

Abstract
Introduction:
Liver Glycogen Storage Disease Type IX (GSD IX) is one of the most common forms of GSD. It is caused by a deficiency in enzyme phosphorylase kinase (PhK), a complex, hetero-tetrameric enzyme comprised of four subunits - α, β, γ, and δ - each with tissue specific isoforms encoded by different genes. Until the recent availability of gene panels and exome sequencing, the diagnosis of liver GSD IX did not allow for differentiation of these subtypes. This study presents the first comprehensive literature review for liver GSD IX subtypes - GSD IX α2, β, and γ2, We aim to better characterize the natural history of liver GSD IX and further investigate if there are subtype-specific differences in clinical presentation.
Methods:
A comprehensive literature review was performed with the help of a medical librarian at Duke University Medical Center to gather all published patients of liver GSD IX. Our refined search yielded 74 articles total. Available patient data were compiled into an excel spreadsheet. Data were analyzed via descriptive statistics. The number of patients with specific symptoms were individually summed and reported as a percentage of the total number of patients for which data were available or were averaged and reported as a mean numerical value. Published pathology reports were scored using the International Association of the Study of the Liver Scale.
Results:
There were a total of 183 GSD IX α2 patients, 17 GSD IX β patients, and 30 GSD IX γ2 patients. Average age at diagnosis was 4 years for GSD IX α2 patients, 2.34 years for GSD IX β patients, and 1.81 years for GSD IX γ2 patients. Hepatomegaly was reported in 164/176 (93.2%) of GSD IX α2 patients, 16/17 (94.1%) of GSD IX β patients, and 30/30 (100%) of GSD IX γ2 patients. Fasting hypoglycemia was reported in 53/121 (43.8%) of GSD IX α2 patients, 8/16 (50%) of GSD IX β patients, and 18/19 (94.7%) of GSD IX γ2 patients. Liver biopsy pathology reports were available and interpreted for 46 GSD IX α2 patients, 3 GSD IX β patients, and 24 GSD IX γ2 patients. 22/46 (47.8%) GSD IX α2 patients, 1/3 (33.3%) GSD IX β patients, and 23/24 (95.8%) GSD IX γ2 patients with available pathology reports documented either some degree of fibrosis or cirrhosis.
Conclusion:
Our comprehensive review demonstrates quantitatively that the clinical presentation of GSD IX γ2 patients is more severe than that of GSD IX α2 or β patients. However, our study also shows the existence of a severe phenotype in GSD IX α2, evidenced by early onset liver pathology in conjunction with clinical symptoms. There is need for a more robust natural history study to better understand the variability in liver pathophysiology within liver GSD IX; in addition, further study of mutations and gene mapping could bring a better understanding of the relationship between genotype and clinical presentation.
Keywords: Glycogen storage disease IX, GSD IX, GSDIX, Liver GSD IX, Liver GSDIX, GSD IX gamma, GSDIX gamma
1. Introduction
With an incidence of 1:100,000 live births, Glycogen Storage Disease Type IX (GSD IX) is one of the most common glycogen storage diseases [1,2]. GSD IX is caused by a deficiency of phosphorylase kinase (PhK), an essential hetero-tetrameric regulatory enzyme in glycogenolysis. The first enzyme in the glycogenolysis pathway, PhK is responsible for phosphorylating and activating muscle and liver glycogen phosphorylase. This facilitates the release of the outer glucose-1-phosphate from glycogen, the first step in debranching of glycogen. Glucose continues to be mobilized through the activity of phosphorylases at first and eventually through the debranching enzyme when the glucose chain is less than five residues, leading to increased free glucose for energy usage [3]. PhK is comprised of four copies of each subunit; alpha, beta, gamma, and delta (αβγδ), each encoded by unique genes [3]. The γ subunit houses the catalytic site which is regulated by the α, β, and δ subunits, as well as extrinsic calmodulin, which is structurally indistinct from the δ subunits [3]. There are muscle and liver specific isomers of PhK, deficiencies of which are caused by pathogenic variants in different genes and lead to different phenotypic manifestations. This review focuses on liver PhK deficiency, also classified broadly as liver GSDIX, and its subtypes [4].
The α subunit is coded by PHKA1 (OMIM *311870) in muscle and PHKA2 (OMIM *300798) in liver, both on the X chromosome; the β subunit is coded by PHKβ the γ subunit is coded by PHKG1 (OMIM *172470) in muscle and PHKG2 (OMIM *172471) in liver; the δ subunit is coded by CALM1 (OMIM *114180), CALM2 (OMIM *114182), and CALM3 (OMIM *114183) [5–7], The genes PHKA1 and PHKG1 encode the muscle specific isoform and are responsible for various subtypes of muscle PhK deficiency [4]. The genes PHKA2, PHKB, and PHKG2 encode the liver isoform α2, β, and γ2 subunits, respectively and are responsible for the various subtypes of liver PhK deficiency [4]. Mutations in the δ subunit and the CALM 1, 2, 3 genes has not yet been definitively linked to a specific phenotype and will not be further addressed in this paper [3].
Mutations in the α2 subunit are most common, X-linked recessive, and responsible for roughly 75% of liver PhK deficiency; mutations in the γ2 subunit are second most common, autosomal recessive, and responsible for almost 25% of liver PhK deficiency; mutations in the β subunit are also autosomal recessive and are far less common [8–11]. In this paper we will focus on liver PhK deficiency, also classified broadly as liver GSDIX, and its subtypes [4].
There is a large amount of variability in the nomenclature of the different liver GSD IX subtypes, with the most recent 2019 American College of Medical Genetics and Genomics guideline proposing utilizing the Roman lettering “a, b, c” to stand for mutations in PHKA2, PHKB, and PHKG2 genes respectively [12]. The Online Mendelian Inheritance in Man Database also classifies the subtype of liver GSD IX utilizing the roman letter “a, b, c” to represent mutations in the PHKA2, PHKB, and PHKG2 genes respectively. For example, GSD IXa refers to a mutation in the PHKA2 gene. However, in this paper we propose referring to the subtype of liver GSD IX utilizing the Greek lettering referring to the mutated subunit of the PhK enzyme, as this is the most concise and clear nomenclature. For example, from now on the disease caused by a mutation in PHKA2 gene, leading to a mutation in the α subunit will be called GSD IX α2. Mutations in PHKB gene encoding the β subunit will be referred to as GSD IX β. Mutations in the PHKG2 gene encoding the γ2 subunit will be referred to as GSD IX γ2.
Until the recent availability of gene panels and exome sequencing, the diagnosis of liver GSD IX did not allow for clear differentiation of these subtypes. However, based on prior case reports, there is growing body of evidence for some genotype-phenotype correlation [8,13–15]. Most liver GSD IX patients present between infancy and two years with hepatomegaly, elevated liver transaminases, elevated triglycerides, motor delay, growth delay, and/or episodes of ketotic hypoglycemia [10]. The clinical presentation of the majority of GSD IX α2 and GSD IX β patients is reported to become milder with age as metabolic demands decrease; however, the symptoms of GSD IX γ2 patients have been shown to persist with age, potentially progressing to cirrhosis, liver failure, hepatocellular carcinoma, and death [4,13–22].
We present the first comprehensive literature review of liver GSD IX in order to characterize the natural history of GSD IX α2, GSD IX γ2 and GSD IX β, and further investigate genotype-phenotype correlations.
2. Methods
2.1. Literature review
To obtain all relevant, published case reports with liver type GSD IX α2, β, and γ2, a comprehensive literature review was conducted through September 2020 using PubMed. Working with a medical librarian at Duke Medical Sciences Research Library, the following search strategy was crafted and performed: “Glycogen storage disease type IX OR GSD IX OR glycogenosis type IX OR Glycogen storage disease type 9 OR Glycogen storage disease type nine” National Library of Medicine Medical Subject Headings (MeSH) terms “glycogen storage disease” was used. Additional literature searches with each of the following terms were performed: “PHKA2”, “PHKG2”, “PHKB”, “GSD IXa”, “GSD IXb”, “GSD IXc”, “GSD alpha”, “GSD beta”, “GSD gamma” to ensure maximal coverage of reported cases.
All papers were reviewed and only papers that specifically contained patient data such as case reports and case series with patients who were reported to have genetically confirmed diagnoses of liver GSD IX were included in the study. These refined criteria yielded 74 total articles. The raw data alongside the specific paper citations is included in the supplementary materials.
All patients cited as previously reported in the literature were noted and the both the initial and recent reference were reviewed and cited to ensure no duplicate patients were recorded.
2.2. Data collection
All relevant clinical data were pulled from the collected literature. For patients reported in multiple sources, data from the most recent source was used. If any data for a particular patient was not mentioned in the most recent report, all older published references citing the patient were searched for the relevant data. Raw data were compiled into an excel spreadsheet by three separate researchers and all data entry was double checked using the original papers by one researcher.
2.2.1. Demographic information
Descriptive information including patient gender and age at diagnosis was extracted. Age at diagnosis was converted to years if previously reported in months. The relationship between patients in the same family was recorded. If available, the genetic variants, amino acid change, and exon location of the mutation for each patient was recorded. The initial presentation, description of the symptoms that the patient was exhibiting which lead to diagnosis/treatment, was also recorded.
2.2.2. Hepatomegaly, growth and development
Patients with documented hepatomegaly, growth retardation, delayed growth, or short stature were reported respectively. All patients with a documented length or height equal to or below the 10th percentile were coded as “delayed” [23]. Any form of developmental delay including gross motor delay, hypotonia, physical deformity, or speech delay were coded as “delayed.” All patients reported as displaying normal development were coded as “normal.” Any parameter with no information reported for a patient was coded as “ND” (no data).
2.2.3. Biochemical laboratory evaluation
Data reported for presence or absence of fasting ketosis, hypertriglyceridemia, hypercholesterolemia, and fasting hypoglycemia, were reported respectively. Enzyme activity was coded based on documented low enzyme activity as measured in either erythrocytes, leukocytes, or hepatocytes, or normal. Aspartate Transaminase (AST) and Alanine Transaminase (ALT) levels were counted as either elevated or normal; patients were also reported as elevated if above the normal reference range of 0–45 units per liter (U/L) for AST or 0–40 U/L for ALT. Numerical values for these parameters were collected when appropriate and averages were reported. Any parameter with no information reported for a patient was coded as “ND” (no data).
2.2.4. Liver biopsy, hepatic adenoma, and transplant
Patients who received liver biopsies or transplants, or were documented to have hepatic adenomas were noted. Age at time of liver biopsy, and age at time of transplant were converted to years if previously reported as months. Pathology reports were coded using the categories denoted by the International Association for Study of the Liver (IASL) [24]. This scale was chosen as the categories were broad while still capturing the progressive nature of liver pathology. As we did not have access to full pathology and histology reports we did not know the most detailed scope of liver disease, thus rendering other histologic grading scales more difficult to use. Pathology reports documenting cirrhosis were coded as stage 4. Severe fibrosis was coded as stage 3. Moderate fibrosis was coded as stage 2. Mild fibrosis was coded as stage 1. No fibrosis was coded as stage 0. Any parameter with no information reported for a patient was coded as “ND” (no data).
2.3. Data analysis
All data were analyzed using descriptive statistics. Descriptive statistical analysis was performed separately for each liver GSD IX subtype. The number of patients with specific symptoms for which data were collected, were individually summed and reported as a percentage of the total number of patients for which data were available, or data were averaged and reported as a mean numerical value. Ranges were also calculated when applicable.
3. Results
3.1. GSD IX α2
3.1.1. Diagnosis and initial presentation
A total of 183 patients with liver type GSD IX α2 were gathered from the literature. Of the 183 patients there were 15 families (34 patients total) with family history of GSD IX α2 of which nine families had sibling pairs with GSD IX α2. The remaining 149 patients were singletons with no family history of GSD IX α2.
Of the 132 patients which reported age at diagnosis, the mean age at diagnosis was 4 years with a range of 0.24–37 years.
Initial presentation was reported for 157 patients. Of the 157 patients, 141 (89.8%) presented with hepatomegaly. 56 (35.7%) patients presented with abnormal liver function. 47 out of 157 (29.9%) patients presented with growth or developmental delays including delay in gross motor milestones, cognitive impairment, short stature, growth retardation, or decrease in growth velocity. 8 out of 157 (5.1%) patients presented with complaints of frequent infections. 6 out of 157 (3.8%) patients presented with frequent hunger. 6 out of 157 (3.8%) patients presented with frequent hypoglycemia. 4 out of 157 (2.5%) patients presented with fatigue. 2 out of 157 (1.3%) patients presented with anemia. 1 out of 157 (0.6%) patient presented with frequent ketosis. 4 out of 157 (2.5%) patients presented with vomiting, diarrhea, or nausea. 4 out of 157 (2.5%) patients presented with splenomegaly. 2 out of 157 (1.3%) patients presented with hypotonia. 1 out of 157 (0.6%) patients presented with osteoporosis. 1 out of 157 (0.6%) presented with excessive weight gain. 1 out of 157 (0.6%) presented with lactic acidosis.
3.1.2. . Clinical presentation
164 (93.2%) patients were reported as having hepatomegaly of the 176 for which it was discussed. AST/ALT levels were reported for 138 patients, and 125 (90.6%) patients displayed elevated levels. Enzyme activity as measured in erythrocytes, leukocytes, or hepatocytes was reported for 116 patients, an enzyme activity was reported as low in 101 (87.1%) patients. Growth was reported for 168 patients, 98 (58.3%) displayed growth retardation. 21 (61.8%) patients displayed fasting ketosis out of the 34 for which it was discussed. Hypertriglyceridemia was present in 64 (65.3%) of the 98 patients for which it was reported. Hypercholesterolemia was present in 41 (45.6%) of the 90 patients for which it was reported. 53 (43.8%) patients displayed fasting hypoglycemia of the 121 patients for which it was discussed. Development was reported for 88 patients and 31 (35.2%) patients displayed delayed development.
Quantitative AST levels were reported for 85 patients, 66 were elevated and 19 were normal. Mean AST was 328.2 U/L. ALT levels were reported for 91 patients, 70 were elevated and 21 were normal. Mean ALT was 274.1 U/L. AST levels ranged from 24 to 2001 U/L. ALT levels ranged from 13 to 1121 U/L. Quantitative triglyceride level was reported for 69 patients. The mean triglyceride level was 6.23 mmol/L with a range of 0.46–43.3 mmol/L. Quantitative total cholesterol level was reported for 66 patients. The mean cholesterol level was 6.52 with a range of 0.57–20.26 mmol/L.
3.1.3. Liver biopsy
68 (37.2%) of the 183 patients underwent a liver biopsy. Mean age at time of biopsy was 3.09 years with a range of 0.5–6.7 years. 60 patients had a documented pathology report. 46 pathology reports were able to be coded under the IASL grading system. 14 patients from China and South Korea utilized wording in the pathology report that was not consistent with our histologic grading scale.
24 of the 46 pathology reports evaluated (52.2%) displayed no fibrosis. 7 (15.2%) displayed mild fibrosis. 4 (8.7%) displayed moderate fibrosis. 7 (15.2%) displayed severe fibrosis. 4 (8.7%) displayed cirrhosis.
Overall, 22 out of 46 evaluable patients with pathology reports (47.8%) displayed some degree of fibrosis (mild, moderate, or severe) or cirrhosis.
The mean IASL score was 1.13.
3.1.4. Hepatic adenoma
Data regarding the presence or absence of a hepatic adenoma was documented for 73 patients. Only one patient was reported to have a hepatic adenoma.
3.1.5. Liver transplant
No patients were reported as receiving a liver transplant.
3.2. GSD IX β
3.2.1. Diagnosis and initial presentation
A total of 17 patients of GSD IX β were gathered from the literature. Initial presentation was reported for all 17 patients. Of the 17 patients there were 2 separate families (4 patients total) with family history of GSD IX β, both families consisted of sibling pairs. The remaining 13 patients did not have family history of GSD IX β.
16 out of 17 patients (94.1%) presented with hepatomegaly. 3 out of 17 patients (17.6%) presented with short stature. 2 out of 17 (11.8%) presented with hypotonia. 1 out of 17 patients (5.9%) presented with abnormal liver function. 1 out of 17 patients (5.9%) presented with hypoglycemia. 1 out of 17 patients (5.9%) presented with splenomegaly.
Of the 15 patients which reported age at diagnosis, the mean age was 2.34 years with a range of 0.5–5.25 years.
3.2.2. Clinical presentation
16 out of 17 patients displayed hepatomegaly (94.1%). Liver enzyme activity as measured through erythrocytes, leukocytes, or hepatocytes was reported for 12 patients. 11 patients displayed low enzyme activity (91.7%). AST/ALT levels were reported for 9 patients, and 7 displayed elevated levels (77.8%). Hypertriglyceridemia was present in 6 (85.7%) out of the 7 patients for which it was reported. Growth was reported for 8 patients, 7 (87.5%) displayed growth retardation. 8 patients of the 16 for which it was reported (50.0%) displayed fasting hypoglycemia. Hypercholesterolemia was present in 1 (50.0%) of the 2 patients for which it was reported. 1 (33.3%) case displayed fasting ketosis out of the 3 for which it was discussed. Development was reported for 12 patients, 3 (25.0%) patients displayed delayed development.
Quantitative AST/ALT levels were reported for 6 patients, 4 were elevated and 2 were normal. Mean AST was 146 U/L. Mean ALT was 115.3 U/L. AST levels ranged from 46 to 274 U/L. ALT levels ranged from 30 to 192 U/L. Quantitative triglyceride level was reported for 1 patient (13.87 mmol/L). Quantitative total cholesterol level was reported for 1 patient (18.87 mmol/L).
3.2.3. Liver biopsy
3 out of 17 patients received a liver biopsy (17.6%). Age at time of biopsy was documented for one patient who received a biopsy at 3.5 years of age. Pathology report was documented for all 3 patients.
2 of the 3 pathology reports (66.7%) displayed no fibrosis. 1 (33.3%) displayed mild fibrosis.
The mean IASL score was 0.33.
3.2.4. Hepatic adenoma
Data regarding the presence or absence of a hepatic adenoma was documented for 3 patients. Only one patient was reported to have a hepatic adenoma.
3.2.5. Liver transplant
No patients were reported as receiving a liver transplant.
3.3. GSD IX γ2
3.3.1. Diagnosis and initial presentation
A total of 30 patients with liver type GSD IX γ2 were gathered from the literature. Initial presentation was reported for 29 out of 30 patients. Of the 30 patients there were 3 separate families (6 patients total) with family history of GSDIX γ2, all families consisted of sibling pairs with GSD IX γ2. The remaining 24 patients did not have family history of GSD IX γ2.
27 out of 29 patients (93.1%) presented with hepatomegaly. 3 out of 29 patients (10.3%) presented with splenomegaly. 10 out of 29 patients (34.5%) presented with frequent hypoglycemia. Additionally, 3 out of 29 patients (10.3%) presented with seizures, 3 out of 29 (10.3%) presented with delayed growth, and 3 out of 29 (10.3%) presented with delayed development. 2 out of 29 patients (6.9%) presented with cholestasis. 2 out of 29 patients (6.9%) presented with “abnormal liver function”.
Of the 26 patients which reported age at diagnosis, the mean age was 1.81 years with a range of 0.3–15 years.
3.3.2. Clinical presentation
All 30 patients (100%) displayed hepatomegaly. AST/ALT levels were reported for 22 patients, and all 22 patients (100%) displayed elevated levels. Fasting ketosis was discussed for 6 patients, and all 6 (100%) displayed fasting ketosis. Fasting hypoglycemia was discussed for 19 patients, and 18 displayed fasting hypoglycemia (94.7%). Liver enzyme activity as measured through erythrocytes, leukocytes, or hepatocytes was reported for 19 patients. 18 patients (94.7%) displayed low enzyme activity. Hypertriglyceridemia was reported in 17 (94.4%) out of the 18 patients for which it was discussed. Growth was documented for 24 patients, 17 patients (70.8%) displayed delayed growth retardation. Development was reported for 20 patients, 10 patients (50%) displayed delayed development. Hypercholesterolemia was documented in 5 patients (45.4%) of the 11 for which it was discussed.
Quantitative AST/ALT levels were reported for 16 patients and all were elevated. Mean AST was 751.1 U/L. Mean ALT was 516.9 U/L. AST levels ranged from 80 to 2000 U/L. ALT levels ranged from 175 to 1125 U/L. Quantitative triglyceride level was reported for 7 patients. The mean triglyceride level was 5.8 mmol/L with a range of 1.75–14.5 mmol/L. Quantitative total cholesterol level was reported for 8 patients. The mean cholesterol level was 9.4 with a range of 37.5–18.4 mmol/L.
3.3.3. Liver biopsy
24 out of 30 patients (80%) received a liver biopsy. Age at time of biopsy was reported for 15 patients. Mean age at time of liver biopsy was 3.03 years with a range of 0.5–20 years. Pathology report was documented for all 24 out of 30 patients.
1 of 24 pathology reports (4.2%) displayed no fibrosis. 5 (20.8%) displayed mild fibrosis. 3 (12.5%) displayed moderate fibrosis. 9 (37.5%) displayed severe fibrosis. 6 (25%) displayed cirrhosis.
23 out of 24 (95.8%) patients displayed either fibrosis (mild, moderate, or severe) or cirrhosis.
The mean IASL score was 2.7.
3.3.4. Hepatic adenoma
Data regarding the presence or absence of a hepatic adenoma was documented for 10 patients. Three patients were reported to have a hepatic adenoma.
3.3.5. Liver transplant
One patient with GSD IX γ2 was reported as receiving a liver transplant at 20 years of age due to cirrhosis of the liver and liver failure. One patient with GSD IX γ2 was reported to have developed hepatocellular carcinoma at 27 years of age and is currently awaiting liver transplant.
3.4. Overall
Table 1 reports the number of patients in each category, for GSD IX α2, β, and γ2 patients. Fig. 1 shows the comparison of age at diagnosis between GSD IX α2, β, and γ2 patients. Fig. 2 show the percentage of reported hypoglycemia, ketosis and hypertriglyceridemia, between GSD IX α2, β, and γ2 patients. Table 2 displays the results from the coding of the pathology reports for GSD IX α2, β, and γ2 patients.
Table 1:
Clinical presentation of all patients with GSD IX published in the literature to date. Symptoms are reflected in fraction format - number of patients with a symptom (numerator) over the total number of patients for which this information was documented (denominator). Symptoms are also reflected as a percentage.
| GSD IX α2 | GSD IX β | GSD IX γ2 | |
|---|---|---|---|
| Total patients (n) | 183 | 17 | 30 |
| Mean age at diagnosis (yrs) | 4 | 2.34 | 1.81 |
| Number of patients with reported age at diagnosis (n) | 132 | 16 | 26 |
| Mean age at liver biopsy (yrs) | 3.09 | 3.5 | 3.03 |
| Number of patients with reported age at biopsy (n) | 34 | 1 | 15 |
| Number of patients with reported Hepatomegaly (n) | 164/176(93.2%) | 16/17 (94.1%) | 30/30 (100%) |
| Number of patients with Fasting hypoglycemia (n) | 53/121 (43.8%) | 8/16 (50%) | 18/19 (94.7%) |
| Number of patients with Fasting ketosis (n) | 21/34 (61.8%) | 1/3 (33.3%) | 6/6 (100%) |
| Number of patients with Hypertriglyceridemia (n) | 64/98 (65.3%) | 6/7 (85.7%) | 17/18 (94.4%) |
| Number of patients with Hypercholesterolemia (n) | 41/90 (45.6%) | 1/2 (50%) | 5/11 (45.4%) |
| Number of patients with Growth delay (n) | 98/168 (58.3%) | 7/8 (87.5%) | 17/24 (70.8%) |
| Number of patients with Developmental delay | 31/88 (35.2%) | 3/12 (25%) | 10/20 (50%) |
| Number of patients with Elevated AST/ALT | 125/138 (90.6%) | 7/9 (77.8%) | 22/22 (100%) |
| Mean AST (U/L) | 328.2 | 146 | 751.1 |
| Mean ALT (U/L) | 274.1 | 115.3 | 516.9 |
Fig. 1.
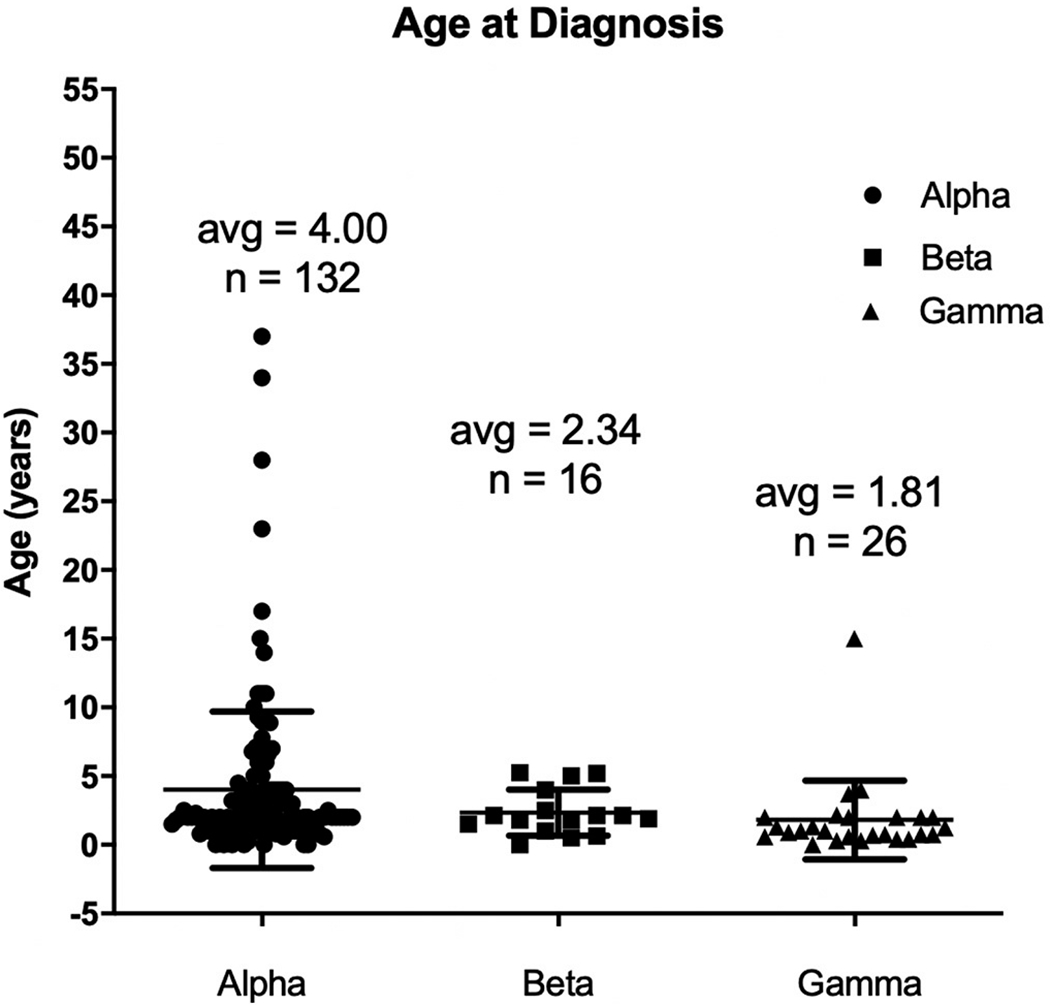
Age at diagnosis for GSD IX patients with GSD IX α2, β, and γ2. Patients with γ2 variant are diagnosed at an earlier age when compared to α2 or β.
Fig. 2.
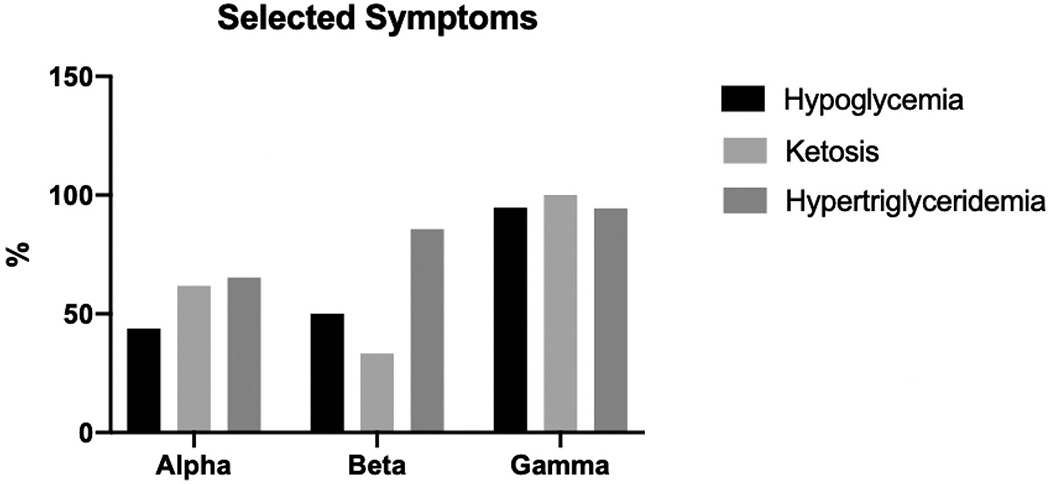
Percentage of GSD IX patients with reported hypoglycemia, ketosis, and hypertriglyceridemia, compared between GSD IX α2, β, and γ2. Patients with γ2 variant have the highest percentage of all three categories, when compared to GSD IX α2 and β patients.
Table 2:
Previously published liver pathology reports were coded using International Association for Study of the Liver (IASL). 95.8% of pathology reports from patients with GSD IX γ2 variant reported liver pathology. Pathology reports from patients with GSD IX α2 and β, were 47.8% and 33% respectively.
| GSD IX α2 | GSD IX β | GSD IX γ2 | |
|---|---|---|---|
| Number of biopsies with path reports | 46 | 3 | 24 |
| Mean score using IASL | 1.13 | 0.33 | 2.7 |
| Percent of cirrhosis [4] | 8.70% | 0.00% | 25.00% |
| Number of cirrhoss | 4 | 0 | 6 |
| Percent of severe fibrosis (3)P | 15.2% | 0.00% | 37.50% |
| Number of severe fibrosis | 7 | 0 | 9 |
| Percent of moderate fibrosis [2] | 8.70% | 0.00% | 12.50% |
| Number of moderate fibrosis | 4 | 0 | 3 |
| Percent of mild fibrosis [1] | 15.20% | 33.33% | 20.80% |
| Number of mild fibrosis | 7 | 1 | 5 |
| Percent of no fibrosis (0) | 52.20% | 66.67% | 4.20% |
| Number of no fibrosis | 24 | 2 | 1 |
| Percent of fibrosis (mild, moderate, severe) or cirrhosis (1,2,3,4) | 47.80% | 33.33% | 95.8% |
| Number of fibrosis (mild, moderate, severe) or cirrhosis (n) | 22 | 1 | 23 |
4. Discussion
Here, we present the first comprehensive literature review including all subtypes of liver GSD IX - GSD IX α2, β, and γ2. From this literature review, we have confirmed previously published findings for liver GSD IX. Presenting signs include hepatomegaly, growth delay, and developmental delay. Common lab findings include ketotic hypoglycemia, hypertriglyceridemia, hypercholesterolemia, elevated AST/ALT, and low enzyme activity [13,20]. Generally, these presenting signs and symptoms have been thought to be benign as they tend to improve with age, especially with GSD IX α2 and β patients. However, our understanding of liver GSD IX is evolving, from a benign disorder to one with significant clinical variability.
The results of our study demonstrated that patients with GSD IX γ2 displayed a more severe phenotype when compared to GSD IX α2 and β. GSD IX γ2 patients on average had a higher percentage of patients with hepatomegaly, fasting hypoglycemia, fasting ketosis, hypertriglyceridemia, and elevated AST/ALT compared to GSD IX α2 and β patients (Table 1, Fig. 2). Quantitative AST/ALT and AST/ALT mean and range were above normal reference values for GSDIX α2, β, and γ2 however, patients with GSD IX γ2 had a higher mean AST/ALT level and range than patients with GSD IX α2 and β (Table 1). In addition, GSD IX γ2 patients were diagnosed at a younger age compared to GSD IX α2 and β patients (Fig. 1). Also, a significantly higher percentage of GSD IX γ2 patients received liver biopsies as compared to GSD IX α2 and β patients (Table 2). All subtypes demonstrated a majority of patients with growth delays. Interestingly, patients with GSD IX β demonstrated the highest percentage of patients with delayed growth. This shows the need for further study regarding the phenotype of a GSD IX β. GSD IX γ2 patients exhibited some form of developmental delay, two times more frequently than GSD IX α2 and β patients (Table 1). These results may have significant implications for future cognitive and physical outcomes for GSD IX γ2 patients later in life.
Patients with GSD IX γ2 demonstrated significant liver fibrosis, when compared to other liver GSD IX subtypes (Table 2). Reported pathology was coded using the categories denoted by the International Association for Study of the Liver (IASL). Of the 24 reported patients of GSD IX γ2 with a recorded liver biopsy, 95.8% of patients had reported fibrosis and/or cirrhosis. This is compared to only 47.8% with fibrosis and or cirrhosis of the 46 GSD IX α2 patients with a liver biopsy we were able to code. In addition, once again, the mean age at diagnosis for GSD IX γ2 patients was younger than GSD IX α2 patients. Overall this provides evidence for the fact that GSD IX γ2 patients suffer from clinically more severe liver symptoms, at an earlier age than patients with mutations in the other subunits.
A potential explanation for this disparity in severity between GSD IX γ2 patients compared to GSD IX α2 and β patients is the location of the mutation and the role of the affected subunit in enzyme functionality. The γ subunit houses the catalytic site which is regulated by the α, β, and δ subunits, and extrinsic calmodulin, which is structurally identical to δ subunit [3]. Phosphorylation of the α and β subunits disinhibits the catalytic site and promotes PhK activation as does the addition of calcium to the δ subunits [3]. Therefore, a mutation in the catalytic site of the PhK enzyme could lead to greater impairment of enzyme activity as compared to a mutation in one of the other three regulatory subunits. However, due to the enzyme’s large size, complexity, and the instability of the subunits when purified, the regulation of the catalytic site has not been fully characterized and more studies are needed to further understand the interaction between the subunits [3].
Based on clinical experience and taking into consideration the young age of most GSD IX γ2 patients at the time of publication, it is likely that with the high prevalence of severe liver disease on biopsy, as liver damage accumulates and progresses many more GSD IX γ2 patients could require a liver transplant in the future [12]. This elucidates the unmet need with regard to treatment of GSD IX γ2. There are currently no non-surgical, minimally invasive, long-term, therapeutic options for any liver GSD IX patients. More studies investigating the clinical presentation and disease progression of liver GSD IX subtypes, specifically GSD IX γ2 will allow for the development of targeted treatment to fill this unmet need.
It is important to note that though GSD IX α2 patients typically display a less severe disease phenotype than those with GSD IX γ2, there is clinical variability within the GSD IX α2 subtype. 47.8% of GSD IX α2 patients with recorded liver biopsies and pathology reports had liver biopsy proven fibrosis and/or cirrhosis. These patients also generally presented with more severe clinical symptoms including growth and/or developmental delay, hepatomegaly, hypoglycemia, ketosis, elevated liver enzymes, hypertriglyceridemia, and hypercholesterolemia compared to others with GSD IX α2. Though no patients with an alpha subunit variant were documented as receiving a liver transplant, taking into account the young mean age at publication, it is possible that patients with a severe liver phenotype may have or will go on to require liver transplantation. Therefore, further follow-up of reported patients is needed in addition to new longitudinal clinical studies.
In this paper we have formalized the nomenclature for the subtypes of liver GSD IX. This method of naming, using the mutated subunit to denote the disease subtype instead of roman lettering or using the mutated gene, allows us to more easily analyze and report information regarding a connection between genotype-and phenotype. We will utilize this subunit centric nomenclature in future literature about liver GSDIX.
Limitations to this review stem from the small number of published patients reports. As a rare disease with nonspecific symptoms that requires genetic testing to confirm diagnosis, liver GSD IX often goes undiagnosed. This review was also limited by the disproportionate number of patients within each subtype. As an X-linked disorder, GSD IX α2 occurs more frequently than autosomal recessive subtypes GSD IX β and GSD IX γ2. This review was also a cross-sectional examination of reported liver GSD IX patients. Most importantly, the published case reports reviewed for this study were written as discrete time points. Longitudinal studies are required to better understand the natural history of liver GSD IX and to understand the variations in liver disease progression over time. In addition, this review is a comprehensive literature review and thus is prone to human error and bias. Our selection criteria excluded articles that were not written in English and articles without genetically confirmed diagnoses of GSD IX. In addition, due to the fact that there is a great amount of variability in the nomenclature of GSD IX currently, it is possible our search strategy might not have covered every single possible case. We mitigated this source of bias by running two separate literature reviews at two different time points with search strategies curated by Duke University Medical Library to ensure we captured the vast majority of the literature.
5. Conclusion
In this paper we present the first comprehensive literature review for liver GSD IX. We provide a standard nomenclature based on mutated subtype, demonstrate evidence for a genotype-phenotype correlation among the subtypes of liver GSD IX, and discuss the clinical variability in GSD IX α2.
Our comprehensive review demonstrates quantitatively that the clinical presentation of GSD IX γ2 patients is generally more severe than that of GSD IX α2 or β patients. We suggest that this is perhaps due to the fact that the γ subunit houses the catalytic site, and damage to this area, as opposed to the regulatory sites, causes greater enzyme impairment and thus more severe disease. However, our study also shows the existence of a severe phenotype, evidenced by early onset liver pathology, in GSD IX α2. A potential hypothesis is that the disease manifestation of liver GSD IX exists on a spectrum. More studies regarding the specific genetic mutations and gene mapping, in conjunction with longitudinal clinical studies, need to be done to investigate this connection.
This comprehensive case review of liver GSD IX demonstrates that there is evidence for a genotype-phenotype connection within liver GSD IX. However, it is also clear that there is a need for a more robust natural history study to better understand the variability in liver pathophysiology within liver GSD IX. In addition, increased study of the mutations and gene mapping could bring a better understanding of the relationship between genotype and clinical presentation. With evidence pointing to the fact that GSD IX γ2 patients suffer from more severe symptoms, it is evident that there is a gap in the literature with regard to specific treatment for this patient population. Lastly, in future literature regarding GSD IX we propose the nomenclature used in this paper, referring to a specific liver GSD IX subtype with the mutated subunit, be standardized and utilized.
Supplementary Material
Acknowledgements
We would like to acknowledge Leticia Flores, Jonathan Stern, and Yajur Sriraman for their contributions.
Abbreviations:
- GSD IX
Glycogen Storage Disease IX
- PhK
Phosphorylase kinase
- AST
Aspartate Transaminase
- ALT
Alanine Transaminase
Footnotes
Declaration of Competing Interest
Priya S. Kishnani has received research/grant support from Sanofi Genzyme, Valerion Therapeutics, and Amicus Therapeutics; consulting fees and honoraria from Sanofi Genzyme, Amicus Therapeutics, Vertex Pharmaceuticals and Asklepios Biopharmaceutical, Inc. (AskBio). She is a member of the Pompe and Gaucher Disease Registry Advisory Board for Sanofi Genzyme, Amicus Therapeutics, and Baebies; and has equity in Actus Therapeutics, which is developing gene therapy for Pompe disease.
References
- [1].Maichele AJ, Burwinkel B, Maire I, Søvik O, Kilimann MW, Mutations in the testis/liver isoform of the phosphorylase kinase γ subunit (PHKG2) cause autosomal liver glycogenosis in the gsd rat and in humans, Nat. Genet. 14 (1996) 337–340 [cited 2020 Mar 10. Available from http://www.ncbi.nlm.nih.gov/pubmed/8896567. [DOI] [PubMed] [Google Scholar]
- [2].Hendrickx J, Willems PJ, Genetic deficiencies of the glycogen phosphorylase system. Vol. 97, Human Genetics, Springer Verlag, 1996, pp. 551–556. [DOI] [PubMed] [Google Scholar]
- [3].Brushia RJ, Walsh DA, Phosphorylase kinase: the complexity of its regulation is reflected in the complexity of its structure, Front. Biosci. J. Virtual Lib. 4 (1999). [DOI] [PubMed] [Google Scholar]
- [4].Herbert M, Goldstein JL, Rehder C, Austin S, Kishnani PS, Bali DS, Phosphorylase Kinase Deficiency, GeneReviews®. University of Washington, Seattle, 1993. [cited 2020 Mar 10]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21634085. [PubMed] [Google Scholar]
- [5].Wüllrich-Schmoll A, Kilimann MW, Structure of the human gene encoding the phosphorylase kinase β subunit (PHKB), Eur. J. Biochem. 238 (2) (1996. Jun 1 [cited 2020 Mar 10]) 374–380. Available from: http://www.ncbi.nlm.nih.gov/pubmed/8681948. [DOI] [PubMed] [Google Scholar]
- [6].Burwinkel B, Hu B, Schroers A, Clemens PR, Moses SW, Shin YS, et al. , Muscle glycogenosis with low phosphorylase kinase activity: mutations in PHKA1, PHKG1 or six other candidate genes explain only a minority of cases, Eur. J. Hum. Genet. 11 (7) (2003. Jul 1) 516–526. [DOI] [PubMed] [Google Scholar]
- [7].Wehner M, Clemens PR, Engel AG, Kilimann MW, Human muscle glycogenosis due to phosphorylase kinase deficiency associated with a nonsense mutation in the muscle isoform of the alpha subunit, Hum. Mol. Genet. 3 (11) (1994. Nov [cited 2020 Mar 10]) 1983–1987. Available from: http://www.ncbi.nlm.nih.gov/pubmed/7874115. [DOI] [PubMed] [Google Scholar]
- [8].Beauchamp NJ, Dalton A, Ramaswami U, Niinikoski H, Mention K, Kenny P, et al. , Glycogen storage disease type IX: high variability in clinical phenotype, Mol. Genet. Metab. 92 (1–2) (2007. Sep) 88–99. [DOI] [PubMed] [Google Scholar]
- [9].Davit-Spraul A, Piraud M, Dobbelaere D, Valayannopoulos V, Labrune P, Habes D, et al. , Liver glycogen storage diseases due to phosphorylase system deficiencies: diagnosis thanks to non invasive blood enzymatic and molecular studies, Mol. Genet. Metab. 104 (1–2) (2011. Sep [cited 2020 Mar 10]) 137–143. Available from http://www.ncbi.nlm.nih.gov/pubmed/21646031. [DOI] [PubMed] [Google Scholar]
- [10].Roscher A, Patel J, Hewson S, Nagy L, Feigenbaum A, Kronick J, et al. , The natural history of glycogen storage disease types VI and IX: long-term outcome from the largest metabolic center in Canada, Mol. Genet. Metab. 113 (3) (2014. Nov 1 [cited 2020 Mar 10]) 171–176. Available from http://www.ncbi.nlm.nih.gov/pubmed/25266922. [DOI] [PubMed] [Google Scholar]
- [11].Pickett-Gies CA, Walsh DA, Phosphorylase Kinase, (1986). [Google Scholar]
- [12].Kishnani PS, Goldstein J, Austin SL, Arn P, Bachrach B, Bali DS, et al. , Diagnosis and Management of Glycogen Storage Diseases Type VI and IX: A Clinical Practice Resource of the American College of Medical Genetics and Genomics (ACMG), ([cited 2019 Nov 2]), 10.1038/s41436- Available from:. [DOI] [PubMed] [Google Scholar]
- [13].Kishnani PS, Goldstein J, Austin SL, Arn P, Bachrach B, Bali DS, et al. , Diagnosis and management of glycogen storage diseases type VI and IX: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG), Genet. Med. 21 (4) (2019. Apr 1 [cited 2020 Mar 10]) 772–789. Available from http://www.ncbi.nlm.nih.gov/pubmed/30659246. [DOI] [PubMed] [Google Scholar]
- [14].Burwinkel B, Amat L, Gray RGF, Matsuo N, Muroya K, Narisawa K, et al. , Variability of biochemical and clinical phenotype in X-linked liver glycogenosis with mutations in the phosphorylase kinase PHKA2 gene, Hum. Genet. 102 (4) (1998. Apr [cited 2020 Mar 10]) 423–429. Available from http://www.ncbi.nlm.nih.gov/pubmed/9600238. [DOI] [PubMed] [Google Scholar]
- [15].Achouitar S, Goldstein JL, Mohamed M, Austin S, Boyette K, Blanpain FM, et al. , Common mutation in the PHKA2 gene with variable phenotype in patients with liver phosphorylase b kinase deficiency, Mol. Genet. Metab. 104 (4) (2011. Dec [cited 2020 Mar 10]) 691–694. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21911307. [DOI] [PubMed] [Google Scholar]
- [16].Wolfsdorf JI, Holm IA, Weinstein DA, Glycogen storage diseases. Phenotypic, genetic, and biochemical characteristics, and therapy, Endocrinol. Metab. Clin. N. Am. 28 (4) (1999. Dec [cited 2020 Mar 10) 801–823. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10609121. [DOI] [PubMed] [Google Scholar]
- [17].Willems PJ, Gerver WJ, Berger R, Fernandes J, The natural history of liver glycogenosis due to phosphorylase kinase deficiency: a longitudinal study of 41 patients, Eur. J. Pediatr. 149 (4) (1990. Jan [cited 2020 Mar 10]) 268–271. Available from http://www.ncbi.nlm.nih.gov/pubmed/2303074. [DOI] [PubMed] [Google Scholar]
- [18].Burwinkel B, Maichele AJ, Aagenaes O, Bakker HD, Lerner A, Shin YS, et al. , Autosomal glycogenosis of liver and muscle due to phosphorylase kinase deficiency is caused by mutations in the phosphorylase kinase beta subunit (PHKB), Hum. Mol. Genet. 6 (7) (1997. Jul [cited 2020 Mar 10]) 1109–1115. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9215682. [DOI] [PubMed] [Google Scholar]
- [19].Van Beurden EACM, De Graaf M, Wendel U, Gitzelmann R, Berger R, Van Den Berg IET, Autosomal recessive liver phosphorylase kinase deficiency caused by a novel splice-site mutation in the gene encoding the liver gamma subunit (PHKG2), Biochem. Biophys. Res. Commun. 236 (3) (1997. Jul 30 [cited 2020 Mar 10]) 544–548. Available from http://www.ncbi.nlm.nih.gov/pubmed/9245685. [DOI] [PubMed] [Google Scholar]
- [20].Bali DS, Goldstein JL, Fredrickson K, Rehder C, Boney A, Austin S, et al. , Variability of disease spectrum in children with liver phosphorylase kinase deficiency caused by mutations in the PHKG2 gene, Mol. Genet. Metab. 111 (3) (2014. Mar [cited 2020 Mar 10]) 309–313. Available from http://www.ncbi.nlm.nih.gov/pubmed/24389071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Burwinkel B, Shiomi S, Al Zaben A, Kilimann MW, Liver glycogenosis due to phosphorylase kinase deficiency: PHKG2 gene structure and mutations associated with cirrhosis, Hum. Mol. Genet. 7 (1) (1998. Jan [cited 2020 Mar 10]) 149–154. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9384616. [DOI] [PubMed] [Google Scholar]
- [22].Li C, Huang L, Tian L, Chen J, Li S, Yang Z, PHKG2 mutation spectrum in glycogen storage disease type IXc: a case report and review of the literature, J. Pediatric Endocrinol. Metab. Walter de Gruyter GmbH 31 (2018. [cited 2020 Mar 10]) 331–338. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29360628. [DOI] [PubMed] [Google Scholar]
- [23].Zachmann M, Sobradillo B, Frank M, Frisch H, Prader A, Bayley-Pinneau, Roche-Wainer-Thissen, and Tanner height predictions in normal children and in patients with various patholgoic conditions, J. Pediatr. 93 (5) (1978. Nov 1) 749–755. [DOI] [PubMed] [Google Scholar]
- [24].Bedossa P, Dargère D, Paradis V, Sampling variability of liver fibrosis in chronic hepatitis C, Hepatology. 38 (6) (2003) 1449–1457. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.