac artery, the external iliac artery is not part of the placental circulation, and the locations of pathology in the G514S probands are at sites of transition of smooth muscle cell origin from neural crest to somatic mesodermal derivation. The more frequent occurrence of aneurysms at branchpoints in the internal iliac artery, and other muscular arteries typically reflects increased susceptibility for aneurysmal development due to altered elastin organization at these points. There was no evidence of arterial dissections leading to isolated mural aneurysms or medial fibrosis which was more diffusely seen in the histopathologic specimens. Surgery was required for dissecting aneurysms in two probands harboring the G514S variant. The remaining aneurysms, including a discrete celiac artery aneurysm, a discrete saccular splenic artery aneurysm, and a pulmonary artery aneurysm, were not associated with locations in which dissections are typical, particularly the pulmonary and splenic artery aneurysms, nor did angiographic appearance suggest antecedent dissection. As arterial mobility and stretch have been hypothesized to mechanically stress the artery in FMD, a relevant hypothesis for the G514S variant is that the extra mobility in arteries with lower tensile strength subjected to pulsatile systolic flow, may be especially prone to undergo fibrotic response and remodeling in response to vascular injury from excessive stretch. In vEDS due to COL3A1 pathogenic variants, the major defect structurally is abnormal fibrillogenesis involving altered incorporation of collagen type 1. A similar mechanism may be hypothesized for the COL5A1 G514S variant, as this is also a fibrillar collagen57. In vEDS, cell signaling alterations have been shown to mediate arterial pathology leading to dissections and ruptures, in phospholipase C/inositol 1,4,5-triphosphate/protein kinase C/extracellular signal–regulated kinase pathways, and have been proposed as potential intervention targets54. Similar modeling and investigations of the COL5A1 G514S genetic variant are warranted. As there is no existing in vivo animal model of FMD, new genetic models would be required to further investigate these hypotheses.
The probands harboring the COL5A1 G514S pathogenic variant in the current report shared remarkable phenotypic similarity, with age of presentation over 40 years and the arterial beds involved, yet the family segregation analyses suggested complete penetrance for a phenotype overlapping with cEDS but incomplete penetrance of vascular phenotype or aneurysms, dissections or stenoses. This may be explained by the relatively early age of assessment of family members or possible unknown modifier genes. Penetrance of cerebrocervical arterial tortuosity was more complete, suggesting the vascular phenotypic assessments for penetrance would best be performed at middle age. The four unrelated families in this study shared a haplotype containing the COL5A1 G514S variant having an estimated population frequency of 0.4% among individuals of European ancestry. A founder effect has implications for screening, as additional individuals harboring this variant are expected to be encountered. Such was confirmed in a query of the TOPMed database, a whole genome sequenced database enriched for individuals with cardiovascular phenotypes. Unfortunately, further clinical data about the two individuals harboring the G514S variant listed in TOPMed was not available to the authors There are currently no clinical means to identify FMD patients predisposed to dissections. Early identification of a predisposition for arterial aneurysms, ruptures or dissections is particularly important to prevent serious vascular complications including death.
The current study suggests that surveillance and management strategies similar to those for vEDS should be considered for patients harboring the COL5A1 p.(Gly514Ser) variant. This would include initial screening imaging, subsequent periodic arterial imaging, close blood pressure monitoring and control, cautious consideration of pregnancy risk, and avoidance of contact sports. Although further study is required to assess surgical risk in patients with this variant, it would seem appropriate that surgical precautions should follow those employed for patients with vEDS. There is insufficient data to suggest similar clinical actions following consensus-based standards3 among patients with sporadic FMD and other COL5A1 low frequency variants predicted to be deleterious by in silico analysis, including those described in this study’s mFMD cohort. The authors consider it prudent to conduct arterial imaging for FMD at least once past the age 40 years, for patients with a COL5A1 variant and clinical features overlapping with those of vEDS or LDS. Moreover, patients with a COL5A1 likely pathogenic or pathogenic variant as per the ACGS and/or ACMG criteria who are also found to have DAAD should be counseled about the signs and symptoms of arterial dissections. Further investigation of the natural history of COL5A1 variants causing arterial disease is warranted. Interventions with antiplatelet agents, beta blockers, or angiotensin receptor blockers to reduce shear stresses and improve arterial remodeling require study. The hypothetical risks of calcium channel blocker and fluoroquinolone medications in promoting dissections in patients with vascular connective tissue disorders presenting as aortic aneurysm and dissection deserve note58,59. Finally, molecular testing for COL5A1 genetic variations should be considered in patients with a clinical phenotype of vEDS in whom no disease-causing COL3A1 variant is identified. It is indeed possible that rare variants in COL5A1 explain part of the incomplete detection rate of molecular testing for vEDS.
Conclusion
The COL5A1 p.(Gly514Ser) substitution is the first COL5A1 variant to be associated in unrelated families with DAAD including arterial dissections and mFMD, and this variant may be the responsible molecular basis of a subset of patients with FMD, particularly suggested by the founder effect in individuals from central Europe. The clinical presentations of the probands had considerable overlap with mFMD, LDS, and vEDS. Molecular testing for COL5A1 G514S and other variants should be considered in individuals presenting with a phenotype of vEDS without an identified likely pathogenic or pathogenic variant in COL3A1 and in individuals with DAAD manifest by cervical arterial tortuosity and FMD, as well as arterial aneurysms and dissections of other arterial beds, especially dissections affecting the iliac and mesenteric arteries. Lastly, this report highlights the value of deep phenotyping to clarify the molecular underpinnings of specific patterns of DAAD.
Supplementary Material
Table 1b. Overview of arterial pathology in the G514S probands.
ICA = internal carotid artery CCA = common carotid artery; Vert = vertebral artery; Pulm = pulmonary artery; CIA = common iliac artery; EIA = external iliac artery; IIA = internal iliac artery; T = Tortuosity, A = Aneurysm, D = Dissection, F = multifocal FMD, E = Ectasia (dilation not meeting criteria for aneurysm).
| Proband | Intracranial ICA | Extracranial ICA | CCA | Vert | Pulm | Celiac | Splenic | Renal | CIA | EIA | IIA | Aortic root (z-score) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | T | T | T | T | D | D, A* | D, A | 1.98 | ||||
| 2 | T | T | T | T | A | F | D, A | E | E | 0.79 | ||
| 3 | F, D, A | F, D, A* | T | D | D | 1.95 | ||||||
| 4 | T | T | T | A | F | A | F | F | −1.64 |
Surgical treatment was required, and operative tissue was available for analysis.
HIGHLIGHTS.
The COL5A1 genetic variant c.1540G>A, p.(Gly514Ser) is associated with a dysplasia-associated arterial disease that encompasses arterial aneurysms, dissections, tortuosity, and multifocal FMD, and the variant lies on a founder haplotype. The implication of a founder effect is that additional carriers exist in the population, which we demonstrate.
COL5A1 p.(Gly514Ser) is the first monogenic factor for multifocal FMD.
COL5A1 genetic variants predicted to be deleterious by in-silico analysis exist in 2.7% of a multifocal FMD cohort, and such COL5A1 variants are associated with arterial dissections, which are known to occur in a subset of patients with FMD but for which no clinical predictors exist. These variants may function as pathogenic variants or disease modifiers.
ACKNOWLEDGEMENTS
The University of Michigan DNA Sequencing Core performed genotyping and Sanger Sequencing. Histology and immunohistochemistry were performed by the In Vivo Animal Core research histology facility at the University of Michigan. The authors acknowledge the University of Michigan Precision Health Initiative and Medical School Central Biorepository for providing biospecimen storage, management, processing and distribution services and the Center for Statistical Genetics in the Department of Biostatistics at the School of Public Health for the Michigan Genomics Initiative genotype data curation and management in support of this research. We thank Eric Wizauer for assistance with radiographic image acquisition. We acknowledge Ken Calderone in the laboratory of Gary Fisher for acquisition of second harmonic generation imaging. We acknowledge Gift of Life Michigan for assistance with procurement of control arterial tissues. We thank the patients who participated in this study and acknowledge the Fibromuscular Dysplasia Society of America for enabling study enrollments at meetings.
Sources of funding: Funding support included grants from NHLBI (R01 HL139672), Doris Duke Charitable Foundation, University of Michigan Frankel Cardiovascular Center, University of Michigan Taubman Institute to S.K.G. Sequencing services were provided through the RS&G Service by the Northwest Genomics Center at the University of Washington, Department of Genome Sciences, under U.S. Federal Government contract number HHSN268201100037C from the National Heart, Lung, and Blood Institute. The Cleveland Clinic FMD Biorepository was supported in part by the National Institutes of Health, National Center for Research Resources, CTSA 1UL1RR024989, Cleveland, Ohio. GeneBank was supported in part by grants from NHLBI and Office of Dietary Supplements (P01 HL076491, P01 HL147823, R01HL128300 and R01HL103866) to S.L.H. Dermal biopsy was supported by P30 AR075043. J.L was supported by NIH T32-HL007853.
ABBREVIATIONS
- COL5A1 G514S
COL5A1 c.1540G>A, p.(Gly514Ser)
- DAAD
dysplasia associated arterial disease
- CADD
phred score: combined annotation dependent depletion score
- FMD
fibromuscular dysplasia
- mFMD
multifocal fibromuscular dysplasia
- EDS
Ehlers-Danlos Syndrome
- cEDS
classical Ehlers-Danlos Syndrome
- vEDS
vascular Ehlers-Danlos Syndrome
- LDS
Loeys-Dietz Syndrome
Footnotes
Disclosures
The authors declare no competing interests. SKG, JCS and HLG are non-compensated members of the Medical Advisory Board of the FMD Society of America (FMDSA). SKG is a non-compensated member of the Scientific Advisory Board of SCAD Alliance. Both organizations are non-profit institutions. G.R.A. is an employee of Regeneron Pharmaceuticals and owns stock and stock options for Regeneron Pharmaceuticals.
Supplemental Materials
REFERENCES
- 1.Harrison EG Jr., McCormack LJ. Pathologic classification of renal arterial disease in renovascular hypertension. Mayo Clin Proc 1971;46:161–7. [PubMed] [Google Scholar]
- 2.Stanley JC, Gewertz BL, Bove EL, Sottiurai V, Fry WJ. Arterial fibrodysplasia. Histopathologic character and current etiologic concepts. Arch Surg 1975;110:561–6. [DOI] [PubMed] [Google Scholar]
- 3.Gornik HL, Persu A, Adlam D, et al. First International Consensus on the diagnosis and management of fibromuscular dysplasia. Vasc Med 2019;24:164–89. [DOI] [PubMed] [Google Scholar]
- 4.Malfait F, Wenstrup R, De Paepe A. Classic Ehlers-Danlos Syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al. , eds. GeneReviews((R)). Seattle (WA)1993. [PubMed] [Google Scholar]
- 5.Symoens S, Syx D, Malfait F, et al. Comprehensive molecular analysis demonstrates type V collagen mutations in over 90% of patients with classic EDS and allows to refine diagnostic criteria. Human mutation 2012;33:1485–93. [DOI] [PubMed] [Google Scholar]
- 6.Borck G, Beighton P, Wilhelm C, Kohlhase J, Kubisch C. Arterial rupture in classic Ehlers-Danlos syndrome with COL5A1 mutation. Am J Med Genet A 2010;152A:2090–3. [DOI] [PubMed] [Google Scholar]
- 7.de Leeuw K, Goorhuis JF, Tielliu IF, et al. Superior mesenteric artery aneurysm in a 9-year-old boy with classical Ehlers-Danlos syndrome. Am J Med Genet A 2012;158A:626–9. [DOI] [PubMed] [Google Scholar]
- 8.Mehta S, Dhar SU, Birnbaum Y. Common iliac artery aneurysm and spontaneous dissection with contralateral iatrogenic common iliac artery dissection in classic ehlers-danlos syndrome. Int J Angiol 2012;21:167–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karaa A, Stoler JM. Ehlers Danlos Syndrome: An Unusual Presentation You Need to Know about. Case Rep Pediatr 2013;2013:764659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yasuda S, Imoto K, Uchida K, et al. Successful endovascular treatment of a ruptured superior mesenteric artery in a patient with EhlersDanlos syndrome. Ann Vasc Surg 2013;27:975 e1–5. [DOI] [PubMed] [Google Scholar]
- 11.Pepin M, Schwarze U, Superti-Furga A, Byers PH. Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N Engl J Med 2000;342:673–80. [DOI] [PubMed] [Google Scholar]
- 12.Malfait F, Francomano C, Byers P, et al. The 2017 international classification of the Ehlers-Danlos syndromes. American journal of medical genetics Part C, Seminars in medical genetics 2017;175:8–26. [DOI] [PubMed] [Google Scholar]
- 13.Devereux RB, de Simone G, Arnett DK, et al. Normal limits in relation to age, body size and gender of two-dimensional echocardiographic aortic root dimensions in persons >/=15 years of age. Am J Cardiol 2012;110:1189–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goldstein SA, Evangelista A, Abbara S, et al. Multimodality imaging of diseases of the thoracic aorta in adults: from the American Society of Echocardiography and the European Association of Cardiovascular Imaging: endorsed by the Society of Cardiovascular Computed Tomography and Society for Cardiovascular Magnetic Resonance. J Am Soc Echocardiogr 2015;28:119–82. [DOI] [PubMed] [Google Scholar]
- 15.Fritsche LG, Gruber SB, Wu Z, et al. Association of Polygenic Risk Scores for Multiple Cancers in a Phenome-wide Study: Results from The Michigan Genomics Initiative. Am J Hum Genet 2018;102:1048–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jun G, Flickinger M, Hetrick KN, et al. Detecting and estimating contamination of human DNA samples in sequencing and array-based genotype data. Am J Hum Genet 2012;91:839–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kent WJ. BLAT--the BLAST-like alignment tool. Genome Res 2002;12:656–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Price AL, Weale ME, Patterson N, et al. Long-range LD can confound genome scans in admixed populations. Am J Hum Genet 2008;83:132–5; author reply 5–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scheet P, Stephens M. A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase. Am J Hum Genet 2006;78:629–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet 2001;68:978–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 2005;21:263–5. [DOI] [PubMed] [Google Scholar]
- 22.Genomes Project C, Auton A, Brooks LD, et al. A global reference for human genetic variation. Nature 2015;526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rueden CT, Schindelin J, Hiner MC, et al. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinformatics 2017;18:529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 2012;9:676–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014;46:310–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 2014;11:361–2. [DOI] [PubMed] [Google Scholar]
- 28.Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res 2012;40:W452–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu X, Wu C, Li C, Boerwinkle E. dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum Mutat 2016;37:235–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010;38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takahara K, Sato Y, Okazawa K, et al. Complete primary structure of human collagen alpha 1 (V) chain. J Biol Chem 1991;266:13124–9. [PubMed] [Google Scholar]
- 32.Paladin L, Tosatto SC, Minervini G. Structural in silico dissection of the collagen V interactome to identify genotype-phenotype correlations in classic Ehlers-Danlos Syndrome (EDS). FEBS Lett 2015;589:3871–8. [DOI] [PubMed] [Google Scholar]
- 33.Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016;536:285–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.2020. Eea. ACGS Best Practice Guidelines for Variant Classification in Rare Disease https://wwwacgsukcom/media/11631/uk-practice-guidelines-for-variant-classification-v4-01-2020pdf. 2020 [DOI] [PubMed] [Google Scholar]
- 36.Lucas SEM, Zhou T, Blackburn NB, et al. Rare, potentially pathogenic variants in 21 keratoconus candidate genes are not enriched in cases in a large Australian cohort of European descent. PLoS One 2018;13:e0199178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schubert JA, Landis BJ, Shikany AR, Hinton RB, Ware SM. Clinically relevant variants identified in thoracic aortic aneurysm patients by research exome sequencing. Am J Med Genet A 2016;170A:1288–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Landis BJ, Schubert JA, Lai D, et al. Exome Sequencing Identifies Candidate Genetic Modifiers of Syndromic and Familial Thoracic Aortic Aneurysm Severity. J Cardiovasc Transl Res 2017;10:423–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wenstrup RJ, Florer JB, Willing MC, et al. COL5A1 haploinsufficiency is a common molecular mechanism underlying the classical form of EDS. Am J Hum Genet 2000;66:1766–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schwarze U, Atkinson M, Hoffman GG, Greenspan DS, Byers PH. Null alleles of the COL5A1 gene of type V collagen are a cause of the classical forms of Ehlers-Danlos syndrome (types I and II). Am J Hum Genet 2000;66:1757–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wenstrup RJ, Florer JB, Davidson JM, et al. Murine model of the Ehlers-Danlos syndrome. col5a1 haploinsufficiency disrupts collagen fibril assembly at multiple stages. J Biol Chem 2006;281:12888–95. [DOI] [PubMed] [Google Scholar]
- 42.Monroe GR, Harakalova M, van der Crabben SN, et al. Familial Ehlers-Danlos syndrome with lethal arterial events caused by a mutation in COL5A1. American Journal of Medical Genetics Part A 2015;167:1196–203. [DOI] [PubMed] [Google Scholar]
- 43.Malfait F Vascular aspects of the Ehlers-Danlos Syndromes. Matrix Biol 2018;71–72:380–95. [DOI] [PubMed] [Google Scholar]
- 44.Perdu J, Boutouyrie P, Bourgain C, et al. Inheritance of arterial lesions in renal fibromuscular dysplasia. J Hum Hypertens 2007;21:393–400. [DOI] [PubMed] [Google Scholar]
- 45.Rushton AR. The genetics of fibromuscular dysplasia. Arch Intern Med 1980;140:233–6. [PubMed] [Google Scholar]
- 46.Kiando SR, Tucker NR, Castro-Vega LJ, et al. PHACTR1 Is a Genetic Susceptibility Locus for Fibromuscular Dysplasia Supporting Its Complex Genetic Pattern of Inheritance. PLoS Genet 2016;12:e1006367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Olin JW, Froehlich J, Gu X, et al. The United States Registry for Fibromuscular Dysplasia: results in the first 447 patients. Circulation 2012;125:3182–90. [DOI] [PubMed] [Google Scholar]
- 48.Poloskey SL, Kim ES, Sanghani R, et al. Low yield of genetic testing for known vascular connective tissue disorders in patients with fibromuscular dysplasia. Vascular Medicine 2012;17:371–8. [DOI] [PubMed] [Google Scholar]
- 49.Ganesh SK, Morissette R, Xu Z, et al. Clinical and biochemical profiles suggest fibromuscular dysplasia is a systemic disease with altered TGF-beta expression and connective tissue features. FASEB J 2014;28:3313–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.O’Connor S, Kim E, Brinza E, et al. Systemic connective tissue features in women with fibromuscular dysplasia. Vasc Med 2015;20:454–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bolen MA, Brinza E, Renapurkar RD, Kim ESH, Gornik HL. Screening CT Angiography of the Aorta, Visceral Branch Vessels, and Pelvic Arteries in Fibromuscular Dysplasia. JACC Cardiovasc Imaging 2017;10:554–61. [DOI] [PubMed] [Google Scholar]
- 52.Kadian-Dodov D, Gornik HL, Gu X, et al. Dissection and Aneurysm in Patients With Fibromuscular Dysplasia: Findings From the U.S. Registry for FMD. J Am Coll Cardiol 2016;68:176–85. [DOI] [PubMed] [Google Scholar]
- 53.Reed NI, Jo H, Chen C, et al. The alphavbeta1 integrin plays a critical in vivo role in tissue fibrosis. Sci Transl Med 2015;7:288ra79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bowen CJ, Calderon Giadrosic JF, Burger Z, et al. Targetable cellular signaling events mediate vascular pathology in vascular Ehlers-Danlos syndrome. J Clin Invest 2020;130:686–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yigit S, Yu H, An B, et al. Mapping the Effect of Gly Mutations in Collagen on alpha2beta1 Integrin Binding. J Biol Chem 2016;291:19196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Qiu Y, Mekkat A, Yu H, et al. Collagen Gly missense mutations: Effect of residue identity on collagen structure and integrin binding. J Struct Biol 2018;203:255–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu X, Wu H, Byrne M, Krane S, Jaenisch R. Type III collagen is crucial for collagen I fibrillogenesis and for normal cardiovascular development. Proc Natl Acad Sci U S A 1997;94:1852–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Daneman N, Lu H, Redelmeier DA. Fluoroquinolones and collagen associated severe adverse events: a longitudinal cohort study. BMJ Open 2015;5:e010077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee CC, Lee MT, Chen YS, et al. Risk of Aortic Dissection and Aortic Aneurysm in Patients Taking Oral Fluoroquinolone. JAMA Intern Med 2015;175:1839–47. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.