

A novel transgenic mouse, in which the transcription factor NFATc1 bears lysine-to-arginine mutations that prevent modification by SUMO, develops normally and is healthy. However, SUMO-insensitive NFATc1 transmits strong tolerogenic signals, thus preventing autoimmune and alloimmune T cell responses.
Abstract
Posttranslational modification with SUMO is known to regulate the activity of transcription factors, but how SUMOylation of individual proteins might influence immunity is largely unexplored. The NFAT transcription factors play an essential role in antigen receptor-mediated gene regulation. SUMOylation of NFATc1 represses IL-2 in vitro, but its role in T cell–mediated immune responses in vivo is unclear. To this end, we generated a novel transgenic mouse in which SUMO modification of NFATc1 is prevented. Avoidance of NFATc1 SUMOylation ameliorated experimental autoimmune encephalomyelitis as well as graft-versus-host disease. Elevated IL-2 production in T cells promoted T reg expansion and suppressed autoreactive or alloreactive immune responses. Mechanistically, increased IL-2 secretion counteracted IL-17 and IFN-γ expression through STAT5 and Blimp-1 induction. Then, Blimp-1 repressed IL-2 itself, as well as the induced, proliferation-associated survival factor Bcl2A1. Collectively, these data demonstrate that prevention of NFATc1 SUMOylation fine-tunes T cell responses toward lasting tolerance. Thus, targeting NFATc1 SUMOylation presents a novel and promising strategy to treat T cell–mediated inflammatory diseases.
Graphical Abstract
Introduction
Nuclear factor of activated T cells (NFAT) proteins belong to a family of transcription factors whose genuine members are designated as NFATc1 (or NFAT2), NFATc2 (NFAT1), NFATc3 (NFAT4), and NFATc4 (NFAT3). In lymphoid cells, NFAT proteins are activated upon antigen receptor engagement, which enables Ca2+/calcineurin-mediated dephosphorylation of preformed cytoplasmic NFATs and their consecutive nuclear translocation. NFATc1 and NFATc2 are highly expressed in peripheral T cells and control, among many other important genes, the expression of subtype-defining lymphokines such as IFN-γ, IL-4, and IL-17, lately visualized by chromatin immunoprecipitation (ChIP) sequencing (Klein-Hessling et al., 2017; Martinez et al., 2015; Serfling et al., 2000). Interleukin 2 (Il2) was the first identified target gene, and substantial amounts of IL-2 are secreted by CD4+ T cells that have been recently activated by antigen. IL-2 binds to a common γ chain–containing receptor, either with intermediate affinity, when paired with the IL-2 receptor β chain CD122, or with high affinity, when present as a trimeric receptor including the IL-2 receptor α unit CD25 (Boyman and Sprent, 2012). In conventional T cells (T conv), expression of CD25 (Il2ra, another known NFAT target gene; Porter and Clipstone, 2002; Schuh et al., 1998) is induced shortly after antigen exposure, enabling activated T conv to respond well to secreted IL-2 as a growth signal. Regulatory T cells (T regs) constitutively express the high-affinity, trimeric IL-2 receptor in line with IL-2 being essential for generation and homeostasis of T regs (Fontenot et al., 2005). Contact of T regs with IL-2 maintains high levels of CD25 as well as expression of forkhead box P3 (Foxp3) as their key transcriptional regulator.
All NFATs share a common DNA binding domain, which is very similar in its conformation to the Rel domain of Rel/NF-κB factors and was therefore designated Rel similarity domain (Hogan et al., 2003; Macian, 2005; Serfling et al., 2000). The N termini of NFAT proteins harbor a strong transactivation domain and a regulatory domain. Calcineurin recognizes several phosphorylation motifs within the regulatory domain and controls the nuclear translocation and, thus, the activity of NFATs. T lymphocytes can express six different NFATc1 isoforms (Serfling et al., 2006). These isoforms are generated by the usage of two different promoters, the inducible P1 and the constitutive P2, which control the expression of the so-called α or β N-terminal peptide, respectively. In addition, alternative splicing and different termination signals generate the C-terminal domains A, B, and C. The longest isoforms NFATc1/αC and NFATc1/βC harbor an extra C-terminal peptide of 246 aa, constituting an additional transactivation domain, TAD-B (Chuvpilo et al., 1999). Notably, this extra C terminus of NFATc1 is homologous to the C terminus of NFATc2 (Chuvpilo et al., 1999). Previously, we demonstrated that SUMO1 posttranslationally modifies NFATc1/C within its C terminus at the lysine residues K702 and K914 (Nayak et al., 2009). SUMOylation at K702 and K917 is requisite for modification at the common site K349, and K702/K914R mutants equal K349/702/917R mutants (Nayak et al., 2009). Although NFATc2 is SUMOylated on homologous sites (Terui et al., 2004), SUMOylation of NFATc1 and NFATc2 results in different functional outcomes. While SUMOylation of NFATc2 is a requisite for nuclear anchorage (Terui et al., 2004), SUMOylation turns NFATc1 into a repressor of Il2 expression (Nayak et al., 2009).
SUMOylation is a posttranslational modification process and can affect a protein’s structure and subcellular localization. SUMOylation is achieved in an enzymatic cascade of E1, E2, and E3 enzymes resembling ubiquitination. However, only a single E2 SUMO-conjugating enzyme, Ubc9, is known (Flotho and Melchior, 2013). The presence of sentrin/SUMO-specific proteases (SENP) ensures reversibility. SUMOylation alters interaction with partner proteins or DNA and orchestrates many biological processes (Flotho and Melchior, 2013). The overall effect of SUMOylation for the immune system has been addressed in conditional Ubc9- or SENP–deficient mice (Decque et al., 2016; Ding et al., 2016; Van Nguyen et al., 2012; Wang et al., 2017; Yu et al., 2018). However, to our knowledge, individual SUMO site–deficient proteins have not been expressed endogenously, i.e., not overexpressed, but under normal locus control in place of the SUMOylatable WT protein.
Here we show that SUMOylation of NFATc1 fine-tunes T cell immune responses in vivo. We generated an Nfatc1 mutant mouse model with lysine-to-arginine point mutations at the two SUMOylation sites K702 and K914. These Nfatc1deltaSUMO mice, solely deficient in NFATc1 SUMOylation, were largely protected from autoimmune and alloimmune diseases such as experimental autoimmune encephalomyelitis (EAE) and acute graft-versus-host disease (aGvHD). Abolished NFATc1 SUMOylation in NFATc1/ΔS+ T cells led to enhanced IL-2 production that promoted T reg expansion and blunted effector lymphokine expression. Thus, posttranslational modification of NFATc1 with SUMO adjusts T cell activation and IL-2 production and presents a novel and promising target to treat T cell–mediated inflammatory diseases.
Results
Endogenous NFATc1/ΔS leads to more IL-2+ but fewer IFN-γ+ and IL-17A+ T cells
Previously, we demonstrated by overexpression studies using cell lines that NFATc1 SUMOylation recruits histone deacetylases, which increases site-specific chromatin condensation and represses the Il2 gene (Nayak et al., 2009). To understand the role of SUMO modification for T cell–mediated immune responses in vivo, we generated a novel mouse strain, which expresses only SUMO-deficient (non-SUMOylatable) NFATc1, termed NFATc1/ΔS here. The codons for lysine K702 and K914, both within exon 10, were mutated to encode arginine replacements, a common substitute with the other positively charged amino acids (Fig. 1 A). The site-specific mutations were confirmed by sequencing (Fig. 1 B), verifying the successful K-to-R mutations of the two SUMOylation sites in Nfatc1deltaSUMO knock-in mice. Immunoblots exemplified comparable expression levels and isoform distribution of NFATc1 and NFATc1/ΔS in total lymphocytes as well as in cultures of naive CD4+ T cells (Fig. 1, C and D). Moreover, nuclear presence of NFATc1/ΔS equaled that of WT NFATc1 (Fig. 1 E). When we isolated CD4+ T cells from Nfatc1deltaSUMO and WT littermates to activate them shortly ex vivo, we observed an enhanced frequency of IL-2–producing cells upon the avoidance of NFATc1 SUMOylation (Fig. 1 F). This did not influence the proliferation behavior in vitro (Fig. 1 G).
Figure 1.

NFATc1 is normally expressed even with successful lysine → arginine mutations or C-terminal deletion. (A) Strategy of creating a mouse expressing NFATc1/ΔS in conjunction with the possibility for NFATc1/ΔBC. Both relevant SUMO sites reside in exon 10, which translates into most of the exclusive amino acids of NFATc1/C, whereas only 15 amino acids are encoded in exon 11 (murine NFATc1/αC = 939 aa). The nondepicted NFATc1/B terminates roughly in the middle of exon 10 (at aa 827). The addition of loxP sites within introns 9 and 10 facilitates the specific loss of the long isoforms B and C. In the case of crossing with a Cre-deleter mouse, only NFATc1/αA, initiated at the inducible promoter P1, and constitutive P2-dependent NFATc1/βA can be expressed. (B) Representative sequencing data of Nfatc1 from mouse tail DNA revealed point mutations leading to lysine → arginine exchanges at position 702 and 914 in Nfatc1deltaSUMO mice (n = 3). (C and D) Immunoblot analysis of whole-cell extracts, prepared from spleen and LNs of WT and Nfatc1deltaSUMO mice, detecting expression of NFATc1 isoforms as well as NFATc2 in comparison to β-actin; total lymphocytes stimulated for 6 h by T/I (C) or CD4+ T cells stimulated by anti-CD3/CD28 plus IL-2 for 48 and 72 h (D). (E) Protein expression of WT NFATc1 and NFATc1/ΔS in nuclear and cytoplasmic extracts. CD4+ T cells from spleen and LNs were stimulated with ConA for 1 or 4 d and restimulated for 6 h with T/I as indicated (immunoblots exist in several further variations). (F) Intracellular IL-2 expression in WT and NFATc1/ΔS+ CD4+ T cells, stimulated for 8 h with T/I. Representative flow cytometry plots and comparison of 11 individual mice/group; Student’s t test ± SEM (**, P < 0.005). (G) Proliferation of NFATc1/ΔS or WT CD4+ T cells, stimulated with either anti-CD3 or anti-CD3/CD28 in the presence or absence of exogenous IL-2, and analyzed by CFSE dilution (n = 3).
Boosted IL-2 expression was still evident after in vitro differentiation of naive NFATc1/ΔS+ CD4+ T cells toward T helper 1 cell (Th1) and Th17 (Fig. 2 A) and in line with enhanced acetylation found at the Il2 promoter in these cells (Fig. 2 B). In contrast, the numbers of cells positive for IFN-γ and IL-17A were reduced (Fig. 2 A). Enhanced IL-2 but diminished IFN-γ expression was in accordance with our former overexpression data in mouse and human T cell lines (Nayak et al., 2009). Interestingly, IL-17A expression was likewise hindered in the presence of NFATc1/ΔS.
Figure 2.

Inhibiting NFATc1 SUMOylation triggers CD4+ T cells to express more IL-2 but less IFN-γ and IL-17A. (A) Th1 and Th17 in vitro differentiation of naive CD4+ T cells from WT and Nfatc1deltaSUMO mice. On day 3, CD4+ T cells were restimulated with T/I followed by intracellular cytokine staining of IL-2 and IL-4 or IL-17A and IFN-γ. Representative flow cytometry plots and comparison of WT versus Nfatc1deltaSUMO CD4+ T cells (Student’s t test ± SEM; n = 10). (B) Chromatin of Th1 and Th17, prepared as for A, was precipitated by anti–H3-Ac. Isolated genomic DNA was subjected to PCR with primers specific for the Il2 and Actb promoters. Student’s two-tailed t test (*, P < 0.05; **, P < 0.005; ***, P < 0.001); mean + SD; n > 3. (C) RT-PCR with primers specific for exon 9 + 11 (Δexon10) showed deletion of the C terminus of Nfatc1 mRNA in CD3+ T cells of Nfatc1deltaBC [Nfatc1deltaSUMO × Cd4cre] but not in CD3− cells nor in WT cells. Primers binding to exon 9 + 10 detected WT Nfatc1 mRNA; n > 3. (D) Immunoblot analysis of unstimulated (w/o) and 6 h stimulated (T/I) CD4+ T cells from WT and Nfatc1deltaBC mice illustrated the differential isoform expression of NFATc1/ΔBC; n > 3. (E) Th1 and Th17 in vitro differentiation of naive CD4+ T cells from WT and Nfatc1deltaBC mice. Analyses as in A; n > 10. (F) ELISA of IL-2 in the supernatant of CD4+ T cells, derived from WT, Nfatc1deltaSUMO, and Nfatc1deltaBC mice and stimulated by anti-CD3 or anti-CD3/CD28 for 48 h. Student’s t test (*, P < 0.05; **, P < 0.005); n = 3.
Deletion of the C-terminal domain of NFATc1 causes increased IL-2 expression and fewer effector cytokines
Both K702 and K914 are encoded in exon 10 and might be responsible for the special features of the long isoform NFATc1/C (Nayak et al., 2009). This “loss of SUMOylation” version of NFATc1 was complemented with the possibility to erase the B and C isoform–specific C terminus by framing exon 10 with loxP sites (Fig. 1 A). By use of Cd4-cre, which is first expressed at the CD4+ CD8+ double-positive stage of thymocytes and presumably ahead of thymic T reg (tT reg) induction (Josefowicz and Rudensky, 2009), a mouse was generated in which T cells can give rise only to the short isoforms NFATc1/αA and NFATc1/βA (Nfatc1deltaBC). Consequently, we sorted CD3+ and CD3− cells from Nfatc1deltaBC mice and analyzed RNA transcripts before and after cell stimulation (Fig. 2 C). Using primer pairs recognizing exon 9 plus exon 11, but not exon 9 plus exon 10 (WT), provoked a small cDNA fragment. This indicated the deletion of the C terminus of NFATc1 in T cells before and after stimulation, but not in other leukocytes derived from the same mouse. Accordingly, as protein extracts of CD4+ T cells from WT mice gave rise to the usual six isoforms running faster after activation-induced dephosphorylation (Serfling et al., 2000), expression of NFATc1 concentrated in αA and βA isoforms in those of Nfatc1deltaBC (Fig. 2 D). This demonstrated the loss of the long isoforms NFATc1/B and NFATc1/C, whereas P1- and P2-mediated α- and β-peptide–containing short isoform expression (Serfling et al., 2012) and their activation via dephosphorylation remained unaltered.
To evaluate whether CD4+ T cells expressing NFATc1/ΔBC behaved similar to NFATc1/ΔS, we also differentiated naive NFATc1/ΔBC+ CD4+ T cells under Th1 or Th17 conditions. Fewer NFATc1/ΔBC+ CD4+ T cells expressed IFN-γ and IL-17A compared with WT T cells, whereas the frequency of IL-2–producing cells increased (Fig. 2 E). To compare NFATc1/ΔBC and NFATc1/ΔS directly, we cultured both NFATc1 mutants and WT CD4+ T cells in the presence of T cell receptor engagement (anti-CD3) with or without costimulation through CD28 and revealed the amount of secreted IL-2. Notably, not only the T cells derived from Nfatc1deltaSUMO, but also those from Nfatc1deltaBC, produced more IL-2 compared with WT T cells (Fig. 2 F). This suggests that the NFATc1/C-specific C terminus functions through SUMOylation to fine-tune cytokine expression.
Nfatc1deltaSUMO mice are largely protected from EAE pathology
Nfatc1deltaSUMO mice revealed similar cell numbers in thymus, spleen, and LNs as in WT mice (Fig. S1 A). Under steady-state conditions, WT and Nfatc1deltaSUMO mice appeared healthy, and T cell subtypes and state of activation and differentiation, including T regs, were parallel between the two genotypes (Fig. S1, B–E). To investigate whether NFAT SUMOylation affects the suppressive capacity of T reg cells, we compared the ability of WT and NFATc1/ΔS+ T regs to suppress proliferation of activated WT T conv in vitro. We found no significant difference in their suppressive capacity, suggesting that NFAT SUMOylation is not required for normal T reg function (Fig. S1 F).
Figure S1.

Lymphoid compartments appear unaltered in Nfatc1deltaSUMO mice in steady state. (A) Total cell number in thymus, spleen, and LNs of WT versus Nfatc1deltaSUMO mice (mean + SD, n > 3). (B) Determination of the number of B and T cells in spleen and LN of WT and Nfatc1deltaSUMO mice by surface staining to B220 and CD3ɛ and flow cytometry. (C and D) Distribution of CD4+ and CD8+ T cells (C) as well as frequency of CD25+ Foxp3+ T reg cells within the CD4+ T cell compartment (D) in thymus, spleen, and LNs of WT versus Nfatc1deltaSUMO mice analyzed by flow cytometry. (E) Detection of the activation status of CD4+ T cells in untreated WT versus Nfatc1deltaSUMO mice, given by representative flow cytometry for resting (CD62L+) and activated (CD69+) CD4+ T cells in spleen or LN. (F) Capacity of CD4+CD25+ T regs, isolated from WT and Nfatc1deltaSUMO mice, to suppress WT CD4+ T cells, stimulated on irradiated splenic cells with anti-CD3. The ratio of T conv over T regs are indicated. Bars show mean + SD of three experiments.
To evaluate whether the observed changes in T cell cytokine production in vitro translate into complex T cell–mediated immune responses in vivo, we challenged Nfatc1deltaSUMO mice by the induction of EAE. EAE serves as an animal model for multiple sclerosis and is initiated by autoreactive CD4+ T cells, predominantly Th17 cells, which infiltrate the central nervous system (CNS). Recognition of myelin-associated antigens leads to activation of NFAT (Dietz et al., 2015; Lodygin et al., 2013). When we subjected Nfatc1deltaSUMO mice to MOG35–55–induced EAE, they were partially protected in comparison with WT littermates (Fig. 3 A). However, peripheral CD4+ T cells from diseased animals proliferated equally well upon recall with MOG35–55 peptide (Fig. 3 B), ruling out that altered T cell expansion is causative for ameliorated EAE pathology. Supernatants of these cultures exhibited strongly reduced levels of IL-17A and IFN-γ (Fig. 3 B). Importantly, this also became evident in CNS, where fewer CD4+ T cells expressed IFN-γ, IL-17, and GM-CSF, culminating in the reduction of pathogenic “Th1/17” cells (Fig. 3 C; Langrish et al., 2005; Peters et al., 2011). In addition, NFATc1/ΔS+ CD4+ T cells from EAE-induced mice synthesized an enhanced level of IL-10, which is reported to counteract the development of pathogenic Th1/17 cells (Fig. 3 D; Huber et al., 2011). Collectively, these data show that as NFATc1 SUMOylation regulated IL-2, IL-10, IFN-γ, and IL-17 production in vitro and in vivo, NFATc1/ΔS protected from cerebral autoimmunity in EAE models.
Figure 3.

Nfatc1deltaSUMO mice are less susceptible to EAE and produce fewer proinflammatory cytokines. (A) WT (n = 4) and Nfatc1deltaSUMO (n = 4) mice were immunized subcutaneously on day 0 with 50 µg MOG35–55 peptide in CFA. Mice were injected i.p. with 200 ng/mouse of pertussis toxin on days 0 and 2. EAE development in mice was assessed by a 0–6 disease scoring system over a period of 18 d. Shown is one representative clinical score of 10 individual EAE experiments. Statistical analysis of mean daily score was evaluated by Mann–Whitney U test followed by Bonferroni posttest (*, P < 0.05; **, P < 0.005). (B) MOG35–55 restimulation (0, 3, 10, and 30 µg/ml) of splenic/inguinal LN cells from EAE-diseased WT or Nfatc1deltaSUMO mice for 3 d. Proliferation was quantified by 3[H]thymidine incorporation. Bars indicate mean of stimulation index (SI) + SD, pooled from three experiments. IL-2, IL-17A, and IFN-γ secretion was analyzed by ELISA. Bars show mean + SD, pooled from two experiments. Statistical differences were determined by two-way ANOVA (**, P < 0.005; ***, P < 0.001). (C) Analysis of cytokine production of CNS-infiltrated CD4+ T cells (CD4 gate) at peak of EAE (day 15 after induction). CNS infiltrates of EAE mice were isolated and restimulated with T/I for 5 h followed by intracellular cytokine staining. Shown are representative plots for WT or Nfatc1deltaSUMO mice and the statistical evaluation of cytokine-positive cells of eight mice per group (IL-17A, IFN-γ, and IL-17A/IFN-γ) and three mice per group (GM-CSF). Statistical differences were determined by Student’s t test; *, P < 0.05; **, P < 0.005; mean ± SEM. (D) Intracellular staining of IL-10 versus IL-17A plus IFN-γ in CD4+ T (gate) cells in CNS infiltrates of individual EAE mice as in C.
NFATc1/ΔS expression favors T reg expansion during EAE
Analyses of CNS infiltrates of acute EAE-diseased Nfatc1deltaSUMO mice revealed the presence of more CD45+ CD11b− lymphocytes, which could be attributed to more CD4+ T cells (Fig. 4, A and B). Notably, however, and in line with the ameliorated disease score, EAE-diseased Nfatc1deltaSUMO mice exhibited more CD4+ CD25+ Foxp3+ T regs that tilted the ratio of T regs over T conv. This phenomenon became more significant over time, especially in the recovery phase after 2 wk (Fig. 4 C). Likewise, absolute numbers of T regs tended toward an increase in the CNS, while those of Th1 and Th17 seemed to be fewer (Fig. 4 D). Cryosections from spinal cords underlined a shifted balance toward CD4+ Foxp3+ T regs over CD4+ T conv in EAE-induced Nfatc1deltaSUMO mice (Fig. 4 E). Therefore, the presence of NFATc1, which cannot be SUMOylated, resulted in T regs that are more protective during EAE.
Figure 4.

EAE-diseased Nfatc1deltaSUMO mice exhibit more T regs in the CNS. (A) Flow cytometric analysis of CNS-infiltrating cells isolated from EAE-diseased WT and Nfatc1deltaSUMO mice: lymphocytes (CD45highCD11b−), macrophages and neutrophils (CD45highCD11bhigh), and microglia cells (CD45lowCD11bhigh). Shown are representative flow cytometry plots and the statistical evaluation of 15 mice/group. Student’s t test; *, P < 0.05. Mean + SEM. (B) Frequency of CD4+ T cells within CNS infiltrates of EAE-diseased WT and Nfatc1deltaSUMO mice. Shown are representative flow cytometry plots and the compilation of 15 mice/group. Student’s t test; **, P < 0.005. Mean + SEM. (C) Detection of T regs in the CNS of WT and Nfatc1deltaSUMO mice after EAE induction by flow cytometry. CNS infiltrates were stained for CD4, CD25, and intracellular Foxp3 on the indicated days after EAE induction. Shown are representative plots and the statistical evaluation. Differences between groups were calculated with Student’s t test; *, P < 0.05; **, P < 0.005. Mean + SEM. (D) Absolute cell numbers (number in CNS-collected cells × percent CD4+ [n = 4] × percent IFN-γ+ [n = 3], IL-17+ [n = 3], or Foxp3+ [n = 4]) were determined on days 22–26. (E) Immunofluorescence staining of spinal cord cryosections of WT and Nfatc1deltaSUMO EAE-induced mice (magnification, 40×; scale bar, 20 µm). Sections were stained with anti-CD4 (Alexa Fluor 488), anti-Foxp3 (Cy3), and DAPI. Shown are two individual spinal cords for both genotypes each. White numbers indicate counted (in a blinded manner) CD4+ and Foxp3+ cells.
Elevated IL-2 production of NFATc1/ΔS+ T cells represses Il17a and enhances Foxp3 expression
IL-2 receptor–mediated signals have been reported to oppose Il17a transactivation (Laurence et al., 2007), which we could verify by adding either hrIL-2 or anti–mIL-2 to Th17 differentiation cultures (Fig. 5 A). Accordingly, when NFATc1/ΔS+ CD4+ T cells were differentiated toward Th17 cells, which resulted in more IL-2–expressing but fewer IL-17A–expressing cells compared with WT Th17 cells, the blockade of IL-2 completely restored IL-17A induction and even augmented the frequency of IL-17A+ cells, as well as the amount of secreted IL-17 (Fig. 5 B). STAT3-dependent IL-17 expression is thought to be halted by IL-2–induced STAT5 phosphorylation. Indeed, we found significantly elevated amounts of STAT5 at the Il17 locus (Fig. 5 C). In contrast to IL-17 expression, the addition of IL-2 enhanced the number of Foxp3+ cells during Th17 differentiation (Fig. 5 D). The potential benefit of elevated levels of IL-2 for peripheral T regs (pT regs), i.e., CD4+ CD25+ Foxp3+ cells induced from peripheral CD4+ T cells, could be demonstrated during in vitro differentiation of naive CD4+ T cells. So-called induced T reg (iT reg) induction in the presence of TGF-β but without exogenous IL-2 resulted in significantly enhanced expression of Foxp3 in NFATc1/ΔS+ CD4+ T cells compared with WT controls (Fig. 5 E). The addition of an anti–mIL-2 antibody during iT reg differentiation not only lowered, but also equalized, Foxp3 induction. These data suggest that the enhanced IL-2 availability caused by NFATc1/ΔS tilts the balance to reduced IL-17A and increased Foxp3 induction in peripheral CD4+ T cells.
Figure 5.

NFATc1/ΔS-induced overexpression of IL-2 limits IL-17A production and favors Foxp3 expression. (A) Th17 differentiation of naive CD4+ WT T cells with the addition of human IL-2 (75 U/ml) and/or anti-murine IL-2–neutralizing antibodies (10 µg/ml). On day 3, Th17 cells were restimulated with T/I followed by intracellular cytokine staining of IL-17A and IFN-γ. Shown are representative flow cytometry plots of Th0 (control) and Th17 cultures of three individual experiments. (B) Th17 differentiation of naive CD4+ T cells gained from WT and Nfatc1deltaSUMO mice under normal and mIL-2–neutralizing conditions as performed for A. Representative flow cytometry plots of intracellular staining of IL-2 and IL-17A of three individual experiments. Amount of secreted IL-17 was determined from the supernatants of Th17 cultures (n = 6) with increasing concentrations of anti–mIL-2. (C) ChIP assay of Th17-differented cells for STAT5 bound to Il17 promoter (site 4) and a nonbinding site (n = 4). (D) Foxp3+ expression was evaluated in WT CD4+ T cells after the addition of IL-2 (75 U/ml rhIL-2) to otherwise Th17-inducing conditions; analyzed by intracellular flow cytometry on day 3. (E) Naive CD4+ T cells from WT and Nfatc1deltaSUMO mice were polarized to iT regs by supplementation with TGFβ, but without the addition of exogenous IL-2; parallel cultures were in the presence of 10 µg/ml anti–mIL-2. On day 3, cells were analyzed by intracellular flow cytometry of Foxp3. Data are shown as representative flow cytometry plots for Foxp3+ cells. Statistics were evaluated from five individual experiments. Statistical evaluation for B, C, and E was achieved by Student’s t test, two-tailed (*, P < 0.05; ***, P < 0.001); mean + SD. (F) CD4+ T cells, isolated from LN and spleen, were differentiated for Th1, Th2, and Th17. On days 2.5 and 3.5, RNA was extracted and subjected to NGS. The heatmap shows 38 differentially expressed genes within all WT versus NFATc1/ΔS+ T cells of both days determined using the edgeR’s glm model.
To gain a broader picture of differential gene expression in Th1 and Th17 cells, we determined the transcriptome of in vitro differentiated Th1 and Th17 while including Th2. After 2.5 and 3.5 d, RNA was extracted and subjected to next-generation sequencing (NGS). On day 2, part of the cells were restimulated and checked for cytokine expression by flow cytometry, showing enhanced IL-2 and less effector cytokine expression (Fig. S2 A). In general, transcriptional changes, measured without restimulation, were mild, more pronounced on day 2.5, and most distinct in Th1 cells (Fig. S2, B, D, and E). Strikingly in Th1, NFATc1/ΔS limited gene expression (and therefore pathways) rather than supporting them (Fig. S2, B and C). Comparisons within the individual T helper subtypes on day 2.5 could not uncover an influence on cytokine expression except for less Il5 in Th2 and less Il13f in Th17 from Nfatc1deltaSUMO mice (Fig. S2, B, D, and E). However, we also contrasted WT Th1, Th2, and Th17 to all NFATc1/ΔS+ ones for both time points and found 38 genes significantly different between the two genotypes (Fig. 5 F). In line with protein data, Il2 RNA appeared transiently increased in NFATc1/ΔS+. In Th17-skewed cells, Il17a and Il17f were fewer from the beginning on, whereas Ifng was surprisingly enriched in early NFATc1/ΔS+ Th1. Although the frequency of IL-13+ cells did not decrease significantly in the sum of our in vitro cultures (Fig. S2 F), mRNAs for Il5 and Il13 were clearly reduced in NFATc1/ΔS+ Th2, verifying our Il13 data with infected EL-4 cells (Nayak et al., 2009). On the other hand, early NFATc1/ΔS+ Th1 expressed some more Il4, Il5, and Il13 in comparison with WT cells. CD4+ T cells can also acquire a cytotoxic phenotype (Juno et al., 2017), and we noticed that distinct Gzma expression in Th2 was less in NFATc1/ΔS+ than in WT cells. Overall, Ctla4 appeared diminished in NFATc1/ΔS+ T cells, characterizing them as less effector-like. Furthermore, Gdpd3 mRNA encoding a glycerophosphodiester phosphodiesterase, which has been described to mark central memory T cells (Kaji et al., 2016), was almost absent in NFATc1/ΔS+ T cells, assuming the lack of a certain metabolic shift. CD24 or heat shock antigen, serving as a costimulatory factor for T cells, has been connected to severity in EAE (Bai et al., 2000) and was distinctly less expressed in NFATc1/ΔS+ T cells, again in line with a protected phenotype of Nfatc1deltaSUMO mice upon EAE induction.
Figure S2.

Expression analyses of in vitro differentiated Th1, Th2, and Th17. (A) For NSG analyses, naive CD4+ T cells from WT and Nfatc1deltaSUMO mice were skewed toward Th1, Th2, and Th17. Differentiation was verified on day 2.5, as some CD4+ T cells were restimulated with T/I followed by intracellular cytokine staining of IL-2 and the key cytokines IFN-γ, IL-4, or IL-17A. (B, D, and E) Heatmaps calculated for Th1 (B), Th2 (D), or Th17 (E) individually between WT and NFATc1/ΔS+ (P = 0.02, log fold-change 1.2). A comparison of the differently expressed genes in the respective subset is given for the two other subsets. (C) Pathway enrichment analysis for NGS data of Th1, day 2.5, conducted using clusterProfiler; top 10 pathways are shown. Red (up-regulation) and blue (down-regulation) dots in the category netplot depict the log fold-changes of gene expression in NFATc1/ΔS+ Th1. (F) In vitro stimulation of CD4+ T cells under Th2 differentiating conditions and intracellular expression of IL-2 and IL-13 after 3 d and additional 5 h of T/I restimulation; n = 5. Student’s t test (*, P < 0.05); mean + SD.
NFATc1/ΔS in Th1 and Th17 ameliorates disease scores during passive EAE
To test the functional difference of WT and NFATc1/ΔS+ Th1 and Th17 in an isolated manner, we transferred MOG35–55-specific CD4+ T cells from 2D2.WT and 2D2.Nfatc1deltaSUMO mice, which had been differentiated in vitro toward Th1 and Th17 (passive EAE; Fig. 6 A). To level out the IL-2 effect, anti–mIL-2 was added to analogous cultures and later to such transplanted animals. Frequencies of IL-2+, IFN-γ+, and IL-17A+ T cells were evaluated after restimulation (Fig. 6 B), parallel to transplantation of mixed non-restimulated Th1 and Th17 cultures. Under Th17-skewing conditions, a restrained amount of iT regs was induced (Fig. 6 C). Expectedly, NFATc1/ΔS+ T cells elicited more Fopx3+ cells. Unlike sole TGF-β cultures (Fig. 5 E), though, anti–mIL-2 did not diminish Foxp3 expression under the influence of TGF-β and IL-6 (Fig. 6 C). Animals that received NFATc1/ΔS+ Th1 plus Th17 experienced ameliorated disease severity in comparison with WT-transplanted animals (Fig. 6 D), whereas the differential cytokine expression was preserved in T cells regained from the CNS (Fig. 6 E). Surprisingly, blocking IL-2 during cell culture and additionally in the EAE-induced animals resulted in less severe clinical scores. This might be due to compensatory enhanced IL-2 expression upon external block with anti–mIL-2 (Fig. 6 B) and the slightly elevated iT reg induction (Fig. 6 C) and/or reduced T cell proliferation in the presence of anti–mIL-2. Nevertheless, anti–mIL-2 equalized disease scores of 2D2.WT versus 2D2.NFATc1/ΔS-transplanted mice (Fig. 6 D), once more suggesting that an NFATc1/ΔS-driven enhanced IL-2 production could be causative for the differential behavior.
Figure 6.

IL-2–dependently, NFATc1/ΔS+ Th1 and Th17 transmit less EAE than WT counterparts. (A) Passive EAE approach by transfer of 2D2+ Th1 and Th17 cells, differentiated in vitro for 3 d and combined for transfer into lymphocyte-deficient mice. 5 × 106 Th1/Th17 cells were injected i.v. into three Rag1−/− mice per group. Parallel cultures and transplanted mice were treated with anti–mIL-2. Three independent experiments were performed. (B) Th1 and Th17 in vitro differentiation, with (w/) and without (w/o) anti–mIL-2, of naive CD4+ T cells from 2D2.WT and 2D2.Nfatc1deltaSUMO mice ahead of transfer. On day 3, a fraction of the cells were restimulated with T/I followed by intracellular cytokine staining of IL-2 and IFN-γ or IL-2 and IL-17A. Representative flow cytometry plots and comparison of WT versus Nfatc1deltaSUMO CD4+ T cells. (C) CD4+ T cells from Th17-skewing conditions were checked for Foxp3 expression by flow cytometry. (D) Weight and clinical scores of mice after passive EAE induction described in A; Mann–Whitney U test; *, P < 0.05. (E) Analysis of IFN-γ versus IL-17 and GM-CSF versus IL-10 expression in CNS-infiltrating CD4+ T cells (CD4 gate) 20 d after induction and restimulated with T/I for 5 h ex vivo. Shown are representative plots and the statistical evaluation of cytokine-positive cells of three mice per group. Statistical differences were determined by Student’s t test; *, P < 0.05.
NFATc1/ΔS renders transplanted T cells less pathogenic during aGvHD
To further evaluate the role of NFATc1 SUMOylation in pathogenic T cell responses, we investigated T cells derived from Nfatc1deltaSUMO mice in aGvHD. GvHD constitutes a major risk for patients undergoing allogeneic hematopoietic stem cell transplantation (allo-HCT). Because of immunological incompatibility between donor T cells and host tissue, T lymphocytes are activated and undergo massive proliferation in the secondary lymphoid organs (SLOs). Then, alloreactive T cells infiltrate the aGvHD main target organs (skin, liver, and intestine), where they cause severe tissue damage and inflammation (Beilhack et al., 2005). We used an MHC major mismatch HCT model to analyze T cell responses during aGvHD (C57BL/6, H-2b into BALB/c, H-2d; Fig. S3 A). Mice receiving NFATc1/ΔS+ T cells survived better, had a less severe clinical score, and lost less weight than WT T cell and bone marrow (BM) recipients (Fig. 7, A and B; and Fig. S3 B). Noninvasive bioluminescence imaging (BLI) revealed reduced proliferation and homing of luciferase transgenic (luc+) CD90.1+ donor T cells from NFATc1/ΔS+ compared with WT donor T cells, correlating with the reduced disease score (Fig. S3 C).
Figure S3.

Transfer of T cells from Nfatc1deltaSUMO mice leads to less acute GvHD compared with WT T cells. (A) Preclinical model of aGvHD after MHC-mismatched allo-HCT. BALB/c (H-2d) mice were lethally irradiated with a single dose of 8.0 Gy and transplanted with 5 × 106 CD90.2+ BM cells and 1.2 × 106 CD90.1+ luc+ T cells from C57BL/6 donors (H-2b). (B) Body weight changes of host BALB/c mice (n = 5), irradiated (irradiation control) and transplanted with allogeneic BM (BM only), BM plus allogeneic WT (BM + WT), or BM plus allogeneic Nfatc1deltaSUMO (BM + ΔS) mice. (C) Representative in vivo BLI of mice transplanted with B6.CD90.2+ BM (BM only), BM plus B6.CD90.1+ luc+ WT T cells, and BM plus B6.CD90.1+ luc+ NFATc1/ΔS+ T cells. Statistical significance was calculated using two-way ANOVA; *, P < 0.05; ***, P < 0.001. (D) Frequency of splenic donor CD4+ and CD8+ T cells after 6 d, analyzed by flow cytometry and quantified from n = 15. (E) Analysis of serum IFN-γ and TNFα on days 2–6 after allo-HCT using cytometric bead array (n = 5, mean + SD).
Figure 7.

NFATc1/ΔS in T cells ameliorates acute GvHD after allogeneic HCT. (A) Cumulative survival of irradiated BALB/c mice (irradiation control), with transfer of B6.BM cells (BM only) or B6.BM cells plus B6.CD90.1+luc+ T cells from WT (BM + WT) or Nfatc1deltaSUMO (BM + ΔS) mice, respectively. Median survival: BM only, undefined; irradiation control, 8 d; BM + WT, 20 d; BM + ΔS, undefined. P = 0.046 between WT and ΔS (log-rank test). (B) Clinical GvHD scoring (score 0–8; Cooke et al., 1996) of the recipient mice listed under A. Mann–Whitney U test; *, P < 0.05. (C) Representative ex vivo BLI data from internal organs and quantification of allogenic luc+ WT versus NFATc1/ΔS+ T cells 6 d after the transfer. Data are compiled from at least two independent experiments with five mice per group per experiment (n = 10). Two-way ANOVA; ***, P < 0.001. (D) Analysis of α4β7 integrin expression on CD90.1+ donor WT versus NFATc1/ΔS+ CD4+ and CD8+ T cells 6 d after allo-HCT in spleen and mesenteric LNs measured by flow cytometry; data from three individual recipient mice are summarized (n = 6). Statistical significance was calculated using Student’s t test; *, P < 0.05. (E) Representative intracellular flow cytometry detecting cytokine expression of splenic CD90.1+CD4+ WT and NFATc1/ΔS+ donor T cells 6 d after allo-HCT (upper). Total spleen cells were restimulated for 6 h with T/I. Quantification of data from (n = 10). Statistical significance was calculated using two-way ANOVA; **, P < 0.01; ***, P < 0.001. (F) In vivo BLI of mice transplanted with B6.CD90.2+ BM plus B6.CD90.1+luc+ WT T conv or plus CD90.1+luc+ NFATc1/ΔS+ T cells. T conv, derived from DT-treated DEREG+ mice, were completely T reg free. Statistical significance was calculated using Mann–Whitney U test; ***, P < 0.001. (G) Mean fluorescent intensity (MFI) and percentage of splenic IL-2–expressing cells after 6 d, analyzed by flow cytometry and quantified from n = 5; Mann–Whitney U test *, P < 0.05. (H) Representative flow cytometry of YFP (DEREG) detecting Foxp3 expression in splenic CD4+ cells 6 d after aGvHD induction. CD4+ T cells from an untreated (healthy) WT DEREG mouse served as control. Mann–Whitney U test; *, P < 0.05.
Limited homing of NFATc1/ΔS+ T cells to GvHD target tissues was especially prominent in the gastrointestinal tract upon ex vivo BLI of the organs (Fig. 7 C). Consistent with former transfer experiments of NFATc1- and/or NFATc2-deficient T cells, also causing less inflammation (Vaeth et al., 2015), this was reflected by less cell surface expression of gut-homing receptor α4β7 integrin on NFATc1/ΔS+ CD4+ T cells, although not on CD8+ T cells, in SLOs (Fig. 7 D). The overall ratio of CD4+ versus CD8+ T cells did not alter between genotypes (Fig. S3 D). As in in vitro cultures and during EAE, effector cytokines were significantly less expressed upon ex vivo restimulation of splenic lymphocytes (Fig. 7 E). Accordingly, serum concentrations of TNF and the dominantly present IFN-γ were less upon transfer of NFATc1/ΔS+ compared with WT T cells (Fig. S3 E). However, the number of IL-2+ CD4+ T cells were not enhanced, hardly reaching WT levels, when NFATc1 could not be SUMOylated (see below). Nevertheless, we conclude that NFATc1/ΔS+ T cells are less pathogenic during aGvHD, correlating with less expansion and less effector cytokine expression.
Transplanted NFATc1/ΔS+ T conv express more IL-2 and more Foxp3 than WT T conv
To reveal the in vivo differentiation and behavior of naive WT versus NFATc1/ΔS+ T conv in an isolated manner, we transplanted MHC-mismatched, T reg–depleted naive T conv. To avoid T regs completely, WT and Nfatc1deltaSUMO were crossed to DEREG mice, which express a diphtheria toxin (DT) receptor-enhanced GFP fusion protein under the control of the Foxp3 gene locus (Lahl et al., 2007). Thus, repetitive DT injections 3 and 2 d before T cell isolation allowed pure T conv acquisition without any contaminating Foxp3+ T regs (Fig. S4 A). Certainly, naive H-2b NFATc1/ΔS+ T conv proliferated and expanded significantly less than WT T conv in H-2d BALB/c mice (Fig. 7 F and Fig. S4, B and C). As before, NFATc1/ΔS+ T conv developed fewer IFN-γ+ than WT T conv (not depicted). In addition, when exclusively monitoring T conv, we could document enhanced IL-2 production per cell and especially an augmented frequency of IL-2+ T conv (Fig. 7 G). Even a subtle induction of pT regs, fostered in NFATc1/ΔS+ T cells, occurred under these conditions (Fig. 7 H). Thus, also in vivo, the inability to SUMOylate NFATc1 in T conv allows them to amplify IL-2 secretion, while halting a strong proinflammatory phenotype, best seen in the absence of tT regs.
Figure S4.

Pure NFATc1/ΔS+ T conv cause less aGvHD than WT T conv. (A) Preclinical model of aGvHD after MHC-mismatched allo-HCT and T conv in the complete absence of T regs. Mice had been intercrossed to DEREG. They were treated by DT 3 and 2 d before sacrifice. BALB/c (H-2d) mice were lethally irradiated with a single dose of 8.0 Gy and transplanted with 5 × 106 BM cells from CD90.2+ Rag1−/− mice and 3 × 105 CD90.1+ luc+ T conv from WT or Nfatc1deltaSUMO C57BL/6 donors (H-2b). (B) Representative in vivo BLI of mice as in A. Quantification was done on days 3 and 5 after aGvHD induction. Statistical significance was calculated using two-way ANOVA; ***, P < 0.001; n = 5. (C) Representative ex vivo BLI of SLOs and target organs of mice treated as indicated in A. Quantification was done on day 6 after aGvHD induction. Statistical significance was calculated using two-way ANOVA; ***, P < 0.001; n = 5. (D) In vivo BLI of mice transplanted with BM cells and luc+ CD90.1+ WT versusluc+ CD90.1+ NFATc1/ΔS+ T cells monitored before sacrifice for NGS (Fig. 9 A) on day 4. (E) Purity of splenic CD4+ CD90.1+ T cells after FACS and before RNA extraction and NGS. (F) Marker selection of genes (∼50 of the first 100) up- and down-regulated each by NFATc1/ΔS upon isolation of allo-HCT recipients.
NFATc1/ΔS+ T cells up-regulate Blimp-1
So far, we had no explanation why IFN-γ was less expressed in NFATc1/ΔS+ T cells. Comparable in vivo chromatin binding of all isoforms irrespective of SUMOylation had been demonstrated by us earlier (Nayak et al., 2009) and could now be verified for the Il17a promoter in Th17 cells (Fig. 8 A). However, the Ifng promoter was significantly less bound by NFATc1/ΔS than by WT NFATc1 in Th1 cells, hinting at a less accessible locus. Because it was reported that IL-2 induces Blimp-1 as well as that Blimp-1 represses IL-2, IFN-γ, and TNF expression (Cimmino et al., 2008; Gong and Malek, 2007; Martins et al., 2008), we speculated whether also in our setting early overexpression of IL-2 led to Blimp-1 induction. Indeed, in vitro stimulated CD3+ T cells expressed Blimp-1 better in the presence of exogenous IL-2 than NFATc1/ΔS+ T cells could do on their own (Fig. 8 B). Equally, when T cells were regained from aGvHD mice after 6 d, CD90.1+ CD4+ NFATc1/ΔS+ T cells exhibited more Blimp-1 in comparison with WT T cells (Fig. 8 C). In keeping with SUMOylation defining a major difference between the short NFATc1/A and the long NFATc1/C factors, NFATc1/ΔBC+ CD4+ T cells also exhibited enhanced Blimp-1 expression that was further up-regulated by the addition of exogenous IL-2 (Fig. 8 D).
Figure 8.

Deficiency of NFATc1 SUMOylation provokes Blimp-1 expression. (A) Th1- and Th17-differentiated cells of WT and Nfatc1deltaSUMO mice were analyzed on day 3 of culture by ChIP with anti-NFATc1. Relative occupancy of NFATc1 at the Ifng promoter region (Th1 cells) or at the Il17a promoter (Th17 cells) was calculated by the ΔΔCt method. Bars show mean + SD of two individual experiments. Two-way ANOVA followed by Bonferroni posttest; **, P < 0.005. (B) Immunoblot of ConA-stimulated CD3+ T cells from WT and Nfatc1deltaSUMO mice, WT cells in parallel with the addition of exogenous IL-2 (n > 3). (C) Blimp-1 expression in CD90.1+ CD4+ T cells, regained after transplantation of BM plus WT or NFATc1/ΔS+ T cells on day 6 and detected by immunoblot (n = 3). (D) Blimp-1 expression in CD4+ T cells from WT and Nfatc1deltaBC mice, stimulated for 1 or 4 d by ConA, with or without the addition of IL-2 (n > 3). Blimp-1, NFATc1, and NFATc1/ΔBC were detected by immunoblot. (E and F) CD4+ T cells from WT, Nfatc1deltaSUMO, Nfatc1deltaBC, and Nfatc1deltaBC.Prdm1fl/fl mice were stimulated under Th1-differentiating conditions without (w/o; E) or with (w/; F) anti–mIL-2 for 2, 3, and 5 d and evaluated for intracellular IL-2 and IFN-γ by flow cytometry. Representative plots of day 3 and quantification of n = 8 for each time point are shown. Student’s t test (comparing every two groups); *, P < 0.05; **, P < 0.005; ***, P < 0.001; ****, P < 0.0001.
To evaluate whether Blimp-1 is necessary to prevent full IFN-γ and late IL-2 expression, we crossed Prdm1fl/fl mice to Nfatc1deltaBC (Nfatc1deltaSUMO.Cd4cre), as conventional Prdm1−/− die in utero. Prdm1 encodes Blimp-1. CD4+ T cells of WT, Nfatc1deltaSUMO, Nfatc1deltaBC, and Nfatc1deltaBC.Prdm1fl/fl were cultured under Th1-inducing conditions. IL-2 and IFN-γ were detected intracellularly after 2, 3, and 5 d upon restimulation (Fig. 8 E). Indeed, without Blimp-1, the frequency of IL-2+ cells was further enhanced, which was still observable after 5 d, when NFATc1/ΔS or NFATc1/ΔBC could not support IL-2 expression better than WT NFATc1 anymore. On the other hand, the repression of NFATc1/ΔS or NFATc1/ΔBC on IFN-γ clearly reverted in the absence of Blimp-1. Proving that the differential amount of IL-2 is causative, the addition of anti–mIL-2 to the cultures abolished all these effects (Fig. 8 F).
NFATc1/ΔS supports and Blimp-1 represses Bcl2A1 expression
To screen for early transcriptional events, possibly differentially regulated by WT versus NFATc1/ΔS directly when cells are challenged in vivo, we isolated CD90.1+ CD4+ T cells, i.e., CD4+ T conv and T reg, during the course of aGvHD. By this, we evaluated the in vivo situation of allogen-activated T cells. We confirmed reduced disease manifestation by BLI and the purity of isolated cells by flow cytometric analyses (Fig. S4, D and E). 4 d after allo-HCT, sufficient RNA could be extracted for NGS (Fig. 9 A). In line with the protein data (Fig. 1, C, D, and E), levels of Nfatc1, Nfatc2, and Nfatc3 RNA were the same (GEO accession no. GSE119313). In general, the influence on the transcriptome was again very subtle by the avoidance of NFATc1 SUMOylation (Fig. S4 F). Cytokines and chemokines were not yet highly expressed, but their RNA was overall less detectable in NFATc1/ΔS+ T cells. Interestingly, RNA of the prosurvival factor Bcl2A1 (also named A1/BFL1) was enhanced in NFATc1/ΔS+ T cells (Fig. 9 A), as had been in in vitro skewed Th1 (Fig. 9 B and Fig. 5 F). This pointed toward a potentially better survival of NFATc1/ΔS+ T cells (Fig. 9 C), as shown for NFATc1/A-overexpressing in comparison with NFATc1/C-overexpressing T cells (Chuvpilo et al., 2002). The murine T cell line EL-4, retrovirally transduced with different NFATc1/C variants, confirmed that the presence of NFATc1/ΔS enhances Bcl2a1 expression (Fig. 9 D). This was true for the K702/914R mutant as well as the triple K349/702/914R mutant. K702/914R had proven to prevent any NFATc1 SUMOylation despite the intact SUMOylation motif around K349 (Nayak et al., 2009), which had been the prerequisite for the design of the Nfatc1deltaSUMO mouse. Justifying the assumption that NFATc1/C’s special function is mediated by SUMOylation, only NFATc1/C, exogenously provided by retroviral infection, repressed Bcl2a1 in EL-4 cells (Fig. S5 A). EL-4 cells mainly express the short isoform NFATc1/A (Fig. S5 B), which could further transactivate Bcl2a1, like its functional homologue NFATc1/ΔS (Fig. S5 A, middle; and Fig. 9 D). In mice, the Bcl2a1 gene has been quadruplicated. All homologues of Bcl2a1 harbor two consensus NFAT sites within their promoters, which were recognized by NFATc1 and NFATc2 in electromobility shift assays (EMSAs; Fig. 9 E). Of note, the NFAT-binding site within Bcl2a1 resembles the dominant one of the Il2 promoter, Pu-box distal, in its composition with an AP1-binding site.
Figure 9.

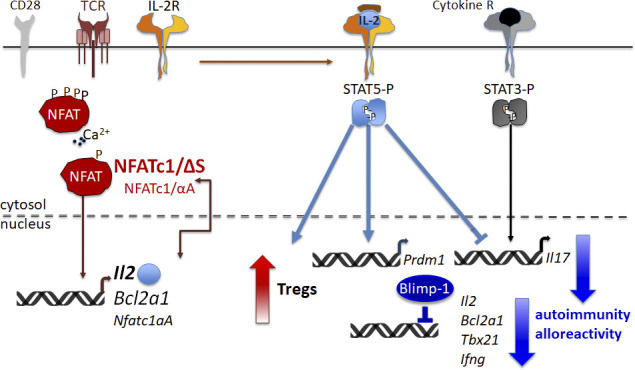
Bcl2A1 is up-regulated by NFATc1/ΔS and repressed by Blimp-1. (A) Heatmap of 200 genes whose expression changed in CD4+ CD90.1+ WT versus NFATc1/ΔS+ T cells (signal to noise, SD adjusted) regained from aGvHD-induced mice on day 4. Shown is the relative difference in RPKM (Morpheus; Broad Institute). (B) Bcl2a1 RNA expression in Th1-skewed cells, measured by qRT-PCR. n = 6, statistical evaluation by Student’s t test; *, P < 0.05. (C) Frequency of 7-AAD+ Annexin+ Th1 cells; n = 4. (D) Detection of Bcl2a1 mRNA in relation to other members of the Bcl2 family and control genes L32 and Gapdh by RNase protection assay using the mApo2 template set (5 µg RNA/lane). EL-4 cells had been retrovirally infected with NFATc1/C-ER and its ΔSUMO and N-terminal SUMO fusion mutants and stimulated with 5-hydroxytamoxifen and T/I for 24 h. 1–6 below phosphoImager values correspond to the lanes of the autoradiography. Shown is one representative from three independent experiments. (E) EMSA with NFAT binding sites-containing probes from the Nfatc1 promoter P1 (tandem site) and Bcl2a1 promoters. Nuclear extracts were prepared from EL-4 cells, stimulated with T/I for 4 h. NFATc1 and NFATc2-specific antibodies supershift the respective proteins (c1ss and c2ss). Consensus nucleotides for NFAT-binding are indicated in red, for AP1 in bold only. (F) Bcl2a1 (n = 9) and Prdm1 (n = 6) RNA expression in CD4+ T cells, collected on day 4 of spleen and intestine of mice induced for aGvHD by WT versus NFATc1/ΔS CD3+ T cells, measured by qRT-PCR. Student’s t test *, P < 0.05. (G) Bcl2a1 promoter activity in response to Blimp-1 was evaluated after transfection with pEYZ/MCS or pEYZ/Blimp-1F along with Bcl2a1 promoter (length indicated)-driven luciferase reporter plasmids in HEK 293T cells. After 36 h, luciferase activity was measured. Data are represented as the mean ± SE. Statistical significance was calculated using Student’s t test; *, P < 0.05. (H) EMSA with oligonucleotides containing Blimp-1 binding from the Myc (PRF) and Bcl2a1 (−50) promoters, using nuclear extracts from HEK 293T cells transiently transfected with Flag-tagged Blimp-1. Blimp-1 was supershifted by either anti-Blimp (α-B) or its tag Flag (α-F). Consensus nucleotides for Blimp-1 binding are indicated in blue. Right: Summary of the hypothesized sequential events downstream of endogenous NFATc1/ΔS expression.
Figure S5.

NFATc1/A transactivates Bcl2a1, whereas Blimp-1 suppresses it. (A) Detection of Bcl2a1 mRNA in relation to other members of the Bcl2 family and control genes L32 and Gapdh by RNase protection assay using the mApo2 template set. EL-4 cells had been retrovirally infected with HA-EGZ, HA-NFATc1/A, or HA-NFATc1/C (see B [anti-HA] for expression), stimulated with T/I for indicated times. Shown is one representative from two independent experiments. (B) Immunoblot of EL-4 cells, stimulated as indicated to detect endogenous NFATc1/αA by anti-NFATc1 or retrovirally expressed (n > 3) exogenous HA-NFATc1/αA and –c1/αC by anti-HA (n = 2). (C) Prdm1 RNA expression in Th1-skewed cells, measured by qRT-PCR. n = 5, Student’s t test; **, P < 0.005. (D) EMSA with radiolabeled oligonucleotide representing a Blimp-1–binding site of the Bcl2a1 promoter (−50), and competition with increasing amounts of cold oligonucleotides representing the other two Blimp-1–binding sites (−70 and −10) as well as an unrelated probe, using nuclear extracts from HEK 293T cells transiently transfected with Flag-tagged Blimp-1. Blimp-1 was supershifted (ss) by using anti-Flag (α-F) antibody (n > 3). Nucleotides of the proximal Bcl2a1 promoter indicate the consensus nucleotides for Blimp-1 binding in blue and for NFAT in red. (E) Bcl2a1 minimal promoter activity in HEK 293T depending on Blimp-1. pEYZ/MCS or pEYZ/Blimp-1F was transiently cotransfected with luciferase reporter construct driven by an intact Bcl2a1 promoter (WT), singly mutated for the −50 site (−50mut) or a variant in which all three Blimp-sites had been erased (triple). After 36 h, luciferase activity was measured. Data are represented as the mean ± SE. Statistical significance was calculated using Student’s t test; *, P < 0.05; **, P < 0.01. (F) CD4+ T cells from WT, Nfatc1deltaSUMO, and Nfatc1deltaBC.Prdm1fl/fl mice were stimulated under Th1-differentiating conditions for 3 and 5 d and evaluated for Bcl2a1 RNA expression by qRT-PCR. Quantification of triplicates of two mice per genotype for each time point are shown; mean + SD. (G) Combined splenic and LN-derived T cells from two WT, two Nfatc1deltaSUMO, or two Nfatc1deltaBC.Prdm1fl/fl mice were stimulated by ConA for 1 and 4 d. Immunoblot analysis of whole-cell extracts, detected Bcl2A1 and ERK (one experiment).
Suppression of Il2, Tnfsf2, and Ifng (including Tbx21) by Blimp-1 has been reported (Cimmino et al., 2008; Martins et al., 2008), and we tested whether this mechanism applies to Bcl2a1 as well. To quantify Blimp-1 expression in Th1 cells, we extracted RNA either 3 d after in vitro differentiation or 4 d after aGvHD induction. Prdm1 RNA was enriched significantly in NFATc1/ΔS+ T cells compared with WT T cells (Fig. S5 C and Fig. 9 F). Ex vivo, Prdm1 levels were still very low in comparison with Bcl2a1 (Fig. 9 F). Overexpressed Blimp-1 could repress the proximal promoter of Bcl2a1 during reporter assays (Fig. 9 G). Accordingly, Blimp-1 binding could be detected to a site (−50) reflecting the one from the Myc promoter. Two additional sites (−10 and −70) were able to partly compete for binding (Fig. 9 H and Fig. S5 D). Mutating all three Blimp-1 binding sites of the Bcl2a1 promoter abolished Blimp-1–mediated repression in a reporter assay (Fig. S5 E). As a result, the absence of Blimp-1 during Th1 differentiation of CD4+ Nfatc1deltaBC T cells prevents down-regulation of Bcl2A1 (Fig. S5, F). Bcl2A1 protein is highly unstable due to ubiquitin-dependent proteasomal degradation (Herold et al., 2006); still, Blimp-1 ablation prevents its loss seen in Nfatc1deltaSUMO CD3+ T cells (Fig. S5 G). With this, we assumed that enhanced IL-2 production leads to enhanced Blimp-1 expression, in turn down-regulating IFN-γ, TNF, and Bcl2A1 as well as IL-2 itself (Fig. 9, model).
NFATc1/ΔS+ T conv support T regs during acute GvHD by enhanced IL-2 expression
EAE-diseased Nfatc1deltaSUMO mice showed an augmented number of T regs (Fig. 4, C–E), and the transcriptome data of allo-transplanted NFATc1/ΔS+ T cells revealed an increase in Foxp3 RNA (Fig. 9 A and Fig. S4 F). Accordingly, a boosted number of T regs was evident in mice receiving NFATc1/ΔS+ T cells (Fig. 10 A). The rate of Foxp3+ cells was clearly higher than that of the observed pT regs (Fig. 7 H), indicating the involvement of tT regs here. As NFATc1/ΔS+ T cells expressed more Tigit RNA (Fig. 9 A), the frequency of T cell immunoreceptor with Ig and ITIM domains (TIGIT)+ T regs increased, indicative of an effector phenotype (Cretney et al., 2018), in mice transplanted with allogeneic NFATc1/ΔS+ T cells versus WT controls (Fig. 10 B). To reveal whether the IL-2–overexpressing NFATc1/ΔS+ T conv were key for the increase in tT reg numbers, we transplanted WT (luc−) tT regs either together with WT or with NFATc1/ΔS+ luc+ T conv. In line with our earlier observations, NFATc1/ΔS+ T conv showed reduced proliferation and target organ infiltration as documented by in vivo and ex vivo BLI (Fig. 10, C and D). To additionally examine the capability of WT versus NFATc1/ΔS+ tT regs, we included tT regs isolated from Nfatc1deltaSUMO mice in parallel groups. It became obvious that NFATc1/ΔS+ tT regs limit the expansion of allogeneic transplanted T conv more efficiently than WT tT regs (Fig. 10, C and D). Especially in the intestine, a prime target organ for aGvHD, the combination of T conv and T regs, both NFATc1/ΔS+, resulted in the least incidence of luc+ T conv. Recollected T regs showed that the presence of NFATc1/ΔS+ T conv has a positive influence on their number, whereas NFATc1/ΔS+ T regs expand better on their own (Fig. 10 E). Interestingly, NFATc1/ΔS+ T regs limit the ratio of IL-2+ CD4+ WT T conv, while NFATc1/ΔS+ T conv withstand this suppressive effect (Fig. 10, F and G). In conclusion, NFATc1/ΔS decisively regulates T conv function, but beyond implications for T conv differentiation, it regulates the frequency and quality of T regs directly and indirectly by NFATc1/ΔS+ T conv.
Figure 10.

Protection from GvHD is encoded in NFATc1/ΔS+ T conv as well as in NFATc1/ΔS+ T regs. (A) Detection of CD25+ Foxp3+ cells as percentage of CD4+CD90.1+ in spleen and mesenteric LNs of aGvHD-induced mice on day 6. Transplanted allogenic luc+ T cells were derived from WT versus Nfatc1deltaSUMO mice. Statistical significance was calculated using Student’s t test; *, P < 0.05; **, P < 0.01. (B) Evaluation of surface expression of TIGIT on CD4+CD90.1+ T conv (no TIGIT detectable) and CD4+CD90.1+ Foxp3+ T regs regained from spleen and mesenteric lymph nodes of aGvHD-induced mice on day 6 by flow cytometry. Statistical significance was calculated with Student’s t test; **, P < 0.01. (C–F) Allogenic pure CD90.1+ luc+ WT versus NFATc1/ΔS+ T conv from pretreated DEREG+ mice were transferred together with BM CD90.2+ luc− and CD90.1+ luc− WT versus NFATc1/ΔS+ T regs in all possible combinations. (C and D) Representative in vivo BLI (C) and ex vivo BLI data from internal organs (D). In vivo data were quantified on days 3 and 5 after aGvHD induction, ex vivo on day 6. Two-way ANOVA; *, P < 0.05; **, P < 0.005; ***, P < 0.001. (E) Estimation of CD25+ Foxp3+ (CD4 gate) T reg frequencies in the presence of WT versus NFATc1/ΔS+ T conv by flow cytometry 6 d after T conv transfer. (F) IL-2 expression per CD4+ T cell (mean fluorescent intensity [MFI]) 6 d after T conv transfer. (G) Frequencies of splenic IL-2+ CD4+ T cells 6 d after T conv transfer, analyzed by flow cytometry and quantified from n = 4; two-way ANOVA; *, P < 0.05.
Discussion
Functional differences between the isoforms of NFATc1 have pointed to NFATc1/C as being similar to NFATc2 and the short isoform NFATc1/A as being special among all NFAT proteins (Chuvpilo et al., 1999, 2002; Serfling et al., 2012). It seemed likely that the NFATc1/C-specific C-terminal peptide serves as a platform for interactions with regulatory proteins, thereby providing functional specificity between the NFATc1 isoforms. Our yeast-2-hybrid screen identified the SUMO-conjugating enzyme Ubc9 and one E3 ligase, PIAS1, as the predominant NFATc1/C-specific interaction partners (Nayak et al., 2009). Accordingly, we could document SUMOylated NFATc1/C in primary CD4+ T cells (Nayak et al., 2009). Having evaluated the C-terminal–specific SUMO consensus sites K702 and K914 as decisive, we went on to mutate them genetically in mice to investigate the role of NFAT SUMOylation in T cell–mediated immune responses in vivo. We found that NFATc1/ΔS, even when expressed under endogenous control, enhances IL-2 and Bcl2A1 expression. Subsequently, less effector cytokine expression and an increase in T reg numbers ameliorated inflammatory conditions such as EAE and aGvHD. Although the shift to more IL-2 was transient, it provided a time window for tolerance installment.
In general, strong IL-2 signals support T conv terminally differentiating into effector T cells, which are then destined for rapid cell death to allow immune responses to cease (Boyman and Sprent, 2012). Low abundance of IL-2, on the other hand, enables CD4+ T cells to differentiate into Th17, T follicular helper, or central memory T cells. During IL-17 induction, IL-2–induced phosphorylation of STAT5 counteracts phosphoSTAT3 binding (Laurence et al., 2007). Accordingly, capturing NFATc1/ΔS-enhanced IL-2 was sufficient to restore IL-17 to WT levels. IL-17 was originally found to play a critical pathogenic role in CNS inflammation in EAE and multiple sclerosis (Langrish et al., 2005) but has been linked to various autoimmune inflammatory diseases since then. Especially, coexpression with IFN-γ promotes chronic inflammation, and such pathogenic Th1/17 cells (Langrish et al., 2005; Lexberg et al., 2010; Peters et al., 2011) were less frequent in EAE-induced Nfatc1deltaSUMO mice.
Contradicting the foreseen effect of elevated IL-2 levels for IFN-γ expression, fewer IFN-γ was produced during EAE as well as during Th1 response-dominated aGvHD. IFN-γ can be repressed by Blimp-1, the latter being induced by IL-2 and, in a negative feedback loop, repressing Il2 (Cimmino et al., 2008; Fu et al., 2017; Gong and Malek, 2007; Martins et al., 2008; Santner-Nanan et al., 2006). Accordingly, we could detect more Blimp-1 RNA and protein in NFATc1/ΔS+ CD4+ T cells regained from aGvHD-challenged mice and in vitro stimulated NFATc1/ΔS+ and NFATc1/ΔBC+ T cells, in which Blimp-1 ablation further enhanced the frequency of IL-2+ cells and reverted the repression on IFN-γ. Additional proof for the connection of NFATc1/ΔS-mediated surplus in IL-2 and subsequent enhanced Blimp-1 expression facilitating repression of IL-2 and IFN-γ, stemmed from the total loss of these effects when anti–mIL-2 was added to the cultures. Consistent with these data, a recent report ensured inhibition of Th1 differentiation by Blimp-1 overexpression (Wang et al., 2018).
Remarkably, Bcl2A1, which harbors a homologous NFAT response element as the IL-2 promoter, was likewise superinduced in NFATc1/ΔS+ T cells and a direct target for Blimp-1 repression. Augmented Bcl2A1 did not provide a significant survival advantage, but this is in line with Bcl2A1-deficient mice and the observation that individual antiapoptotic proteins contribute only quantitatively to survival outcome (Carrington et al., 2017; Schenk et al., 2017). In contrast with other antiapoptotic Bcl2 family members, however, Bcl2A1 agrees with proliferation, mediating survival of T cells after activation (Gonzalez et al., 2003). Thus, genes immediately up-regulated after TCR activation, Il2 and Bcl2a1, are positively regulated by NFATc1/ΔS. Persistent TCR signaling leads to the abundant expression of promoter P1-driven NFATc1/αA, which supports proliferation and opposes activation-induced cell death (Serfling et al., 2012). Here we revealed that NFATc1/ΔS functionally resembles NFATc1/αA or, conversely, that NFATc1/C (and maybe all long NFATs) need to be SUMOylated for pronounced effector cytokine production. Our data suggest the level of transactivated Il2 to be decisive for the dichotomy between NFATc1/A and NFATc1/C. In addition, differentially regulated Bcl2A1 could have a direct impact, as one report demonstrated that Bcl2A1 overexpression in T cells impaired Th17 differentiation (Iglesias et al., 2016).
Recently, analysis of Ubc9-deficient thymocytes revealed that late positive selection is compromised by complete loss of SUMOylation in CD4+ and CD8+ thymocytes/T cells and suggested that this is mediated via disturbed retention of NFATc1 in the nucleus (Wang et al., 2017). We had not been able to detect an influence of SUMOylation on its nuclear presence before (Nayak et al., 2009), nor in mature T cells, but we did not address thymocytes. More importantly, our Nfatc1deltaSUMO mice developed normally without any change in frequency of any kind of lymphocytes, in our opinion implying that NFATc1 is not the only player in Ubc9-deficient thymocytes. Possibly, however, it would be sufficient to block SUMOylation of all NFAT proteins to achieve the phenotype of Ubc-9 ablation. Of note, SUMOylation sites are conserved between NFATc1/C, NFATc2, and NFATc3.
The same group (Ding et al., 2016) published a severe autoimmune phenotype when SUMOylation is specifically hindered in T regs, showing that a high percentage of differentially expressed genes were NFAT target genes. In Nfatc1deltaSUMO (and Nfatc1deltaBC) mice, T regs appeared to function at least normally, if not better. Even aged mice did not succumb to any autoimmune disease, again bringing into question whether all NFAT proteins have to be prevented from SUMOylation and/or whether IRF4, as implied by the authors, is the more important SUMO target in T regs (Ding et al., 2016).
In Nfatc1deltaSUMO mice, we observed an increase in the number of T regs upon challenge, most likely owing to the elevated IL-2 levels, i.e., in a cell-extrinsic manner. Support of T regs by NFATc1/ΔS+ T conv could be demonstrated after aGvHD induction. In turn, in vivo, not only is the heightened IL-2 expression of NFATc1/ΔS+ T conv intrinsically repressed by enforced Blimp-1, but the boosted number of T regs also counteracts IL-2+ NFATc1/ΔS+ T conv more efficiently than the lower number supported by WT T conv. This notion could explain why differential IL-2 expression was detected best in the absence of T regs in vitro as well as in vivo in T conv–transplanted animals.
We found previously that thymus-derived T regs develop and function normally without one or two NFAT proteins. Also, peripherally or in vitro induced pT regs and iT regs exhibit full repressive capacity upon NFAT ablation, although their differentiation surely hinges on the level of NFAT proteins present (Dietz et al., 2015; Vaeth et al., 2012, 2014, 2015). Nevertheless, T regs depend on continuous TCR signaling, most likely involving NFAT proteins, to acquire their effector phenotype, for example to up-regulate CTLA-4, CD103, inducible T cell costimulator, and TIGIT (Levine et al., 2014; Vahl et al., 2014). Accordingly, we showed that differentiation of follicular T regs, i.e., expression of CXCR5 and therefore homing to germinal centers, strictly depends on NFATc1 (Vaeth et al., 2014). Therefore, the heightened level of TIGIT on T regs in allogeneic transplanted mice could indicate a cell-intrinsic effect of NFATc1/ΔS in T regs, which became evident in the superior suppressiveness during our criss-cross GvHD experiments. TIGIT+ T regs resemble a subtype of T regs specifically suppressing Th1 and Th17, but not Th2 (Joller et al., 2014), supporting the notion that NFATc1/ΔS expression ameliorates autoimmune diseases and alloreactivity.
Thus, it might be worthwhile to consider targeting NFATc1 SUMOylation for therapeutic intervention, and the outcome might resemble protocols of low-dose IL-2 or related IL-2 therapies (Abbas et al., 2018). Such protolerogenic therapeutic approaches are mostly meant to support T regs. Nonetheless, the risk of activating unwanted cell types and thereby exacerbating the disease is a prevailing worry. Blocking NFATc1 SUMOylation or expressing NFATc1/ΔS or NFATc1/ΔBC would resemble low-dose IL-2 therapy without any complexing or modification of IL-2. Additionally, IL-2 would be secreted very locally by T conv being activated by antigen-presenting cells and often in a trimeric complex with the IL-2–consuming T regs. Then, instead of an overshooting response, IL-2–induced STAT5 activation counteracts IL-17 induction, whereas Blimp-1 limits IL-2 and Th1 responses. At least for the preclinical animal models applied in our study, this was enough to install long-lasting tolerance.
Materials and methods
Construction of the conditional point mutation targeting vector for the Nfatc1 gene
This combined transgenic mouse strain was created by inGenious Targeting Laboratory. A 9.22-kb genomic DNA used to construct the targeting vector was first subcloned from a positively identified B6 BAC clone (RP23: 12N8). The region was designed such that the long homology arm extends ∼6.3 kb 5′ to the first point mutation in exon 10. The LoxP-flanked Neo cassette was inserted downstream of exon 10 and is 915 bp away from the first point mutation and 291 bp away from the second point mutation. The short homology arm extends 2.04 kb to the 5′ end of the Neo cassette. The targeting vector was confirmed by restriction analysis and sequencing after each modification. The boundaries of the two homology arms were confirmed by sequencing with P6 and T7 primers that read through both sides of the backbone vector. The floxed Neo cassette was confirmed by sequencing with N1 and N2 primers that read from the 5′ and 3′ ends of the Neo cassette, respectively, into the genomic sequences. The point mutations were confirmed by sequencing with N2 and PT1 primers. The sequencing results also confirmed that no other mutations were introduced into the modified region.
The two AAG→CGG mutations in exon 10 were generated by three-step PCR mutagenesis. Eight mutagenesis primers, PT1–8, were designed to amplify a fragment with a size of 1.12 kb, which incorporates the mutations at the desired locations. PT1–4 were used for engineering the first point mutation, and PT5–8 were used for engineering the second point mutation. The point mutations were engineered into the primers PT2, PT3, PT6, and PT7. The PCR fragment carrying the mutations was then used to replace the correspondent WT sequence using conventional cloning methods. Oligonucleotides used to generate the point mutations are as follows (mutations are underlined): PT1 (BsiWI), 5′-AACTTCGTACGTCCTCTTCGTATTAGAACCAGCTG-3′; PT2/LOX1, 5′-CCGTATAATTGGAACTGTGGAGAGGAAAG-3′; PT3, 5′-TCCTCTCCACAGTTCCAATTATACGGACAGAACCCACGGACGACTTTGAG-3′; PT4, 5′-ACAGCTACTGTTCAGATGTGGAC-3′; PT5, 5′-TGAGTCCACATCTGAACAGTAGC-3′; PT6, 5′-CCGGATCACTACAGGAATAGGGGCCAAG-3′; PT7, 5′-TGGCCCCTATTCCTGTAGTGATCCGGCAAGAGCCTGAGGAATTGGACC-3′; and PT8 (BsiWI), 5′-ACAGTCGTACGACTGTCTGAGGTTGACATGG-3′.
Primers Ex9for (5′-CCTGGTGGTTGAGATACC-3′) and Ex11rev (5′-GCTGGAGAGGTCGTGCTA-3′) were used to confirm the point mutations by sequencing and DL1 (5′-ACTGGCAGAACTTACCAGAGTC-3′) plus LOX1 for genotyping to indicate the 5′ loxP site. Deletion of exon 10 was proven by Ex9for plus Ex11rev in comparison to Ex9for plus PT4.
Mice
Nfatc1deltaSUMO were received as C57BL6 × BALB/c F1 hybrids. For all in vivo experiments, Nfatc1deltaSUMO were crossed back to pure genetic C57BL/6 background (H-2b; CD90.2+) for >12 generations. To express NFATc1/ΔBC in T cells, Nfatc1deltaSUMO were bred with B6-Tg (Cd4cre)1Cwi/Cwilbcm (Lee et al., 2001), obtained from the European Mouse Mutant Archive (Nfatc1deltaSUMO × Cd4cre). To delete Blimp-1 in NFATc1/ΔBC+ T cells, Prdm1fl/fl was crossed in (Martins et al., 2006), Nfatc1deltaBC.Prdm1fl/fl. A MOG35–55-specific TCR was introduced by crossings with 2D2 mice (Bettelli et al., 2003). To generate Nfatc1deltaSUMO.luc.Cd90.1 mice, Nfatc1deltaSUMO were bred to B6.L2G85.CD90.1 transgenic mice ubiquitously expressing firefly luciferase and CD90.1 as a congenic marker (Beilhack et al., 2005). Additionally, DEREG was crossed in, i.e., a BAC-transgene expressing a DT receptor-eGFP fusion protein under the control of the Foxp3 locus (Lahl et al., 2007). Mice, including Rag1−/− (Mombaerts et al., 1992b), were bred in the pathogen-free animal facility of the Center for Experimental Molecular Medicine or the Department of Neurology at the University of Wuerzburg. BALB/c (H-2d) WT recipient mice were purchased from Charles River. Animals were mostly used for experiments between 8 and 12 wk of age. All animal experiments were approved by the respective authorities Regierung von Unterfranken and complied with German animal protection law.
MOG35–55 EAE induction
Myelin oligodendrocyte glycoprotein is a minor component of CNS myelin. The peptide MOG35–55 is highly encephalitogenic and produces a relapsing-remitting neurological disease. For active MOG35–55 EAE induction, mice were immunized s.c. in the tail base with 50 µg MOG35–55 (MEVGWYRSPFSRVVHLYRNGK; Research Genetics) in CFA (Difco) containing 550 µg/ml Mycobacterium tuberculosis H37RA (Difco) as previously published (Dietz et al., 2015). Pertussis toxin (200 ng per mouse; Sigma-Aldrich) was administered i.p. on days 0 and 2. For passive EAE, Rag1−/− received a mixture (5 × 106 in 200 µl) of in vitro differentiated, PBS-washed Th1/Th17 2D2+ CD4+ T cells retroorbitally. 2D2+ CD4+ T cells were differentiated for Th1 and Th17 with or without 10 µg/ml anti-mouse IL-2 (BE0043, clone JES6-1A12; Bio X Cell) for 2 d and cultivated on fresh plates for 2 additional days. Transplanted mice were treated with 200 µg anti–mIL-2 or IgG control (BE0089; Bio X Cell) i.p. on days 3 and 6. Clinical signs were assessed daily over the total period and scored in a single-blinded fashion as previously described (Dietz et al., 2015).
Isolation of CNS infiltrates
EAE mice were perfused with PBS, and the spinal cord and brain were dissected and digested with PBS containing 20 units of DNaseI (Thermo Fisher Scientific) and 2 mg/ml collagenase type II (Gibco) for 20 min at 37°C (Dietz et al., 2015). For separation of myelin from the infiltrated cells, the homogenate was loaded onto a 30%:37%:70% Percoll (Sigma-Aldrich) gradient and density centrifuged. The enriched CNS infiltrates were harvested and used for flow cytometry of CD4+ T cell cytokine production and cell composition.
MOG35–55 recall assay
For analysis of MOG35–55-specific cytokine production and proliferation of CD4+ T cells, EAE mice were sacrificed, and the spleen and draining (inguinal) LNs were dissected and mashed through a cell strainer (Dietz et al., 2015). The total spleen/LN cell suspension was cultured with different amounts of MOG35–55 peptide in RPMI 1640 (5% FCS, β-mercaptoethanol, and penicillin/streptomycin; Invitrogen) for 3 d. For quantification of cytokine secretion, supernatants were used in ELISA assays. For MOG35–55-specific proliferation, 3[H]thymidine was added to the culture 18 h before measurement, and incorporation was measured with a Mach 2 Harvester (Tomtech).
BM and T cell isolation
BM cells were isolated by flushing femur and tibia bones of Rag1−/− mice with PBS containing 0.1% BSA and passing through a 70-µm cell strainer (Vaeth et al., 2015). Spleens were directly passed through a 70-µm cell strainer with erythrocyte lysis buffer (168 mM NH4Cl, 10 mM KHCO3, and 0.1 mM EDTA) and washed twice with PBS containing 0.1% BSA. T cells were enriched from B6J and B6J.Nfatc1deltaSUMO spleens using the Dynal Mouse T cell Negative Isolation Kit (Life Technologies) according to the manufacturer’s instructions. B6J.CD4+CD25+ tT regs were enriched by staining negatively isolated T cells with anti-CD25-PE (PC61; BD PharMingen), and separation of CD4+ CD25+ and CD4+ CD25– cells was performed using anti-PE MicroBeads (Miltenyi Biotech) following the manufacturer’s instructions. Purity of the CD4+ CD25+ or CD4+ CD25– population was typically >90% or 95%, respectively.
Allo-HCT
BALB/c host mice (H-2d CD90.2+) were conditioned by myeloablative total body irradiation at a dose of 8.0 Gy with a Faxitron CP-160 x-ray system and injected retroorbitally with sex- and age-matched 5 × 106 B6 BM cells (H-2b CD90.2+) with or without 1.2 × 106 B6 T cells (H-2b CD90.1+ luc+) within 2 h after irradiation (Vaeth et al., 2015). For tT reg–mediated suppression of aGvHD, BALB/c host mice were conditioned as before, but injected only with BM cells and 6 × 105 B6 CD4+ CD25high tT regs (H-2bCD90.2+ luc–). 24 h later, 1.2 × 106 B6 CD4+ CD25– T conv (H-2b CD90.1+ luc+) were injected retroorbitally (Vaeth et al., 2015). Mice were treated with antibiotic drinking water (Baytril; Bayer) for 7 d after allo-HCT to prevent opportunistic infections. Transplanted mice were assessed daily for weight loss and clinical scoring for aGvHD symptoms; adapted from Cooke et al. (1996) and Vaeth et al. (2015).
In vivo and ex vivo BLI
Mice were anesthetized by i.p. injection of 80 mg/kg body weight ketamine hydrochloride (Pfizer) and 16 mg/kg xylazine (CP Pharma). Mice were injected with 300 mg/kg D-luciferin (Biosynth) together with anesthetics. After 10 min, BLI signals of the anesthetized mice were imaged with an IVIS Spectrum imaging system (Caliper Life Sciences; Vaeth et al., 2015). For ex vivo imaging of internal organs 6 d after allo-HCT, mice were injected with D-luciferin and euthanized 10 min later. Internal organs were removed and subjected to BLI. All pictures were taken with a maximum of 5-min exposure time and analyzed with Living Image 4.0 software (Caliper Life Sciences).
ELISA and cytometric bead array
Supernatants were subjected to ELISA for secreted cytokines IL-17A, IFN-γ, and IL-2 and quantified by cytokine-specific ELISA (BD PharMingen) according to the manufacturer’s guidelines. Serum cytokine concentrations were determined using BioLegend’s LEGENDplex bead-based immunoassays according to the manufacturer’s protocol 6 d after allo-HCT. Data were acquired on a FACS Canto II (BD Biosciences) flow cytometer and analyzed with LEGENDplex Data Analysis software.
Flow cytometry
Total CNS infiltrates, spleen, and LN cells were washed once in FACS buffer (PBS containing 0.1% BSA) before Fc receptor blocking with anti-CD16/CD32 (93; eBioscience, or 2.4G2; BD PharMingen). Staining of surface molecules was performed at room temperature using antibodies against CD4 (RM4-5), CD8α (53-6.7), CD90.1 (OX-7), α4β7 (LPAM-1, DATK32), CD25 (PC61), CD62L (MEL-14), CD69 (H1.2F3), CD45 (30-F11), CD11b (M1/70), PD1 (J43), CD27 (LG.3A10), and TIGIT (Vstm3; differently conjugated; all eBioscience, BioLegend, or BD) in FACS buffer for 15 min in the dark. Intracellular Foxp3 (FJK-16s, PE- or APC-conjugated; eBioscience) staining was performed using the Foxp3 staining kit (eBioscience) according to the manufacturer’s instructions. Intracellular cytokine staining of splenocytes with APC-conjugated IL-2 (JES6-5H4), IFN-γ (XMG1.2), and IL-10 (JES5-16E3) and PE-conjugated IL-4 (11B11), IL-17 (eBio17B7), GM-CSF (MP1-22E9), and TNFα (MP6-XT22; all eBioscience) was performed after 3–6-h restimulation with T/I (tetradecanoyl phorbol acetate [10 ng/ml; Sigma-Aldrich] plus ionomycin [5 nM; Merck Biosciences]) in the presence of GolgiStop and GolgiPlug (both BD Pharmingen) using the IC Fixation Buffer Kit (eBioscience). Data were acquired on a FACS Canto II (BD Biosciences) flow cytometer and analyzed with FlowJo software (TreeStar).
In vitro CD4+ Th differentiation
A mouse CD4+ T cell isolation kit (Miltenyi Biotec or BioLegend) was used to isolate CD4+ T cells from LN and spleen. The isolated CD4+ T cells were stimulated for 3 d with plate-bound anti-hamster IgG (12.5 µg/ml), soluble anti-CD3ɛ (145-2C11; BD PharMingen), and anti-CD28 (37.51; BD PharMingen). On day 2, cells were transferred to a fresh plate, without or with the addition of 75 U/ml hIL-2 (Peprotech) and 10 µg/ml anti–mIL-2 (Bio X Cell). The culture conditions for Th0 (no differentiation) were 2.5 µg ConA; for Th1, Th2, and iT reg, 0.5 µg/ml anti-CD3 and 1 µg/ml anti-CD28; and for Th17, 0.25 µg/ml anti-CD3 and 1 µg/ml anti-CD28. For skewing, the following cytokines were added to the culture: Th0, 2.5 µg/ml anti–mIL-4 (Bio X Cell) and 2.5 µg/ml anti–mIFN-γ (Bio X Cell); Th1, 10 ng/ml mIL-12 (PeproTech) and 2.5 µg/ml anti–mIL-4; Th2, 50 ng/ml mIL-4 (PeproTech) and 2.5 µg/ml anti–mIFN-γ; Th17, 0.5 ng/ml mTGFβ, 20 ng/ml mIL-6 (both Peprotech), 2.5 µg/ml anti–mIFN-γ, and 2.5 µg/ml anti–mIL-4; and iT reg, 5 ng/ml mTGFβ, 5 µg/ml anti–mIFN-γ, and 5 µg/ml anti–mIL-4. The cell culture medium was RPMI 1640 for Th1, Th2, and iT reg or IMDM for Th17 cell differentiation and supplemented with 5% FCS, β-mercaptoethanol, and penicillin/streptomycin (Invitrogen).
Protein extraction, immunoblots, and EMSAs
Whole-cell or nuclear extracts from primary murine T cells were prepared with the M- or N-PER Kit (Thermo Fisher Scientific), respectively. Proteins were resolved by 8–12% SDS-PAGE followed by immunoblotting. The primary antibodies used were rat anti-Bcl2A1 (#11470; Lang et al., 2014), mouse anti-NFATc1 (7A6; BD PharMingen), rabbit anti-NFATc2 (IG-209; ImmunoGlobe), and goat anti–Blimp-1 (C-21), mouse anti–β-actin (C-4), goat anti-lamin B (C-20), and anti-ERK (H-72; all Santa Cruz Biotechnology) with anti-mouse, -rabbit, or -goat peroxidase-coupled secondary antibodies. 3 µg of nuclear extracts from EL-4 cells or pEYZ/Blimp-1F (Schmidt et al., 2008)-transfected HEK 293T cells were prepared and subjected to EMSA as described (Berberich-Siebelt et al., 2000, 2006). Antibodies for supershifts were anti-NFATc1 (AB1–205; ImmunoGlobe), anti-NFATc2 and –Blimp as above, and anti-Flag mAb (M2; Sigma-Aldrich).
Viral infection, RNA purification, RNase protection assay, and PCR
Retroviral infection of EL-4 cells with NFATc1/C-ER and its ΔSUMO and N-terminal SUMO fusion mutants was done as previously (Nayak et al., 2009). RNA was extracted with Trizol (Ambion/Life Technologies). RNase protection assays (PharMingen Riboquant) were performed with the multiprobe template set mApo2 according to the manufacturer’s instructions (Nayak et al., 2009). cDNA was synthesized with the iScript II Kit (Bio-Rad), and PCR was performed with primers listed in the first paragraph.
ChIP
CD4+ T cells were activated and, if indicated, differentiated for 3 d. For STAT5 detection, cells were restimulated by 100 U/ml hIL-2 for 1 h (Laurence et al., 2007), followed by chromatin cross-linking (10 min with 1% formaldehyde); sonication of cells (14 min); and immunoprecipitation with anti-NFATc1 (7A6; BD PharMingen), anti-IgG (BD PharMingen), rabbit polyclonal H3-Ac (39139; Active Motif), rabbit anti-STAT5 (AF2168; R&D Systems), or rabbit IgG (02-6102; Thermo Fisher Scientific) overnight at 4°C and protein A/G Magnetic Beads (88803; Thermo Fisher Scientific; 2 h at 4°C). Cross-links were reversed at 65°C for 4 h, and the eluted purified DNA was analyzed by quantitative RT-PCR (qRT-PCR). For NFATc1, ChIP-IT Express kit (Active Motif) was used according to the manufacturer’s instructions. For NFATc1 and H3-Ac evaluation, the following primers were used: Ifng promoter, forward 5′-CACAAACAAAGGCTCCCTGT-3′, reverse 5′-CCACCTGTGCCATTCTTGT-3′; Il17a promoter, forward 5′-AATAGATTCTCAATGGTAGCC-3′, reverse 5′-GAAAATTCTTACTTTTGTAAACAG-3′; Il2 promoter, forward 5′-ATGGGGGTGTCACGATGTTTT-3′, reverse 5′-AACCCGACCAAGAGGGATTTC-3′; and Actb, forward 5′-TGCAAAGAAGCTGTGCTCGC-3′, reverse 5′-GCCGTTCCGAAAGTTGCCTT-3′; and for STAT5 binding at the Il17a promoter (Laurence et al., 2007): P4, forward 5′-TCATCGGCTCCCACACAGA-3′, reverse 5′-GGCAGTACCGAAGCTGTTTCA-3′; and no binding site Nb1, forward 5′-CACCAGCCAACCCAATTAAAA-3′, reverse 5′-CCTTCCTCATGTGATATGGCAAA-3′). The ΔΔCt value for each sample was normalized to the corresponding input value and subtracted from the normal rabbit IgG control.
Transient transfection and reporter gene assay
HEK 293T cells were transfected with pBcl2a1-luc and pEYZ/MCS (mock with multiple cloning sites) or pEYZ/Blimp-1F (Schmidt et al., 2008) by using the conventional calcium phosphate technique. Luciferase assays were performed as previously described (Nayak et al., 2009). Bcl2a1 promoter-regulated luciferase reporters were made with the pGL3 vector. A putative Blimp-1 binding site present in the pGL3 backbone was removed by Ssp1 cleavage. A murine Bcl2a1 proximal promoter fragment comprising ∼2 kb specific promoter sequences was obtained by PCR using the primers 5A1pro/2011, 5′-AGATCTTAAGGCAATAGGTGGAGCGTGAAG-3′, plus 3A1Pr.Nco-ATG, 5′-CCATGGTCCCTGGCAGAGCTGACTG-3′. 5A1pro/211, 5′-AGATCTACTGCCAGAGTGACTTTGAGG-3′, or 5A1pro/79, 5′-AGATCTCATTTTCCTCCTTCACAGTCTG-3′ plus 3A1Pr.Nco-ATG gave rise to pGL3-211_Bcl2a1 and pGL3-79_Bcl2a1, respectively. Blimp-1 site-deficient pGL3-79_Bcl2a1 was generated by site-specific mutagenesis. Each of the Blimp-1 binding sites was replaced with the previously reported mutant site 5′-CTAACCAGCT-3′ (Kuo and Calame, 2004).
RNA sequencing assays
In vitro differentiated Th1, Th2, and Th17 were collected on days 2.5 and 3.5. Cells from aGvHD-diseased mice were enriched by flow cytometry using Zombie Aqua Fixable Viability Kit, anti-CD90.1 (OX-7), and anti-CD4 (RM4-5) and directly collected in RNA buffer. Quantity of total RNA was assessed with Qubit 2.0, and quality was checked using an RNA 6000 Pico chip on Agilent’s bioanalyzer. Barcoded mRNA sequencing libraries were prepared from 700 ng (in vitro differentiated cells) or 10 ng (ex vivo) total RNA using a NEBNext Poly(A) mRNA Magnetic Isolation Module and NEBNext Ultra II RNA Library Prep Kit for Illumina according to the manual. All quality control was done using Invitrogen’s Qubit HS assay, and fragment size was determined on Agilent’s 2100 Bioanalyzer HS DNA assay. Barcoded RNA sequencing libraries were onboard clustered using HiSeq Rapid SR Cluster Kit v2 with 8pM, and 59 nucleotides were sequenced on the Illumina HiSeq2500 using HiSeq Rapid SBS Kit v2. Analysis of raw sequencing data and calculation of RPKM (reads per kilobase of transcript per million mapped reads) was performed using the CLC Genomics Workbench v7.5 (Qiagen) with CLC’s default settings for RNA sequencing analysis. Quality control was accessed using FASTQC, and low-quality ends and leftover adapter sequences were trimmed. Afterward, reads were aligned to Mus musculus genome assembly GRCm38 (99) using STAR (Dobin and Gingeras, 2016), and a two-pass mode of alignment strategy was applied to obtain higher alignment sensitivity. The mapped reads distribution was evaluated using RseQC. Sequence counting and feature assignment was then performed with subread-featureCounts (Liao et al., 2019). The resulting matrix was imported into R for differential gene expression analysis. The batch effect was minimized by removing unwanted variation normalization, taking both housekeeping 12 ribosomal protein gene expression values and residuals obtained from a first-pass regression of the counts as two normalization steps into account. A common dispersion value was estimated by the variation of housekeeping genes from different cell libraries. The differential expression genes were then determined with edgeR. We compared significantly regulated genes in ΔSUMO defined by a threshold of P value 0.02 and log fold-change 1.4, together with six other interesting genes (Bcl2a1b, Bcl2a1d, Tbx21, Prdm1, Il4, and Ctla4) that were slightly below the threshold but also differentially expressed, plotted into a heatmap.
Statistical analyses
All data are shown as mean ± SD (or SEM when indicated) and represent combined data from at least three independent experiments unless otherwise noted. Figures were prepared using GraphPad Prism 5, Adobe Photoshop and Illustrator, and Corel Draw.
The results were analyzed with Prism software (GraphPad) using two-tailed paired or unpaired Student’s t test, Mann–Whitney U test, and two-way ANOVA. Differences with P values < 0.05 were considered significant: *, P < 0.05; **, P < 0.005, and ***, P < 0.001. Replicates, as indicated, are individual mice or experiments.
Data availability
RNA sequencing data have been deposited in GEO, accession nos. GSE148087 and GSE119313.
Online supplemental materials
Fig. S1 shows that lymphoid compartments appear unaltered in Nfatc1deltaSUMO mice in steady state. Fig. S2 shows expression analyses of in vitro differentiated Th1, Th2, and Th17. Fig. S3 shows that transplantation of allogeneic T cells from Nfatc1deltaSUMO donor mice leads to less-acute GvHD compared with WT T cells. Fig. S4 shows that pure NFATc1/ΔS+ T conv cause less GvHD than WT T conv. Fig. S5 shows that NFATc1/A transactivates Bcl2a1, whereas Blimp-1 suppresses it.
Acknowledgments
The generation of the Nfatc1deltaSUMO mouse was outsourced to inGenious Targeting Laboratory. We are indebted to Benjamin Lunz, Nadine Winter, and Melanie Schott for excellent experimental assistance, and thank Sophia Hochrein for support with T-cell differentiation, as well as Marco J. Herold for an aliquot of the anti-Bcl2A1 serum.
This work was mainly supported by grants from the Deutsche Forschungsgemeinschaft (SPP1365 to F. Berberich-Siebelt; 324392634 TRR 221 to F. Berberich-Siebelt, A. Rosenwald, and A. Beilhack; and TRR 221 INF to C. Liang) as well as from the Wilhelm Sander-Stiftung (2012.047.2 to F. Berberich-Siebelt) and the Interdisciplinary Center for Clinical Research, University of Würzburg, provided by the Bundesministerium für Bildung und Forschung (A-167 to M. Buttmann and F. Berberich-Siebelt). The Fritz Thyssen Stiftung (10.13.2.215 to M. Vaeth and F. Berberich-Siebelt) and the Else Kröner-Fresenius-Stiftung (2015_A232 to F. Berberich-Siebelt) provided additional funding. M. Qureischi held a fellowship by the Graduate School of Life Sciences, University of Würzburg. Y. Xiao receives a fellowship by the China Scholarship Council.
Author contribution: Y. Xiao, L. Dietz, M. Qureischi, and M. Vaeth designed and performed research and analyzed the data; S. Klein-Hessling analyzed and discussed data; S.D. Vallabhapurapu, M. Klein, A. Mottok, and A. König did experiments and analyzed data; C. Liang analyzed data; I. Berberich performed research and discussed data; E. Serfling, T. Bopp, A. Rosenwald, M. Buttmann, and A. Beilhack discussed the data and provided financial support; F. Berberich-Siebelt conceptualized the research goals, acquired major funding, designed and performed research, analyzed and discussed the data, and wrote the manuscript.
References
- Abbas, A.K., Trotta E., Simeonov D.R., Marson A., and Bluestone J.A.. 2018. Revisiting IL-2: Biology and therapeutic prospects. Sci. Immunol. 3. eaat1482. 10.1126/sciimmunol.aat1482 [DOI] [PubMed] [Google Scholar]
- Bai, X.F., Liu J.Q., Liu X., Guo Y., Cox K., Wen J., Zheng P., and Liu Y.. 2000. The heat-stable antigen determines pathogenicity of self-reactive T cells in experimental autoimmune encephalomyelitis. J. Clin. Invest. 105:1227–1232. 10.1172/JCI9012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beilhack, A., Schulz S., Baker J., Beilhack G.F., Wieland C.B., Herman E.I., Baker E.M., Cao Y.A., Contag C.H., and Negrin R.S.. 2005. In vivo analyses of early events in acute graft-versus-host disease reveal sequential infiltration of T-cell subsets. Blood. 106:1113–1122. 10.1182/blood-2005-02-0509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berberich-Siebelt, F., Berberich I., Andrulis M., Santner-Nanan B., Jha M.K., Klein-Hessling S., Schimpl A., and Serfling E.. 2006. SUMOylation interferes with CCAAT/enhancer-binding protein beta-mediated c-myc repression, but not IL-4 activation in T cells. J. Immunol. 176:4843–4851. 10.4049/jimmunol.176.8.4843 [DOI] [PubMed] [Google Scholar]
- Berberich-Siebelt, F., Klein-Hessling S., Hepping N., Santner-Nanan B., Lindemann D., Schimpl A., Berberich I., and Serfling E.. 2000. C/EBPbeta enhances IL-4 but impairs IL-2 and IFN-gamma induction in T cells. Eur. J. Immunol. 30:2576–2585. [DOI] [PubMed] [Google Scholar]
- Bettelli, E., Pagany M., Weiner H.L., Linington C., Sobel R.A., and Kuchroo V.K.. 2003. Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J. Exp. Med. 197:1073–1081. 10.1084/jem.20021603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyman, O., and Sprent J.. 2012. The role of interleukin-2 during homeostasis and activation of the immune system. Nat. Rev. Immunol. 12:180–190. 10.1038/nri3156 [DOI] [PubMed] [Google Scholar]
- Carrington, E.M., Zhan Y., Brady J.L., Zhang J.G., Sutherland R.M., Anstee N.S., Schenk R.L., Vikstrom I.B., Delconte R.B., Segal D., et al. 2017. Anti-apoptotic proteins BCL-2, MCL-1 and A1 summate collectively to maintain survival of immune cell populations both in vitro and in vivo. Cell Death Differ. 24:878–888. 10.1038/cdd.2017.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuvpilo, S., Avots A., Berberich-Siebelt F., Glöckner J., Fischer C., Kerstan A., Escher C., Inashkina I., Hlubek F., Jankevics E., et al. 1999. Multiple NF-ATc isoforms with individual transcriptional properties are synthesized in T lymphocytes. J. Immunol. 162:7294–7301. [PubMed] [Google Scholar]
- Chuvpilo, S., Jankevics E., Tyrsin D., Akimzhanov A., Moroz D., Jha M.K., Schulze-Luehrmann J., Santner-Nanan B., Feoktistova E., König T., et al. 2002. Autoregulation of NFATc1/A expression facilitates effector T cells to escape from rapid apoptosis. Immunity. 16:881–895. 10.1016/S1074-7613(02)00329-1 [DOI] [PubMed] [Google Scholar]
- Cimmino, L., Martins G.A., Liao J., Magnusdottir E., Grunig G., Perez R.K., and Calame K.L.. 2008. Blimp-1 attenuates Th1 differentiation by repression of ifng, tbx21, and bcl6 gene expression. J. Immunol. 181:2338–2347. 10.4049/jimmunol.181.4.2338 [DOI] [PubMed] [Google Scholar]
- Cooke, K.R., Kobzik L., Martin T.R., Brewer J., J. Delmonte, Jr., Crawford J.M., and Ferrara J.L.. 1996. An experimental model of idiopathic pneumonia syndrome after bone marrow transplantation: I. The roles of minor H antigens and endotoxin. Blood. 88:3230–3239. 10.1182/blood.V88.8.3230.bloodjournal8883230 [DOI] [PubMed] [Google Scholar]
- Cretney, E., Leung P.S., Trezise S., Newman D.M., Rankin L.C., Teh C.E., Putoczki T.L., Gray D.H., Belz G.T., Mielke L.A., et al. 2018. Characterization of Blimp-1 function in effector regulatory T cells. J. Autoimmun. 91:73–82. 10.1016/j.jaut.2018.04.003 [DOI] [PubMed] [Google Scholar]
- Decque, A., Joffre O., Magalhaes J.G., Cossec J.C., Blecher-Gonen R., Lapaquette P., Silvin A., Manel N., Joubert P.E., Seeler J.S., et al. 2016. Sumoylation coordinates the repression of inflammatory and anti-viral gene-expression programs during innate sensing. Nat. Immunol. 17:140–149. 10.1038/ni.3342 [DOI] [PubMed] [Google Scholar]
- Dietz, L., Frommer F., Vogel A.L., Vaeth M., Serfling E., Waisman A., Buttmann M., and Berberich-Siebelt F.. 2015. NFAT1 deficit and NFAT2 deficit attenuate EAE via different mechanisms. Eur. J. Immunol. 45:1377–1389. 10.1002/eji.201444638 [DOI] [PubMed] [Google Scholar]
- Ding, X., Wang A., Ma X., Demarque M., Jin W., Xin H., Dejean A., and Dong C.. 2016. Protein SUMOylation Is Required for Regulatory T Cell Expansion and Function. Cell Rep. 16:1055–1066. 10.1016/j.celrep.2016.06.056 [DOI] [PubMed] [Google Scholar]
- Dobin, A., and Gingeras T.R.. 2016. Optimizing RNA-Seq Mapping with STAR. Methods Mol. Biol. 1415:245–262. 10.1007/978-1-4939-3572-7_13 [DOI] [PubMed] [Google Scholar]
- Flotho, A., and Melchior F.. 2013. Sumoylation: a regulatory protein modification in health and disease. Annu. Rev. Biochem. 82:357–385. 10.1146/annurev-biochem-061909-093311 [DOI] [PubMed] [Google Scholar]
- Fontenot, J.D., Rasmussen J.P., Gavin M.A., and Rudensky A.Y.. 2005. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat. Immunol. 6:1142–1151. 10.1038/ni1263 [DOI] [PubMed] [Google Scholar]
- Fu, S.H., Yeh L.T., Chu C.C., Yen B.L., and Sytwu H.K.. 2017. New insights into Blimp-1 in T lymphocytes: a divergent regulator of cell destiny and effector function. J. Biomed. Sci. 24:49. 10.1186/s12929-017-0354-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong, D., and Malek T.R.. 2007. Cytokine-dependent Blimp-1 expression in activated T cells inhibits IL-2 production. J. Immunol. 178:242–252. 10.4049/jimmunol.178.1.242 [DOI] [PubMed] [Google Scholar]
- Gonzalez, J., Orlofsky A., and Prystowsky M.B.. 2003. A1 is a growth-permissive antiapoptotic factor mediating postactivation survival in T cells. Blood. 101:2679–2685. 10.1182/blood-2002-04-1229 [DOI] [PubMed] [Google Scholar]
- Herold, M.J., Zeitz J., Pelzer C., Kraus C., Peters A., Wohlleben G., and Berberich I.. 2006. The stability and anti-apoptotic function of A1 are controlled by its C terminus. J. Biol. Chem. 281:13663–13671. 10.1074/jbc.M600266200 [DOI] [PubMed] [Google Scholar]
- Hogan, P.G., Chen L., Nardone J., and Rao A.. 2003. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 17:2205–2232. 10.1101/gad.1102703 [DOI] [PubMed] [Google Scholar]
- Huber, S., Gagliani N., Esplugues E., W. O’Connor, Jr., Huber F.J., Chaudhry A., Kamanaka M., Kobayashi Y., Booth C.J., Rudensky A.Y., et al. 2011. Th17 cells express interleukin-10 receptor and are controlled by Foxp3− and Foxp3+ regulatory CD4+ T cells in an interleukin-10-dependent manner. Immunity. 34:554–565. 10.1016/j.immuni.2011.01.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglesias, M., Augustin J.J., Alvarez P., Santiuste I., Postigo J., Merino J., and Merino R.. 2016. Selective Impairment of TH17-Differentiation and Protection against Autoimmune Arthritis after Overexpression of BCL2A1 in T Lymphocytes. PLoS One. 11. e0159714. 10.1371/journal.pone.0159714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joller, N., Lozano E., Burkett P.R., Patel B., Xiao S., Zhu C., Xia J., Tan T.G., Sefik E., Yajnik V., et al. 2014. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity. 40:569–581. 10.1016/j.immuni.2014.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josefowicz, S.Z., and Rudensky A.. 2009. Control of regulatory T cell lineage commitment and maintenance. Immunity. 30:616–625. 10.1016/j.immuni.2009.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juno, J.A., van Bockel D., Kent S.J., Kelleher A.D., Zaunders J.J., and Munier C.M.. 2017. Cytotoxic CD4 T Cells-Friend or Foe during Viral Infection? Front. Immunol. 8:19. 10.3389/fimmu.2017.00019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaji, T., Hijikata A., Ishige A., Kitami T., Watanabe T., Ohara O., Yanaka N., Okada M., Shimoda M., Taniguchi M., et al. 2016. CD4 memory T cells develop and acquire functional competence by sequential cognate interactions and stepwise gene regulation. Int. Immunol. 28:267–282. 10.1093/intimm/dxv071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein-Hessling, S., Muhammad K., Klein M., Pusch T., Rudolf R., Flöter J., Qureischi M., Beilhack A., Vaeth M., Kummerow C., et al. 2017. NFATc1 controls the cytotoxicity of CD8+ T cells. Nat. Commun. 8:511. 10.1038/s41467-017-00612-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo, T.C., and Calame K.L.. 2004. B lymphocyte-induced maturation protein (Blimp)-1, IFN regulatory factor (IRF)-1, and IRF-2 can bind to the same regulatory sites. J. Immunol. 173:5556–5563. 10.4049/jimmunol.173.9.5556 [DOI] [PubMed] [Google Scholar]
- Lahl, K., Loddenkemper C., Drouin C., Freyer J., Arnason J., Eberl G., Hamann A., Wagner H., Huehn J., and Sparwasser T.. 2007. Selective depletion of Foxp3+ regulatory T cells induces a scurfy-like disease. J. Exp. Med. 204:57–63. 10.1084/jem.20061852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang, M.J., Brennan M.S., O’Reilly L.A., Ottina E., Czabotar P.E., Whitlock E., Fairlie W.D., Tai L., Strasser A., and Herold M.J.. 2014. Characterisation of a novel A1-specific monoclonal antibody. Cell Death Dis. 5. e1553. 10.1038/cddis.2014.519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langrish, C.L., Chen Y., Blumenschein W.M., Mattson J., Basham B., Sedgwick J.D., McClanahan T., Kastelein R.A., and Cua D.J.. 2005. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 201:233–240. 10.1084/jem.20041257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurence, A., Tato C.M., Davidson T.S., Kanno Y., Chen Z., Yao Z., Blank R.B., Meylan F., Siegel R., Hennighausen L., et al. 2007. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 26:371–381. 10.1016/j.immuni.2007.02.009 [DOI] [PubMed] [Google Scholar]
- Lee, P.P., Fitzpatrick D.R., Beard C., Jessup H.K., Lehar S., Makar K.W., Pérez-Melgosa M., Sweetser M.T., Schlissel M.S., Nguyen S., et al. 2001. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity. 15:763–774. 10.1016/S1074-7613(01)00227-8 [DOI] [PubMed] [Google Scholar]
- Levine, A.G., Arvey A., Jin W., and Rudensky A.Y.. 2014. Continuous requirement for the TCR in regulatory T cell function. Nat. Immunol. 15:1070–1078. 10.1038/ni.3004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lexberg, M.H., Taubner A., Albrecht I., Lepenies I., Richter A., Kamradt T., Radbruch A., and Chang H.D.. 2010. IFN-γ and IL-12 synergize to convert in vivo generated Th17 into Th1/Th17 cells. Eur. J. Immunol. 40:3017–3027. 10.1002/eji.201040539 [DOI] [PubMed] [Google Scholar]
- Liao, Y., Smyth G.K., and Shi W.. 2019. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 47. e47. 10.1093/nar/gkz114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodygin, D., Odoardi F., Schläger C., Körner H., Kitz A., Nosov M., van den Brandt J., Reichardt H.M., Haberl M., and Flügel A.. 2013. A combination of fluorescent NFAT and H2B sensors uncovers dynamics of T cell activation in real time during CNS autoimmunity. Nat. Med. 19:784–790. 10.1038/nm.3182 [DOI] [PubMed] [Google Scholar]
- Macian, F. 2005. NFAT proteins: key regulators of T-cell development and function. Nat. Rev. Immunol. 5:472–484. 10.1038/nri1632 [DOI] [PubMed] [Google Scholar]
- Martinez, G.J., Pereira R.M., Äijö T., Kim E.Y., Marangoni F., Pipkin M.E., Togher S., Heissmeyer V., Zhang Y.C., Crotty S., et al. 2015. The transcription factor NFAT promotes exhaustion of activated CD8+ T cells. Immunity. 42:265–278. 10.1016/j.immuni.2015.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins, G.A., Cimmino L., Shapiro-Shelef M., Szabolcs M., Herron A., Magnusdottir E., and Calame K.. 2006. Transcriptional repressor Blimp-1 regulates T cell homeostasis and function. Nat. Immunol. 7:457–465. 10.1038/ni1320 [DOI] [PubMed] [Google Scholar]
- Martins, G.A., Cimmino L., Liao J., Magnusdottir E., and Calame K.. 2008. Blimp-1 directly represses Il2 and the Il2 activator Fos, attenuating T cell proliferation and survival. J. Exp. Med. 205:1959–1965. 10.1084/jem.20080526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mombaerts, P., Iacomini J., Johnson R.S., Herrup K., Tonegawa S., and Papaioannou V.E.. 1992b. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 68:869–877. 10.1016/0092-8674(92)90030-G [DOI] [PubMed] [Google Scholar]
- Nayak, A., Glöckner-Pagel J., Vaeth M., Schumann J.E., Buttmann M., Bopp T., Schmitt E., Serfling E., and Berberich-Siebelt F.. 2009. Sumoylation of the transcription factor NFATc1 leads to its subnuclear relocalization and interleukin-2 repression by histone deacetylase. J. Biol. Chem. 284:10935–10946. 10.1074/jbc.M900465200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters, A., Lee Y., and Kuchroo V.K.. 2011. The many faces of Th17 cells. Curr. Opin. Immunol. 23:702–706. 10.1016/j.coi.2011.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter, C.M., and Clipstone N.A.. 2002. Sustained NFAT signaling promotes a Th1-like pattern of gene expression in primary murine CD4+ T cells. J. Immunol. 168:4936–4945. 10.4049/jimmunol.168.10.4936 [DOI] [PubMed] [Google Scholar]
- Santner-Nanan, B., Berberich-Siebelt F., Xiao Z., Poser N., Sennefelder H., Rauthe S., Vallabhapurapu D.S., Berberich I., Schimpl A., Kreth H.-W., et al. 2006. Blimp-1 is expressed in human and mouse T cell subsets and leads to loss of IL-2 production and to defective proliferation. Signal Transduct. 6:268–279. 10.1002/sita.200500062 [DOI] [Google Scholar]
- Schenk, R.L., Tuzlak S., Carrington E.M., Zhan Y., Heinzel S., Teh C.E., Gray D.H., Tai L., Lew A.M., Villunger A., et al. 2017. Characterisation of mice lacking all functional isoforms of the pro-survival BCL-2 family member A1 reveals minor defects in the haematopoietic compartment. Cell Death Differ. 24:534–545. 10.1038/cdd.2016.156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt, D., Nayak A., Schumann J.E., Schimpl A., Berberich I., and Berberich-Siebelt F.. 2008. Blimp-1Deltaexon7: a naturally occurring Blimp-1 deletion mutant with auto-regulatory potential. Exp. Cell Res. 314:3614–3627. 10.1016/j.yexcr.2008.09.008 [DOI] [PubMed] [Google Scholar]
- Schuh, K., Twardzik T., Kneitz B., Heyer J., Schimpl A., and Serfling E.. 1998. The interleukin 2 receptor alpha chain/CD25 promoter is a target for nuclear factor of activated T cells. J. Exp. Med. 188:1369–1373. 10.1084/jem.188.7.1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serfling, E., Avots A., Klein-Hessling S., Rudolf R., Vaeth M., and Berberich-Siebelt F.. 2012. NFATc1/αA: The other Face of NFAT Factors in Lymphocytes. Cell Commun. Signal. 10:16. 10.1186/1478-811X-10-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serfling, E., Berberich-Siebelt F., Chuvpilo S., Jankevics E., Klein-Hessling S., Twardzik T., and Avots A.. 2000. The role of NF-AT transcription factors in T cell activation and differentiation. Biochim. Biophys. Acta. 1498:1–18. 10.1016/S0167-4889(00)00082-3 [DOI] [PubMed] [Google Scholar]
- Serfling, E., Klein-Hessling S., Palmetshofer A., Bopp T., Stassen M., and Schmitt E.. 2006. NFAT transcription factors in control of peripheral T cell tolerance. Eur. J. Immunol. 36:2837–2843. 10.1002/eji.200536618 [DOI] [PubMed] [Google Scholar]
- Terui, Y., Saad N., Jia S., McKeon F., and Yuan J.. 2004. Dual role of sumoylation in the nuclear localization and transcriptional activation of NFAT1. J. Biol. Chem. 279:28257–28265. 10.1074/jbc.M403153200 [DOI] [PubMed] [Google Scholar]
- Vaeth, M., Bäuerlein C.A., Pusch T., Findeis J., Chopra M., Mottok A., Rosenwald A., Beilhack A., and Berberich-Siebelt F.. 2015. Selective NFAT targeting in T cells ameliorates GvHD while maintaining antitumor activity. Proc. Natl. Acad. Sci. USA. 112:1125–1130. 10.1073/pnas.1409290112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaeth, M., Müller G., Stauss D., Dietz L., Klein-Hessling S., Serfling E., Lipp M., Berberich I., and Berberich-Siebelt F.. 2014. Follicular regulatory T cells control humoral autoimmunity via NFAT2-regulated CXCR5 expression. J. Exp. Med. 211:545–561. 10.1084/jem.20130604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaeth, M., Schliesser U., Müller G., Reissig S., Satoh K., Tuettenberg A., Jonuleit H., Waisman A., Müller M.R., Serfling E., et al. 2012. Dependence on nuclear factor of activated T-cells (NFAT) levels discriminates conventional T cells from Foxp3+ regulatory T cells. Proc. Natl. Acad. Sci. USA. 109:16258–16263. 10.1073/pnas.1203870109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vahl, J.C., Drees C., Heger K., Heink S., Fischer J.C., Nedjic J., Ohkura N., Morikawa H., Poeck H., Schallenberg S., et al. 2014. Continuous T cell receptor signals maintain a functional regulatory T cell pool. Immunity. 41:722–736. 10.1016/j.immuni.2014.10.012 [DOI] [PubMed] [Google Scholar]
- Van Nguyen, T., Angkasekwinai P., Dou H., Lin F.M., Lu L.S., Cheng J., Chin Y.E., Dong C., and Yeh E.T.. 2012. SUMO-specific protease 1 is critical for early lymphoid development through regulation of STAT5 activation. Mol. Cell. 45:210–221. 10.1016/j.molcel.2011.12.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, A., Ding X., Demarque M., Liu X., Pan D., Xin H., Zhong B., Wang X., Dejean A., Jin W., et al. 2017. Ubc9 Is Required for Positive Selection and Late-Stage Maturation of Thymocytes. J. Immunol. 198:3461–3470. 10.4049/jimmunol.1600980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, A.Y.L., Loh C.Y.Y., Chen S.J., Kao H.K., Lin C.H., Chuang S.H., Lee C.M., Sytwu H.K., and Wei F.C.. 2018. Blimp-1 prolongs allograft survival without regimen via influencing T cell development in favor of regulatory T cells while suppressing Th1. Mol. Immunol. 99:53–65. 10.1016/j.molimm.2018.04.004 [DOI] [PubMed] [Google Scholar]
- Yu, X., Lao Y., Teng X.L., Li S., Zhou Y., Wang F., Guo X., Deng S., Chang Y., Wu X., et al. 2018. SENP3 maintains the stability and function of regulatory T cells via BACH2 deSUMOylation. Nat. Commun. 9:3157. 10.1038/s41467-018-05676-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
RNA sequencing data have been deposited in GEO, accession nos. GSE148087 and GSE119313.