

Abstract
The lack of stringent regulations regarding raw materials for herbal supplements used for medicinal purposes has been a constant challenge in the industry. Ginkgo biloba L. leaf extracts attract consumers because of the supposed positive effect on mental performance and memory. Supplements are produced using dried leaf materials and standardized leaf extracts such as EGb 761. Adulteration of Ginkgo biloba L. plants and extracts are becoming more and more common practice due to economically driven motivation from increasing demand in the market and the high cost of raw materials and production. Reinforcement in quality control (QC) to avoid adulterations is necessary to ensure the efficacy of the supplements. In this study, liquid chromatography-high resolution mass spectrometry (LC-HRMS) was used with principal component analysis (PCA) as an unsupervised exploratory method to analyze, identify, and evaluate the adulterated Ginkgo biloba L. plant materials and dried leaf extracts using the PCA scores and loadings obtained and compound identification.
Keywords: nontargeted analysis, Ginkgo biloba L., adulteration, LC-HRMS, PCA
Introduction
Ginkgo biloba L. (Ginkgoaecae) or the Maidenhair tree is the sole living specie of the Ginkgophyta division and is considered a living fossil dating back to 180 million years based on the fossil records where its genus was once a diverse taxon [1]. Ginkgo biloba is native to China but, can be found as ornamental trees in countries with warm temperate climates such as Japan, Korea, Australia, some parts of Europe and North America [1–3]. Most of the scientific and medicinal research of Ginkgo biloba L. focuses on the leaves and the extracts, because these contain the active constituents such as flavonoids and terpene trilactones (TTLs), to which the supposed health benefits are attributed. The use of ginkgo leaf extracts started in the 1960s in Germany for the improvement of the blood circulation, to fight fatigue, as an aid for early dementia, for memory improvement, and as a cure for tinnitus [2, 4, 5]. The antioxidant effects of gingko leaves were attributed to flavonol glycosides, which are the most prevalent group of flavonoids in gingko leaves especially the derivatives of quercetin, kaempferol, and isorharmnetin [1, 4, 6, 7]. Terpene trilactones, including ginkgolide A (GA), ginkgolide B (GB), ginkgolide C (GC), a minor TTL named ginkgolide J (GJ), and bilobalide, are considered the true markers of a pure ginkgo leaves as they are uniquely attributed to Ginkgo biloba [2, 4, 5, 7].
The demand for natural products has increased in the dietary supplement industry resulting in the large-scale cultivation of ginkgo in several parts of the world. According to a review by S. Gafner in 2018 [8], a consistent growth in the global demand in leaf extracts was observed from US $118 million in 2013 to US $162 million in 2016. The estimated global demand for dried ginkgo leaves was 60,000 metric tons in 2014. Manufacturers from ConsumerLab.com reported that the cost for a Ginkgo biloba extract varied between US $35 per kg and US $90 per kg, while the price of rutin, a known adulterant isolated from buckwheat, is approximately US $10 per kg. Canadian Phytopharmaceutical Corp. also reported that the ginkgo extracts from Chinese manufacturers in 2015 cost between US $150 per kg and US $240 per kg, while Japanese sophora flower extracts (another widely used adulterant) were sold for US $30 per kg [8]. For this reason, ginkgo products are susceptible to adulteration specifically to boost the flavonol glycoside content using lower-cost sources.
The roles of quality assurance (QA) and quality control (QC) can be significant in the industry in assuring proper plant parts and correct botanical taxon are used in manufacturing the finished product, and consistent quality of ginkgo leaves and extracts, despite their inherent natural variability and chemical complexity. With the numerous studies conducted over the years, the established ratio for ginkgo leaf extracts is 6% or greater terpene trilactones, 24% or greater flavonol glycosides, and less than 5 mg/kg of ginkgolic acids [1, 2, 5, 9]. Pharmacopoeias, such as the Chinese Pharmacopoeia and USP, establish testing methods to ensure standardization of raw herbal materials by providing monographs [10]. Most manufacturers of ginkgo leaf extracts comply with the different pharmacopoeias in their regions to improve quality control. Chinese Pharmacopoeia and USP directives include monitoring a quercetin/kaempferol/isorhamnetin (Q/K/I) ratio of the hydrolyzed extract based on the respective peak areas using HPLC methods with an acceptable range of 1/0.8–1.2/≥1 [8, 9]. Authentic composition is critical in quality control of herbal supplements. However, adulteration of botanicals is still common due to economical motivation, and can jeopardize not only the quality but also the safety of the finished products [4, 11].
Ginkgo leaf extracts can be adulterated in numerous ways. Spiking the original plant extracts or product formulations with pure flavonols or flavonol glycosides is the most common form of adulteration, manufacturers use less expensive materials to achieve the target chemical specification of 24% flavonol glycosides. The pure flavonols found in ginkgo products like rutin, quercetin and kaempferol are the typical compounds used in spiking as they are highly effective in inflating the assay values of flavonol glycosides. However, it was observed that as the total flavonol content increases, the authenticity of ginkgo decreases for these adulterated samples [11]. The other parts of Ginkgo biloba (roots, stem, and seeds) were also used to reduce the cost of manufacture but, since these plant parts contain a different set of active components, may contribute to different physiological effects that could be harmful to the consumers. Furthermore, fortifications using other flavonol glycoside-rich extracts such as Styphnolobium japonicum (Fabaceae) and Fagopyrum esculentum M. (Polygonaceae) of the original Ginkgo biloba L. plant extracts have been reported [8]. It was also noted that with this method of adulteration, additional compounds might be present as other plant extracts have their own set of active components [8, 10].
A useful tool for benchmark comparison in the prevention of adulteration of botanicals is the use of Certified Reference Materials (CRMs). CRMs are homogeneous, stable materials that have been well-characterized for one or more property values and provide associated uncertainties and traceabilities using validated procedures [5]. Analytical techniques such as chromatography and spectroscopy have been used extensively to detect, characterize, and estimate both quantitively and qualitatively the different bioactive components in Ginkgo biloba L. leaves and leaf extracts to meet the required specifications especially for the flavonol glycosides. Among the most commonly used techniques are high performance liquid chromatography (HPLC) and gas chromatography (GC) coupled with various detectors such as mass spectrometry (MS), thin-layer chromatography (TLC), inductively coupled plasma mass spectrometry (ICP-MS), nuclear magnetic resonance spectroscopy (NMR), and near infrared spectroscopy (NIR) [2, 7, 12–16]. Liquid chromatography coupled with high resolution mass spectrometry (LC-HRMS) is a useful tool for a nontargeted approach as the full scan acquisition mode allows retrospective analysis without further injections and without limitations in the number of monitored compounds [7]. Nontargeted MS provides a holistic approach in which known and unknown compounds are detected, quantified, and all the obtained variables are considered simultaneously as the synergic or total effect between variables are not possible to be examined individually. This type of approach requires multivariate techniques since univariate and classical statistical approaches are unfeasible [17].
In this study, an LC-HRMS was utilized as a tool to investigate the authenticity of Ginkgo biloba L. samples including dried plant material parts (leaves and stems) and a variety of dried leaf extracts (different water-solvent preparations) from different manufacturers, by a nontargeted approach with the aid of NIST Standard Reference Materials and subsequent data analysis. Principal component analysis (PCA), an unsupervised exploratory technique for multivariate analysis, was used to discriminate and discern patterns in each resulting large dataset to create models that will aid effective detection and identification of adulterated ginkgo samples for quality control purposes.
Materials and Methods
Ginkgo Samples
A total of 32 samples of Ginkgo biloba L. plant materials and dried leaf extracts were used in the study and labelled as datasets A (plant materials) and B (dried leaf extracts). Ginkgo leaves were classified into two types, untreated and steam-treated, and were obtained from the same supplier (source A) while the stem samples were from a different source (source B). Dried leaf extracts were collected from different commercial manufacturers and were prepared using a variety of water-solvent ratios. These ginkgo samples were then adulterated at NIST, randomly labeled A1 through A16 for plant materials or B1 through B16 for extract materials and are shown according to the adulteration scheme in Table 1. This table shows the summary of the classification of samples by adulteration and by material source. The samples that were duplicated in the study served as a blind check for the reproducibility of the chemometric analysis. During the LC-HRMS analysis, the samples’ identities were not used for the nontargeted analysis and were only used to aid data analysis.
Table 1.
Summary of Ginkgo biloba L. sample classification used in the study
| Source of material | Adulteration Level | |||
|---|---|---|---|---|
| 1: 0% | 2: 3% | 2: 7% | 3: 15% | |
| Dataset A - Plant parts | ||||
| Source A: Ginkgo leaves | ||||
| A1 - untreated | A9 | A3 | A16 | A12, A4 |
| A2 - steam-treated | A5 | A14 | A7, A13 | A8 |
| Source B: Ginkgo stem | A15 | A2, A10 | A1 | A11 |
| Source C: SRM 3246 Ginkgo biloba L. leaves | A6, SRM 3246 | |||
| Dataset B - Dried leaf extracts | ||||
| Source D: Ginkgo extract aqueous | B10 | B5 | B13 | B7 |
| Source E: Ginkgo extract ethanol:water | B3, B8 | B12, B16 | B1 | B9 |
| Source F: Ginkgo extract acetone:water | B6 | B14 | B11 | B4, B15 |
| Source G: Ginkgo extract acetone:water with lecithin | B2 | |||
| Source H: SRM 3247 Ginkgo biloba L. extract | SRM 3247 | |||
The SRMs used in this study were NIST SRM 3246 (Ginkgo biloba (Leaves)) for the leaf samples and NIST SRM 3247 (Ginkgo biloba (Extract)) for the commercial raw leaf extract samples. The NIST SRM 3247 was prepared according to the German Pharmacopoeia (non-clinical) and was acquired from the manufacturer. Further storage preparations were done at ChromaDex Inc. as stated in the certificate of analysis. The SRMs served as analytical quality control materials to aid in the evaluation of the authenticity of these samples.
Chemicals
All solvents used for LC-HRMS analysis were Optima™ LC-MS grade and were purchased from Fisher Chemical, Fisher Scientific Company L.L.C, Pittsburgh, PA, USA. The extraction solvent was prepared by mixing methanol, water, and formic acid to achieve a concentration of 90:9:1 (volume fraction). Mobile phases A and B for gradient elution were prepared using 0.1% (v/v) formic acid in water and 0.1% (v/v) formic acid in acetonitrile, respectively. Previous studies had reported poor peak shape for terpenoid (−)-bilobalide, a main component of Ginkgo biloba, with the use of formic acid in the mobile phase, however, the extraction procedure performed in this study was not meant to identify specific compounds (e.g. bilobalide and other terpene trilactones), but rather to broadly profile the compounds in the sample extracts [7].
LC-HRMS Analysis
A 0.3 g to 0.6 g sample was weighed into pre-weighed 15 mL polypropylene (PP) centrifuge tubes. Approximately 5 mL of extraction solvent was added, the tubes re-weighed, and the mixtures were vortexed to ensure there were no dry sample at the bottom. The samples, including the SRMs, were sonicated for 15 minutes and were centrifuged at 50 Hz for 15 minutes. The supernatant was collected and filtered through a 0.45 μm nylon filter (Phenomenex, Torrance, CA, USA) into a new set of centrifuge tubes. All samples were extracted in duplicate on different days and refrigerated until ready for analysis. The duplicate extracts were not combined subsequently but were run as individual samples. Blank samples were also prepared in duplicate for both sample sets. Separate pooled samples for plant material and leaf extract samples were prepared in a similar way. Using a micropipette, 100 μL of each plant or leaf extract sample was placed into a vial then mixed thoroughly using a vortex. All sample extracts were placed in HPLC vials and consequently positioned in the autosampler for LC-MS analysis.
The chromatographic separation was performed using a Thermo Ultimate 3000 Liquid Chromatograph coupled with Q-Exactive Hybrid Orbitrap Mass Spectrometer which was controlled with Thermo Scientific Chromeleon Chromatography Data System version 6.80 SR11 (Thermo Fisher Scientific, Waltham, MA, USA). The analyses were conducted in reversed phase using a Halo C18 column (2.1 mm × 100 mm, 2.7 μm particle size, MAC-MOD Analytical Inc., Chadds Ford, PA, USA). Gradient elution was used in the LC separation, because the polarity of the main components present in Ginkgo biloba varies. The mass spectrometer was operated using electrospray ionization (ESI) in full scan mode for positive and negative ionization modes, independently. Table 2 shows the detailed chromatographic and mass spectrometer conditions used in the analysis.
Table 2.
Chromatographic parameters used in LC-HRMS for Ginkgo biloba L. samples
| Instrument Conditions | ||
|---|---|---|
| LC Conditions | ||
| Injection volume | 5 μL | |
| Flow Rate | 0.3 mL/min | |
| Mobile Phase | A: 0.1% formic acid in water, B: 0.1% formic acid in acetonitrile (As 100% water for A, 100% acetonitrile for B, and 50:50 water:acetonitrile with 0.1% formic acid for C) | |
| Gradient | Time | %B |
| 0 | 5 | |
| 1 | 5 | |
| 15 | 95 | |
| 25 | 95 | |
| Equilibration time | 10 min | |
| Column Temperature | 25 °C | |
| MS Conditions | ||
| Ionization | Electrospray | |
| Polarity Ionization | Positive/Negative (separately) | |
| Voltage | 3000(+)/2500(−) | |
| Capillary Temperature | 350 °C | |
| Sheath Gas | 35 | |
| Auxiliary Gas | 10 | |
| Probe Heater Temperature | 300 °C | |
| MS1 Scan Range | 100 – 1500 m/z | |
| MS1 Resolution | 70,000 | |
| MS1 AGC Target | 3e6 | |
| MS1 Maximum IT | 100 ms | |
| MS2 Experiment: | TopN (5) | |
| MS2 Resolution | 17,500 | |
| MS2 AGC Target: | 1e5 | |
| MS2 Maximum IT: | 50 ms | |
| Dynamic exclusion: | 5 s | |
| Collision Energy: | 30 with 50% stepped NCE (15, 30, 45) | |
Data Analysis for PCA
Experimental data were collected in Microsoft Excel™ 2016 (Microsoft Corporation, Redmond, WA, USA) and processed using the PLS_Toolbox 8.6.2 (Eigenvector Research, Inc., Manson, WA, USA) running in MATLAB R2018a (The Mathworks Inc, Natick, MA, USA). MZmine 2 Version 2.36 software (http://mzmine.github.io/), a Java-based open source software used for data processing, feature extraction, and differential profiling, was also used to pre-process the MS/MS data before importing it to MATLAB [18, 19].
Preprocessing using MZmine 2 software was performed based on approaches for nontargeted metabolomics and lipidomics datasets optimized internally at NIST as shown in Figure 1. The workflow is composed of several data processing stages and requires different sets of criteria to be optimized. The LC-MS1 data of the instrument full scan raw data were converted to an open source format (.mzxml file) using ProteoWizard MS convert tool before importing into the MZmine. Datasets for LC-HRMS were divided into four groups: A negative, A positive, B negative and B positive, with A and B describing the plant materials and leaf extracts, respectively, while the terms positive and negative designate the ionization polarity modes used in the analysis. Each dataset was processed and analyzed separately.
Fig. 1.

Workflow of LC-MS feature extraction based on MZmine 2.0
The nontargeted batch file steps described in Figure 1 were performed first to create the feature peak list that will be used for the samples. Pooled, blank, and SRM samples were imported and a mass list was built using the mass detection step. An appropriate noise level setting was used based on the sensitivity of the instrument and on the original chromatograms and mass spectra of the samples. Ion chromatograms were constructed for each of the masses in the mass list using the chromatogram builder to produce a peak list containing the extracted ion chromatograms (EICs or XICs) for masses that have been detected by mass spectrometer continuously over a certain duration of time. After the EICs were built, peak detection by chromatogram deconvolution was performed using a local minimum search algorithm which aims to find the local minima in the chromatogram as border points between individual peaks and can set restrictions on minimum absolute and relative intensities, or a minimum ratio of peak top or edge [19]. Construction of EICs and detection of chromatographic peaks from the EICs are considered important steps as an ion chromatogram may contain multiple peaks and these functions are useful for the identification and relative quantitation of compounds. Also, errors produced at this stage can spread throughout the data preprocessing and succeeding statistical analysis to be performed [20]. Isotopic peaks grouper was then used to combine the features corresponding to the same analyte with different charge states and isotopomers. Once the data were deisotoped, join aligner was performed to align and combine the peaks based on the retention time and m/z tolerance settings. The final step was filling the gaps by using two functions, the peak finder and the same tR and m/z range gap filler. Areas without peaks in some scans will be filled in and the peaks with the same tR and m/z range that were not detected due to being close to the detection limit in the original peak window can be identified.
The peak list extracted from MZmine consisting of the column features of row ID, row retention time, row m/z, and the peak areas of the blank, pooled samples, and SRM samples were exported into a comma-separated value (.csv) file. The row ID is defined as the number that identifies the peak list row and this peak list row can have one or several peaks that have the same mass range and retention time range but originating from a different raw data. The row retention time is the representative retention time value (average retention time of all peaks) and the row m/z is the representative m/z value (average m/z value of all peaks) for a row peak. The retention time value or m/z value of each peak is dependent on peak detection method [21]. This peak list was the transformed data matrix after preprocessing using the nontargeted batch file steps. Peak areas of EICs that were higher in the blank than the pooled samples or SRMs were removed manually using Excel. The feature list (1) was created from this peak list in a new .csv file containing only the selected data with the following features in the sequence of row m/z, row retention time, and row ID. The targeted batch file steps in Figure 1 were then performed by importing the samples and SRMs and by using the feature list (1) as the “targeted peak list” in the targeted peak detection in constructing the EICs for the samples. Additional steps including peak list rows filter and duplicate filter were done apart from the similar steps in the nontargeted batch file procedure. The final feature list (2) was then extracted and saved in a similar manner as the first feature list. This is the final preprocessed dataset that was used for multivariate analysis.
Preprocessing prior to the use of a chemometric technique is necessary to transform the measured data into a more suitable form for the data analysis as variables measured can have different units and systematic effects and interferences may be present which can make the data analysis difficult. Each data processing step was performed multiple times with different values to obtain the optimized parameters. The final parameters used in data processing are summarized in Table 3 for LC-HRMS. The .CSV files of the final feature list from MZmine 2.0 were imported to MATLAB R2018a for further multivariate analysis. With the PLS Toolbox, unsupervised exploratory analysis using the PCA with some preprocessing methods was performed on the extracted data from the Ginkgo biloba samples and SRMs. Using a preprocessing step to transform the data into a suitable form for data analysis can make data analysis less difficult.
Table 3.
Parameters used in MZmine for Ginkgo biloba LC-HRMS datasets A and B
| MZmine Parameters | LC-MS1 setting |
|---|---|
| Mass Detection | |
| Noise level | *1×107 |
| Chromatogram Builder | |
| Type of scans | MS level 1 |
| Minimum time span | 0.15 min |
| Minimum height | 1×107 |
| m/z tolerance | 0.005 m/z or 10 ppm |
| Chromatogram Deconvolution | |
| Algorithm | Local minima search |
| Chromatographic threshold | 10% |
| Minimum retention time range | 0.1 Min |
| Minimum relative height | 10% |
| Minimum absolute height | *1×107 |
| Minimum ratio of peak top/edge | 1 |
| Peak duration range | 0–10 min |
| Isotopic Peaks Grouper | |
| m/z tolerance | 0.005 m/z or 10.0 ppm |
| Retention time tolerance | 0.1 absolute min |
| Maximum charge | 1 |
| Representative Isotope | Most intense |
| Join Aligner | |
| m/z tolerance | 0.005 to 10.0 ppm |
| Weight for m/z | 20 |
| Retention time tolerance | 0.1 absolute min |
| Weight for RT | 20 |
| Peak finder | |
| Intensity tolerance | 100.0% |
| m/z tolerance | 0.005 m/z or 10.0 ppm |
| Retention time tolerance | 0.1 absolute min |
| Same RT and m/z range gap filler | |
| m/z tolerance | 0.005 m/z or 10.0 ppm |
| Targeted Peak Detection | |
| Peak list file | Select Targeted peak list created |
| Intensity tolerance | 100.0% |
| Noise level | *1×107 |
| m/z tolerance | 0.005 m/z or 10.0 ppm |
| Retention time tolerance | 0.1 absolute min |
| Peak List Rows Filter | 0.0000 to 80.0000 |
| m/z range | Remove rows that match all criteria |
| Keep or Remove rows | |
| Duplicate Peak Filter | |
| Algorithm | NEW AVERAGE |
| m/z tolerance | 0.005 m/z or 10.0 ppm |
| RT Tolerance | 0.1 absolute min |
dataset A both modes: 1×107; dataset B negative ion mode: 5×107; and dataset B positive ion mode: 2×107
All nontargeted results were normalized by sample and extraction solvent masses (for a relative sample concentration) using the Equation 1 below which is further elaborated in the Supplemental Information.
| Equation 1 |
Normalizing the data by reducing the peak area to relative sample concentration can minimize the within-replicate variability and incorporate the discrepancy from sample preparation into the concentration values. This calculation assumes that the extraction efficiency (i.e. recovery) for each individual compound is equal across all samples given the similar nature of the sample matrices. Peak identification was done using R scripts [22] linked to the NIST MS Search program (v2.3; https://chemdata.nist.gov) by scanning the final feature list (2) for both positive and negative ion modes obtained from MZMine as these lists were assumed to contain all the detected peaks in the samples and SRMs.
Results and Discussion
LC-HRMS analysis was performed using the sample set of 16 plant materials, 16 dried leaf extracts, 2 SRMs of leaves and dried leaf extracts, and a pooled sample for each set (plant material and leaf extracts). The nontargeted approach for LC-HRMS was carried out using a full scan mode for both negative and positive ion modes creating four datasets: A negative, A positive, B negative, and B positive, with A and B describing the plant materials and leaf extracts, respectively [23]. These four final data matrices were analyzed as some compounds only appear in one mode or another due to their pH.
For the adulteration classification, the data analysis using PCA showed that there were no significant differences in the adulterated samples of groups 3% and 7%. The original PCA results of the entire dataset showed that the groups 3% and 7% were clustered together which may be assumed that the adulteration was significant enough to separate the samples. Thus, the adulteration levels were grouped as 0 % adulteration, 3 % to 7 % adulteration, and 15 % adulteration for both plant materials and dried leaf extract samples to give a more visual presentation of the adulteration screening in the PCA score plots.
PCA of Plant Material Samples
The final matrices for dataset A, plant material samples, are summarized in Table 4. Using the adulteration level classification, the PCA score plots of the plant material samples in Fig. 2 shows a separation trend among three levels of adulteration (0 % adulteration, 3 % to 7 % adulteration, and 15 % adulteration) with only mean-centering as the preprocessing method. Loadings have information about variables, in this case, m/z, peak area, and retention time values. Analyzing the results without using a strong preprocessing method can be useful to examine the raw loadings that will enable identification of the significant peaks responsible for the separation. The mean-centered results of the plant material (dataset A) for both positive and negative ion modes using the first four principal components had total variation explained of 98.56 % and 97.02 %, respectively. The best separation of samples by adulteration level was obtained using a combination of PCs 1 and 3 for the A positive ion mode and PCs 2 and 3 for the A negative ion mode as shown in Figure 2. The number of principal components for all the score plots created was selected based on the variance captured (%) plot with the principal component containing a percent variance greater than 1 %.
Table 4.
Data matrices for plant material samples
| Data matrix | samples × variables |
|---|---|
| A negative ion mode | 34 × 77 |
| A positive ion mode | 34 × 175 |
| Combined negative and positive ion modes of A (normalized results) | 34 × 252 |
| Combined negative and positive ion modes of A (principal component results) | 34 × 8 |
Fig. 2.

PCA scores and loadings of plant materials (dataset A) using mean centering and classification by adulteration level: (A) score plot of dataset A positive ion mode, encircled samples: adulterated and A6 samples (B) loadings plot of dataset A positive ion mode, (C) score plot of dataset A negative ion mode, encircled samples: adulterated and A6 samples, and (D) loadings plot of dataset A negative ion mode; the encircled scores in 2A and 2C plots pertain to the adulterated samples and the encircled loadings in 2B and 2D plots are the variables correlated with the adulterated samples
Using NIST-MS Search, a summary of the identified compounds present in the positive ion mode and the negative ion mode are presented in Table 5. For high-resolution mass spectrometry, any compound with a match factor (MF) higher than 500 is considered a “Good Match”, which is a tentative identification but not definitive. As mentioned on the Data Analysis for PCA under the Materials and Methods Section, the feature list (2), containing all the sample features, was scanned on the database instead of the 68 individual sample results. It was a more efficient way to identify the compounds for all the samples as the data tool used was PCA and the same feature list was used to build the PCA plots. The only disadvantage of scanning the feature list was that samples containing the identified compounds cannot be presented in this study. However, the score plots (samples) and the loading plots (variables) were found to have a strong correlation on the adulterated samples and their corresponding loadings which were the variables (m/z, peak area, and retention time values) as shown in Figure 2. This also shows how LC-HRMS plays a role in terms of its high sensitivity by detecting high mass accuracies, in this case, it detected up to 4 decimal places for the identified compounds especially for sophoricoside and genistein, which can be strong evidences of adulteration.
Table 5.
List of peaks identified for normalized unprocessed dataset A (plant materials)
| Positive ion mode | |||||
|---|---|---|---|---|---|
| Loadings ID from PCA | m/z | RT | Compound Name | MF | Prob. |
| 42 | 579.1694 | 6.8944 | Isorhoifolin | 718 | 97.35 |
| 47 | 433.1119 | 7.0029 | Sophoricoside | 658 | 96.98 |
| 24 | 611.1590 | 6.3266 | Rutin | 460 | 93.13 |
| 25 | 611.1589 | 6.3550 | Rutin | 460 | 93.13 |
| 150 | 282.2783 | 17.2641 | Oleamide | 872 | 92.49 |
| Negative ion mode | |||||
| 19 | 609.1468 | 6.3919 | Luteolin-7,3’-di-o-glucoside | 345 | 96.77 |
| 20 | 593.1521 | 6.7255 | Tiliroside | 801 | 94.80 |
| 29 | 269.0456 | 8.9310 | Genistein | 706 | 83.89 |
| 22 | 431.0987 | 7.0047 | Sophoricoside | 908 | 83.40 |
| 30 | 285.0406 | 9.0730 | Kaempferol | 742 | 74.47 |
| 17 | 755.2057 | 6.0245 | - | - | - |
Figures 2A and 2C show that the plant material samples were separated along PC 3 for both modes and the encircled loadings (Figs. 2B and 2D) on the loadings plot suggest a correlation on the adulterated samples. The separations were not distinct however, the score plots exhibited a clear trend with respect to the different adulteration levels that did not appear from other methods using the same samples in the master thesis study conducted by Cruz, M., including ICP-MS, NIR, and GC-MS [23]. For the positive ion mode, loadings ID 47 on the positive quadrant along PC3 (Fig. 2B), identified as sophoricoside, was one of the variables with the highest loadings with respect to differentiating the adulterated samples. For the negative ion mode, loading IDs 22 and 29 correspond to the sophoricoside and genistein, respectively, which contribute significantly to the discrimination of the adulterated samples along PC2 (Figs. 2C and 2D). For the positive and negative ion modes (Figs. 2A and 2C), the repeatability of the SRM 3246 was observed to be slightly different in the MS1 normalized dataset and PCA results. The differences of the position of two SRMs may be attributed to the sample preparation as it was done on different days. It was also observed that one of the SRM samples had a different behavior, SRM3246–1 was clustered with the other unadulterated leaves samples, while SRM3246–2 and sample A6 behaved in a similar manner. The differences between SRM 3246 leaves and the ginkgo plant material samples may be attributed to the provenance of the leaves, sample heterogeneity, and storage preparation.
Based on the literature, sophoricoside is not an innate compound in Ginkgo biloba L. and is specifically found in the dried fruits and flower buds of Styphnolobium japonicum (L.) Schott (syn. Sophora japonica L., Fabaceae) or the Japanese Pagoda tree. This tree is a known medicinal plant and one of the commonly alleged adulterants of ginkgo extracts used to boost the flavonol glycoside contents due to its lower cost compared to the authentic Ginkgo biloba extracts [8]. Glycitein, a common compound found from several plants from the family Fabaceae including Japanese sophora, was also detected, but was not reported in the study [8]. Upon closer examination of the loadings plot of the positive ion of dataset A, the loading ID 80, identified as glycitein, was not clearly separated and was clustered with the other loadings that were positioned just below the reported loadings (encircled loadings in Fig. 2B). This might be due to the differences on the concentration levels of sophoricoside and glycitein present in the adulterated samples for this study.
In the case of genistein, there was a question over whether genistein is a component of Ginkgo biloba L. since according to the review and studies of H. Wohlmuth et al. and S. Gafner [8, 11], few reports were published concerning authors claiming that genistein was a genuine constituent of G. biloba even though only low concentrations were detected. In one paper, genistein was purified from a commercial leaf extract however, the authenticity of the raw material used to manufacture the ginkgo extract in that study was not demonstrated [24]. In another publication, quantification of flavonoids using ginkgo plant parts such as leaf, stem, and fruit from three authentic ginkgo trees in India was detailed and the authors noted that genistein was absent in female ginkgo tree leaves but, was identified in the leaf and stem of male ginkgo trees [25]. The reported genistein by Yao et al. [26] had concentrations between 5–28 μg/g dry leaf using a validated HPLC-UV method with detection at 350 nm.
However, from the data in this study, it suggested that at the levels that genistein was detected, it was an indicator of the adulteration. This compound was also directly correlated to the adulterated samples based on the PCA results. If genistein was present in the unadulterated samples, then it was below the detection limit of the qualitative technique. In the study of López-Gutierrez [7], the isoflavone genistein was detected in low concentrations (between 0.02 and 2.41 mg/g) together with the remarkably high concentrations of rutin (27.2–38.2 mg/g) in three products. They also reported the presence of glycitein in two products which clearly an indicator of adulteration[7, 8]. Genistein has been reported to be native to the pericarp of fruits and flowers of Styphnolobium japonicum L., and consequently, researchers have proposed that genistein can be used as a marker to detect adulteration with extracts of Japanese sophora [8, 11, 24].
Figure 3 shows that autoscaling was the best fit for both positive and negative dataset A. Autoscaling compares the variables based on correlations and these variables become equally important. A disadvantage of this approach is that measurement errors will increase as noise and interferences are also adjusted at the same level as those of relatively large variables [27]. Figs. 3A and 3B show the score plots of both ion modes and showed consistent separations between PC2 and PC3. Using the first three principal components, a total of 94.21 % cumulative variance for positive ion mode and 90.26 % for negative ion mode using adulteration level classifications were obtained for LC-HRMS method and these % cumulative variances were not observed from other analytical techniques [23]. Concatenated plant material (dataset A) of the normalized positive and negative ion modes data extracted from MZmine and the concatenated principal components of both ion modes were also observed and are shown in Figs. 3C and 3D with the separations between PC2 and PC3, and PC1 and PC3, respectively. LC-HRMS provided adequate results in comparison with other aforementioned methods based on the same plant part material samples especially for small adulterations [23]. However, a model based on the LC-HRMS results still needs additional resources, such as additional authentic samples, to improve the robustness of the PCA models obtained.
Fig. 3.
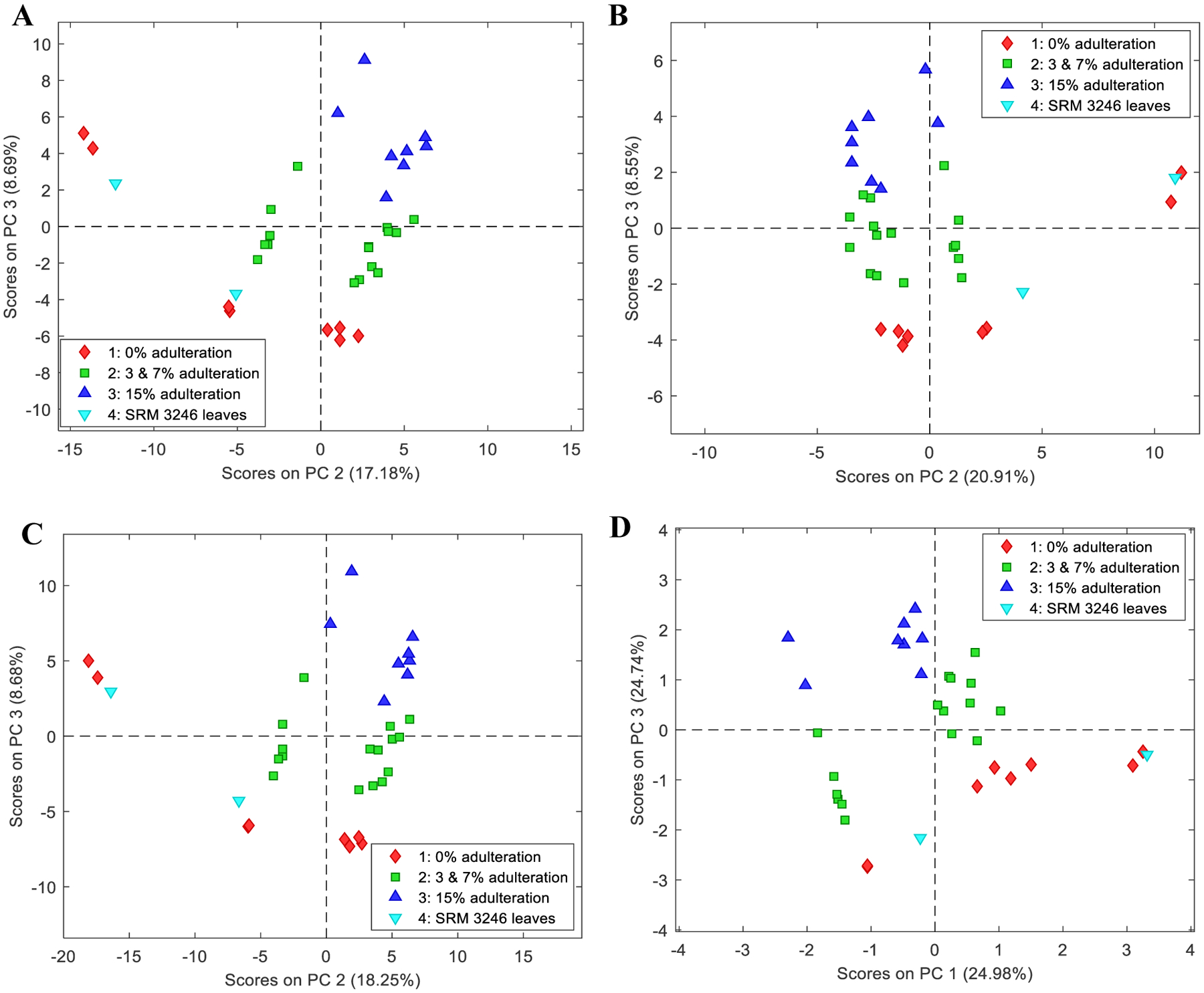
PCA scores of plant materials (dataset A) and the combined dataset A results using autoscaling and classification by adulteration level: (A) score plot of dataset A positive ion mode (B) score plot of dataset A negative ion mode (C) Score plot of total dataset A (positive and negative ion modes) using the normalized data extracted from MZmine; and (D) score plot of total dataset A using the concatenated principal components
The PCA score plots for the plant part materials by source classification were found to have a distinct separation among samples for both ion modes with and without preprocessing, as well as the combined results as shown in the Figure, Supplemental Information 2.
PCA of Dried Leaf Extract Samples
For the dried leaf extracts (dataset B), 34 samples by 58 variables yielded dataset B negative, and 34 samples by 163 variables yielded dataset B positive. Results for dataset B were also combined however, are not reported as trends or separation for adulteration level were not observed. All score plots of dataset B used class centroid centering and scaling, a class-aware type of autoscaling that is useful for samples in subsets identified by a row class set, as the preprocessing method. In this approach, the data are centered by class centroid method to avoid the mean being dominated by the most populous subset, and then scaled by the pooled standard deviation of the classes. Samples that belong to an unknown class are not used in the calculation of centroid or pooled variance [28].
The dried leaf extract samples (dataset B) were examined based on adulteration level per material source as the material source variation appeared to overshadow the adulteration level which may be attributed to the origin of leaves, solvent extracts and composition used during the manufacturing processes of the dried leaf. Table 6 shows the different extraction solvents and ratios based on the COA of dried leaf extracts. Similar to SRM 3246 leaves and plant material samples in dataset A, SRM 3247 also behaved differently with respect to the rest of the ginkgo leaf extract samples as observed in the PCA score plots. This might be attributed to the differences in the provenance of the leaves, dried leaf extract preparation and sample heterogeneity. Score plots in Fig. 4A and 5A represent the complete dataset identified by material source for both positive and negative ion modes which were separated between PC 1 and PC2 with a total cumulative variance of 87.84 %, and PC1 and PC3 with 89.24 %, respectively. The remaining score plots for dataset B show the adulteration level for each material source.
Table 6.
Solvent extraction ratios and preparations of dried leaf samples based on COA
| Ginkgo dried leaf extract samples | Process | Solvent Ratio | Native Extract Ratio | Excipients |
|---|---|---|---|---|
| Ginkgo extract aqueous | Water extraction; spray dry | Not specified | Not specified | Not specified |
| Ginkgo extract ethanol:water | Not specified | Ethanol (60–80%)/Water (20–40%) | Not specified | Not specified |
| Ginkgo extract acetone:water | Not specified | 35–67:1 | Syrup, Corn, Dehydrated | |
| Ginkgo extract acetone:water with lecithin | Not specified | 35–67:1 | Lecithin (origin: Soy) |
Fig. 4.

PCA scores for dried leaf extracts (dataset B) using class centroid scaling and centering, and classification by adulteration level for each material source in positive ion mode: (A) score plot of whole dataset based on the material source, encircled groups represent the leaf extract types analyzed individually (B) score plot of the source D (extract aqueous) samples, (C) score plot of the source E (extract ethanol:water) samples and (D) score plot of the source F (extract acetone:water) samples
Fig. 5.

PCA scores for dried leaf extracts (dataset B) using class centroid scaling and centering, and classification by adulteration level for each material source in negative ion mode: (A) score plot of whole dataset based on the material source, encircled groups represent the leaf extract types analyzed individually (B) score plot of the source D (extract aqueous) samples, (C) score plot of the source E (extract ethanol:water) samples, and (D) score plot of the source F (extract acetone:water) samples
Conclusions
The use of LC-HRMS and PCA to determine the authenticity of Ginkgo biloba L. samples enabled transformation of the results into a new set of data containing principal components and projection into PCA models aided the visualization and evaluation of the data. Determination of adulterated samples among the plant materials (dataset A) and the dried leaf extracts (dataset B) was possible using the score plots obtained in PCA. The ginkgo results of LC-HRMS were transformed into datasets which were divided in two separate ionization modes, negative and positive, creating a total of four datasets: A positive, A negative, B positive, and B negative. The obtained score plots and loadings plots for dried leaf materials on both negative and positive ion modes showed promising results using the adulteration level classification as separation of adulterated samples from unadulterated ones were visible in the score plots even only using mean centering as the preprocessing method. Furthermore, a clear correlation between the adulterated samples and the variables that influences the sample behavior was also observed using the loadings. Consequently, these loadings were inspected showing significant variables that were selected and identified using NIST-MS Search which include the presence of sophoricoside, for both negative and positive ion modes, and genistein for positive ion mode. Based on the phytochemical investigations and literature searches, these compounds are not known to be native in Ginkgo biloba L. and in the case of genistein, if it is a genuine component, it will be only be detected in very low concentrations. This could be an indication that these ginkgo plant samples were indeed contaminated or adulterated, possibly with the extracts from Styphnolobium japonicum L. or Sophora japonica L. plant which is known to have the said compounds.
The LC-HRMS results for the ginkgo extract samples did not obtain initial separations based on adulteration level but by observing the trends for each individual source using the adulteration level classification, it showed a possibility of using the method for investigation purposes as the unadulterated samples were separated from the adulterated extracts. With the LC-HRMS results, a different tool such as NMR or NIR may be used to further explore these extract samples and it can be an easier and more efficient method especially for the manufacturers. Compared to the leaf samples, extract samples are initially processed and thus, the origins and manufacturing processes may have contributed to its complexity. The extract samples may have become too similar with each other that the nontargeted approach using LC-HRMS and the type of sample preparation used may not be appropriate and enough to fully discriminate adulterated from unadulterated samples. It must also be considered that the composition of Ginkgo biloba L. leaves and dried leaf extracts may vary due to provenance, heterogeneity, and manufacturing processes. For the SRMs, the results are not very useful in this study since their chemical compositions were either too similar or too different with the samples. However, if SRMs are used repeatedly, as in QC purposes for example, consistent results from assay performance can be monitored and consequently the method can be evaluated to be performing as expected.
Overall, LC-HRMS method was capable of detecting small adulterations for the plant part material (dataset A). However, from the quality control point of view, LC-HRMS may be a difficult instrument to maintain and to handle for routine purposes. Further improvement of the study can be done such as development of other instrumental techniques as screening methods and the addition of more authentic samples to evaluate and validate the robustness of the PCA models obtained using other chemometric techniques.
Supplementary Material
Acknowledgements
This project was part of a research master thesis supported by the Education, Audiovisual, and Culture Executive Agency (EACEA) under the program Erasmus Mundus Masters in Quality in Analytical Laboratories (EMQAL 10th edition), Gdansk University of Technology (GUT), and its collaboration with the National Institute of Standards and Technology (NIST Gaithersburg, USA) and the National Institute of Metrology, Quality and Technology (INMETRO Brazil). This project would not be possible without the help and overwhelming support of NIST and GUT supervisors, colleagues and program coordinators.
Footnotes
Conflict of Interest
The authors declare that they have no conflict of interest.
Certain commercial equipment, instruments or materials may be identified in this report to adequately specify the experimental procedure. Such identification does not imply recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the materials or equipment identified are necessarily the best available for the purpose.
To obtain up-to-date official values for NIST reference materials, consult the NIST Standard Reference Material web site at https://www.nist.gov/srm.”
References
- [1].van Beek TA. Volume 12: GINKGO BILOBA. In: Hardman R, editor. Medicinal and Aromatic Plants - Industrial Profiles. Amsterdam, Netherlands: Harwood Academic Publishers; 2006. pp. 1–523. [Google Scholar]
- [2].van Beek TA, Montoro P. Chemical analysis and quality control of Ginkgo biloba leaves, extracts, and phytopharmaceuticals. J Chromatogr A. 2009;1216:2002–2032. [DOI] [PubMed] [Google Scholar]
- [3].Demirezer LÖ, Büyükkaya A, Uçaktürk E, Kuruüzüm-Uz A, Güvenalp Z, Palaska E. Adulteration determining of pharmaceutical forms of Ginkgo biloba extracts from different international manufacturers. Rec Nat Prod. 2014;8(4):394–400. [Google Scholar]
- [4].Liu XG, Wu SQ, Li P, Yang H. Advancement in the chemical analysis and quality control of flavonoid in Ginkgo biloba. J Pharm Biomed. Anal 2015;113:212–225. [DOI] [PubMed] [Google Scholar]
- [5].Rimmer CA, et al. Characterization of a suite of ginkgo-containing standard reference materials. Anal Bioanal Chem. 2007;389(1):179–196. [DOI] [PubMed] [Google Scholar]
- [6].Ding XP, Qi J, Chang YX, Mu LL, Zhu DN, Yu BY. Quality control of flavonoids in Ginkgo biloba leaves by high-performance liquid chromatography with diode array detection and on-line radical scavenging activity detection. J Chromatogr A. 2009;1216(11)2204–2210). [DOI] [PubMed] [Google Scholar]
- [7].López-Gutiérrez N, Romero-González R, Vidal JLM, Frenich AG. Quality control evaluation of nutraceutical products from Ginkgo biloba using liquid chromatography coupled to high resolution mass spectrometry. J Pharm Biomed. Anal 2016;121;151–160. [DOI] [PubMed] [Google Scholar]
- [8].Gafner S. Adulteration of Ginkgo biloba Leaf Extract. Bot Adulterants Bull. 2018;January:1–8. [Google Scholar]
- [9].Chandra A, et al. Qualitative categorization of supplement grade Ginkgo biloba leaf extracts for authenticity. J Funct Foods. 2011;3(2):107–114. [Google Scholar]
- [10].Ma YC, et al. An effective identification and quantification method for Ginkgo biloba flavonol glycosides with targeted evaluation of adulterated products. Phytomedicine. 2016;23(4):377–387. [DOI] [PubMed] [Google Scholar]
- [11].Wohlmuth H, Savage K, Dowell A, Mouatt P. Adulteration of Ginkgo biloba products and a simple method to improve its detection. Phytomedicine. 2014;21(6):912–918. [DOI] [PubMed] [Google Scholar]
- [12].Tokalıoglu S. Determination of trace elements in commonly consumed medicinal herbs by ICP-MS and multivariate analysis. Food Chem. 2012;134:2504–2508. [DOI] [PubMed] [Google Scholar]
- [13].Zhao L, et al. Determination of Total Flavonoids Contents and Antioxidant Activity of Ginkgo biloba Leaf by Near-Infrared Reflectance Method. Int J Anal Chem. 2018;2018:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Harnly JM, Luthria D, Chen P. Detection of adulterated ginkgo biloba supplements using chromatographic and spectral fingerprints. J AOAC Int. 2012;95(6):1579–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Li C-Y, Lin C-H, Wu C-C, Lee K-H, Wu T-S. Efficient 1 H Nuclear Magnetic Resonance Method for Improved Quality Control Analyses of Ginkgo Constituents. J Agric Food Chem. 2004;52:3721–3725. [DOI] [PubMed] [Google Scholar]
- [16].Agnolet S, Jaroszewski JW, Verpoorte R. et al. 1H NMR-based metabolomics combined with HPLC-PDA-MS-SPE-NMR for investigation of standardized Ginkgo biloba preparations. Metabolomics. 2010;6:292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Commiso M, Strazzer P, Toffali K, Stocchero M, Guzzo F. Untargeted metabolomics: an emerging approach to determine the composition of herbal products. Comput Struct Biotechnol. J 2013;4(5):1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Katajamaa M, Miettinen J, Orešič M. Processing methods for differential analysis of LC/MS profile data. BMC Bioinformatics. 2006;22(5):634–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Pluskal T, Castillo S, Villar-Briones A, Orešič M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinformatics. 2010;11:395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Myers OD, Sumner SJ, Li S, Barnes S, and Du X. One Step Forward for Reducing False Positive and False Negative Compound Identifications from Mass Spectrometry Metabolomics Data: New Algorithms for Constructing Extracted Ion Chromatograms and Detecting Chromatographic Peaks. Anal Chem. 2017;89:2. [DOI] [PubMed] [Google Scholar]
- [21].MZmine Development Team. MZmine 2.3 Manual. 2005–2011. http://mzmine.sourceforge.net/manual.pdf. Accessed 28 Nov 2019.
- [22].R Core Development Team. R: A language and environment for statistical computing. In: R Foundation for Statistical Computing, Vienna, Austria. 2013. http://www.R-project.org/. Accessed 30 July 2019. [Google Scholar]
- [23].Cruz MB. Determination of the authenticity of Ginkgo biloba L. plant part materials and dry leaf extracts using different analytical methods and chemometric techniques [master’s thesis] Gdansk, Poland: Gdansk University of Technology; 2019. [Google Scholar]
- [24].Wang F, Jiang K, Li Z, Purification and Identification of Genistein in Ginkgo biloba Leaf Extract. Chinese J Chromatogr. 2007;25(4):509–513. [DOI] [PubMed] [Google Scholar]
- [25].Pandey R, Chandra P, Arya KR, Kumar B. Development and validation of an ultra high performance liquid chromatography electrospray ionization tandem mass spectrometry method for the simultaneous determination of selected flavonoids in Ginkgo biloba. J Sep Sci. 2014;37(24):3610–3618. [DOI] [PubMed] [Google Scholar]
- [26].Yao JB et al. Seasonal variability of genistein and 6-hydroxykynurenic acid contents in Ginkgo biloba leaves from different areas of China. Nat Prod Commun. 2017;12(8):1241–1244. [Google Scholar]
- [27].van den Berg RA, Hoefsloot H-CJ, Westerhuis JA, Smilde AK, van der Werf MJ. Centering, scaling, and transformations: improving the biological information content of metabolomics data. BMC Genomics. 2006;7:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Eigenvector Research Documentation. Advanced Preprocessing: Variable Centering - Eigenvector Documentation Wiki. http://wiki.eigenvector.com/index.php?title=Advanced_Preprocessing:_Variable_Centering. Accessed 18 Apr 2019.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.