

Abstract
The nutraceutical Nicotinamide Riboside (NR), an efficacious biosynthetic precursor to NAD, is readily metabolized by the purine nucleoside phosphorylase (PNP). Access to the PNP-stable versions of NR is difficult because the glycosidic bond of NR is easily cleaved. Unlike NR, NRH, the reduced form of NR, offers sufficient chemical stability to allow the successful functionalisation of the ribosyl-moiety. Here, we report on a series of NRH and NR derived amino acid conjugates, generated in good to excellent yields and show that O5’-esterification prevents the PNP-catalyzed phosphorolysis of these NR prodrugs.
Introduction
Nicotinamide Riboside (NR) is a pyridine-containing nucleoside derived from Nicotinamide (Nam), and a component of vitamin B3. Like Nam, NR, and its phosphorylated form, Nicotinamide MonoNucleotide (NMN) are naturally occurring and biosynthetic precursors to the redox cofactor Nicotinamide Adenine Dinucleotide (NAD) (Fig. 1).1–4 Vitamin B3-derived cofactors also include NAD’s reduced form NADH, its phosphorylated form, NADP, and the reduced form of NADP, NADPH.5 Through the interplay of these cofactors, vitamin B3-derived biology is central to cellular homeostasis, metabolism, growth, and proliferation.6–9 Critically, NAD levels change in response to dietary and environmental cues and external stressors.10 In turn, depletion of NAD levels impacts health and associates with the emergence and progression of metabolic diseases and aging.11–15
Fig. 1.
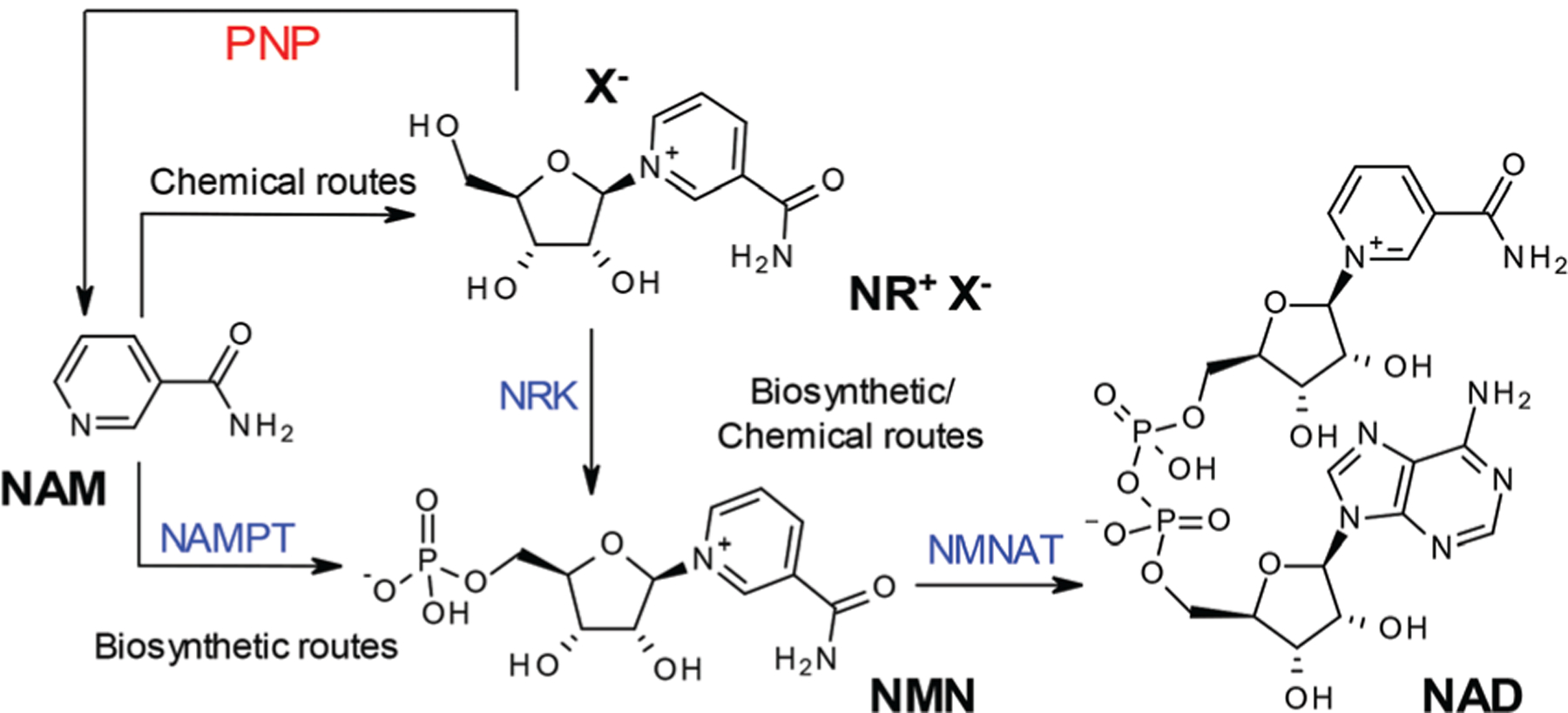
Chemical and biosynthetic routes to nicotinamide adenine dinucleotide (NAD) from nicotinamide (NAM) via nicotinamide riboside (NR) and nicotinamide mononucleotide (NMN). Biosynthetic enzymes: NAMPT: nicotinamide phosphoribosyl transferase; NRK: nicotinamide riboside kinase; NMNAT: Nicotinamide mononucleotide adenylyl transferase. Hydrolytic enzyme: PNP: purine nucleoside phosphorylase.
Since NR is capable of raising the levels of NAD,16 it has come to the forefront of NAD biology. In cell-based work, it has become a versatile biochemical tool to manipulate NAD levels with the view of investigating NAD function.18–20 As NR becomes more widely available, more biological and clinical studies in mammals have emerged.21–24 These employ NR either orally or through parenteral injections. The beneficial outcomes that these studies have evidenced have propelled NR as a functional nutraceutical supplement.
However, evidence supporting bolus NR’s bioavailability and systemic bio-distribution upon oral or parenteral administration is becoming tenuous.25 There is growing evidence that the gut microbiome metabolizes a large proportion of oral NR to nicotinic acid.26 NR is also a substrate for plasma hydrolases, such as purine nucleoside phosphorylase (PNP), which converts NR to nicotinamide and 1-phospho-riboside.27,28 These developments could challenge the premise that oral NR impacts directly and systemically NAD, as it becomes difficult for biologists to associate beneficial effects of NR supplementation, solely to NR over that of nicotinic acid and Nam.19,29,30 Yet, NR’s ability to restoring intracellular NAD levels through the nicotinamide riboside kinase (NRK) pathway has been supported by numerous studies (Fig. 1).17–24 Therefore, tools that provide a low and yet sustained delivery of NR, which better reflects the abundance of nucleosides’ in circulation, might shed new light on the importance of NR in cell biology.
Ultimately, NR-derivatives, which release NR upon specific conditions, would enable the targeted regulation of NAD levels via the NRK pathway, in a given tissue or cell-type. With the view of developing entities that would be resistant to phosphorolysis, we have sought to mask the C5′-OH position of NR temporarily. Ester functionalities at this position would decrease NR’s affinity for PNP and thus temporarily reduce NR’s availability for a PNP-catalysed degradation to Nam when used in cell-based work19 or delivered parenterally.25 Amino ester conjugates were selected as the ubiquitous distribution of amino acids, and dedicated transporters have been shown to play a significant role in the improvement of nucleosidederived drugs’ efficacy.31,32 Such reversible modification of the C5′-hydroxyl moiety offers means to generate PNP-stable versions of NR with an increased half-life that could be used in cell-based assays in the first instance.30
However, unlike other nucleosides, NR is difficult to functionalize. This difficulty arises from the lability of the glycosidic bond of NR, which is also readily cleaved when NR is exposed to nucleophiles.33 Most conditions required for the introduction of an amino acid on a nucleosidic scaffold include the transient protection of hydroxyl moieties and strong nucleophilic or basic reagents.34 Thus, these conditions are unsuitable in the manipulation of the NR-scaffold. Furthermore, the C5′-OH of NR is significantly less reactive towards electrophiles than the C5′-OH of purine and pyrimidine nucleosides and requires longer reaction time and harsher reaction conditions for its conversion to derivatives such as NMN,35 with conversions often achieved in low yields. Factors that influence such outcomes include the solubility of the ribosylated species, the chemical instability of key synthetic intermediates, and the reaction times.5 Unlike NR, the reduced form of NR, NRH,36 is stable to nucleophiles and amenable to the conjugation conditions that are usually applied to canonical nucleosides, and therefore a suitable synthetic intermediate.
Liquid-assisted mechano-chemical reaction conditions allow for the efficient production of NR and NRH.37 Therefore, we sought first to develop solution-phase chemistry to generate C5′-O-amino acid conjugates of NR via a synthetic sequence that bypasses the reduced reactivity of the C5′-OH of NR and then employ liquid-assisted mechanochemistry with the view of enhancing overall yields. In a buffered solution, even the simplest amino acid conjugate was not substrate for PNP and slowly released NR upon the chemical hydrolysis of the ester linkage.
Results and discussion
In the first instance, we sought to synthesize the O-5′-CBz protected glycine-NR conjugate 5 via glycosylation of a pre-functionalized riboside (Scheme 1). The 2,3-O-acetonide ribose 2, generated from d-ribose 1, was doubly esterified with the CBz-protected amino acid in a reaction catalysed by CDI to give the diester 4 in 80% isolated yield. Vorbrüggen-type glycosylation conditions were then explored to generate 5. Regardless of the reaction conditions (e.g., solution phase, milling, Lewis acid equivalency), only unreacted nicotinamide and decomposition materials were detected by NMR in the crude mixture (Scheme 1).
Scheme 1.
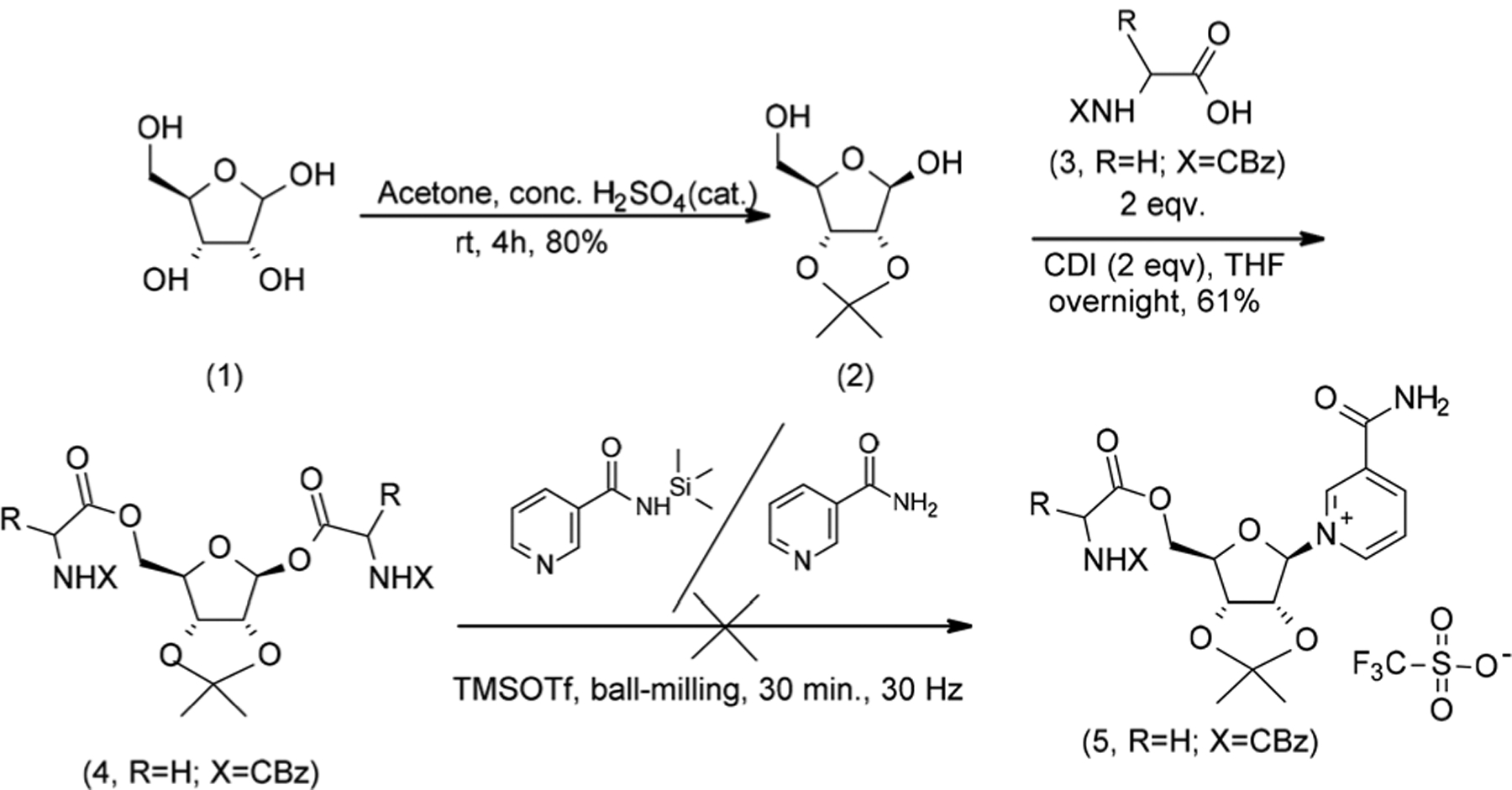
Glycosylation of CBz-protected glycine 2,3-acetonide riboside diester to generate the 5’-O-glycine NR ester conjugate.
Therefore, to access 5, we sought to perform the glycosylation step prior to the conjugation step and use NR chloride as a synthetic precursor. NR chloride 6 was converted to its acetonide 7 under acidic conditions, which required careful neutralization with powdered Na2CO3 before solvent removal. While basic conditions promoted the rapid C–N bond breakage, residual acid promoted product decomposition and compromised subsequent chemistry.
Coupling of a series of Boc protected amino acids 9a–9g with the C5′ hydroxyl group of 7 was then explored. Coupling conditions included CDI, DCC, EDCI, HATU, and HOBT both in solution (THF, CH3CN, etc.)38 and under liquid-assisted milling.39 Regardless of the coupling reagents and conditions applied, all attempts of direct esterification of 7 failed and yielded the release of Nam. In hindsight, it is likely that the 2,3-acetonide introduces conformational changes in the ribosyl scaffold of 7,40 which increases the reactivity of the C1′ position towards nucleophiles, and favours the release of Nam. Furthermore, the reduced nucleophilicity of the C5′-OH in 7 is likely to stem from increased interactions between the O5′ and the positively charged pyridinium moiety.
In light of these challenges, we sought to stabilize the glycosidic linkage and increase the nucleophilicity of the C5′ OH by generating the 2,3-diol protected NRH 8 (Scheme 2) as the key synthetic intermediate for the generation of the NR-conjugates 12a–12g. To achieve this, 7 was converted to its reduced form 8 in the presence of NaHCO3 and Na2S2O4 using a biphasic system.37,41 The reaction crude was sufficiently pure for use in coupling reactions with the partially protected amino acids 9a–9g.
Scheme 2.
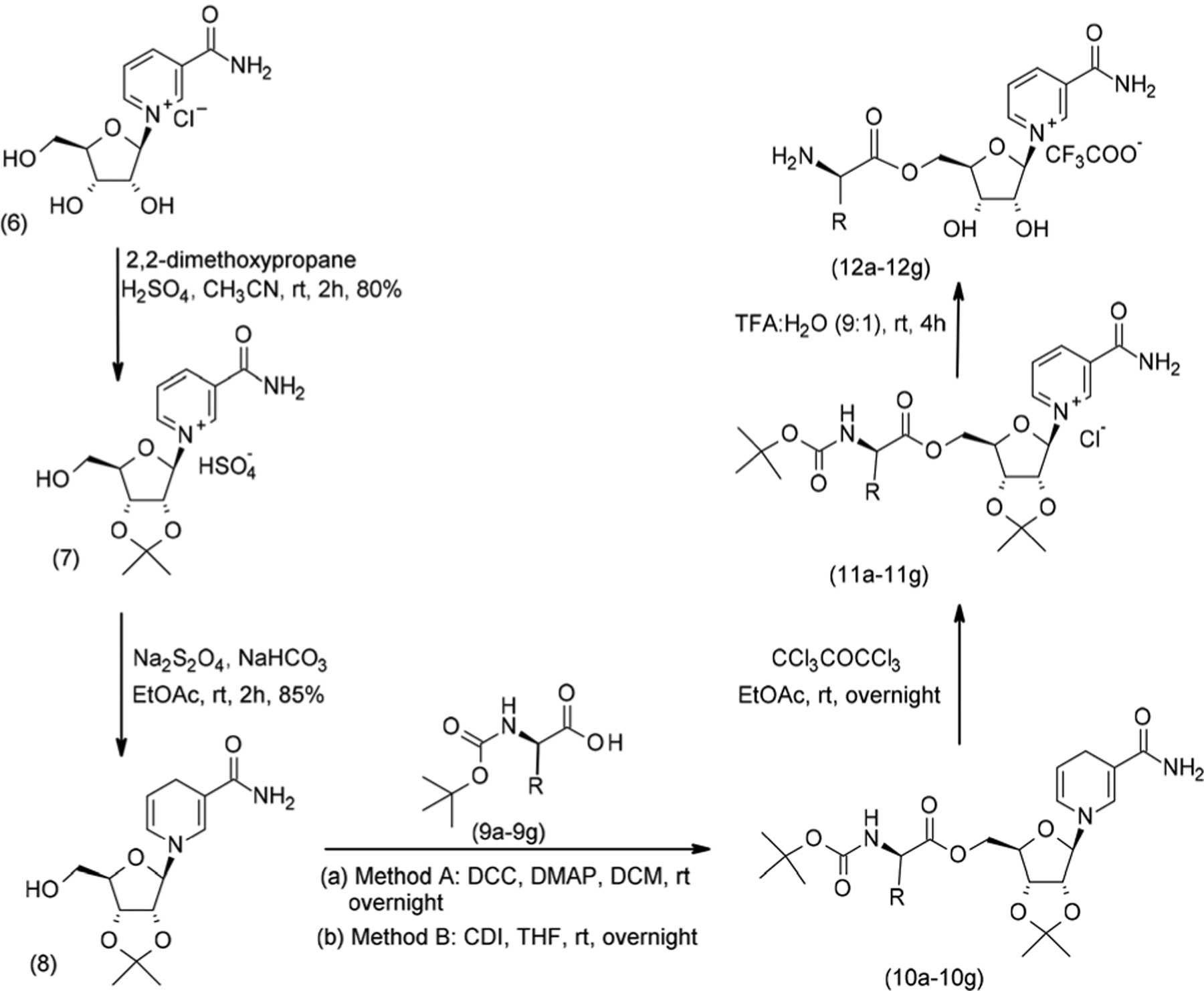
Solvent-based conjugation of 2’,3’-O-acetonide NR to Boc-protected amino acids.
Esters 10a–10g (Table 1 entries A1–G1) were synthesized using seven different Boc-protected amino acids (N-Boc-glycine, N-Boc-l-leucine hydrate, N-Boc-l-valine, N-Boc, tryptophan, N-Boc-l-phenylalanine, N-Boc-l-isoleucine, and N-Boc-l-methionine) in the presence of five different coupling agents, that included CDI, DCC, EDCI, HATU, and HOBT. Among them, DCC and CDI proved to be the best reagents to generate 10a–g. Compounds 10b, 10c, 10e, and 10f were best synthesized by DCC coupling in medium to good yields (43%–71%, Table 1; entries B1, C1, E1, and F1), while compounds 10a, 10d, and 10g were best obtained (30%–66%) by the CDI coupling method (Table 1; entries A1, D1 and G1). Critically, the NRH-scaffold is particularly sensitive to oxidation. As the reaction conditions employed an excess of coupling reagents, extensive workup and purification processes were required. This greatly affected isolated yields.
Table 1.
Multistep conversion of NR+Cl− to 5’-O-conjugated NR+ CF3COO− (12a–g). Method A: DCC coupling; method B: CDI coupling
| Entry | Boc protected amino acids | R | Conjugation product | Conjugation (yield = %) | Entry | Oxidation/deprotection product | Oxidation/deprotection (yield = %) |
|---|---|---|---|---|---|---|---|
| A1 | 9a | H | 10a | Method: B (66%) | A2 | 12a | 84% |
| B1 | 9b | 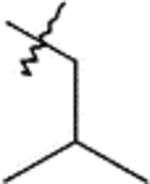 |
10b | Method: A (43%) | B2 | 12b | 84% |
| C1 | 9c | 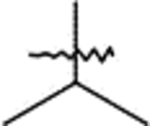 |
10c | Method: A (54%) | C2 | 12c | 61% |
| D1 | 9d | 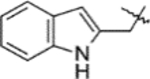 |
10d | Method: B (52%) | D2 | 12d | 60% |
| E1 | 9e | 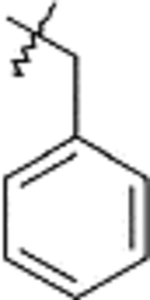 |
10e | Method: A (71%) | E2 | 12e | 73% |
| F1 | 9f | 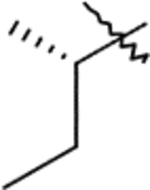 |
10f | Method: A (46%) | F2 | 12f | 60% |
| G1 | 9g | 10g | Method: B (30%) | G2 | 12g | 88% |
Seeking further yield optimisation for the generation of 10a–10g from 9a–9g, we investigated mechanochemical approaches and examined a greater number of solvent and reagents’ combinations (Scheme 3). The results are summarized in Table 2. Having explored five coupling reagents under different coupling conditions (Table 2; examples of attempted conditions: A–H), we observed that EDCI in the presence of DMAP and DMF used in catalytic amounts showed the most significant improvements across all the combinations which were attempted. Significantly, the yields improved when the equivalencies of coupling reagents and reaction time were increased, offering conjugates 10a–10g in 79 to 95% yields (Table 2; entries: L–R; and Scheme 3). Compared to solution-phase conditions, the milling protocol allowed for a reduction in the overall reaction time and the reagent equivalency. The later enabled a more efficient purification protocol and thus minimized the uncontrolled oxidation of the NRH scaffold in air.
Scheme 3.
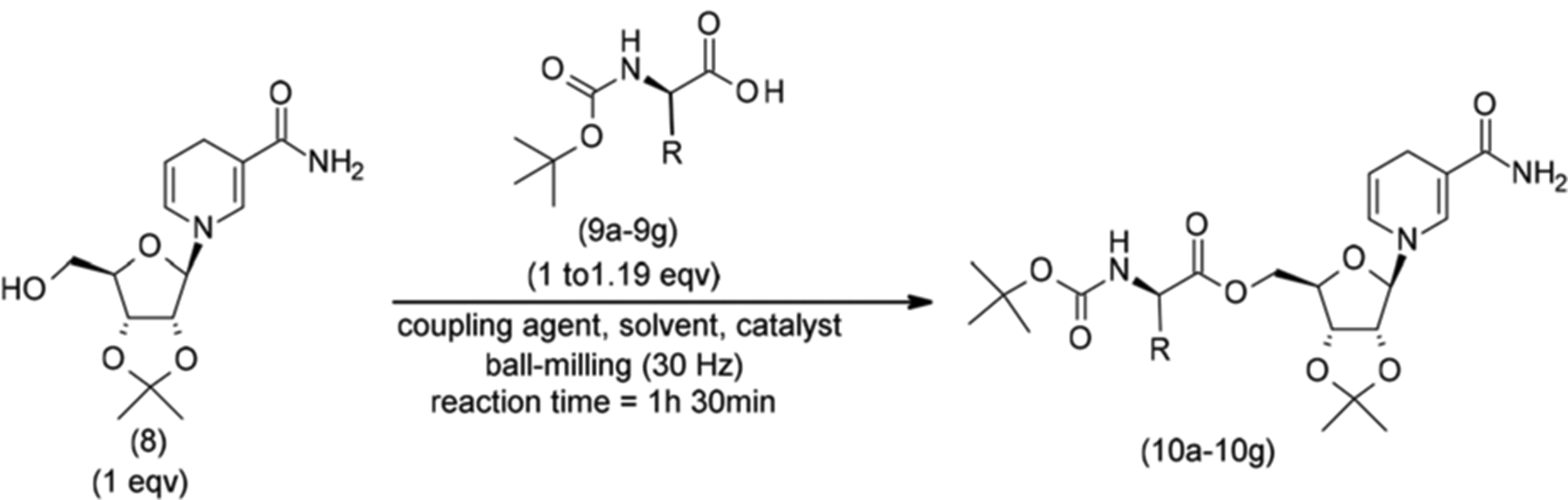
Synthesis of 2,3-diol protected NRH amino acid conjugates 10a–10g via mechanochemistry.
Table 2.
Conditions explored for the coupling of 8 with 9a–9g
| Entry | Amino acid | Equiv. | Coupling agent | Solvent (cat.) | BM (30 Hz) | Yielda, % (pdt) |
|---|---|---|---|---|---|---|
| A | 9a | 1 | CDI (1 eq.) | No solvent | 30 min | NoR |
| B | 9a | 1 | HOBt (1 eq.) | No solvent | 30 min | NoR |
| C | 9a | 1 | EDCI (1eq.) + HOBt (1 eq.) | No solvent | 30 min | NoR |
| D | 9a | 1 | CDI (1 eq.) | DMF | 30 min | Trace |
| E | 9a | 1 | HOBt (1 eq.) | DMF | 30 min | NoR |
| F | 9a | 1 | EDCI (1eq.) + HOBt (1 eq.) | DMF | 30 min | Trace |
| G | 9a | 1 | HATU(1eq.) + DIEA (1 eq.) | DMF | 30 min | Trace |
| H | 9b | 1.19 | EDCI (1eq.) + DMAP (5 mol%) | No solvent | 30 min | NoR |
| I | 9b | 1.19 | EDCI (1eq.) + DMAP (10 mol%) | DMF | 45 min | 33 (10b) |
| J | 9b | 1.19 | EDCI (2eq.) + DMAP (1 eq.) | DMF | 45 min | 58 (10b) |
| K | 9b | 1.19 | EDCI (1eq.) + DMAP (1 eq.) | H2O | 90 min | 46 (10b) |
| L | 9b | 1.19 | EDCI (1eq.) + DMAP (1 eq.) | DMF | 90 min | 79 (10b) |
| M | 9a | 1.19 | EDCI (1eq.) + DMAP (1 eq.) | DMF | 90 min | 95 (10a) |
| N | 9c | 1.19 | EDCI (1eq.) + DMAP (1 eq.) | DMF | 90 min | 95 (10c) |
| O | 9d | 1.19 | EDCI (1eq.) + DMAP (1 eq.) | DMF | 90 min | 88 (10d) |
| P | 9e | 1.19 | EDCI (1eq.) + DMAP (1 eq.) | DMF | 90 min | 82 (10e) |
| Q | 9f | 1.19 | EDCI (1eq.) + DMAP (1 eq.) | DMF | 90 min | 85 (10f) |
| R | 9g | 1.19 | EDCI (1 eq.) + DMAP (1 eq.) | DMF | 90 min | 78 (10g) |
Yield determined by 1H-NMR; NoR: no reaction
The NRH based products 10a–10g were then oxidized to their respective NR parents 11a–11g in the presence of hexachloroacetone33,37 (Scheme 2). The reaction conditions were optimized with ethyl acetate, as it facilitated the partial removal of hexachloroacetone. Yet, purification by silica gel column chromatography was necessary to obtain the individual reaction intermediates sufficiently pure to be used in the final two-step one-pot deportation. The simultaneous removal of the Boc protecting group and the acetonide in derivatives 11a–11g (Table 1, entries A2–F2) was conducted without loss of the O5′-ester or that of nicotinamide and required the combination of TFA : H2O (9 : 1).42,43 This deprotection was achieved for all conjugates (60–88%) in good to very good yields (Table 1; entries A2–G2). Of note, compounds 12a–12g hydrolyse to NR under aqueous conditions and therefore had to be used promptly.
Having gained access to the conjugated forms of NR, we sought to establish whether the masking at the C5′ position reduced its affinity for the purine nucleoside phosphorylase, PNP, while at the same time, being sufficiently labile to release NR in solution (Scheme 4). In an NMR-based enzymatic assay, NR+ Cl− was incubated with PNP (ESI) in KH2PO4 containing HEPES buffer at room temperature, and the reaction monitored over time by 1H NMR spectroscopy. PNP was then incubated with the simplest amino acid conjugate of NR, 12a (ESI†) under the same conditions. The production of nicotinamide over time provided information on the chemical and enzymatic hydrolysis of NR, while the generation of d-riboside-1-phosphate informed on the rate of the PNP-catalysed release of nicotinamide. The glycine ester bond got hydrolysed over time, releasing NR, which was then converted to nicotinamide and d-riboside-1-phosphate by PNP (Chart 1). The presence of the ester at the C5-position of NR prevented the PNP-catalysed phosphorolysis.
Scheme 4.
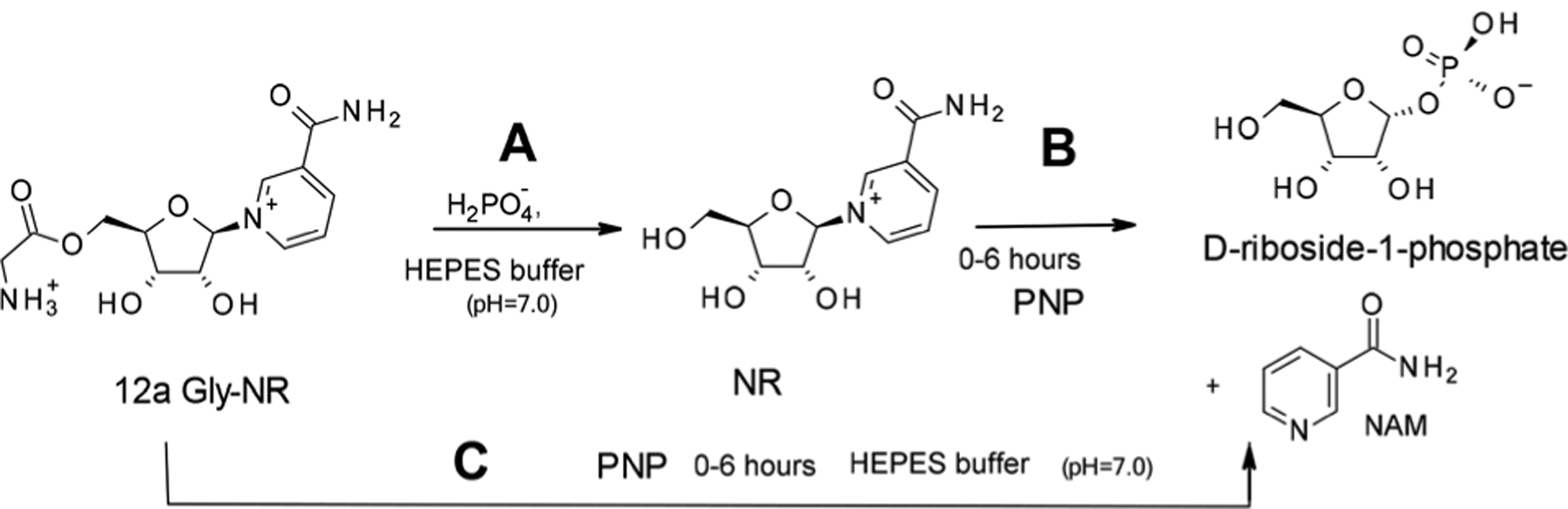
Effect of PNP on NR+ and its glycine conjugate, Gly-NR, 12a. A: Stability of Gly-NR in buffer (ESI†); B: phosphorolysis of NR by PNP in buffer; C: chemical hydrolysis of Gly-NR to NR followed by PNP-catalyzed released of NAM and formation of d-riboside 1-phosphate.
Chart 1.
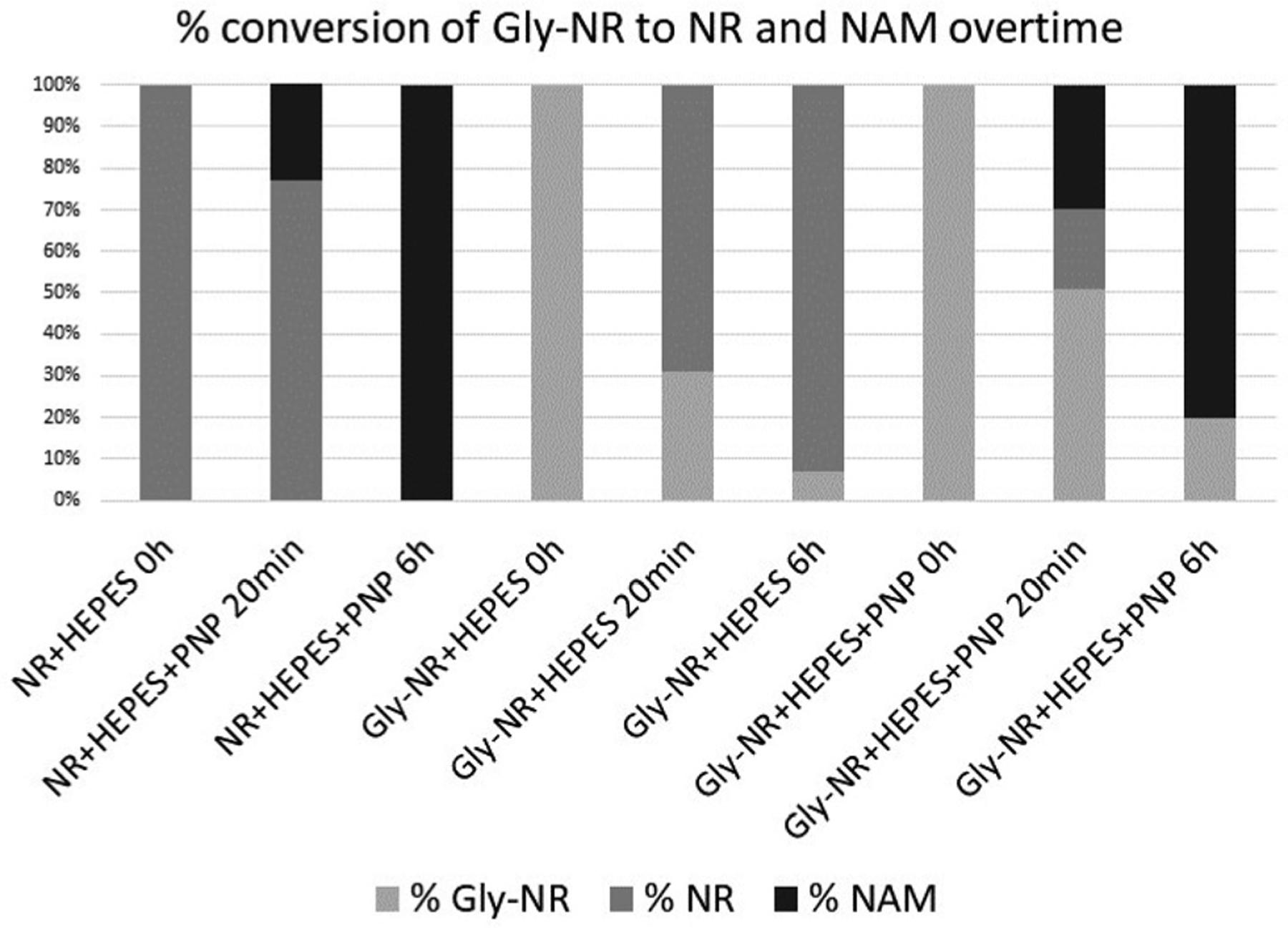
% conversion of NR and 12a to NAM when incubated with PNP in KH2PO4-containing HEPES buffer at room temperature.
Experimental
General information
All the chemicals and solvents were purchased from Alfa-Aesar, Sigma-Aldrich, Merck, and TCI (USA). All the solvents were dried following standard distillation procedures. Pre-coated silica gel coated glass plates (0.2 mm silica gel F254 Merck plates) were used for thin-layer chromatography (TLC), and the spots were visualized either in UV lamp or p-anisaldehyde stain. Melting points were determined on a capillary point apparatus equipped with a digital thermometer (digimilt) and are uncorrected. 1H NMR, 13C NMR, and 19F NMR experiments were recorded on Bruker 400 MHz spectrometer, and the chemical shifts were reported in δ (ppm), respectively. The coupling constants J were reported in Hz, and the following abbreviations were used to describe peak splitting patterns in 1H NMR spectra s = singlet, d = doublet, t = triplet, dd = doublet of doublet, m = multiplet, and brs = broad singlet. High-resolution mass spectrometry (HRMS) was performed with a Waters synapt G2 HDMS instrument using time-of-flight (TOF-MS) with ESI/APCI- hybrid quadrupole. Compounds 4–10 were purified using a Biotage medium pressure liquid chromatography (MPLC) system with a UV detection wavelength set at 254 nm. The purification was conducted on normal phase (SNAP KP-SIL 50 g) equilibrated for approximately 40 min before sample loading and elution. The elution gradient was performed by using hexane : ethyl acetate (100 : 0 to 0 : 100) at a flow rate of 30 mL min−1. The final products 12a–12g were purified by standard column chromatography by using methanol : water (3 : 7) as an eluent. FT-IR spectra were recorded on model FT/IR-4100 type A, using the KBr disk technique.
Experimental information
Chemistry
General procedure for the synthesis of diester of 2,3-O-isopropylidene-d-ribose (4).
2,3-O-Isopropylidene-d-ribose (2) was prepared according to reported procedure.44 To a stirred solution of 2,3-O-isopropylidene-d-ribose 2 (1.00 mmol) in anhydrous THF (10 mL), N-carbobenzyloxyglycine (2.00 mmol) and 1,1′-carbonyldiimidazole CDI (2.00 mmol) were added, and the reaction mixture was stirred at room temperature until complete disappearance of the starting material (TLC/NMR). Upon completion of the reaction, the reaction mixture was concentrated under reduced pressure, and the crude product was purified by flash column chromatography (n-hexane : ethyl acetate = 1 : 1) to give compound 4 as a colourless oil. Treatment of this oily product with diethyl ether and n-hexane gave a white solid.
Yield 61%, 1H NMR (400 MHz; acetone-d6) 7.35–7.33 (m, 10H, Ar–H), 6.75–6.70 (m, 2H, 2 × NH), 6.14 (s, 1H, H-1), 5.08 (s, 4H, 2 × Ar–CH2), 4.81–4.80 (m, 2H, H-2 & H-4), 4.43 (t, J = 6.54 Hz 1H, H-3), 4.28–4.23 (m, 1H, H5a), 4.15–4.11 (m, 1H, H5b), 3.98–3.83 (m, 4H, 2 × NHCH2), 1.42 (s, 3H, −CH3), 1.29 (s, 3H, −CH3); 13C NMR (100 MHz; acetone-d6) 169.80 (COOCH2), 168.74 (COOCH2), 156.70 (COONH), 156.62 (COONH), 137.16 (Ar–C), 128.30 (Ar–C), 127.85 (Ar–C), 127.78 (Ar–C), 112.46 (C-1), 102.64 (C-4), 85.14 (C-2), 81.28 (C-3), 66.06 (Ar–CH2), 64.41 (C-5), 42.54 (−CH2NH2), 42.30 (−CH2NH2) 25.73 (CH3), 24.06 (CH3); MS(ES): m/z 573.08 [M+ + H]; m/z 595.11 [M+ + Na].
General procedure for the synthesis of 2,3-diol protected NR (7).
In a flame dried flask under an argon atmosphere, oleum H2SO4 (0.43 mL, 8.00 mmol) was slowly added to dry acetonitrile (25.00 mL) at 0 °C. After 5 minutes, 2,2-dimethoxypropane (14.40 mL, 117.53 mmol) was added to the stirred acetonitrile solution at the same temperature. NR chloride (3.00 g, 11.75 mmol) was added to the reaction mixture at 0 °C, and the reaction was warmed up to room temperature. The progress of the reaction was monitored by thin-layer chromatography (TLC) and NMR analysis. Within 2 hours, complete conversation was observed. The reaction mixture was cooled again to 0 °C in an ice bath and was quenched by the addition of powdered solid Na2CO3 (0.64 g, 6.00 mmol) and stirred for 1 h. 0.3 mL of water was slowly added. Upon complete neutralization of acid (pH = 6–7), residual solids were filtered off, and the filtrate was concentrated under reduced pressure. The crude product was dissolved in a minimum volume of DCM and purified by silica gel chromatography (60 Å) using DCM/MeOH (9 : 1) as eluent to obtain (7) as a white solid.
Yield 80%, 1H NMR (400 MHz; D2O) 9.44 (s, 1H, Ar–H), 9.17 (d, J = 6.24 Hz, 1H, Ar–H), 8.92 (d, J = 7.96 Hz, 1H, Ar–H), 8.22 (t, J = 7.16 Hz, 1H, Ar–H), 6.41 (s, 1H, H-1), 5.22 (d, J = 6.53 Hz 1H, H-4), 5.00 (d, J = 5.76 Hz, 1H, H-2), 4.81 (s, 1H, H-3), 3.92 (d, J = 12.6 Hz, 1H, H5a), 3.77 (dd, J = 12.6 Hz & 3.24 Hz, 1H, H5b), 1.61 (s, 3H, −CH3), 1.38 (s, 3H, −CH3); 13C NMR (100 MHz; D2O) 165.69 (CONH2), 145.51 (Ar–C), 142.56 (Ar–C), 140.26 (Ar–C), 133.62 (Ar–C), 128.17 (Ar–C), 115.18 (CH3–C–CH3), 102.56 (C-1), 89.23 (C-4), 86.70 (C-2), 81.53 (C-3), 61.05 (C-5), 25.92 (CH3), 24.18 (CH3); HRMS calcd for C14H19N2O5 [M]+ 295.1294 found 295.1279.
General procedure for the synthesis of 2,3-diol protected NRH (8).
To an RBF containing NaHCO3 (2.77 g, 33.00 mmol) and Na2S2O4 (2.26 g, 13.00 mmol) was added 2 g of the protected diol (7) dissolved in a minimum amount of water. Following 1 min stirring, 20 mL of ethyl acetate were added. The resulting biphasic solution was stirred at rt for 2 h, and the reaction was monitored by TLC with the appearance of a fluorescent material. Upon completion of the reaction, the EtOAc layer was separated, and the aqueous layer was extracted with 15 mL EtOAc (2×). All the organic layers were combined, dried over Na2SO4, and concentrated under reduced pressure. The light yellow oily product was deemed of sufficient purity for subsequent chemistry.
Yield 85%, 1H NMR(400 MHz; MeOD) 7.01 (s, 1H,  ), 5.95 (d, J = 6.56 Hz, 1H,
), 5.95 (d, J = 6.56 Hz, 1H, 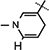 ), 4.97 (d, J = 4.08 Hz, 1H, H-1), 4.92–4.88 (m, 1H, H-4), 4.77–4.75 (m, 1H, H-2), 4.714.66 (m, 1H,
), 4.97 (d, J = 4.08 Hz, 1H, H-1), 4.92–4.88 (m, 1H, H-4), 4.77–4.75 (m, 1H, H-2), 4.714.66 (m, 1H, 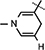 ), 4.01–3.98 (m, 1H, H-3), 3.70–3.61 (m, 2H, H5a & 5b), 2.96 (br s, 2H,
), 4.01–3.98 (m, 1H, H-3), 3.70–3.61 (m, 2H, H5a & 5b), 2.96 (br s, 2H, 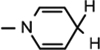 ), 1.49 (s, 3H, CH3), 1.30 (s, 3H, CH3); 13C NMR (100 MHz; MeOD) 172.68 (CONH2), 137.45 (N–CH=C-CONH2), 125.17 (N–CH=CH), 115.39 (N–CH=C–CONH2), 105.92 (N–CH=CH–CH2), 101.90 (C-1), 96.40 (C-4), 82.85 (C-2), 80.93 (O–C(CH3)3, 79.78 (C-3), 61.25 (C-5), 26.02 (CH3), 24.30 (CH3), 21.83 (N–CH=CH–CH2); HRMS calcd for C14H21N2O5[M + H]+ 297.1450 found 297.1436.
), 1.49 (s, 3H, CH3), 1.30 (s, 3H, CH3); 13C NMR (100 MHz; MeOD) 172.68 (CONH2), 137.45 (N–CH=C-CONH2), 125.17 (N–CH=CH), 115.39 (N–CH=C–CONH2), 105.92 (N–CH=CH–CH2), 101.90 (C-1), 96.40 (C-4), 82.85 (C-2), 80.93 (O–C(CH3)3, 79.78 (C-3), 61.25 (C-5), 26.02 (CH3), 24.30 (CH3), 21.83 (N–CH=CH–CH2); HRMS calcd for C14H21N2O5[M + H]+ 297.1450 found 297.1436.
General methods of coupling of 2,3-diol protected NRH with different Boc-protected amino acids (10a–10g).
Two different coupling methods were used for the synthesis of the protected NRH-amino acid adducts 10a–g.
Method A
DCC coupling.
Compounds 10b, 10c, 10e, and 10f were synthesized using DCC as coupling reagents and were obtained in good yields when compared to their conversion using CDI as a coupling reagent. With this method, a well-stirred solution of 1.00 mmol Boc-protected amino acids in 10 ml anhydrous CH2Cl2 placed under argon was added 10 mol% DMAP and 1.20 mmol 2,3-protected diol NRH (8), dissolved in a minimum amount of DCM. DCC was added once the solution had been cooled to 0 °C, stirred for 5 min at 0 °C before being warmed up to 25 °C and stirred for 24 h under argon. The precipitated urea was filtered off, and the filtrate was concentrated under reduced pressure. The crude was re-suspended in DCM, washed with saturated NaHCO3 and brine, and dried over Na2SO4. The solvent was removed by evaporation, and the ester products were purified by silica column chromatography (MPLC) by using 1 : 1 hexane : ethyl acetate as eluent.
Method B
CDI coupling.
Compounds 10a, 10d, and 10g were synthesized by this method in good quantity as compared to DCC coupling. According to this method, CDI (0.16 g, 1.00 mmol) was added to a well-stirred solution of Boc protected amino acids (1.00 mmol) in THF (10.0 mL) kept at 0 °C under argon. Once the solution had been warmed up to rt, it was stirred for 4 hours under argon. 2,3-Diol protected NRH (8) (0.30 g, 1.00 mmol) dissolved in THF (2 mL) was added to this solution, and stirring was continued for 24 hours. The reaction was monitored by TLC. Upon completion, the solvent was removed under vacuum, and the crude was purified by silicabased column chromatography (MPLC) using hexane and ethyl acetate as eluents.
Reduced nicotinamide N-tert-butyloxycarbonyl glycine riboside 10a.
Yield 66%, mp 90.1–90.7 °C. 1H NMR(400 MHz; MeOD) 7.13 (s, 1H, 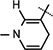 ), 5.99 (d, J = 7.04 Hz, 1H,
), 5.99 (d, J = 7.04 Hz, 1H, 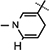 ), 4.89 (d, J = 3.6 Hz, 1H, H-1), 4.86–4.82 (m, 1H, H-4), 4.68–4.66 (m, 2H, H-5), 4.28 (d, J = 3.4 Hz, 2H, N–CH2), 4.19–4.17 (m, 1H, H-2), 3.85 (d, J = 4.68 Hz, 1H, H-3), 3.06 (br s, 2H,
), 4.89 (d, J = 3.6 Hz, 1H, H-1), 4.86–4.82 (m, 1H, H-4), 4.68–4.66 (m, 2H, H-5), 4.28 (d, J = 3.4 Hz, 2H, N–CH2), 4.19–4.17 (m, 1H, H-2), 3.85 (d, J = 4.68 Hz, 1H, H-3), 3.06 (br s, 2H, 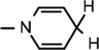 ), 1.52 (s, 3H, CH3), 1.43 (s, 9H, 3 × CH3), 1.32 (s, 3H, CH3); 13C NMR (100 MHz; MeOD) 171.61 (COO), 170.44 (CONH2), 157.08 (NHCOO), 135.96 (N–CH=C–CONH2), 126.36 (N–CH=CH), 114.35 (N–CH=C–CONH2), 103.37 (N–CH=CH–CH2), 101.90 (C-1), 97.22 (C-2), 81.81 (C-3), 80.4–80.2 (O–C(CH3)3), 64.31 (C-5), 41.73 (NH–CH2–COO), 27.3–23.8 (5 × CH3), 22.18 (N–CH=CH–CH2); IR (KBr) νmax: 1743.33 cm−1 (νcoo); HRMS calcd for C21H32N3O8 [M + H]+ 454.2189 found 454.2186.
), 1.52 (s, 3H, CH3), 1.43 (s, 9H, 3 × CH3), 1.32 (s, 3H, CH3); 13C NMR (100 MHz; MeOD) 171.61 (COO), 170.44 (CONH2), 157.08 (NHCOO), 135.96 (N–CH=C–CONH2), 126.36 (N–CH=CH), 114.35 (N–CH=C–CONH2), 103.37 (N–CH=CH–CH2), 101.90 (C-1), 97.22 (C-2), 81.81 (C-3), 80.4–80.2 (O–C(CH3)3), 64.31 (C-5), 41.73 (NH–CH2–COO), 27.3–23.8 (5 × CH3), 22.18 (N–CH=CH–CH2); IR (KBr) νmax: 1743.33 cm−1 (νcoo); HRMS calcd for C21H32N3O8 [M + H]+ 454.2189 found 454.2186.
Reduced nicotinamide N-tert-butyloxycarbonyl leucine riboside 10b.
Yield 43%, mp 90.2–90.8 °C. 1H NMR(400 MHz; MeOD) 7.15 (s, 1H, 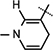 ), 5.98 (d, J = 6.8 Hz, 1H,
), 5.98 (d, J = 6.8 Hz, 1H, 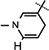 ), 4.90 (brs, 1H, H-1), 4.86–4.82 (m, 1H, H-4), 4.63 (brs, 2H, H-5), 4.34–4.30 (m, 1H, H-2), 4.22–4.16 (m, 4H, H-3, NH, NH–CH,
), 4.90 (brs, 1H, H-1), 4.86–4.82 (m, 1H, H-4), 4.63 (brs, 2H, H-5), 4.34–4.30 (m, 1H, H-2), 4.22–4.16 (m, 4H, H-3, NH, NH–CH, 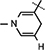 ), 3.07 (brs, 2H,
), 3.07 (brs, 2H, 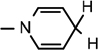 ), 1.76–1.65 (m, 1H, (CH3)2CHCH2), 1.61–1.57 (m, 2H, (CH3)2CHCH2), 1.52 (s, 3H, CH3), 1.43 (s, 9H, 3 × CH3), 1.31(s, 3H, CH3), 0.946 (d, J = 6.56 Hz, 3H, CH3), 0.914 (d, J = 6.5 Hz, 3H, CH3); 13C NMR (100 MHz; MeOD) 173.15 (COO), 171.43 (CONH2), 156.68 (NHCOO), 135.96 (N–CH=C–CONH2), 126.52 (N–CH=CH), 114.39 (N–CH=C–CONH2), 103.38 (N–CH=CH–CH2), 101.90 (C-1), 97.20 (C-2), 81.81 (C-3), 80.3–80.2 (O–C(CH3)3), 79.21 (C-4), 64.50 (C-5), 53.50 (NH–CH–COO), 39.97 [(CH3)2CH–CH2], 27.4–24.38 (5 × CH3), 22.03 (N–CH=CH–CH2), 20.50 (2 × CH3); IR (KBr) νmax: 1739.48 cm−1 (νcoo); HRMS calcd for C25H40N3O8 [M + H]+ 510.2815 found 510.2817.
), 1.76–1.65 (m, 1H, (CH3)2CHCH2), 1.61–1.57 (m, 2H, (CH3)2CHCH2), 1.52 (s, 3H, CH3), 1.43 (s, 9H, 3 × CH3), 1.31(s, 3H, CH3), 0.946 (d, J = 6.56 Hz, 3H, CH3), 0.914 (d, J = 6.5 Hz, 3H, CH3); 13C NMR (100 MHz; MeOD) 173.15 (COO), 171.43 (CONH2), 156.68 (NHCOO), 135.96 (N–CH=C–CONH2), 126.52 (N–CH=CH), 114.39 (N–CH=C–CONH2), 103.38 (N–CH=CH–CH2), 101.90 (C-1), 97.20 (C-2), 81.81 (C-3), 80.3–80.2 (O–C(CH3)3), 79.21 (C-4), 64.50 (C-5), 53.50 (NH–CH–COO), 39.97 [(CH3)2CH–CH2], 27.4–24.38 (5 × CH3), 22.03 (N–CH=CH–CH2), 20.50 (2 × CH3); IR (KBr) νmax: 1739.48 cm−1 (νcoo); HRMS calcd for C25H40N3O8 [M + H]+ 510.2815 found 510.2817.
Reduced nicotinamide N-tert-butyloxycarbonyl valine riboside 10c.
Yield 54%, mp 89.3–92.0 °C. 1H NMR (400 MHz; MeOD) 7.13 (s, 1H, 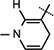 ), 5.99 (d, J = 6.4 Hz, 1H,
), 5.99 (d, J = 6.4 Hz, 1H, 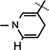 ), 4.90 (d, J = 3.2 Hz, 1H, H-1), 4.86–4.83 (m, 1H, H-4), 4.64 (brs, 2H, H-5), 4.35–4.24 (m, 1H, H-2), 4.23 4.17 (m, 2H, H-3,
), 4.90 (d, J = 3.2 Hz, 1H, H-1), 4.86–4.83 (m, 1H, H-4), 4.64 (brs, 2H, H-5), 4.35–4.24 (m, 1H, H-2), 4.23 4.17 (m, 2H, H-3, 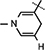 ), 4.04 (d, J = 5.84 Hz, 1H, NH–CH), 3.07 (brs, 2H,
), 4.04 (d, J = 5.84 Hz, 1H, NH–CH), 3.07 (brs, 2H, 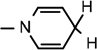 ), 2.17–2.05 (m, 1H, (CH3)2CH), 1.52 (s, 3H, CH3), 1.43 (s, 9H, 3 × CH3), 1.31(s, 3H, CH3), 0.947 (d, J = 5.86 Hz, 6H, 2 × CH3); 13C NMR (100 MHz; MeOD) 172.00 (COO), 171.41 (CONH2), 156.80 (NHCOO), 135.06 (N–CH=C–CONH2), 126.52 (N–CH=CH), 114.46 (N–CH=C–CONH2), 103.48 (N–CH=CH–CH2), 101.98 (C-1), 97.11 (C-2), 81.77 (C-3), 80.33 (O–C(CH3)3), 79.21 (C-4), 64.25 (C-5), 60.13 (NH–CH–COO), 59.52 (CH3–C–CH3), 33.37 (CH–NH), 30.23 (3 × CH3), 27.44 (CH3), 24.31 (CH3), 22.27 (N–CH=CH–CH2), 19.58 (CH3), 18.25 (CH3), 17.23 (CH3), 13.16 (CH3); IR (KBr) νmax: 1738.51 cm−1 (νcoo); HRMS calcd for C24H38N3O8 [M + H]+ 496.2659 found 496.2657.
), 2.17–2.05 (m, 1H, (CH3)2CH), 1.52 (s, 3H, CH3), 1.43 (s, 9H, 3 × CH3), 1.31(s, 3H, CH3), 0.947 (d, J = 5.86 Hz, 6H, 2 × CH3); 13C NMR (100 MHz; MeOD) 172.00 (COO), 171.41 (CONH2), 156.80 (NHCOO), 135.06 (N–CH=C–CONH2), 126.52 (N–CH=CH), 114.46 (N–CH=C–CONH2), 103.48 (N–CH=CH–CH2), 101.98 (C-1), 97.11 (C-2), 81.77 (C-3), 80.33 (O–C(CH3)3), 79.21 (C-4), 64.25 (C-5), 60.13 (NH–CH–COO), 59.52 (CH3–C–CH3), 33.37 (CH–NH), 30.23 (3 × CH3), 27.44 (CH3), 24.31 (CH3), 22.27 (N–CH=CH–CH2), 19.58 (CH3), 18.25 (CH3), 17.23 (CH3), 13.16 (CH3); IR (KBr) νmax: 1738.51 cm−1 (νcoo); HRMS calcd for C24H38N3O8 [M + H]+ 496.2659 found 496.2657.
Reduced nicotinamide N-tert-butyloxycarbonyl tryptophan riboside 10d.
Yield 52%, mp 92.5–94.3 °C. 1H NMR (400 MHz; MeOD) 8.08 (d, J = 7.92 Hz, 1H, Ar–H), 7.58 (d, J = 7.6 Hz, 1H, Ar–H), 7.50 (s, 1H, Ar–H), 7.28 (t, J = 7.42 Hz, 1H, Ar–H), 7.22 (t, J = 7.34 Hz, 1H, Ar–H), 7.12 (s, 1H, 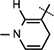 ), 5.89 (d, J = 7.92 Hz, 1H,
), 5.89 (d, J = 7.92 Hz, 1H, 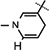 ), 4.84–4.82 (m, 2H, H-1 & H4), 4.51 (t, J = 7.2 Hz, 1H, NH–CH), 4.42 (t, J = 5.04 Hz, 1H, H-2), 4.32–4.30 (m, 1H, H-3), 4.20 (brs, 2H, H-5), 3.25–3.05 (m, 4H, NH–CH–CH2 &
), 4.84–4.82 (m, 2H, H-1 & H4), 4.51 (t, J = 7.2 Hz, 1H, NH–CH), 4.42 (t, J = 5.04 Hz, 1H, H-2), 4.32–4.30 (m, 1H, H-3), 4.20 (brs, 2H, H-5), 3.25–3.05 (m, 4H, NH–CH–CH2 & 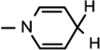 ), 1.64 (s, 9H, 3 × CH3), 1.48 (s, 3H, CH3), 1.37 (s, 9H, 3 × CH3), 1.27 (s, 3H, CH3); 13C NMR (100 MHz; MeOD) 171.97 (COO), 171.54 (CONH2), 156.32 [NHCOO(CH3)3], 149.53 [NHCOO(CH3)3], 136.24 (N–CH=C–CONH2), 135.38 (Ar–C), 125.98 (Ar–C), 124.10 (N–CH=CH), 123.46 (Ar–C), 122.37 (Ar–C), 118.62 (Ar–C), 116.07 (Ar–C), 114.82 (Ar–C), 114.33 (N–CH=C–CONH2), 103.63 (N–CH=CH–CH2), 101.71 (C-1), 97.15 (C-2), 83.41 (C-3), 81.90 (C-3), 80.28–80.08 (O–C(CH3)3), 79.30 (C-4), 64.53 (C-5), 60.11 (NH–CH), 27.33–26.27 (8 × CH3) 24.42 (N–CH=CH–CH2), 22.23 (NH–CH–CH2); IR (KBr) νmax: 1733.69 cm−1 (νcoo); HRMS calcd for C35H47N4O10 [M + H]+ 683.3292 found 683.3276.
), 1.64 (s, 9H, 3 × CH3), 1.48 (s, 3H, CH3), 1.37 (s, 9H, 3 × CH3), 1.27 (s, 3H, CH3); 13C NMR (100 MHz; MeOD) 171.97 (COO), 171.54 (CONH2), 156.32 [NHCOO(CH3)3], 149.53 [NHCOO(CH3)3], 136.24 (N–CH=C–CONH2), 135.38 (Ar–C), 125.98 (Ar–C), 124.10 (N–CH=CH), 123.46 (Ar–C), 122.37 (Ar–C), 118.62 (Ar–C), 116.07 (Ar–C), 114.82 (Ar–C), 114.33 (N–CH=C–CONH2), 103.63 (N–CH=CH–CH2), 101.71 (C-1), 97.15 (C-2), 83.41 (C-3), 81.90 (C-3), 80.28–80.08 (O–C(CH3)3), 79.30 (C-4), 64.53 (C-5), 60.11 (NH–CH), 27.33–26.27 (8 × CH3) 24.42 (N–CH=CH–CH2), 22.23 (NH–CH–CH2); IR (KBr) νmax: 1733.69 cm−1 (νcoo); HRMS calcd for C35H47N4O10 [M + H]+ 683.3292 found 683.3276.
Reduced nicotinamide N-tert-butyloxycarbonyl phenylalanine riboside 10e.
Yield 71%, mp 93.7–95.8 °C. 1H NMR (400 MHz; MeOD) 7.29–7.18 (m, 5H Ar–H), 7.14 (s, 1H, 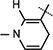 ), 5.97 (d, J = 7.88 Hz, 1H,
), 5.97 (d, J = 7.88 Hz, 1H, 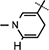 ), 4.88–4.84(m, 2H, H-1 & H-4).4.52 (t, J = 5.26 Hz, 1H, NH–CH), 4.43–4.39 (m, 2H, H-2 & H-3), 4.20 (brs, 2H, H-5), 4.13 (d, J = 3.24 Hz, 1H, NH–CH), 3.12–2.94 (m, 4H, NH–CH–CH2 &
), 4.88–4.84(m, 2H, H-1 & H-4).4.52 (t, J = 5.26 Hz, 1H, NH–CH), 4.43–4.39 (m, 2H, H-2 & H-3), 4.20 (brs, 2H, H-5), 4.13 (d, J = 3.24 Hz, 1H, NH–CH), 3.12–2.94 (m, 4H, NH–CH–CH2 & 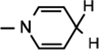 ), 1.51 (s, 3H, CH3), 1.37 (s, 9H, 3 × CH3), 1.31 (s, 3H, CH3); 13C NMR (100 MHz; MeOD) 172.03 (COO), 171.56 (CONH2), 156.29 (NHCOO), 137.07 (N–CH=C–CONH2), 136.25 (Ar–C), 128.93–126.04 (Ar–C), 114.34 (N–CH=C–CONH2), 103.61 (N–CH=CH–CONH2), 101.75 (C-1), 97.23 (C-2), 81.87 (C-3), 80.29–80.15 (O–C(CH3)3), 79.25 (C-4), 64.53 (C-5), 55.41 (NH–CH), 37.26 (Ar–CH2), 27.32 (3 × CH3), 26.24 (CH3), 24.36 (CH3), 22.25 (N–CH=CH–CH2); IR (KBr) νmax: 1743.33 cm−1 (νcoo); HRMS calcd for C28H38N3O8 [M + H]+ 544.2653 found 544.2669.
), 1.51 (s, 3H, CH3), 1.37 (s, 9H, 3 × CH3), 1.31 (s, 3H, CH3); 13C NMR (100 MHz; MeOD) 172.03 (COO), 171.56 (CONH2), 156.29 (NHCOO), 137.07 (N–CH=C–CONH2), 136.25 (Ar–C), 128.93–126.04 (Ar–C), 114.34 (N–CH=C–CONH2), 103.61 (N–CH=CH–CONH2), 101.75 (C-1), 97.23 (C-2), 81.87 (C-3), 80.29–80.15 (O–C(CH3)3), 79.25 (C-4), 64.53 (C-5), 55.41 (NH–CH), 37.26 (Ar–CH2), 27.32 (3 × CH3), 26.24 (CH3), 24.36 (CH3), 22.25 (N–CH=CH–CH2); IR (KBr) νmax: 1743.33 cm−1 (νcoo); HRMS calcd for C28H38N3O8 [M + H]+ 544.2653 found 544.2669.
Reduced nicotinamide N-tert-butyloxycarbonyl isoleucine riboside 10f.
Yield 46%, mp 88.6–89.9 °C. 1H NMR (400 MHz; MeOD) 7.13 (s, 1H,  ), 5.98 (d, J = 6.64 Hz, 1H,
), 5.98 (d, J = 6.64 Hz, 1H, 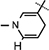 ), 4.89 (d, J = 3.4 Hz, 1H, H-1), 4.86–4.83 (m, 1H, H-4), 4.66–4.64 (m, 2H, H-5), 4.35–4.32 (m, 1H, H-2), 4.22–4.18 (m, 2H, H-3,
), 4.89 (d, J = 3.4 Hz, 1H, H-1), 4.86–4.83 (m, 1H, H-4), 4.66–4.64 (m, 2H, H-5), 4.35–4.32 (m, 1H, H-2), 4.22–4.18 (m, 2H, H-3, 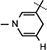 ), 4.10 (d, J = 6.04 Hz, 1H, NH–CH), 3.07 (brs, 2H,
), 4.10 (d, J = 6.04 Hz, 1H, NH–CH), 3.07 (brs, 2H, 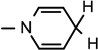 ), 1.90–1.84 (m, 1H, CH2CH2CHNH), 1.52 (s, 3H, CH3), 1.43 (s, 9H, 3 × CH3), 1.32(s, 3H, CH3), 1.28–1.20 (m, 2H, CH2), 0.933–0.880 (m, 6H, 2 × CH3); 13C NMR (100 MHz; MeOD) 172.01 (COO), 171.42 (CONH2), 156.76 (NHCOO), 135.07 (N–CH=C–CONH2), 126.26 (N–CH=CH), 114.46 (N–CH=C–CONH2), 103.50 (N–CH=CH–CH2), 101.97 (C-1), 97.14 (C-2), 81.74 (C-3), 80.3 (O–C(CH3)3), 79.23 (C-4), 64.50 (C-5), 58.56 (NH–CH), 36.84 CH3CH2CHCH3, 27.43 (3 × CH3), 26.26 (CH3), 24.86 (CH3), 24.29 (CH3CH2), 22.27 (N–CH=CH–CH2), 14.47 (CH3), 10.49 (CH3); IR (KBr) νmax: 1737.55 cm−1 (νcoo); HRMS calcd for C25H40N3O8 [M + H]+ 510.2815 found 510.2821.
), 1.90–1.84 (m, 1H, CH2CH2CHNH), 1.52 (s, 3H, CH3), 1.43 (s, 9H, 3 × CH3), 1.32(s, 3H, CH3), 1.28–1.20 (m, 2H, CH2), 0.933–0.880 (m, 6H, 2 × CH3); 13C NMR (100 MHz; MeOD) 172.01 (COO), 171.42 (CONH2), 156.76 (NHCOO), 135.07 (N–CH=C–CONH2), 126.26 (N–CH=CH), 114.46 (N–CH=C–CONH2), 103.50 (N–CH=CH–CH2), 101.97 (C-1), 97.14 (C-2), 81.74 (C-3), 80.3 (O–C(CH3)3), 79.23 (C-4), 64.50 (C-5), 58.56 (NH–CH), 36.84 CH3CH2CHCH3, 27.43 (3 × CH3), 26.26 (CH3), 24.86 (CH3), 24.29 (CH3CH2), 22.27 (N–CH=CH–CH2), 14.47 (CH3), 10.49 (CH3); IR (KBr) νmax: 1737.55 cm−1 (νcoo); HRMS calcd for C25H40N3O8 [M + H]+ 510.2815 found 510.2821.
Reduced nicotinamide N-tert-butyloxycarbonyl methionine riboside 10g.
Yield 30%, mp 91.6–92.9 °C. 1H NMR (400 MHz; MeOD) 7.14 (s, 1H, 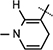 ), 5.99 (d, J = 6.6 Hz, 1H,
), 5.99 (d, J = 6.6 Hz, 1H,  ), 4.90 (brs, 1H, H-1), 4.86–4.83 (m, 1H, H-4), 4.65 (brs, 2H, H-5), 4.35–4.20 (m, 5H, H-2, H-3, Boc-NH–CH, Boc-NH–CH,
), 4.90 (brs, 1H, H-1), 4.86–4.83 (m, 1H, H-4), 4.65 (brs, 2H, H-5), 4.35–4.20 (m, 5H, H-2, H-3, Boc-NH–CH, Boc-NH–CH,  ), 3.29 (brs, 2H,
), 3.29 (brs, 2H,  ), 2.64–2.51 (m, 4H, S–CH2–CH2), 2.07 (s, 3H, CH3), 1.52 (s, 3H, CH3), 1.43(s, 9H, 3 × CH3), 1.31 (s, 3H, CH3); 13C NMR (100 MHz; MeOD) 172.44 (COO), 171.54 (CONH2), 156.66 (NHCOO), 135.91 (N–CH=C–CONH2), 126.51 (N–CH=CH), 114.39 (N–CH=C–CONH2), 103.35 (N–CH=CH–CH2), 101.98 (C-1), 97.14 (C-2), 81.80 (C-3), 80.35–80.16 (O–C(CH3)3), 79.32 (C-4), 64.64 (C-5), 60.12 (NH–CH–COO), 52.58 (acetonide–OCHO–), 30.57 (CH2), 29.80 (CH2), 27.39–24.30 (5 × CH3), 22.26 (N–CH=CH–CH2), 14.47 (CH3), 13.87 (SCH3); IR (KBr) νmax: 1738.51 cm−1 (νcoo); HRMS calcd for C24H38N3O8S [M + H]+ 528.2380 found 528.2387.
), 2.64–2.51 (m, 4H, S–CH2–CH2), 2.07 (s, 3H, CH3), 1.52 (s, 3H, CH3), 1.43(s, 9H, 3 × CH3), 1.31 (s, 3H, CH3); 13C NMR (100 MHz; MeOD) 172.44 (COO), 171.54 (CONH2), 156.66 (NHCOO), 135.91 (N–CH=C–CONH2), 126.51 (N–CH=CH), 114.39 (N–CH=C–CONH2), 103.35 (N–CH=CH–CH2), 101.98 (C-1), 97.14 (C-2), 81.80 (C-3), 80.35–80.16 (O–C(CH3)3), 79.32 (C-4), 64.64 (C-5), 60.12 (NH–CH–COO), 52.58 (acetonide–OCHO–), 30.57 (CH2), 29.80 (CH2), 27.39–24.30 (5 × CH3), 22.26 (N–CH=CH–CH2), 14.47 (CH3), 13.87 (SCH3); IR (KBr) νmax: 1738.51 cm−1 (νcoo); HRMS calcd for C24H38N3O8S [M + H]+ 528.2380 found 528.2387.
General procedure for the synthesis of NR-amino acid conjugates under ball-milling conditions (5).
A metallic jar was charged with the components according to Scheme 1 or Scheme 3/Table 2 (entries A–R) and was vibrated at a rate of 1800 rpm (30 Hz) at room temperature as per described in each entry. After that time, 1H NMR was used to assess whether the reaction had reached completion. The crude material was then re-suspended in DCM and extracted for purification by silica gel chromatography.
General procedure of oxidation of 2,3-diol protected NRH-amino acid esters (10a–10g) to 2,3-diol protected NR-amino acid esters (11a–11g).
1.00 mmol of compounds (10a–10g) was solubilized in 15 mL of EtOAc to which was added hexachloroacetone (5.00 mmol). The flask then was sealed and left stirring vigorously overnight. The reaction was monitored by 1H NMR analysis. Upon completion, EtOAc was removed under reduced pressure, and the crude oily products (11a–11g) were used for the next step without further purification.
General procedure for the deprotection of Boc and 2,3-diol ring simultaneously from 2,3-diol protected NR-amino acid esters (11a–11g).
1 mmol of respective ester product was dissolved in a mixture of TFA : H2O (9 mL : 1 mL) and the resulting solution was stirred at rt for 3–4 hours. Reaction progress was monitored by NMR analysis. Upon completion of the reaction, the solvent was removed under reduced pressure at low temperature, and the crude was purified by standard column chromatography by using H2O : MeOH (7 : 3) as an eluent to afford the desire products (12a–12g) in good yields.
Nicotinamide glycine riboside trifluoroacetate salt 12a.
Yield 84%, mp 92.6–93.0 °C. 1H NMR (400 MHz; D2O) 9.33 (s, 1H, Ar–H), 9.09 (d, J = 6.4 Hz, 1H, Ar–H), 8.93 (d, J = 8.4 Hz, 1H, Ar–H), 8.22 (t, J = 8.0 Hz, 1H, Ar–H), 6.19 (d, J = 4.0 Hz, 1H, H-1), 4.62–4.60 (m, 3H, 2H-5, H-4), 4.42 (t, J = 4.74 Hz, 1H, H-2), 4.28 (t, J = 4.62 Hz, 1H, H-3), 3.93 (d, J = 5.6 Hz, 2H, NH2–CH2–COO); 13C NMR (100 MHz; D2O) 167.93 (COO), 165.54 (CONH2), 162.89 (q, JC–F = 35.0 Hz, CF3CO), 145.59 (Ar–C), 142.59 (Ar–C), 140.00 (Ar–C), 134.03 (Ar–C), 128.48 (Ar–C), 116.30 (q, JC–F = 290 Hz, CF3CO), 99.54 (C-1), 84.15 (C-4), 77.06 (C-2), 69.54 (C-3), 64.72 (C-5), 40.01 (NH2–CH2–COO); 19F NMR (377 MHz, MeOD) δF: −76.94 (s); IR (KBr) νmax: 1747.19 cm−1 (νcoo); HRMS calcd for C13H18N3O6 [M]+ 312.1196 found 312.1197.
Nicotinamide leucine riboside trifluoroacetate salt 12b.
Yield 84%, mp 92.1–92.7 °C. 1H NMR (400 MHz; D2O) 9.33 (s, 1H, Ar–H), 9.10 (d, J = 6.2 Hz, 1H, Ar–H), 8.94 (d, J = 8.12 Hz, 1H, Ar–H), 8.24 (t, J = 7.14 Hz, 1H, Ar–H), 6.21 (d, J = 2.2 Hz, 1H, H-1), 4.66–4.61 (m, 3H, H4 & H5), 4.39 (t, J = 4.76 Hz, 1H, H-2), 4.25 (t, J = 4.72 Hz, 1H, H-3), 4.17 (t, J = 7.0 Hz, 1H, NH–CH), 1.73–1.58 (m, 2H, (CH3)2CHCH2), 0.890–0.863 (m, 1H, (CH3)2CHCH2), 0.792 (t, J = 6.72 Hz, 6H, 2 × CH3); 13C NMR (100 MHz; D2O) 170.55 (COO), 165.36 (CONH2), 162.87 (q, JC–F = 35.15 Hz, CF3CO), 145.70 (Ar–C), 142.63 (Ar–C), 139.83 (Ar–C), 134.04 (Ar–C), 128.59 (Ar–C), 116.32 (q, JC–F = 290.13 Hz, CF3CO), 99.37 (C-1), 84.01 (C-4), 77.13 (C-2), 69.54 (C-3), 65.10 (C-5), 51.33 (NH2–CH), 38.81 [(CH3)2 CH CH2], 23.95 [(CH3)2CH], 21.32 (CH3), 20.95 (CH3); 19F NMR (377 MHz, MeOD) δF: −76.93 (s); IR (KBr) νmax: 1752.01 cm−1 (νcoo); HRMS calcd for C17H26N3O6 [M]+ 368.1822 found 368.1816.
Nicotinamide valine riboside trifluoroacetate salt 12c.
Yield 61%, mp 89.3–92.0 °C. 1H NMR (400 MHz; D2O) 9.33 (s, 1H, Ar–H), 9.09 (d, J = 6.08 Hz, 1H, Ar–H), 8.93 (d, J = 7.96 Hz, 1H, Ar–H), 8.23 (t, J = 7.16 Hz, 1H, Ar–H), 6.21 (d, J = 4.04 Hz, 1H, H-1), 4.63 (brs, 3H, H4 & 2H5), 4.41 (t, J = 4.56 Hz, 1H, H-2), 4.25 (t, J = 4.4 Hz, 1H, H-3), 4.09 (d, J = 4.6 Hz, 1H, NH2–CH), 2.24–2.20 (m, 1H, (CH3)2CH), 0.943 (d, J = 7.0 Hz, 3H, CH3), 0.913 (t, J = 10.16 Hz, 3H, CH3); 13C NMR (100 MHz; D2O) 169.63 (COO), 165.45 (CONH2), 163.22 (q, JC–F = 34.84 Hz, CF3CO), 145.65 (Ar–C), 142.68 (Ar–C), 139.90 (Ar–C), 134.04 (Ar–C), 128.54 (Ar–C), 116.31 (q, JC–F = 289.72 Hz, CF3CO), 99.43 (C-1), 83.79 (C-4), 76.97 (C-2), 69.38 (C-3), 64.94 (C-5), 58.24 (NH2–CH), 29.33 [(CH3)2 CH], 17.23 (CH3), 16.89 (CH3); 19F NMR (377 MHz, MeOD) δF: −76.94(s); IR (KBr) νmax: 1747.19 cm−1 (νcoo); HRMS calcd for C16H24N3O6 [M]+ 354.1665 found 354.1661.
Nicotinamide tryptophane riboside trifluoroacetate salt 12d.
Yield 60%, mp 93.1–94.8 °C. 1H NMR (400 MHz; D2O) 8.94 (s, 1H, Ar–H), 8.83 (d, J = 8.08 Hz, 1H, Ar–H), 8.78 (d, J = 6.4 Hz, 1H, Ar–H), 8.08 (t, J = 7.18 Hz, 1H, Ar–H), 7.31 (d, J = 8.0 Hz, 1H, Ar–H), 7.24 (d, J = 7.96 Hz, 1H, Ar–H), 7.14 (s, 1H, Ar–H), 6.97 (t, J = 7.48 Hz, 1H, Ar–H), 6.81 (t, J = 7.28 Hz, 1H, Ar–H), 5.90 (d, J = 3.4 Hz, 1H, H-1), 4.46–4.33 (m, 5H, H2, H3, H4 & 2H5), 3.44–3.34 (m, 2H, Ar–CH2), 3.07 (t, J = 4.92 Hz, 1H, NH2–CH); 13C NMR (100 MHz; D2O) 170.23 (COO), 164.92 (CONH2), 145.19, 141.94, 139.09, 135.96, 133.32, 128.28, 125.96, 124.72, 122.08, 119.74, 117.69, 112.01, 106.89, 99.27 (Ar–C), 83.77 (C-4), 77.15 (C-2), 68.55 (C-3), 64.65 (C-5) 53.31 (NH2–CH), 26.70 (NH2–CH2); 19F NMR (377 MHz, D2O) δF: −75.37 (s); IR (KBr) νmax: 1746.23 cm−1 (νcoo); HRMS calcd for C22H25N4O6 [M]+ 441.1774 found 441.1773.
Nicotinamide phenylalanine riboside trifluoroacetate salt 12e.
Yield 73%, mp 95.8–97.1 °C. 1H NMR (400 MHz; D2O) 9.18 (s, 1H, Ar–H), 9.05 (d, J = 6.32 Hz, 1H, Ar–H), 8.97 (d, J = 8.12 Hz, 1H, Ar–H), 8.24 (t, J = 7.18 Hz, 1H, Ar–H), 7.16–7.12 (m, 5H, Ar–H), 6.09 (d, J = 3.92 Hz, 1H, H-1), 4.67–4.42 (m, 4H, H3, H4 & 2H5), 3.98 (t, J = 4.66 Hz, 1H, H-2), 3.49 (t, J = 5.08 Hz, 1H, NH2–CH), 3.24–3.19 (m, 1H, Ar–CH2) 3.09 (dd, J = 9.04 Hz & 9.0 Hz, 1H, Ar–CH2); 13C NMR (100 MHz; D2O) 169.66 (COO), 165.31 (CONH2), 162.90 (q, JC–F = 35.31 Hz, CF3CO), 145.58–127.85 (Ar–C), 116.32 (q, JC–F = 289.89 Hz, CF3CO), 99.39 (C-1), 83.61 (C-4), 77.06 (C-2), 68.70 (C-3), 64.75 (C-5), 54.05 (NH2–CH), 36.04 (Ar–CH2); 19F NMR (377 MHz, MeOD) δF: −76.94 (s); IR (KBr) νmax: 1749.12 cm−1 (νcoo); HRMS calcd for C20H24N3O6 [M]+ 402.1665, found 402.1663.
Nicotinamide isoleucine riboside trifluoroacetate salt 12f.
Yield 60%, mp 89.3–91.2 °C. 1H NMR (400 MHz; D2O) 9.32 (s, 1H, Ar–H), 9.10 (d, J = 5.88 Hz, 1H, Ar–H), 8.94 (d, J = 7.76 Hz, 1H, Ar–H), 8.24 (t, J = 6.94 Hz, 1H, Ar–H), 6.21 (d, J = 3.96 Hz, 1H, H-1), 4.62 (brs, 2H, 2 × H-5), 4.42 (t, J = 4.46 Hz, 1H, H-2), 4.24 (brs, 1H, H-3), 4.15 (d, J = 3.92 Hz, 1H, NH2–CH), 1.95–1.93 (m, 1H, CH3CH), 1.37–1.18 (m, 2H, CH3–CH2), 0.913 (d, J = 6.92 Hz, 3H, CH3), 0.781 (t, J = 7.36 Hz, 3H, CH3); 13C NMR (100 MHz; D2O) 169.62 (COO), 165.46 (CONH2), 162.89 (q, JC–F = 35.31 Hz, CF3CO), 145.72–128.58 (Ar–C), 116.31 (q, JC–F = 289.97 Hz, CF3CO), 99.41 (C-1), 83.81 (C-4), 76.97 (C-2), 69.45 (C-3), 64.98 (C-5), 57.21 (NH2–CH), 36.08 (CH3 CH2 CH), 24.90 (CH3 CH2 CH), 14.04 (CH3), 10.81 (CH3CH2); 19F NMR (377 MHz, MeOD) δF: −76.95 (s); IR (KBr) νmax: 1747.19 cm−1 (νcoo); HRMS calcd for C17H26N3O6 [M]+ 368.1822 found 368.1820.
Nicotinamide methionine riboside trifluoroacetate salt 12g.
Yield 88%, mp 92.3–94.5 °C. 1H NMR (400 MHz; D2O) 9.33 (s, 1H, Ar–H), 9.10 (d, J = 6.2 Hz, 1H, Ar–H), 8.94 (d, J = 8.12 Hz, 1H, Ar–H), 8.24 (t, J = 8.0 Hz, 1H, Ar–H), 6.20 (d, J = 4.32 Hz, 1H, H-1), 4.63–4.57 (m, 3H, H1 & 2H5), 4.41 (t, J = 4.68 Hz, 1H, H-2), 4.33 (t, J = 6.5 Hz, 1H, H-3), 4.26 (t, J = 4.7 Hz, 1H, NH2–CH), 2.56 (q, J = 6.06 Hz, 2H, SCH2CH2), 2.14 (t, J = 6.0 Hz, 2H, SCH2CH2), 1.93(s, 3H, CH3); 13C NMR (100 MHz; D2O) 169.74 (COO), 165.43 (CONH2), 162.91 (q, JC–F = 35.33 Hz, CF3CO), 145.63–128.57 (Ar–C), 116.32 (q, JC–F = 290.05 Hz, CF3CO), 99.41 (C-1), 83.95 (C-4), 77.11 (C-2), 69.53 (C-3), 65.21 (C-5), 51.50 (NH2–CH), 28.83 (SCH2CH2), 28.44 (SCH2CH2), 13.89 (CH3); 19F NMR (377 MHz, MeOD) δF: −76.96 (s); IR (KBr) νmax: 1747.19 cm−1 (νcoo); HRMS calcd for C16H24N3O6S [M]+ 386.1386 found 386.1376.
PNP enzyme activity assay.
Phosphorolysis of NR+ Cl− and its glycine conjugate (12a, Gly-NR) by PNP (Purine Nucleoside Phosphorylase, Sigma Aldrich) was performed in HEPES buffer, containing KH2PO4 and 10% D2O at 25 °C and was monitored by 1H NMR. All spectra were obtained at 300 K on a Bruker AscendTM 400 MHz ultra-shielded spectrometer (Bruker Biospin) operating at 400.13 MHz for protons. TopSpin 3.2 (Bruker BioSpin) was used for all NMR spectral acquisition and pre-processing. The automation of sample submission was performed using ICON-NMR (Bruker BioSpin). Incubations were conducted in the NMR tube which contained a final volume of 505 μl : 450 μl HEPES buffer (100.0 mM, pH 7.0), containing 100 mM KH2PO4, 50.0 μl NR-Cl or NR glycine conjugate (50.0 mM in 1 mL D2O) and when appropriate 5 μl PNP (1 mg dissolved in 50 μl HEPES buffer) and measurements taken at t = 0, 20 min and 6 h (ns = 128). For each independent experiment, freshly prepared solutions of NR-Cl and NR glycine conjugate in HEPES buffer, containing 100 mM KH2PO4 and 50 μl D2O, were used.
Conclusions
A novel series of NR-amino acids conjugates (12a–12g) have been generated in good to excellent yields, using a synthetic sequence that is easily scalable. The use of a protected NRH derivative as synthetic intermediate allows for the manipulation of the nicotinoyl riboside scaffold and modification of the C5′-OH position under basic or neutral conditions. Critically, this work is the first evidence that simple man-made modifications at the C5-position of NR can extend the half-life of NR when it is exposed to PNP-like phosphorylases. This approach offers new strategies for the development of slow-releasing formulations of NR, that minimize nicotinamide generation before NR’s conversion to NAD. This work is relevant as it offers tasks-specific tools to explore the complexity of systemic NAD metabolism, a complexity just recently revealed.
Supplementary Material
Acknowledgements
We thank ChromaDex and the Mitchell Cancer Institute for financial support for M. E. M. and F. H.
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ob00134a
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.Redeuil K, Vulcano J, Prencipe FP, Benet S, Campos-Gimenez E and Meschiari M, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci, 2019, 1110–1111, 74–80. [DOI] [PubMed] [Google Scholar]
- 2.Ummarino S, Mozzon M, Zamporlini F, Amici A, Mazzola F, Orsomando G, Ruggieri S and Raffaelli N, Food Chem, 2017, 221, 161–168. [DOI] [PubMed] [Google Scholar]
- 3.Trammell SA, Yu L, Redpath P, Migaud ME and Brenner C, J. Nutr, 2016, 146, 957–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tempel W, Rabeh WM, Bogan KL, Belenky P, Wojcik M, Seidle HF, Nedyalkova L, Yang T, Sauve AA, Park HW and Brenner C, PLos Biol, 2007, 5, e263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Makarov MV, Trammell SAJ and Migaud ME, Biochem. Soc. Trans, 2019, 47, 131–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berger F, Ramirez-Hernandez MH and Ziegler M, Trends Biochem. Sci, 2004, 29, 111–118. [DOI] [PubMed] [Google Scholar]
- 7.Chiarugi A, Dolle C, Felici R and Ziegler M, Nat. Rev. Cancer, 2012, 12, 741–752. [DOI] [PubMed] [Google Scholar]
- 8.Dolle C, Skoge RH, Vanlinden MR and Ziegler M, Curr. Top. Med. Chem, 2013, 13, 2907–2917. [DOI] [PubMed] [Google Scholar]
- 9.Imai S and Guarente L, Trends Cell Biol, 2014, 24, 464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kirkland JB and Meyer-Ficca ML, Adv. Food Nutr. Res, 2018, 83, 83–149. [DOI] [PubMed] [Google Scholar]
- 11.Frederick DW, Loro E, Liu L, Davila A Jr., Chellappa K, Silverman IM, Quinn WJ 3rd, Gosai SJ, Tichy ED, Davis JG, Mourkioti F, Gregory BD, Dellinger RW, Redpath P, Migaud ME, Nakamaru-Ogiso E, Rabinowitz JD, Khurana TSand Baur JA, Cell Metab, 2016, 24, 269–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fessel JP and Oldham WM, Antioxid. Redox Signal, 2018, 28, 180–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hershberger KA, Martin AS and Hirschey MD, Nat. Rev. Nephrol, 2017, 13, 213–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mouchiroud L, Houtkooper RH and Auwerx J, Crit. Rev. Biochem. Mol. Biol, 2013, 48, 397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garrido A and Djouder N, Trends Cancer, 2017, 3, 593–610. [DOI] [PubMed] [Google Scholar]
- 16.Ratajczak J, Joffraud M, Trammell SA, Ras R, Canela N, Boutant M, Kulkarni SS, Rodrigues M, Redpath P, Migaud ME, Auwerx J, Yanes O, Brenner C and Canto C, Nat. Commun, 2016, 7, 13103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ear PH, Chadda A, Gumusoglu SB, Schmidt MS, Vogeler S, Malicoat J, Kadel J, Moore MM, Migaud ME, Stevens HE and Brenner C, Cell Rep, 2019, 26, 969–983. [DOI] [PubMed] [Google Scholar]
- 18.Fouquerel E, Goellner EM, Yu Z, Gagne JP, Barbi de Moura M, Feinstein T, Wheeler D, Redpath P, Li J, Romero G, Migaud M, Van Houten B, Poirier GG and Sobol RW, Cell Rep, 2014, 8, 1819–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilk A, Hayat F, Cunningham R, Li J, Garavaglia S, Zamani L, Ferraris DM, Sykora P, Andrews J, Clark J, Davis A, Chaloin L, Rizzi M, Migaud M and Sobol RW, Sci. Rep, 2020, 10, 651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu HW, Smith CB, Schmidt MS, Cambronne XA, Cohen MS, Migaud ME, Brenner C and Goodman RH, Proc. Natl. Acad. Sci. U. S. A, 2018, 115, 10654–10659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trammell SA, Schmidt MS, Weidemann BJ, Redpath P, Jaksch F, Dellinger RW, Li Z, Abel ED, Migaud ME and Brenner C, Nat. Commun, 2016, 7, 12948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dellinger RW, Santos SR, Morris M, Evans M, Alminana D, Guarente L and Marcotulli E, npj Aging Mech. Dis, 2017, 3, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Canto C, Houtkooper RH, Pirinen E, Youn DY, Oosterveer MH, Cen Y, Fernandez-Marcos PJ, Yamamoto H, Andreux PA, Cettour-Rose P, Gademann K, Rinsch C, Schoonjans K, Sauve AA and Auwerx J, Cell Metab, 2012, 15, 838–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang H, Ryu D, Wu Y, Gariani K, Wang X, Luan P, D’Amico D, Ropelle ER, Lutolf MP, Aebersold R, Schoonjans K, Menzies KJ and Auwerx J, Science, 2016, 352, 1436–1443. [DOI] [PubMed] [Google Scholar]
- 25.Liu L, Su X, Quinn WJ 3rd, Hui S, Krukenberg K, Frederick DW, Redpath P, Zhan L, Chellappa K, White E, Migaud M, Mitchison TJ, Baur JA and Rabinowitz JD, Cell Metab, 2018, 27, 1067–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shats I, Williams JG, Liu J, Makarov MV, Wu X, Lih FB, Deterding LJ, Lim C, Xu X, Randall TA, Lee E, Li W, Fan W, Li J-L, Sokolsky M, Kabanov AV, Li L, Migaud ME, Locasale JW and Li X, Cell Metab, 2020, 31, 564–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wielgus-Kutrowska B, Kulikowska E, Wierzchowski J, Bzowska A and Shugar D, Eur. J. Biochem, 1997, 243, 408–414. [DOI] [PubMed] [Google Scholar]
- 28.Belenky P, Christensen KC, Gazzaniga F, Pletnev AA and Brenner C, J. Biol. Chem, 2009, 284, 158–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shabalin K, Nerinovski K, Yakimov A, Kulikova V, Svetlova M, Solovjeva L, Khodorkovskiy M, Gambaryan S, Cunningham R, Migaud ME, Ziegler M and Nikiforov A, Int. J. Mol. Sci, 2018, 19(12), 3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kulikova V, Shabalin K, Nerinovski K, Yakimov A, Svetlova M, Solovjeva L, Kropotov A, Khodorkovskiy M, Migaud ME, Ziegler M and Nikiforov A, Metabolites, 2019, 9(12), 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vrettos EI, Mező G and Tzakos AG, Beilstein J. Org. Chem, 2018, 14, 930–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Derudas M, Carta D, Brancale A, Vanpouille C, Lisco A, Margolis L, Balzarini J and McGuigan C, J. Med. Chem, 2009, 52, 5520–5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Makarov MV and Migaud ME, Beilstein J. Org. Chem, 2019, 15, 401–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu JA, Guo XP, Liang S, An F, Shen HY and Xu YJ, Tetrahedron, 2015, 71, 1409–1412. [Google Scholar]
- 35.Lee J, Churchil H, Choi WB, Lynch JE, Roberts FE, Volante RP and Reider PJ, Chem. Commun, 1999, 729–730, DOI: 10.1039/a809930h. [DOI] [Google Scholar]
- 36.Yang Y, Mohammed FS, Zhang N and Sauve AA, J. Biol. Chem, 2019, 294, 9295–9307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Makarov MV, Harris NW, Rodrigues M and Migaud ME, Org. Biomol. Chem, 2019, 17, 8716–8720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsakos M, Schaffert ES, Clement LL, Villadsen NL and Poulsen TB, Nat. Prod. Rep, 2015, 32, 605–632. [DOI] [PubMed] [Google Scholar]
- 39.Ravalico F, James SL and Vyle JS, Green Chem, 2011, 13, 1778–1783. [Google Scholar]
- 40.Amigues EJ, Armstrong E, Dvorakova M, Migaud ME and Huang M, Nucleosides, Nucleotides Nucleic Acids, 2009, 28, 238–259. [DOI] [PubMed] [Google Scholar]
- 41.Giroud-Gerbetant J, Joffraud M, Giner MP, Cercillieux A, Bartova S, Makarov MV, Zapata-Perez R, Sanchez-Garcia JL, Houtkooper RH, Migaud ME, Moco S and Canto C, Mol. Metab, 2019, 30, 192–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chevallier OP and Migaud ME, Beilstein J. Org. Chem, 2006, 2, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chevallier OP and Migaud ME, Nucleosides, Nucleotides Nucleic Acids, 2008, 27, 1127–1143. [DOI] [PubMed] [Google Scholar]
- 44.Srihari P, Kumaraswamy B and Yadav J, Tetrahedron, 2009, 65, 6304–6309. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.