

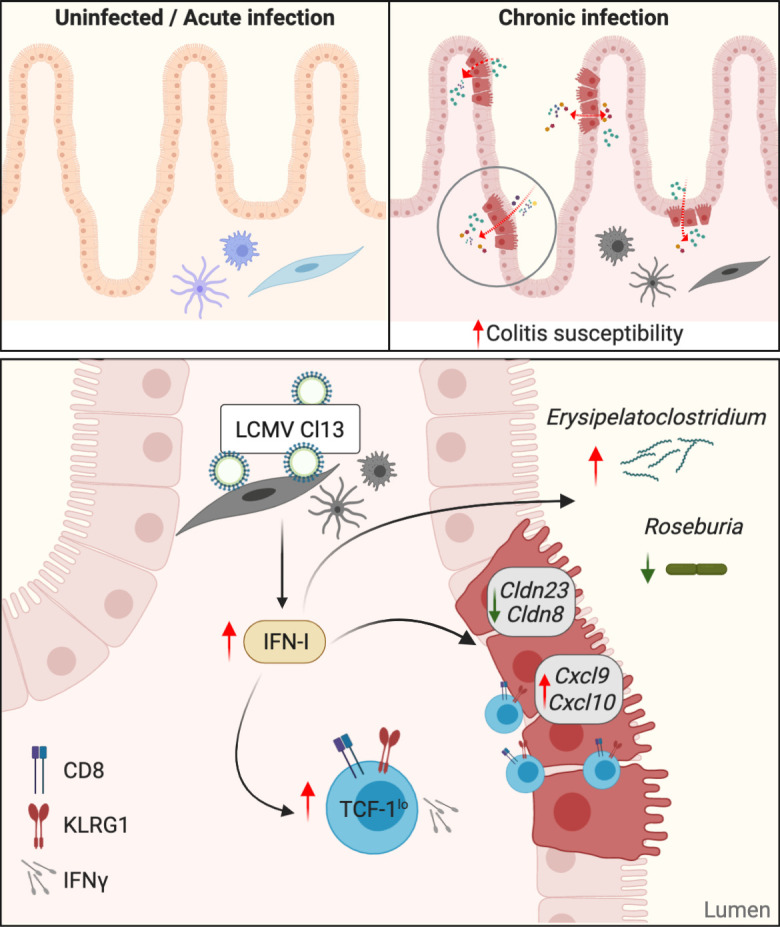
Intestinal barrier dysfunction is a marker of disease progression during chronic viral infections, but its causes remain mostly unknown. This study demonstrates essential roles for type I IFN signaling and CD8 T cell responses in increasing intestinal permeability during chronic viral infection.
Abstract
Intestinal barrier leakage constitutes a potential therapeutic target for many inflammatory diseases and represents a disease progression marker during chronic viral infections. However, the causes of altered gut barrier remain mostly unknown. Using murine infection with lymphocytic choriomeningitis virus, we demonstrate that, in contrast to an acute viral strain, a persistent viral isolate leads to long-term viral replication in hematopoietic and mesenchymal cells, but not epithelial cells (IECs), in the intestine. Viral persistence drove sustained intestinal epithelial barrier leakage, which was characterized by increased paracellular flux of small molecules and was associated with enhanced colitis susceptibility. Type I IFN signaling caused tight junction dysregulation in IECs, promoted gut microbiome shifts and enhanced intestinal CD8 T cell responses. Notably, both type I IFN receptor blockade and CD8 T cell depletion prevented infection-induced barrier leakage. Our study demonstrates that infection with a virus that persistently replicates in the intestinal mucosa increases epithelial barrier permeability and reveals type I IFNs and CD8 T cells as causative factors of intestinal leakage during chronic infections.
Graphical Abstract
Introduction
Chronic infections such as those caused by HIV, Mycobacterium tuberculosis, or Plasmodium falciparum constitute a global health burden and induce host adaptations that allow for long-term host–pathogen coexistence (Stelekati and Wherry, 2012; Virgin et al., 2009; Zuniga et al., 2015). This is in contrast to nonequilibrium outcomes, such as pathogen eradication or host death (Virgin et al., 2009). Immune adaptations are a major determinant of host–pathogen equilibrium, and are driven by the extracellular environment, whereby antigen and oxygen levels, cytokine sensing, or pattern recognition receptor engagement have been shown to imprint cell-intrinsic transcriptional and epigenetic rewiring (Virgin et al., 2009; Zuniga et al., 2015). Some of the best-studied long-term host adaptations to chronic pathogens were initially found in the mouse model of infection with the rodent-borne arenavirus lymphocytic choriomeningitis virus (LCMV) and include loss of selected functions of the immune system (Hashimoto et al., 2018; Zuniga et al., 2015). This may have evolved to ensure host fitness in the face of widespread pathogen replication (Virgin et al., 2009; Zuniga et al., 2015). An exhausted or hypofunctional state of pathogen-specific CD8 T cells was, for instance, first observed in mice infected with persistent LCMV variants, which continuously replicate in systemic circulation and most tissues for ∼60–90 d (Gallimore et al., 1998; Zajac et al., 1998). Such a T cell exhaustion state, in which CD8 T cells progressively lose effector functions (Wherry et al., 2003) and up-regulate surface expression of coinhibitory receptors such as PD1 (Barber et al., 2006; Wherry et al., 2007), was later extended to human infections and cancer (Hashimoto et al., 2018).
The gastrointestinal (GI) tract harbors the gut microbiota and food components as well as their derived metabolites and a large number of immune cells in which crosstalk ensures intestinal homeostasis (Belkaid and Harrison, 2017; Rooks and Garrett, 2016). To prevent pathology, gut luminal contents need to be compartmentalized away from the host (Odenwald and Turner, 2017). This is achieved by the intestinal epithelium, which acts as a physical, immunological, and selectively permeable barrier (Peterson and Artis, 2014) that enables bidirectional flux of ions, water, nutrients, and waste (Buckley and Turner, 2018). There are three described routes of solute transport across the intestinal epithelium: the size and charge-restricted pore pathway, which allows paracellular ion and water exchange and is regulated by claudin proteins (Odenwald and Turner, 2017); the leak pathway, which permits paracellular passage of solutes ≤90 Å in diameter and requires activated myosin light chain kinase activity (Graham et al., 2019; Su et al., 2013; Weber et al., 2010); and the unrestricted pathway, which allows passage of particles >100 Å in diameter at sites of epithelial damage (Shen et al., 2011; Turner, 2009).
Infections with chronic pathogens such as HIV or P. falciparum in humans and simian immunodeficiency virus (SIV) in rhesus macaques (RMs) are associated with intestinal barrier dysfunction (Ponte et al., 2016; Tincati et al., 2016; Wilairatana et al., 1997). For instance, peripheral immune activation, a strong predictor of disease progression in HIV-infected individuals, was proposed to stem from translocation of gut luminal components into the peripheral circulation as a result of an increasingly permeable intestinal epithelium (Brenchley et al., 2006). Epithelial barrier dysfunction was also described in SIV-infected RMs, which develop immune activation and AIDS-related morbidity (Brenchley et al., 2006; Estes et al., 2010). In spite of the prevalence of barrier dysfunction upon chronic viral infections, its underlying mechanisms remain largely undefined, in part due to the lack of small animal models in which interventional experiments can be performed.
In this study, we employed infection of mice with the acute (Armstrong [ARM]) and persistent (Cl13) variants of LCMV to identify GI tract adaptations in the context of chronic infections. We found that a persistent, but not an acute, LCMV isolate sustainably replicated in hematopoietic and mesenchymal, but not in epithelial cells within the small intestine. Chronic LCMV infection also resulted in long-term, size-selective increase in permeability of small-molecular-weight molecules, typically associated with the leak pathway of paracellular flux (Shen et al., 2011; Turner, 2009). The transcriptional tight junction dysregulation associated with this barrier leakage required type I IFN signaling, which also influenced intestinal epithelial cell (IEC) proliferative capacity and glycolytic metabolism. In addition, type I IFN signaling increased IEC expression of the CXCR3+ effector T cell chemoattractants Cxcl9 and Cxcl10 (Groom and Luster, 2011) and promoted CD8 T cell accumulation and effector functions within the small intestine. Type I IFN signaling also promoted the overrepresentation of Erysipelatoclostridium and reduced the relative abundance of Roseburia in the intestinal microbiome of chronically infected mice, which associated with the induction of intestinal leakage. Importantly, both type I IFN receptor (IFNAR) blockade and CD8 T cell depletion fully abrogated the intestinal permeability increase after LCMV Cl13 infection. Lastly, we observed that viral persistence and increased barrier permeability associated with enhanced morbidity and mortality upon secondary chemical or microbial intestinal insults. Overall, our data establish the existence of a previously unrecognized role for the type I IFN–CD8 T cell axis in the modulation of intestinal epithelial barrier function during chronic viral infection and raise the possibility that type I IFN represents a cause, and not necessarily a consequence, of microbial translocation during other infections. Finally, our work puts forward the usage of chronic LCMV infection in mice as a cost-effective model to study the biology of leaky gut at the whole-organism level.
Results
LCMV Cl13 persists in mesenchymal and hematopoietic cells, but not in epithelial cells within intestinal tissues
To investigate host adaptations to persistent LCMV infection in the GI tract, we first assessed the intestinal localization and replicating capacity of the persistent LCMV Cl13 isolate in comparison to the acute LCMV ARM strain (Ahmed et al., 1984; Wherry et al., 2003). Immunofluorescence with anti-LCMV antibodies (Abs) revealed the presence of virus in the distal small intestines of Cl13-, but not ARM-infected, mice, on days 9 and 24 postinfection (p.i.; Fig. 1 A), consistent with a previous report (Beura et al., 2015). Importantly, plaque assays in ileum and colon homogenates indicated that the Cl13 (but not ARM) isolate productively replicated in this compartment at days 9 and 24 p.i. (Fig. 1 B, left graph) and up to day 300 p.i., a time point after systemic viremia was cleared (Fig. 1 B, right graph). We next investigated the cellular tropism of LCMV Cl13 in the intestine via immunofluorescence staining with anti-LCMV Ab in combination with Abs against CD45 and vimentin, which label hematopoietic and mesenchymal cells, respectively (Coulombe and Wong, 2004). We detected a significant overlap between LCMV signal and staining for both CD45 and vimentin at days 9 (Fig. 1, C and E) and 24 (Fig. 1 E and Fig. S1 A) p.i. In contrast, co-staining of LCMV and EpCAM, a pan-epithelial cell marker (Trzpis et al., 2007), did not overlap at any of the two time points assessed (Fig. 1, D and E; and Fig. S1 B). Overall, these results indicate that systemically inoculated LCMV Cl13 virus persistently replicated along the ileum and colon, even after systemic viremia was cleared, and that its cellular targets include hematopoietic and mesenchymal, but not epithelial, cells within the intestinal mucosa.
Figure 1.

LCMV Cl13 persists in small intestinal mesenchymal and hematopoietic, but not epithelial, cells and increases intestinal permeability to small molecules. See also Fig. S1 and Fig. S2.C57BL/6 mice were infected with LCMV ARM or Cl13 or left uninfected and analyzed at indicated time points p.i. (A, C, and D) Representative immunofluorescence staining of ileum sections with anti-LCMV Abs (red) and DAPI (blue), as well as anti-Vimentin (green) or anti-CD45.2 (cyan) Abs (C) or anti-EpCAM (green) Ab (D). Arrowheads highlight regions of overlap between markers. Scale bars represent 230 µm (A) or 36 µm (C and D). (B) LCMV PFUs in ileum and colon; limit of detection (LOD) is indicated by dotted lines. (E) Mander’s overlap coefficient between LCMV and EpCAM, Vimentin, or CD45 markers. (F) In vivo intestinal permeability to FD4 was measured in ARM- and Cl13-infected mice and normalized to levels in uninfected mice. Averages (E) and averages ± SEM (B and F) are shown. Data are representative of two independent experimental repeats (A–E) or pooled from two independent experimental repeats (F) with n = 3–8 mice/group. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; Kolmogorov–Smirnov test (F). n.s., not significant.
Figure S1.

LCMV Cl13 infects intestinal mesenchymal and hematopoietic cells, but not epithelial cells. Related to Fig. 1.C57BL/6 mice were infected with LCMV ARM or Cl13 or left uninfected. (A and B) Representative immunofluorescence staining of ileum sections with anti-LCMV Abs (red) and DAPI (blue) as well as anti-Vimentin (green; A, top), anti-CD45.2 (cyan; A, bottom), or anti-EpCAM (green; B) Abs at indicated time points p.i. Arrowheads highlight regions of overlap between markers. All scale bars correspond to 36 µm. Data are representative of two independent experimental repeats with n = 4 mice/group.
LCMV Cl13 infection persistently increases intestinal permeability to small molecules
We next sought to identify functional adaptations to persistent LCMV Cl13 infection along the GI tract. Among these, increased intestinal permeability has been reported during chronic viral infections in humans and nonhuman primates (Ponte et al., 2016; Tincati et al., 2016). To test whether LCMV Cl13 infection also caused gut leakage, we performed an in vivo intestinal permeability assay with small-molecular-weight FITC-labeled 4-kD dextran (FD4; Volynets et al., 2016) on days 3, 8, 20, 30, 45, and 60 after ARM or Cl13 infection and in uninfected mice. FD4 flux from the gut lumen to the serum was not affected by ARM infection but was significantly increased in Cl13-infected mice (Fig. 1 F). The FD4 permeability increase was detected, and peaked, at day 8 after Cl13 infection and persisted until day 45 p.i. (Fig. 1 F). In contrast, permeability of the larger FITC-labeled 40-kD dextran (FD40) was not increased in Cl13- versus ARM-infected or uninfected mice (Fig. S2 A). Consistent with this, histological examination failed to identify epithelial damage within the small and large intestines from Cl13- or ARM-infected mice at days 9 or 24 p.i. (Fig. S2, B and C). Together, the lack of apparent epithelial cell damage and/or ulceration and the unchanged FD40 permeability ruled out involvement of the unrestricted pathway of solute exchange (Shen et al., 2011; Turner, 2009) at the time points studied. Instead, our data indicated that infection with Cl13, but not ARM, caused a sustained increase in intestinal permeability of small-molecular-weight molecules typically associated with the leak pathway of paracellular flux (Shen et al., 2011; Turner, 2009).
Figure S2.

LCMV Cl13 infection neither increases intestinal permeability to FD40 nor causes epithelial layer disruption. Related to Fig. 1. C57BL/6 mice were infected with LCMV ARM or Cl13 or left uninfected (Un) and analyzed at indicated time points p.i. (A) In vivo intestinal permeability to FD40 was measured in ARM- and Cl13-infected mice and normalized to levels in uninfected mice. (B and C) Hematoxylin and eosin staining of small (B) and large (C) intestinal sections. All scale bars correspond to 72 µm. (A) Averages ± SEM are shown. Data in A are pooled from two experimental repeats where statistical significance was achieved in one out of two experiments or upon pooling. Data in B and C are representative of two independent experimental repeats. *, P < 0.05; Kruskal–Wallis with Dunn’s multiple comparisons correction (A).
LCMV Cl13 infection induces dysregulation in tight junction gene expression as well as a type I IFN signature in IECs
To elucidate the mechanisms driving increased intestinal barrier permeability during LCMV Cl13 infection, we next performed transcriptomic analysis of FACS-purified IECs (CD45−EpCAM+) from the small intestine of mice left uninfected or infected with ARM or Cl13 at day 9 p.i. IEC purity was assessed via FACS (average purity = 96.4% ± 4.4%). Notably, while both ARM and Cl13 infections induce a vigorous immune response at this time point (Wherry et al., 2003), Cl13 infection exerted much greater IEC-intrinsic transcriptional perturbations than ARM infection. Specifically, the number of differentially expressed genes (DEGs; with cutoffs at adjusted P value <0.05 and minimal fold change = 2) was 41 in ARM-infected versus uninfected mice and 642 in Cl13-infected versus uninfected mice (Fig. 2 A, Table S1, and Table S2). In addition, Cl13-infected mice clustered more distantly from uninfected mice than ARM-infected mice by principal component 1 on a principal-component analysis (PCA) plot (Fig. 2 B). Given that the size-selective increase in paracellular transport upon Cl13, but not ARM, infection was consistent with engagement of the leak pathway that is caused by tight junction dysregulation (Shen et al., 2011; Turner, 2009), we next assessed expression of several tight junction–related genes extracted from MSigDB or previous studies (Liberzon et al., 2011; Subramanian et al., 2005; Tsai et al., 2017). We did not observe differences in any of the 29 genes analyzed in ARM-infected versus uninfected mice (Table S3). In contrast, we detected significant down-regulation in Cl13- versus ARM-infected and uninfected mice of Cldn23 and Patj (Fig. 2 C), which encode for claudin-23 and Pals-associated tight junction protein, respectively. We also observed significant down-regulation of Cldn3 and Cldn8 in Cl13-infected versus uninfected mice and observed the same tendency but without reaching significance when comparing Cl13-infected with ARM-infected mice (Fig. 2 C). Overall, these data indicated that LCMV Cl13 infection resulted in IEC down-modulation of tight junction gene expression, which was consistent with the aforementioned increase in intestinal barrier permeability.
Figure 2.

LCMV Cl13 infection induces dysregulation in tight junction gene expression as well as a type I IFN signature in IECs. See also Fig. S3, Table S1, Table S2, Table S3, Table S4, and Table S5.C57BL/6 mice were infected with LCMV ARM or Cl13 or left uninfected (Un) and RNA-sequencing analysis was performed on FACS-purified IECs on day 9 p.i. (A) Number of DEGs with adjusted P value (p.adj) < 0.05 and fold change (FC) ≥ 2 between IECs from ARM- or Cl13-infected versus uninfected mice. (B) PCA plot with IEC transcriptomes from uninfected and ARM- and Cl13-infected mice. PC, principal component. (C) Gene counts by DESeq2 for indicated genes were normalized to average counts in the uninfected group. (D) Differentially enriched pathways (P < 0.05; FDR < 0.25) in ARM- versus Cl13-infected mice by GSEA. (E) DEGs in ARM- versus Cl13-infected mice. RNA sequencing was performed in a total of three independent experiments, in which each sequenced sample consists of IECs pooled from three mice. Averages ± SEM are shown (C). DEG (A and E), PCA (B), and FDR-adjusted P values (C) were computed by DESeq2. *, adjusted P value < 0.05.
To identify signals underlying tight junction dysregulation as well as increased paracellular transport between ARM- and Cl13-infected mice, we performed gene set enrichment analysis (GSEA) of the IEC transcriptomes with Hallmarks (Subramanian et al., 2005), Kyoto Encyclopedia of Genes and Genomes (Kanehisa and Goto, 2000), and Reactome (Fabregat et al., 2018) databases. This analysis identified positive enrichment for inflammatory pathways such as IFN-γ and IFN-α responses as well as IL-6 and IL-2 signaling, among others (Fig. 2 D and Table S4). Cl13-infected mice also up-regulated pathways related to cell cycle (Myc and E2F targets and G2M checkpoint) and metabolism (mTORC1 signaling and pancreas β cells) compared with ARM-infected mice (Fig. 2 D). We further detected down-modulation of pathways related to xenobiotic, bile acid, and heme metabolism; estrogen signaling (Fig. 2 D); and “Slc-mediated transmembrane transport” or “digestion and absorption” (Table S4). On the other hand, direct comparison of IEC transcriptomes from ARM- versus Cl13-infected mice identified 394 DEGs, 150 of which were down-regulated and 244 of which were up-regulated by Cl13 infection (Fig. 2 E and Table S5). Consistent with pathway analysis, a notable fraction of down-regulated genes were related to nutrient transport, such as solute carrier family genes Slc36a1, Slc2a2, Slc9a3, Slc46a1, and Slc15a1 (Fig. 2 E and Table S5), which encode for amino acid, glucose, ion, folate, and peptide transporters, respectively (Liang et al., 1995; Pathak et al., 1996; Qiu et al., 2007; Sagné et al., 2001; Wheeler and Hinkle, 1985). Other down-regulated genes were related to immune defense, such as Reg3a and Reg3b, which encode for C-type lectins with bactericidal activity (Miki et al., 2012; Mukherjee et al., 2014), and Il33, which is involved in tissue repair responses (Molofsky et al., 2015; Fig. 2 E). In contrast, the vast majority of genes and pathways up-regulated in IECs from Cl13- versus ARM-infected mice were related to the antiviral immune response. Specifically, we observed increased expression of IFN-stimulated genes (ISGs) Mx1, Isg15, Mx2, Tap1, Cxcl9, and Cxcl10, among others (Fig. 2 E and Table S5).
Overall, our data demonstrated that sustained LCMV replication in intestinal tissues is associated with worse bactericidal response and tissue repair and transcellular nutrient transport capacities of the intestinal epithelium, which could have implications for limiting close contact with the microbiota, protection against secondary insults, and/or whole-body nutrition. Viral persistence also promoted an antiviral transcriptional program in IECs consistent with enhanced type I IFN signaling that is associated with tight junction dysregulation and induction of a leaky gut phenotype.
Type I IFN signaling increases intestinal permeability and immune cell recruitment genes while down-regulating tight junction–related genes in IECs from LCMV-Cl13–infected mice
To investigate the potential causal role of type I IFNs in the enhancement of intestinal barrier permeability during chronic viral infection, we blocked signaling through its receptor via i.p. injection of anti-IFNAR-1 or isotype control Ab from day −1 through day 9 p.i. with Cl13 (Teijaro et al., 2013; Wilson et al., 2013). While anti-IFNAR-1 Ab treatment did not alter FD4 translocation in uninfected mice (compared with isotype control mice), we observed a significant reduction in serum FD4 levels in anti-IFNAR-1– versus isotype-treated Cl13-infected mice, with FD4 permeability returning to uninfected mouse levels in the Cl13-infected mice that received the IFNAR-1 Ab treatment (Fig. 3 A). Thus, type I IFN signaling was essential to increase paracellular flux of small molecules through the intestinal epithelium during Cl13 infection.
Figure 3.

Type I IFN signaling increases intestinal permeability and immune cell recruitment genes while down-regulating tight junction–related genes in IECs from LCMV-Cl13–infected mice. See also Fig. S3, Table S6, Table S7, Table S8, and Table S9.C57BL/6 mice were infected with LCMV Cl13 or left uninfected (Un) and injected with isotype (IgG1) or anti-IFNAR-1 Ab (αIFNAR) i.p. and analyzed at day 9 p.i. (A) In vivo intestinal permeability to FD4. (B–E) RNA sequencing was performed in IECs. (B) PCA plots showing individual samples from IgG1- versus αIFNAR-treated Cl13-infected mice. (C and D) Venn diagram overlapping pathways (C) or DEGs (D) from Cl13 versus ARM (data from Fig. 2) and Cl13-infected plus IgG1 versus αIFNAR treatment comparisons. (E) DEGs in IEC transcriptomes from IgG1- versus αIFNAR-treated Cl13-infected mice. Averages ± SEM are shown (A). Data are pooled from three independent experimental repeats with n = 3–4 mice/group (A). (B–E) RNA sequencing was performed in a total of two independent experiments, in which each sequenced sample consists of IECs pooled from three mice. PCA (B) and DEGs (C–E) were calculated by DESeq2. (A) Kruskal–Wallis test with Dunn’s correction for multiple comparisons. (C) Pathway enrichment was determined by GSEA (P < 0.05; FDR < 0.25). *, P <0.05; **, P <0.01; ***, P <0.001; ****, P <0.0001. FC, fold change; p.adj, adjusted P value; PC, principal component.
To identify the mechanism employed by type I IFN to increase intestinal barrier leakage, we next performed RNA-sequencing analysis of FACS-purified IECs from isotype- or anti-IFNAR-1–treated Cl13-infected mice on day 9 p.i. IEC purity was assessed via FACS (average purity = 96.5% ± 3.2%). This treatment had a significant impact on IEC transcriptomes, since IECs from isotype-treated mice clustered away from anti-IFNAR-1–treated Cl13-infected mice by principal component 1 (Fig. 3 B). In addition, Ab treatment readily reduced type I IFN signaling in IECs, since the hallmark IFN-α response pathway and expression of several ISGs was significantly enriched in isotype- versus anti-IFNAR-1–treated Cl13-infected mice (Fig. S3, A and B; Table S6; and Table S7). By analyzing overlap of all differential pathways and DEGs from the aforementioned Cl13 versus ARM comparison (Fig. 2, D and E; Table S4; and Table S5) with those altered in isotype- versus anti-IFNAR-1–treated Cl13-infected mice, we found that type I IFN signaling explained 34% and 37% of altered pathways and DEGs in Cl13-infected mice, respectively (Fig. 3, C and D). At the pathway level, we found that type I IFN down-modulated xenobiotic metabolism but promoted mTORC1 metabolism, glycolysis, cell cycle, and several proinflammatory pathways (Fig. 3 C and Table S8). Among transcripts significantly down-regulated by type I IFN signaling, we found genes encoding for proteins related to cellular metabolism (Acsm3 and Sord, among others), solute transport (Abcg8, Abcg5, and Slc9a3) and immune defense (Il33, Tcim, and Socs1). Most importantly, we found that type I IFN signaling also down-regulated expression of the tight junction–encoding genes Cldn8, Cldn15, and Cldn23 (Fig. 3 E and Table S7), two of which we found to be down-regulated in Cl13- versus ARM-infected and/or uninfected mice (Fig. 2 C). In contrast, the vast majority of genes up-regulated by type I IFN (Fig. 3 E and Table S9) were ISGs and genes related to immune processes such as Cxcl10, Cxcl9, Icam1, or Csf1, whose products can contribute to T cell recruitment (Groom and Luster, 2011) as well as adhesion (Sumagin et al., 2014) and maturation of innate immune cell types (Stanley et al., 1978).
Figure S3.

Antibody-mediated blockade of IFNAR-1 reduces IFN signaling in IECs after LCMV Cl13 infection. C57BL/6 mice were infected with LCMV ARM or Cl13 or left uninfected (Un; A) or injected with isotype (IgG1) or anti-IFNAR-1 Ab (αIFNAR) i.p. (A and B), and RNA-sequencing analysis was performed on FACS-purified IECs at day 9 p.i. (A) Venn diagram (left) shows overlapping GSEA results from the “interferon α hallmarks” gene signature between IEC from Cl13- versus ARM-infected mice and IECs from IgG1- versus anti-IFNAR-1–treated Cl13-infected mice. 10 highly enriched and overlapping ISGs (i.e., highest rank metric scores, FDR < 0.1) were selected for the heatmap (right) depicting Z-scores computed from DESeq2 normalized counts (Table S2, Table S5, and Table S7). (B) Fold reduction in the expression of selected ISG in IECs from IgG1-treated versus anti-IFNAR-1–treated Cl13-infected mice is shown. Averages ± SEM are shown. FDR-adjusted P values (p-adj) were computed by DESeq2.
Overall, our data indicated that type I IFN signaling was responsible for the increased intestinal permeability and for the induction of metabolic, proliferative, and proinflammatory transcriptional pathways in IECs after chronic LCMV infection. Remarkably, type I IFN signaling was responsible for tight junction transcriptional dysregulation and promoted expression of the immune cell recruitment genes Cxcl9 and Cxcl10 in IECs from chronically infected mice.
Intestinal CD8 T cell responses are enhanced by type I IFNs and are essential for barrier leakage during LCMV Cl13 infection
Type I IFN acts as a third signal to CD8 T cells during viral infections and promotes T cell accumulation and effector functions (Swiecki and Colonna, 2011). In addition, type I IFNs induce a cell-intrinsic antiviral state (Swiecki and Colonna, 2011) and can modulate immune cell recruitment to mucosal tissues in response to infections (Abe et al., 2007; Dorhoi et al., 2014; Neil et al., 2019; Shahangian et al., 2009). We found that IECs from Cl13- versus ARM-infected mice expressed higher levels of Cxcl9 and Cxcl10 in a type I IFN–dependent manner (Fig. 2 E and Fig. 3 E). Since CXCL9 and CXCL10 can be responsible for the recruitment of CXCR3-expressing effector CD4 and CD8 T cells (Groom and Luster, 2011), we hypothesized that type I IFNs could be mediating recruitment of T cell populations relevant for the induction of barrier dysfunction. To test this hypothesis, we first investigated CD4 T cell responses in the small intestine of Cl13-infected mice treated with isotype- or anti-IFNAR-1 Ab. Analysis of CD4 T cells specific for the immunodominant GP67–77 LCMV epitope (I-AbGP67–77+; Homann et al., 2007; Oxenius et al., 1995) revealed no differences in proportions but increased numbers of I-AbGP67–77+ CD4 T cells infiltrating the intraepithelial lymphocyte (IEL) and lamina propria (LP) compartments of the small intestine in anti-IFNAR-1– versus isotype-treated Cl13-infected mice at day 9 p.i. (Fig. S4 A). Expression levels of the coinhibitory receptor PD-1 on LCMV-specific CD4 T cells were increased upon anti-IFNAR-1 treatment (Fig. S4 B). Furthermore, we observed reduced proportions of IFN-γ–producing CD4 T cells upon ex vivo restimulation with PMA and ionomycin or with GP67–77 peptide in the LP of anti-IFNAR-1– versus isotype-treated Cl13-infected mice (Fig. S4, C and E), although the total numbers of IFN-γ–producing CD4 T cells (Fig. S4, C and E) or their potential to coproduce TNF-α (Fig. S4 D) were unchanged by anti-IFNAR-1 treatment. Contrary to our hypothesis, these data showed that type I IFNs restricted (rather than enhanced) I-AbGP67–77+ CD4 T cell accumulation in the small intestine, uncoupling Cxcl9 and Cxcl10 expression by IECs to CD4 T cell recruitment to this tissue. Consistently, i.p. treatment of Cl13-infected mice with anti-CD4 or isotype Ab on days −2, −1, 0, and 5 p.i. did not counteract the increased barrier permeability observed at day 9 p.i. (Fig. S4 F), even though this treatment was very efficient at reducing CD4 T cell accumulation in spleen and small intestine (Fig. S4 G). These data indicated that increased barrier permeability after Cl13 infection occurred to the same extent in the absence of antiviral CD4 T cell responses.
Figure S4.

Type I IFN signaling restricts small intestinal virus-specific CD4 T cell accumulation and CD4 T cells are dispensable for barrier dysfunction during LCMV Cl13 infection. Related to Fig. 4. C57BL/6 mice were infected with LCMV Cl13 and treated with isotype (IgG1) or anti-IFNAR-1 Ab (αIFNAR; A–E) or anti-CD4 (αCD4) or isotype Ab (IgG2a) i.p. on days −2, −1, 0, and 5 p.i. (F and G) and analyzed on day 9 p.i. (A–F) FACS analysis of the small intestinal IEL or LPL compartments was done on day 9 p.i. (A and B) Frequencies and numbers (A) and PD1 mean fluorescence intensity (MFI; B) of I-AbGP67–77+ CD4 T cells. (C–E) Cytokine production upon ex vivo PMA/ionomycin (C and D) or GP67–77 peptide (E) stimulation of LPL cells. (F) In vivo intestinal permeability to FD4. (G) Numbers of I-AbGP67–77+ in indicated organs. (A–G) Averages ± SEM are shown. Data are pooled from two (A and B) or three (F and G) independent experimental repeats or representative of two independent experimental repeats (C–E) with n = 3–5 mice/group. Although a tendency was consistently observed in all independent experiments, statistical significance was reached in one out of two experiments in A (right) and E (left, white versus pink squares). Statistical significance between Cl13 + IgG2a and Cl13 + αCD4 was only reached in one out of three experiments (F). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; two-tailed t test (A–C), Mann–Whitney test (G), and Kruskal–Wallis test with Dunn’s multiple comparisons’ correction (E–G).
We next profiled intestinal antiviral CD8 T cell responses in the same Cl13-infected mice treated with isotype- or anti-IFNAR-1 Ab. In contrast to the aforementioned observations in the CD4 T cell compartment, we detected reduced numbers of CD8 T cells specific for the LCMV immunodominant epitope GP33–41 (DbGP33–41+; Wherry et al., 2003) in both IELs and LP lymphocytes (LPLs) from anti-IFNAR-1– versus isotype-treated mice, although frequencies were only reduced in the IEL compartment (Fig. 4 A). DbGP33–41 tetramer+ cells from anti-IFNAR-1– versus isotype-treated mice expressed higher levels of PD-1 (Fig. 4 B), which might be related to a tendency (albeit not statistically significant) toward higher viral loads in the anti-IFNAR-1–treated group (Fig. 4 C). In addition, lower frequencies of the DbGP33–41 tetramer+ cells were positive for inhibitory killer cell lectin-like receptor G1 (KLRG1), a cytolytic-associated molecule that marks CD8 T cells with an effector fate (Angelosanto et al., 2012; Fig. 4 D). We detected higher proportion and number of stem-like TCF-1hiTim3lo DbGP33–41+ cells (Im et al., 2016; Utzschneider et al., 2016; Wu et al., 2016) in the IEL of anti-IFNAR-1 versus isotype-treated mice (Fig. 4 E), which was remarkable, as this stem-like subset is typically absent from this tissue compartment (Im et al., 2016). Conversely, frequencies and numbers of TCF-1lo DbGP33–41+, which includes cells with increased cytolytic potential (Hudson et al., 2019; Zander et al., 2019), were reduced in anti-IFNAR-1–treated Cl13-infected mice (Fig. 4 E). At the functional level, we detected significantly lower frequencies and numbers of CD8 T cells with the capacity to coproduce IFN-γ/TNF-α upon ex vivo restimulation with PMA/ionomycin and with cognate antigens in anti-IFNAR-1– versus isotype-treated Cl13-infected mice (Fig. 4, F and H). Also, CD8 T cells from anti-IFNAR-1–treated mice produced lower levels of IFN-γ on a per-cell basis after PMA/ionomycin restimulation (Fig. 4 G).
Figure 4.

Intestinal CD8 T cell responses are enhanced by type I IFNs and are essential for barrier leakage during chronic LCMV Cl13 infection. See also Fig. S4. (A–H) C57BL/6 mice were infected with LCMV Cl13 or left uninfected (Un) and injected with isotype (IgG1, red) or anti-IFNAR-1 Ab (αIFNAR, white) i.p. (A, B, and D–H) FACS analysis of the small intestinal IEL or LPL compartments was done on day 9 p.i. Shown are frequencies and numbers of DbGP33–41+ CD8 T cells (A), as well as PD1 mean fluorescence intensity (MFI; B), frequency of KLRG1+ (D), and frequencies and numbers of Tim3loTCF-1hi or TCF-1lo, within DbGP33-41+ CD8 T cells (E), and their corresponding representative FACS plots. (C) Viral RNA in ileum was quantified by quantitative PCR. (F–H) Frequencies, numbers (F–H), and representative FACS plots (F), as well as IFN-γ mean fluorescence intensity (G) of LPL CD8 T cells upon 5 h of ex vivo PMA/ionomycin stimulation (F and G) or with GP33–41, GP276–286, and NP396–404 peptides (H). (I–K) C57BL/6 mice were infected with LCMV Cl13 or left uninfected (Un) and injected with isotype (IgG2a) or CD8-depleting Abs (αCD8) i.p. In vivo intestinal permeability to FD4 (I), as well as levels of viral RNA (J) and Ifnb (K) in the small intestine, was determined on day 9 p.i. Averages ± SEM are shown (A–K). Data are pooled from two (A–E, J, and K) or three (I) or representative of three (F and G) or two (H) independent experimental repeats. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; Mann–Whitney t test (A–E, G, H, J, and K) or Kruskal-Wallis with Dunn’s multiple comparisons correction (F and I). n.s., not significant.
Since type I IFN–mediated increase in intestinal CD8 T cell accumulation and effector CD8 T cell phenotype associated with enhanced intestinal permeability in Cl13-infected mice, we next hypothesized that CD8 T cells may be contributing to intestinal leakage. To test this hypothesis, we treated Cl13-infected mice with CD8-depleting or isotype Ab on days −2, −1, 0, and 5 p.i. This treatment was highly efficient at reducing CD8 T cells from both the spleen and the small intestine (spleen; 3.2-fold ± 1.1-fold; LP, 6.6-fold ± 0.01-fold; IEL, 4.7-fold ± 3.9-fold), and it significantly reduced FD4 permeability, reaching uninfected levels in CD8-depleted Cl13-infected mice (Fig. 4 I), in spite of increased intestinal viral loads and Ifnb levels (Fig. 4, J and K).
Together, these data demonstrated that CD8 T cell responses, whose effector functions, accumulation, and TCF-1lo phenotype were reinforced by type I IFNs, were essential causative factors of the increased intestinal permeability observed during chronic LCMV infection. Our results also uncoupled the induction of intestinal leakage from direct effects of viral loads and showed that increased Ifnb levels could not increase intestinal permeability in the absence of CD8 T cells during chronic LCMV infection.
Type I IFN signaling promotes overrepresentation of Erysipelatoclostridium and reduces Roseburia in the intestinal microbiome after LCMV Cl13 infection
Increased intestinal permeability has been associated with compositional shifts of the intestinal microbiome (Harbison et al., 2019; Martinez-Medina et al., 2014; Thaiss et al., 2018). To investigate whether the induction of intestinal leakage was coupled with microbiome shifts after Cl13 infection, we performed two independent experiments in which we treated 10 mice per group with isotype or anti-IFNAR-1 Ab, as in Fig. 3 and Fig. 4, followed by 16S rRNA amplicon (Fig. 5) or shotgun metagenomics (Fig. S5) sequencing, of DNA obtained from colonic and cecal contents on day 9 p.i. Sampling of cecal and colonic contents was chosen over stool pellets, as it allows detection of taxa abundant inside the large intestine, which may not necessarily be eliminated in stools (Gu et al., 2013; Tanca et al., 2017). As shown in Fig. 5 A and Fig. S5 A, we observed no significant differences in intra-individual α-diversity between isotype-treated and anti–IFNAR1-treated Cl13-infected mice, as determined by the Shannon diversity index, which combines richness and evenness (Shannon, 1997). We next assessed intragroup taxonomic diversity via phylogenetic and nonphylogenetic β-diversity computation followed by principal-coordinates analysis (PCoA). Phylogenetic and nonphylogenetic analysis of the 16S data revealed differential separation between isotype-treated and anti-IFNAR-1–treated groups by the first principal coordinate and statistically significant differences between both groups by permutational ANOVA (PERMANOVA; Fig. 5 B). This difference was, however, not confirmed by nonphylogenetic shotgun metagenomics analysis of an independent cohort of mice from the same vendor (Fig. S5, B and C). Moreover, analysis of phylum abundances indicated that the ratios of Firmicutes over Bacteroidetes as well as Verrucomicrobia over Firmicutes and Bacteroidetes were not significantly different in isotype- versus anti-IFNAR-1–treated Cl13-infected mice in both cohorts, despite the biological and technical differences in these analyses (Fig. 5, C–E; and Fig. S5, D–F). Taken together, these data demonstrated that type I IFN–dependent intestinal leakage did not associate with large-scale microbiome shifts early after Cl13 infection.
Figure 5.

Type I IFN signaling promotes overrepresentation of Erysipelatoclostridium and reduces Roseburia in the intestinal microbiome after LCMV Cl13 infection. See also Fig. S5 and Table S10. C57BL/6 mice were infected with LCMV Cl13 and injected with isotype (IgG1) or anti-IFNAR-1 Ab (αIFNAR) i.p. and sacrificed at day 9 p.i. 16S rRNA gene amplicon sequencing was performed on colonic and cecal contents. (A) Alpha diversity by the Shannon diversity index. (B) Beta diversity PCoA with Unweighted UniFrac, Jaccard and Bray–Curtis distances. (C) Frequency of sequences at the phylum level at indicated time points. (D and E) Frequency of phylum Firmicutes divided by Bacteroidetes (D) or phylum Verrucomicrobia divided by Firmicutes and Bacteroidetes (E) for each individual mouse. (F) Songbird multinomial regression analysis of genera in IgG1-treated versus αIFNAR-treated Cl13-infected mice. X axes correspond to individual taxa. Taxa highlighted in red were consistently perturbed, in the same direction and with rank cutoffs of −0.5 and 0.5, after shotgun metagenomics analysis of an independent cohort of mice. UP indicates taxa up-regulated in αIFNAR-treated versus IgG1-treated Cl13-infected mice. Data in A–F are representative of one independent repeat by 16S rRNA amplicon sequencing, with n = 9–10 mice/group. (A and C–E) Averages ± minimum/maximum (A) or ± SEM (C–E). (B) Significance was computed with PERMANOVA (999 permutations). ns, not significant; PC, principal component.
Figure S5.

Type I IFN signaling promotes overrepresentation of Erysipelatoclostridium and reduces Roseburia species in the intestinal microbiome after LCMV Cl13 infection. C57BL/6 mice were infected with LCMV Cl13 and injected with isotype (IgG1) or anti-IFNAR-1 Ab (αIFNAR) i.p., and shotgun metagenomic sequencing was performed on colonic and cecal contents on day 9 p.i. (A) Alpha diversity by the Shannon diversity index. (B and C) Beta diversity PCoA with Jaccard (B) and Bray–Curtis (C) distances. (D) Frequency of sequences at the phylum level at indicated time points. (E and F) Frequency of phylum Firmicutes divided by Bacteroidetes (E) or phylum Verrucomicrobia divided by Firmicutes and Bacteroidetes (F) for each individual mouse. (G) Songbird multinomial regression analysis of species in IgG1-treated versus αIFNAR-treated Cl13-infected mice. X axes correspond to individual taxa. The genera of the taxa highlighted in red were consistently perturbed, in the same direction and with rank cutoffs of −0.5 and 0.5, after 16S shotgun metagenomics analysis of an independent cohort of mice. UP indicates taxa up-regulated in αIFNAR-treated versus IgG1-treated Cl13-infected mice. Data are representative of one independent repeat by shotgun metagenomics with n = 9–10 mice/group. Data are shown as averages ± minimum/maximum (A) or ± SEM (D–F). Significance in B and C was computed with PERMANOVA (999 permutations). ns, not significant; PC, principal component.
We next performed songbird multinomial regression analysis (Morton et al., 2019) to identify specific taxa perturbed by IFNAR-1 blockade. Among the taxa with cutoff ranks at 0.5 and −0.5, which indicate a moderate to high degree of perturbation, we found that type I IFN signaling decreased the relative abundance of Roseburia (rank 1.14; Fig. 5 F and Table S10) and one of its species (i.e., Roseburia hominis, rank 0.54; Fig. S5 G and Table S10). Conversely, type I IFN signaling promoted the overrepresentation of Erysipelatoclostridium (rank −1.79; Fig. 5 F and Table S10) and one of its species (i.e., Clostridium cocleatum, rank −1.60; Fig. S5 G and Table S10).
Overall, these data demonstrated that type I IFN signaling drove overrepresentation of Erysipelatoclostridium commensals as well as reductions in Roseburia early after chronic viral infection. Although these data do not demonstrate a causal relationship between intestinal barrier leakage and microbiome composition, they do increase our understanding of the role of type I IFNs in modulating the composition of the intestinal microbiome early after chronic viral infection and provide an association between the induction of intestinal leakage and specific taxa perturbations.
LCMV Cl13 infection enhances susceptibility to colitis induced by chemical or secondary pathogen insults
Barrier dysfunction precedes disease relapses in humans afflicted by Crohn’s disease (D’Incà et al., 1999; Tibble et al., 2000; Wyatt et al., 1993), and in mouse models of immune-, chemically, and bacterially induced colitis, increased solute flux consistent with the leak pathway promotes host morbidity (Conlin et al., 2009; Graham et al., 2019; Sturgeon et al., 2017). In addition, overrepresentation of Erysipelatoclostridium commensals and reductions in Roseburia spp. are associated with human IBD (Hedin et al., 2014; Machiels et al., 2014; Mancabelli et al., 2017) and the induction of intestinal leakage after Cl13 infection (Fig. 5).
To test the effects of chronic viral infection on host tolerance to chemically induced colitis, we performed a 10-d course of treatment with 2% dextran sulfate sodium (DSS; Okayasu et al., 1990) between days 21 and 31 p.i. with Cl13 or ARM and in uninfected mice. As expected with the low dose of dextran sulfate sodium used (Eichele and Kharbanda, 2017), we did not observe any perceptible effects on the body weight of uninfected and ARM-infected treated mice compared with untreated controls (Fig. 6 A). In contrast, Cl13-infected mice suffered reductions in body weight of 7% on average after 10 d of treatment, which was significantly lower from that of ARM-infected and uninfected treated mice (Fig. 6 A), indicating that chronic viral infection with Cl13 conferred increased susceptibility to acute, chemically induced colitis.
Figure 6.

LCMV Cl13 infection enhances susceptibility to colitis induced by chemical or secondary pathogen insults. C57BL/6 mice were infected with LCMV Cl13 or ARM or left uninfected. At days 21 or 30 p.i., mice were treated with 2% wt/vol dextran sulfate sodium for 10 d consecutively (A) or i.o. infected with 3.5 × 109 CFUs of C. rodentium (+C.rod) or left untreated (untx; B and C). Percentage of initial body weight (A and B) and survival curves (C) are shown. Averages (C) or averages ± SEM (A and B) are shown. Data are pooled from two (A) and representative of two (B and C) independent experiment with n = 3–5 mice/group. (A) One-way ANOVA with Tukey’s multiple comparisons correction on day 31 p.i. (B) One-way ANOVA with Dunnett’s multiple comparisons test with ARM+C.rod and Cl13+C.rod versus Un+C.rod at every time point p.i. (C) Log-rank Mantel–Cox survival test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
We next evaluated the impact of Cl13 infection on host tolerance to bacterially induced colitis with the rodent-borne enteropathogen Citrobacter rodentium (Schauer et al., 1995). As expected, C. rodentium challenge caused mild weight loss and no effect in survival in the absence of LCMV coinfection (Bouladoux et al., 2017), and similar effects were observed 10 d after C. rodentium was inoculated in mice previously infected with ARM (ARM+C.rod; Fig. 6 B). In contrast, we detected profound body weight loss (≤20% of initial weight) that preceded 100% mortality when C. rodentium was inoculated into Cl13-infected mice (Fig. 6, B and C).
In summary, we found that viral persistence rendered Cl13-infected mice more susceptible to intestine-specific stressors, such as colitis induced by chemicals or bacteria, suggesting that chronic infection–induced adaptations in the intestinal tract have implications for host tolerance to secondary enteric insults.
Discussion
Intestinal barrier dysfunction constitutes a marker of disease progression during chronic viral infection, yet the mechanisms leading to an altered gut barrier in this context have not been causally defined. In this study, we found that in contrast to its acute parental strain, the persistent LCMV variant Cl13 continuously replicated in hematopoietic and mesenchymal, but not epithelial, cells within the small intestine and caused long-term increase in intestinal paracellular permeability to small molecular weight molecules. Such intestinal barrier alteration associated with type I IFN–mediated down-regulation of genes encoding tight junction–related proteins in IECs. Type I IFN signaling also promoted cell cycle, glycolytic metabolism, and inflammation-associated transcriptional pathways on IECs, including increased expression of CXCR3+-effector T cell chemoattractants (i.e., Cxcl9 and Cxcl10) in chronically infected mice. Importantly, IFNAR blockade restricted CD8 T cell accumulation, effector functions, and the abundance of TCF-1lo versus TCFhi CD8 T cell subsets within the small intestine, and both type I IFN signaling blockade and CD8 T cell depletion prevented intestinal leakage after chronic infection. Type I IFNs also promoted Erysipelatoclostridium and Roseburia microbiome shifts. Finally, we found that long-term viral persistence and increased intestinal barrier permeability associated with enhanced susceptibility to chemical and bacterially induced colitis in the chronic infection setting.
Our study demonstrated that systemic inoculation with a persistent, but not acute, LCMV isolate resulted in long-term viral replication in hematopoietic and mesenchymal, but not epithelial, cells of the small intestine that was sustained even after systemic viremia was resolved. Within the hematopoietic compartment, it is most likely that dendritic cells and macrophages, but not T cells or B cells, are targeted by LCMV in intestinal mucosa, as these cells are productively infected by arenaviruses in other tissues (Macal et al., 2012; Mahanty et al., 2003; Matloubian et al., 1993; Sevilla et al., 2000). On the other hand, the most likely targets among mesenchymal cells may include vimentin-expressing fibroblasts, myofibroblasts, pericytes, and stromal stem cells (Mueller et al., 2007; Pinchuk et al., 2010). Intriguingly, while LCMV and other Old World arenaviruses productively infect human IEC lines in vitro (Warner et al., 2018), we failed to detect viral proteins in IEC from LCMV Cl13–infected mice, despite high and persistent viral titers in intestinal mucosa. Thus, our study highlights important discrepancies between in vitro and in vivo LCMV infection and suggest that infection of IEC lines may not necessarily reflect arenavirus tropism in vivo.
As mentioned in the Introduction, there are three described routes of solute transport across the intestinal epithelium: the pore, leak, and unrestricted pathways (Odenwald and Turner, 2017). Given that the causative mechanisms and implications of each of these routes of solute transport can be different, it is important to identify the specific pathway that is altered in a given context. Claudin-2–mediated flux through the pore pathway, for instance, accelerated bacterial clearance (Tsai et al., 2017), while flux via the myosin light chain kinase–dependent leak pathway exacerbated disease (Conlin et al., 2009) after C. rodentium infection. Our work revealed that chronic, but not acute, LCMV infection persistently enhanced intestinal barrier permeability, selectively increasing the flux of small-molecular-weight (∼4 kD) molecules, which is consistent with alteration of the leak pathway. We did not, however, detect increased leakage of larger molecules (∼40 kD) or macroscopic epithelial layer disruption, indicating the absence of frank epithelial damage or ulceration and an intact unrestricted transport route in this setting. These results highlight chronic LCMV infection in mice as an additional model system to study the causative factors, implications and effects of potential drug therapies on the intestinal leak transport pathway in the context of persistent inflammation. It is important to remark that, although the intestinal transport route that leads to increased microbial product translocation during other chronic infections has not yet been defined, previous studies documented the presence of enterobacterial DNA and bacterial lipopolysaccharide in plasma from HIV-infected humans and SIV-infected RM (Brenchley et al., 2006; Marchetti et al., 2008). In addition, increased ratio of lactulose-to-mannitol in urine, which is indicative of enhanced paracellular solute transport (i.e., leak or pore pathway), were observed in humans infected with P. falciparum (Wilairatana et al., 1997). It should also be noted that LCMV ARM spreads at a significant slower rate than LCMV Cl13 (Ahmed and Oldstone, 1988; Li et al., 2009) and that we detected the greatest intestinal leakage as early as 8 d after Cl13 infection, raising the possibility that infections with acute pathogens that sustain replication for at least 1 wk may also increase intestinal permeability, likely in a more transient manner.
By performing transcriptomic analysis of IECs from acutely versus chronically infected mice, we found that the aforementioned intestinal leakage associated with down-regulation of genes encoding for tight junction–related proteins. Among them, PATJ has been involved in the formation and polar distribution of tight junction complexes (Shin et al., 2005), while claudin-23 expression is significantly reduced in tumors versus healthy intestines during colorectal cancer (Cherradi et al., 2019), whereby increased tight junction–dependent permeability associates with development of this disease (Soler et al., 1999). Claudin-3, on the other hand, is recruited to the tight junction (Lei et al., 2012), and its increased expression is associated with intestinal barrier maturation in neonate mice (Patel et al., 2012), while claudin-8 can limit Na+ flux in kidney cell lines (Yu et al., 2003). Overall, down-regulation of genes encoding for PATJ, claudin-3, and claudin-23, which are involved in tight junction modulation (Cherradi et al., 2019; Lei et al., 2012; Patel et al., 2012; Shin et al., 2005; Soler et al., 1999), were consistent with alteration of the leak pathway after chronic LCMV infection. On the other hand, decreased claudin-8 transcript levels in IECs from chronically versus acutely infected mice suggested that the pore pathway might also be altered after chronic LCMV infection.
Our study further identified a type I IFN transcriptional signature in IECs from chronically versus acutely infected mice. Indeed, we found that signaling through IFNAR accounted for 34% of altered pathways and 37% of DEGs in IECs from LCMV Cl13– versus ARM-infected mice. Type I IFN signaling not only down-regulated the aforementioned genes encoding for claudin-8 and claudin-23 but also inhibited a xenobiotic-related transcriptional signature. Interestingly, engagement of the xenobiotic sensor pregnane X receptor prevented increase in intestinal permeability under homeostatic conditions (Venkatesh et al., 2014). Type I IFN signaling also promoted expression of cell cycle–related genes, consistent with enhanced type I IFN–mediated IEC proliferation previously reported during latent murine cytomegalovirus infection (Sun et al., 2015), and glycolysis-related metabolic signatures, which are characteristic of proliferating cells (Lunt and Vander Heiden, 2011).
Importantly, we demonstrated that type I IFN signaling was essential for increasing intestinal permeability after chronic LCMV infection. In the same line, an association between type I IFN signaling and barrier dysfunction has been previously established in chronic infections caused by HIV in humans and SIV in RM (Ponte et al., 2016; Tincati et al., 2016). In addition, SIV infection of sooty mangabeys, a natural viral reservoir, does not induce a sustained type I IFN signature nor increases microbial product translocation into the systemic circulation (Bosinger et al., 2009; Brenchley et al., 2006; Estes et al., 2010; Harris et al., 2010). These association studies led to the proposal that increased intestinal permeability allows translocation of microbial products, which in turn causes downstream type I IFN signaling and immune activation (Brenchley et al., 2006; Estes et al., 2010). While our transcriptome data are consistent with the previously reported association between type I IFNs and increased intestinal permeability during chronic viral infection, our type I IFN blocking experiments compellingly demonstrated that type I IFNs can be a cause, and not necessarily a consequence, of intestinal leakage. On the other hand, type I IFN signaling was shown to preserve intestinal integrity during chemically induced colitis (Canesso et al., 2018; Neil et al., 2019) and to accelerate wound healing via increasing IEC proliferation during latent viral infection (Sun et al., 2015). It is therefore conceivable that type I IFNs may exert dual roles on the intestinal barrier, on the one hand promoting IEC proliferation to favor wound healing and opposing the unrestricted pathway and, on the other hand, down-regulating tight junction–related proteins, thereby increasing translocation of microbial products via the leak pathway. Importantly, our results do not demonstrate that IEC-intrinsic type I IFN signaling is required for the induction of intestinal leakage after chronic viral infection, and it is therefore possible that type I IFNs may act indirectly, via other cell types, to increase intestinal permeability. Interestingly, a single-nucleotide polymorphism predicted to be in the IFNAR1 gene was recently associated with humans afflicted by Crohn’s disease (Jostins et al., 2012), further supporting potential unappreciated roles for type I IFNs in modulating intestinal barrier dysfunction.
We also demonstrated that type I IFN signaling promoted IEC expression of Cxcl9 and Cxcl10, whose products can contribute to the recruitment of CXCR3-expressing T cells to the intestine (Groom and Luster, 2011). Consistently, IFNAR blockade in chronically infected mice limited accumulation, effector features, and the abundance of TCF-1lo populations of virus-specific CD8 T cells in the small intestines. This is in line with previous studies indicating that type I IFN signaling promotes clonal expansion and survival of CD8 T cells in secondary lymphoid organs, liver, brain, and lung (Kolumam et al., 2005), as well as their IFN-γ production (Nguyen et al., 2002; Teijaro et al., 2013), and is consistent with its role in repressing the generation of TCF-1hi virus-specific CD8 T cells (Wu et al., 2016) in the spleen. On the other hand, we observed that type I IFNs limited (rather than promoted) accumulation of virus-specific CD4 T cells in small intestines from chronically infected mice, which is consistent with the type I IFN inhibition of splenic CD4 T cell accumulation that was previously reported (Teijaro et al., 2013; Wilson et al., 2013).
Importantly, we also demonstrated that CD8 (but not CD4) T cell responses were essential to enhance intestinal leakiness after chronic LCMV infection. This is consistent with increased CD8 T cells observed in the intestinal mucosa of SIV-infected RM (Schmitz et al., 2001; Veazey et al., 1998) and HIV-infected individuals (Allers et al., 2016; Shacklett et al., 2003), as well as a positive correlation of perforin-expressing CD8 T cells with microbial translocation that was reported in acutely HIV-infected individuals (Allers et al., 2017). Causal evidence linking CD8 T cells and enhanced intestinal permeability in vivo was, however, previously lacking. Instead, reduced numbers of intestinal Th17 CD4+ T cells have been extensively proposed as the cause of barrier dysfunction in both SIV-infected RM and HIV-infected humans (Ponte et al., 2016; Tincati et al., 2016). While our results do not contradict a potential role for reduced Th17 responses in causing microbial translocations during HIV or SIV infections, they raise the possibility that infiltrating CD8 T cells may also play an essential role. Our CD8 T cell depletion experiments further demonstrated that increased viral loads or Ifnb levels are insufficient to increase intestinal permeability in the absence of CD8 T cells. This suggests that CD8 T cells act downstream of type I IFN signaling to induce intestinal leakage after chronic infection and that the leaky gut does not result from direct viral effects.
We also uncovered a causal role for type I IFNs in oppositely modulating the abundance of two intestinal Firmicute taxa (i.e., Erysipelatoclostridium and Roseburia) early after chronic LCMV infection. Importantly, while the intestinal microbiome as a whole provides tonic type I IFN signals to innate immune cells and allows timely control of a chronic virus (Abt et al., 2012), a bidirectional relationship between type I IFNs and microbiome composition had not been previously demonstrated in this context. Our rigorous analysis also provides a valuable resource that could guide future studies on the microbiome–immune system crosstalk during viral infections.
Last but not least, our data indicated that chronic LCMV infection enhanced susceptibility to chemically and C. rodentium–induced intestinal colitis, which associated with increased intestinal permeability via the leak pathway. It has previously been shown that increased solute flux consistent with the leak pathway promoted host morbidity in mouse models of immune-, chemically, and bacterially induced colitis (Conlin et al., 2009; Graham et al., 2019; Sturgeon et al., 2017) and that barrier dysfunction preceded disease relapses in humans afflicted by Crohn’s disease (D’Incà et al., 1999; Tibble et al., 2000; Wyatt et al., 1993). Besides barrier dysfunction, factors such as diet, gut microbiome composition, or preexisting immune deficiencies can also confer susceptibility to experimental colitis (Desai et al., 2016; Eichele and Kharbanda, 2017; Mullineaux-Sanders et al., 2019). Along the same line, we found that the induction of intestinal leakage associated with increased abundance of Erysipelatoclostridium commensals and underrepresentation of Roseburia spp. in the intestinal microbiome, both of which are linked to inflammatory bowel disease (Hedin et al., 2014; Machiels et al., 2014; Mancabelli et al., 2017). Thus, while our data suggest a potential role for the enhanced intestinal leakage as a promorbidity factor in the context of colitis-inducing stressors in chronically infected mice, a role for other virus-induced intestinal adaptations in mediating this effect cannot be ruled out.
Overall, our findings propose a previously unrecognized causal role for type I IFN signaling in driving intestinal barrier leakage during viral infection, which could be explained by tight junction dysregulation and enhanced intestinal CD8 T cell responses. Importantly, we also demonstrated an unappreciated essential role for CD8 T cells as causative factors of increased intestinal permeability and established an association between enhanced leak pathway, specific intestinal microbiome perturbations, and susceptibility to colitis during chronic viral infection. These new mechanistic insights on intestinal barrier regulation might have implications for other chronic infections (Ponte et al., 2016; Tincati et al., 2016; Wilairatana et al., 1997), inflammatory diseases (Odenwald and Turner, 2013), and/or certain cancers (Bindels et al., 2018; Puppa et al., 2011; Sundström et al., 1998), where intestinal leakage has been reported. In addition, the possibility that type I IFNs and/or CD8 T cells may induce increased intestinal permeability after infections with acute pathogens that exhibit slow clearance should also be considered.
Materials and methods
Mice, infections, and Ab treatments
6- to 10-wk-old female C57BL/6 mice were purchased from The Jackson Laboratory and housed under specific pathogen–free conditions. Mice were infected i.v. with 2 × 106 PFUs of LCMV ARM or Cl13. Viral stocks were grown, identified, and quantified as reported previously (Ahmed et al., 1984). Viral loads were determined by standard plaque assay of serum or tissue homogenates (Ahmed et al., 1984). For CD8 and CD4 T cell depletion studies, mice were i.p. injected with anti-CD8α (53–6.72; BioXcell), anti-CD4 (GK1.5; BioXcell), or IgG2a (2A3; BioXcell) Abs on days −2, −1, and 5 p.i. (250 µg/mouse) as well as on the day of infection (200 µg/mouse). For IFNAR in vivo blockade, mice were i.p. injected with IFNAR neutralizing Ab (MAR1-5A3; BioXCell) or mouse IgG1 isotype control (MOPC-21; BioXcell) on days −1 and 0 p.i. (500 µg/mouse) as well as on days 2, 4, and 6 p.i. (250 µg/mouse) with Cl13 (Teijaro et al., 2013; Wilson et al., 2013). Uninfected control mice were treated alongside.
For secondary infection experiments, C. rodentium DBS100 obtained from Dr. Lifan Lu’s laboratory (University of California, San Diego [UCSD], La Jolla, CA) was streaked out from a frozen stock on a Luria broth (LB; Sigma) plate supplemented with nalidixic acid (50 µg/ml; Sigma) and incubated at 37°C overnight (Bouladoux et al., 2017). An individual colony was then inoculated into liquid LB + nalidixic acid media and grown overnight. Mice were infected with 4 × 109 CFUs intraorally (i.o.; Bouladoux et al., 2017).
For chemically induced colitis experiments, we supplemented mouse drinking water with 2% wt/vol DSS (MP Biochemicals) and treated mice for 10 d consecutively.
Mice were maintained in a closed breeding facility at the Biomedical Sciences Building, UCSD, and housed in ventilated cages containing HEPA filters. Mouse handling conformed to the requirements of the National Institutes of Health and the Institutional Animal Care and Use Guidelines of UCSD.
Histopathological analysis and immunofluorescence
For histopathological analysis, mice were sacrificed, and tissues were harvested and placed in 10% formalin solution within 5 min. Tissues remained in fixing solution for 24 h, after which they were washed twice with deionized water and later placed in 70% ethanol. The Tissue Technology Shared Resource at UCSD performed downstream paraffin embedding, sectioning, hematoxylin/eosin staining, and histopathological evaluation of intestinal tissues.
For immunofluorescence analysis, mice were individually sacrificed and their intestines placed in optimal cutting temperature compound (Akura) within 5 min. 10-µm sections were cut, fixed in 4% paraformaldehyde for 10 min, washed three times in 1× PBS (Fischer), blocked for 1 h in gelatin buffer (3% normal donkey serum, 40 mg/ml BSA, 0.1% fishscale gelatin, 0.05% Tween20, and 0.1% Triton-X-100 in PBS 1×) plus Fc block, and stained with primary Ab overnight in gelatin buffer. After three washes, sections were stained with DAPI. Sections were mounted and images were acquired with a Leica SP5 confocal microscope at the Microscopy Core at UCSD. Primary Abs used were EpCAM-APC (clone G8.8; Thermo Fisher Scientific), CD45.2-FITC (clone 104; BioLegend), rabbit anti-Vimentin (clone D21H3; CST), and guinea pig anti-LCMV polyclonal Ab (Tinoco et al., 2009). Secondary Abs were goat anti–guinea pig–Rhodamine Red-X Fab (polyclonal AB_2632433; Jackson ImmunoResearch) and AffiniPure F(ab′)2 fragment goat anti-rabbit-APC (polyclonal AB_2337987; Jackson ImmunoResearch).
In vivo intestinal permeability assay
Mice were water-deprived for 4 h, followed by intraoral inoculation with FD4 (Sigma) or FD40 (Sigma) at 44 mg/100 g body weight (Volynets et al., 2016). 4 h later, mice were bled, and serum was collected and stored at −80°C for future quantification. Fluorescence at 488 nm was determined with a plate reader (BMG Labtech). Background was subtracted using values from serum samples belonging to uninoculated mice. Absolute FD4 or FD40 amounts per milliliter of blood were determined via standard curve extrapolation.
Lymphocyte isolation from blood, spleen, and small intestine
Spleens were harvested, digested for 20 min at 37°C in 1 mg/ml collagenase D (Roche) and mashed through a 100-µm filter. Cells were then centrifuged at 1,500 rpm at 4°C for 5 min. RBCs were lysed by incubating pellets in 1 ml RBC-lysis buffer (150 mM NH4Cl, 10 mM KHCO3, and 0.1 mM Na2EDTA in deionized water, at pH 7.4) for 4 min at room temperature. After RBC lysis, 10 ml FACS buffer (PBS 1× + 3% FBS) was added, followed by centrifugation as stated above. Pellets were resuspended in 5 ml complete RPMI (Gibco), which consisted of RPMI supplemented with 10% heat-inactivated FBS (Atlanta Biologicals), 2% penicillin/streptavidin (P/S; BioWhittaker), 1 mM sodium pyruvate (Na-pyr; Thermo Fisher Scientific), 1 mM L-glutamine (BioWhittaker), 20 mM Hepes (Thermo Fisher Scientific), and 55 mM β-mercaptoethanol (Life Technologies). Absolute leukocyte numbers were determined via forward scatter/side scatter gating in a Guava Easycyte automated cell counter (MilliporeSigma).
Small intestines were harvested, cleaned from adipose tissue, and flushed, and macroscopically visible Peyer’s patches were excised. To isolate the epithelial layer, intestines were longitudinally and transversely cut into 1- to 2-cm pieces, placed in Pyrex conical flask containing a stirring magnet and 30 ml IEL buffer, and stirred on a stirring plate (Scilogex) set at 37°C for a total of 15 min. IEL buffer consisted of 1× PBS supplemented with 3% FBS, 2% P/S, 1 mM Na-pyr, 20 mM Hepes, and 10 mM EDTA (Thermo Fisher Scientific). Soluble fraction was then collected, diluted in RPMI supplemented with 3% FBS, and pelleted by centrifugation at 1,500 rpm for 5 min at 4°C. Afterward, to obtain LP, tissue pieces were minced and stirred at 37°C for 30 min in 15 ml LPL enzyme media consisting of RPMI, supplemented with 3% FBS, 2% P/S, 1 mM Na-pyr, 20 mM Hepes, 0.4 mg/ml collagenase D (Roche), and DNase I 10 µg/ml (Roche). Suspension was then filtered through a 100-µm strainer and pelleted by brief centrifugation 1,500 rpm for 5 min at 4°C. Finally, epithelial or LP pellets were resuspended in 5 ml 44% Percoll solution, and 2.5 ml 67% Percoll solution were added underneath with a Pasteur pipette. Tubes were then spun down at 2,000 rpm (with no brake set) at room temperature for 20 min. Percoll solution was prepared by adding 1 part of 10× PBS (Gibco) to 9 parts of Percoll (GE Healthcare Biosciences) and further mixed with IEL buffer to obtain a 44% or 67% solution. The interphase layers containing IELs or LPLs were collected after centrifugation and resuspended in 1 ml complete RPMI, and leukocyte counts were determined with a Guava Easycyte (MilliporeSigma) as indicated above.
Quantitative PCR
For quantification of transcripts in the small intestine, tissues were collected from sacrificed mice, snap frozen, and stored at −80°C. Later, tissues were thawed, bead homogenized in RLT buffer (Qiagen) and centrifuged for 10 min at 10,000 rpm to pellet debris. Supernatants were used for total RNA extraction using RNeasy kits (Qiagen), digested with DNase I (RNase-free DNase set; Qiagen), and reverse transcribed into cDNA using Moloney Murine Leukemia Virus Reverse Transcriptase (M-MLV RT; Promega). The expression of various genes was quantified using Fast SYBR Green Master Mix (Thermo Fisher Scientific) or TaqMan Fast Universal PCR Master Mix with probe sets from the Universal Probe Library (Roche) in technical triplicates, followed by real-time PCR using a CFX96 Touch Detection System (Bio-Rad). Relative transcript levels were normalized against murine Gapdh or Actb as indicated. Graphs depicting quantitative RT-PCR analysis of murine genes represent biological replicates of individual mice. SYBR Green quantitative PCR primer sequences are as follows: LCMV nucleoprotein: forward 5′-GCATTGTCTGGCTGTAGCTTA-3′, reverse 5′-CAATGACGTTGTACAAGCGC-3′; LCMV glycoprotein: forward 5′-CATTCACCTGGACTTTGTCAGACTC-3′, reverse 5′-GCAACTGCTGTGTTCCCGAAA-3′; Actb: forward 5′-AGGGAAATCGTGCGTGACAT-3′, reverse 5′-GAACCGCTCGTTGCCAATAG-3′. TaqMan quantitative PCR primer sequences are as follows: Ifnb: forward 5′-CTGGCTTCCATCATGAACAA-3′, reverse 5′-AGAGGGGGTGGTGGAGAA-3′ (probe 18); Gapdh: forward 5′-TCCCACTCTTCCACCTTCGA-3′, reverse 5′-AGTTGGGATAGGGCCTCTCTT-3′ (probe 9).
Flow cytometry
Cells were stained at a maximal concentration of 2 × 108 cells/ml in FACS buffer or as indicated below. For surface staining of splenocytes, IELs and LP cells were first stained with a fixable viability dye (Ghost Dye; Tonbo) in 1× PBS for 10 min at 4°C followed by staining with MHC class I tetramer H2-Db/GP33–41-BV421 (provided by National Institutes of Health Tetramer Core Facility, Atlanta, GA) in FACS buffer for 1 h at room temperature, followed by staining for 20 min at 4°C with remaining surface Abs. Alternatively, cells already stained with the viability dye were stained with MHC class II tetramer I-Ab/GP67–77-APC (provided by National Institutes of Health Tetramer Core Facility) in FACS buffer for 2 h at 37°C followed by staining for 20 min at 4°C with Abs against cell surface markers. For intracellular staining after ex vivo peptide restimulation, cells were fixed in 1% paraformaldehyde for 10 min at room temperature and stained with Abs in 1× perm/wash buffer (Thermo Fisher Scientific) for 30 min at 4°C. Alternatively, for intranuclear staining, cells were fixed with the Foxp3 Transcription Factor Staining Buffer Set Kit (Thermo Fisher Scientific) per the vendor’s recommendation and stained with Abs in 1× perm/wash buffer for 1 h at room temperature. The following Abs used in this study were purchased from Thermo Fisher Scientific, BD Biosciences, or BioLegend: CD8a BV786 or APC efluor 780 (53–6.7), CD4 BV650 or BV711 (RM4-5), PD-1 BV650 (29F.1A12), IFN-γ APC (XMG1.2), TNF-α FITC (MP6-XT22), IL-2 PE (JES6-5H4), KLRG1 PeCy7 or PE-TR (2F1), and TIM3 PeCy7 (RMT3-23). Anti-TCF-1 (C63D9) conjugated with Alexa Fluor 647 or Alexa Fluor 488 was purchased from Cell Signaling Technologies. Cells were acquired using a Digital LSR II flow cytometer (Becton Dickinson) or ZE5 Cell Analyzer (Bio-Rad). Flow cytometric data were analyzed with FlowJo software v9.9.6 and v10.
Ex vivo T cell stimulation
Small intestinal LPLs were cultured at 2.5 × 106 cells/ml in round-bottom 96-well plates for 5 h in complete RPMI supplemented with brefeldin A (1 µg/ml; Sigma), PMA (50 ng/ml; Sigma) combined with ionomycin (1 µg/ml; Sigma), or 1 µg/ml of the MHC class I–restricted LCMV NP396–404, GP33–41, GP276-286, and 10 µg/ml of the MHC class II–restricted peptide GP67–77 (all >99% pure; Synpep). Cells were then stained with a fixable viability dye (Tonbo Ghost Dye) and surface staining with the anti-CD4 or anti-CD8 mAbs described above, fixed in 1% paraformaldehyde, permeabilized, and stained with the aforementioned Abs against IFN-γ, TNF-α, and IL-2 in 1× perm/wash buffer for 30 min at 4°C. Unstimulated controls in which cells were cultured without peptide or PMA/ionomycin were performed in parallel and showed background levels of cytokine staining (data not shown).
IEC isolation, FACS purification, and RNA extraction
We obtained the intestinal epithelial pellets by separating the epithelial layer in IEL buffer, as described above for lymphocyte isolation. Pellets were resuspended and digested by stirring in 10 ml IEC enzyme media, which consisted of RPMI supplemented with 5% FBS, 1% P/S, 0.4 mg/ml collagenase D (Roche), 0.3 U/ml Dispase (Roche), and 10 µg/ml DNaseI (Roche) for 20 min at 37°C. Suspension was filtered through a 100-µm strainer and spun down at 1,500 rpm for 5 min at 4°C. Three mice per group were pooled and stained with Abs against EpCAM-APC (clone G8.8; Thermo Fisher Scientific) and CD45.2-FITC (clone 104; BioLegend) as well as propidium iodide (Sigma) before FACS purification in FACSAria III (BD). 50,000 cells were sorted, washed with PBS 1×, resuspended in RLT (Qiagen) + β-mercaptoethanol (10 µl/ml; Life Technologies), and stored at −80°C. The following day, RNA was extracted with RNAeasy Micro Kit (Qiagen) by following the manufacturer’s protocol and resuspended in molecular grade distilled H2O and stored at -−80°C until library preparation.
mRNA library preparation and RNA sequencing
Total RNA was extracted from 50,000 IECs and downstream processing was performed by the UCSD Institute for Genomic Medicine. mRNA stranded libraries were constructed by using TruSeq RNA Library Prep Kit v2 (Illumina) followed by single-end sequencing of 75 base-pair fragments with a HiSeq 4000 (Illumina). Raw sequencing files were processed with CASAVA 1.8.2 (Illumina) to generate .fastq files.
RNA-sequencing processing and data analysis
FASTQ files were uploaded onto the Galaxy project portal (https://usegalaxy.org/). We first obtained read quality reports by running the Fastqc module (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/), which gave us overall high-quality scores. Next, we used STAR (Spliced Transcripts Alignment to a Reference; Dobin et al., 2013) to align RNA-sequencing reads to the mouse genome with the following parameters: mm10, GRCm38, 75 bp length of the genomic sequence around annotated junctions and STAR Galaxy version 2.7.2b default parameters for seed, alignment, limits, and chimeric alignment. To assemble RNA-sequencing alignments into potential transcripts and to obtain gene abundance estimation files, we used StringTie (Pertea et al., 2015) with mouse GTF file (https://www.gencodegenes.org/mouse/) as reference to guide assembly. The following default parameters were used: minimum isoform fraction, 0.15; minimum anchor length for junctions, 10; minimum junction coverage, 1; minimum bundle reads per base pair coverage to consider for assembly, 2; gap between read mappings triggering a new bundle, 50; fraction of bundle allowed to be covered by multihit reads, 0.95; trimming of predicted transcripts based on coverage, disabled; multimapping correction, disabled. Next, differentially expressed features were determined by DESeq2 (Love et al., 2014). We defined DEGs by false discovery rate (FDR)–adjusted P < 0.05 and fold change ≥ 2 throughout the manuscript. GSEA was performed through the software version 4.0.3 (Subramanian et al., 2005). We defined differentially enriched pathways with P < 0.05 and FDR < 0.25 cutoffs.
Sample collection for microbiome studies
Mice were sacrificed and the peritoneal cavity opened with one set of tools. A new sterile set of tools was then used to retrieve colonic and cecal contents, which were placed in sterile tubes, frozen on dry ice, and stored at −80°C. Both sets of tools were submerged in 70% ethanol between animals belonging to the same group, and a new set of sterile tools was used when changing experimental groups.
DNA extraction for 16S rRNA V4 amplicon sequencing and shotgun metagenomic sequencing
We followed the protocol described previously (Marotz et al., 2017). Briefly, to extract DNA from colonic and cecal contents, frozen samples were thawed and transferred into 96-well plates containing garnet beads. DNA was extracted using Qiagen PowerSoil DNA kit adapted for magnetic bead purification and eluted in 100 µl Qiagen elution buffer. DNA quantification was performed via Quant-iT PicoGreen Assay (Invitrogen).
Library generation and sequencing
Sequencing was done following standard protocols from the Earth Microbiome Project (Caporaso et al., 2012; Gilbert et al., 2014). Extracted DNA was amplified by using barcoded primers. Each sample was PCR amplified in triplicate and V4 pair-end sequencing or whole-genome sequencing using a miniaturized version of the KAPA HyperPlus kit was performed using Illumina MiSeq or HiSeq.
16S rRNA gene amplicon data analysis
Processing of our 16S rRNA raw data was done with Qiita (https://qiita.ucsd.edu/; Gonzalez et al., 2018) and involved splitting of FASTQ libraries, demultiplexing, and trimming of sequences to 150 bp of length (Caporaso et al., 2010), followed by deblur 1.1.0 (Amir et al., 2017) with the following Qiita default parameters: indel probability, 0.01; mean per nucleotide error rate, 0.005; minimum per-sample read threshold, 2; Greengenes_13.8 as reference phylogeny for SATe-enabled phylogenetic placement; and one thread per sample. Features with a minimum frequency of 10 were retained and reference hit table obtained was rarefied to 10,000 counts, which was in the plateau region of α-rarefaction curves (Gotelli and Colwell, 2001). To assign taxonomy, we used the prefitted sklearn-based taxonomy classifier (classify_sklearn) with the Green Genes classifier (gg-13-8-99-515-806-nb-classifier; Bokulich et al., 2018; DeSantis et al., 2006). Beta diversity was computed with the “β_phylogenetic” module using Unweighted UniFrac. This was followed by PCoA (“pcoa” module), and significance was calculated for “infection” with the “β_group_significance” module using a PERMANOVA test with 999 permutations. Alpha diversity was computed using the α module for Shannon diversity index. All the aforementioned analyses were done with Qiita platform version 052020 5f09f46.
To identify differentially abundant genera among groups, we collapsed our rarefied .biom tables to the genus level and used songbird multinomial regression analysis (Morton et al., 2019) with default parameters (epochs of 10,000, differential prior of 0.5; https://github.com/biocore/songbird). We applied cutoff rank values of 0.5 and −0.5 to focus our analysis on moderately to highly perturbed genera. Validation of the model was performed for all analyses with tensor board.
Shotgun metagenomics data analysis
To preprocess shotgun metagenomics sequencing data, we performed adapter trimming with Atropos v1.1.15 (Didion et al., 2017), followed by quality control filtering (QC_Filter) to bowtie2/Mus_musculus, followed by genome alignment of adapter-trimmed files with bowtie2 (with Rep94 database; Langmead and Salzberg, 2012) by running SHOGUN 0.1.5 (Hillmann et al., 2018) via Qiita (https://qiita.ucsd.edu/; Gonzalez et al., 2018). A table in .biom format with taxonomic predictions at the species level was then rarefied to a depth of 200,000 or 450,000 counts. Alpha diversity was computed using the Shannon diversity index, as above, while β-diversity was computed with the β module using Jaccard and Bray–Curtis metrics. PCoA representation and PERMANOVA were calculated as for the 16S rRNA gene amplicon data. To identify differentially abundant species between groups, we collapsed our rarefied .biom tables to the species level and used songbird multinomial regression analysis (Morton et al., 2019) with parameters described above for 16S rRNA gene amplicon data analysis.
Statistical analysis
An unpaired two-tailed Student's t test was used to compare two groups. If variances were not equal by F-test, data were tested using the nonparametric Mann–Whitney U test. Significant differences among more than two groups were determined based on one-way ANOVA with Tukey’s multiple comparison correction or, in the case of unequal variances, nonparametric Kruskal–Wallis test with Dunn’s multiple comparison correction. Statistical tests were performed using GraphPad Prism v8.
Data availability
All RNA-sequencing data have been deposited to the Gene Expression Omnibus (accession no. GSE155120) and the Sequence Read Archive (accession no. PRJNA648708). All 16S rRNA gene amplicon and shotgun metagenomics sequencing data are publicly available on the European Nucleotide Archive (https://ebi.ac.uk/ena; accession no. ERP123227) and Qiita (https://qiita.ucsd.edu, study ID 11043). On Qiita, processed 16S .fastq and metagenomics .fastq files can be found under preparation ID 31428 and preparation ID 92095, respectively. Detailed metadata for all samples can be found under the “Sample Information” tab.
Online supplemental material
Fig. S1 shows LCMV Cl13 infects vimentin+ and CD45+ cells in the small intestine at day 24 p.i., but not EpCAM+ cells at day 9 p.i. Fig. S2 shows that LCMV Cl13 infection does not increase intestinal permeability to FD40 or causes epithelial layer disruption. Fig. S3 shows that Ab-mediated blockade of IFNAR-1 reduces IFN signaling in IECs after LCMV Cl13 infection. Fig. S4 shows that type I IFN signaling restricts small intestinal virus-specific CD4 T cell accumulation and shows that CD4 T cell responses are dispensable for the induction of intestinal barrier leakage during LCMV Cl13 infection. Fig. S5 shows that type I IFN signaling promotes overrepresentation of Erysipelatoclostridium and reduces Roseburia species in the intestinal microbiome after LCMV Cl13 infection. Table S1 and Table S2 list all DEGs in IECs between ARM-infected or Cl13-infected versus uninfected mice, respectively. Table S3 compares the expression levels of tight junction–related genes among uninfected, ARM-infected, and Cl13-infected mice. Table S4 and Table S5 list enriched pathways and DEGs, respectively, in IECs from Cl13 versus ARM-infected mice. Table S6 and Table S7 list enriched pathways and DEGs, respectively, in IECs from isotype-treated versus anti-IFNAR-1–treated Cl13 infected mice. Table S8 and Table S9 list pathways and DEGs, respectively, regulated by type I IFNs among those significantly altered in Cl13-infected versus ARM-infected mice. Table S10 shows songbird multinomial regression results comparing microbiomes from Cl13-infected mice treated with isotype control versus anti–IFNAR-1–blocking Abs.
Supplementary Material
lists DEGs in IECs between ARM-infected and uninfected mice on day 9 p.i. (see also Fig. 2).
shows DEGs in IECs between Cl13-infected and uninfected mice on day 9 p.i. (see also Fig. 2).
outlines tight junction–related genes in IECs from Cl13- or ARM-infected and uninfected mice on day 9 p.i. (see also Fig. 2).
shows pathways enriched in IEC transcriptomes from Cl13- versus ARM-infected mice on day 9 p.i. (see also Fig. 2).
lists differentially expressed genes in IECs between Cl13- versus ARM-infected mice on day 9 p.i. (see also Fig. 2).
displays pathways enriched in IEC transcriptomes from isotype-treated versus anti-IFNAR-1–treated Cl13-infected mice on day 9 p.i. (see also Fig. 3).
lists DEGs in IECs between isotype-treated versus anti-IFNAR-1–treated Cl13-infected mice on day 9 p.i. (see also Fig. 3).
shows differentially enriched pathways regulated by type I IFNs from IECs obtained from Cl13- versus ARM-infected mice on day 9 p.i. (see also Fig. 3).
displays DEGs regulated by type I IFNs among DEGs by IECs from Cl13- versus ARM-infected mice on day 9 p.i. (see also Fig. 3).
shows songbird multinomial regression ranks by comparing microbiomes from Cl13-infected mice treated with isotype controls (IgG1) versus IFNAR-1 blocking Abs (anti-IFNAR-1; see also Fig. 5).
Acknowledgments
We thank members of the Zúñiga laboratory for discussions and critical reading of the manuscript. We thank Dr. Li-fan Lu and Dr. Chia-Hao Lin (UCSD, San Diego, CA) for providing C. rodentium DBS100 and Dr. Manuela Raffatellu (UCSD, San Diego, CA) for protocol on how to prepare stocks. We thank Dr. Gail Ackermann for data upload and metadata curation on Qiita. We thank personnel at the Animal Facility, IGM Genomics center sequencing core, Moore’s Cancer Center Histopathology core, and the UCSD Microscopy core (National Institute of Neurological Disorders and Stroke grant NS047101). The graphical abstract was created with BioRender.com.
Research was supported by National Institutes of Health grants AI081923, AI113923, and AI132122, as well as a UCSD Center for Microbiome Innovation seed grant to E.I. Zúñiga. Research was further supported by National Institute of Diabetes and Digestive and Kidney Diseases grants R01DK61931, R01DK068271, R24DK099803, and P30DK034854 to J.R. Turner. L. Labarta-Bajo was partly supported by a La Caixa fellowship for studies in North America and the UCSD Frontiers of Innovation Scholars Program.
Author contributions: L. Labarta-Bajo designed, performed, analyzed, and interpreted all experiments and wrote the manuscript. S.P. Nilsen and J.R. Turner provided feedback for interpreting data related to intestinal permeability and tight junction modulation after infection. G. Humphrey, T. Schwartz, and K. Sanders performed all preprocessing and 16S/shotgun metagenomic sequencing. A. Swafford assisted with experimental design of microbiome studies and provided guidance in -omics data analysis. R. Knight supervised microbiome analysis and provided advice. E.I. Zúñiga conceived and supervised the project, designed and interpreted experiments, and wrote the manuscript. All authors revised the manuscript.
References
- Abe, K., Nguyen K.P., Fine S.D., Mo J.H., Shen C., Shenouda S., Corr M., Jung S., Lee J., Eckmann L., et al. 2007. Conventional dendritic cells regulate the outcome of colonic inflammation independently of T cells. Proc. Natl. Acad. Sci. USA. 104:17022–17027. 10.1073/pnas.0708469104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abt, M.C., Osborne L.C., Monticelli L.A., Doering T.A., Alenghat T., Sonnenberg G.F., Paley M.A., Antenus M., Williams K.L., Erikson J., et al. 2012. Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity. 37:158–170. 10.1016/j.immuni.2012.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed, R., and Oldstone M.B.. 1988. Organ-specific selection of viral variants during chronic infection. J. Exp. Med. 167:1719–1724. 10.1084/jem.167.5.1719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed, R., Salmi A., Butler L.D., Chiller J.M., and Oldstone M.B.. 1984. Selection of genetic variants of lymphocytic choriomeningitis virus in spleens of persistently infected mice. Role in suppression of cytotoxic T lymphocyte response and viral persistence. J. Exp. Med. 160:521–540. 10.1084/jem.160.2.521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allers, K., Puyskens A., Epple H.J., Schürmann D., Hofmann J., Moos V., and Schneider T.. 2016. The effect of timing of antiretroviral therapy on CD4+ T-cell reconstitution in the intestine of HIV-infected patients. Mucosal Immunol. 9:265–274. 10.1038/mi.2015.58 [DOI] [PubMed] [Google Scholar]
- Allers, K., Puyskens A., Epple H.J., Schürmann D., Hofmann J., Moos V., and Schneider T.. 2017. Distribution and Activation of CD8+ T Cells in the Duodenal Mucosa before and after HIV Seroconversion. J. Immunol. 198:481–491. 10.4049/jimmunol.1601278 [DOI] [PubMed] [Google Scholar]
- Amir, A., McDonald D., Navas-Molina J.A., Kopylova E., Morton J.T., Zech Xu Z., Kightley E.P., Thompson L.R., Hyde E.R., Gonzalez A., et al. 2017. Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. mSystems. 2. e00191-16. 10.1128/mSystems.00191-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelosanto, J.M., Blackburn S.D., Crawford A., and Wherry E.J.. 2012. Progressive loss of memory T cell potential and commitment to exhaustion during chronic viral infection. J. Virol. 86:8161–8170. 10.1128/JVI.00889-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber, D.L., Wherry E.J., Masopust D., Zhu B., Allison J.P., Sharpe A.H., Freeman G.J., and Ahmed R.. 2006. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 439:682–687. 10.1038/nature04444 [DOI] [PubMed] [Google Scholar]
- Belkaid, Y., and Harrison O.J.. 2017. Homeostatic Immunity and the Microbiota. Immunity. 46:562–576. 10.1016/j.immuni.2017.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beura, L.K., Anderson K.G., Schenkel J.M., Locquiao J.J., Fraser K.A., Vezys V., Pepper M., and Masopust D.. 2015. Lymphocytic choriomeningitis virus persistence promotes effector-like memory differentiation and enhances mucosal T cell distribution. J. Leukoc. Biol. 97:217–225. 10.1189/jlb.1HI0314-154R [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindels, L.B., Neyrinck A.M., Loumaye A., Catry E., Walgrave H., Cherbuy C., Leclercq S., Van Hul M., Plovier H., Pachikian B., et al. 2018. Increased gut permeability in cancer cachexia: mechanisms and clinical relevance. Oncotarget. 9:18224–18238. 10.18632/oncotarget.24804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokulich, N.A., Kaehler B.D., Rideout J.R., Dillon M., Bolyen E., Knight R., Huttley G.A., and Gregory Caporaso J.. 2018. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome. 6:90. 10.1186/s40168-018-0470-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosinger, S.E., Li Q., Gordon S.N., Klatt N.R., Duan L., Xu L., Francella N., Sidahmed A., Smith A.J., Cramer E.M., et al. 2009. Global genomic analysis reveals rapid control of a robust innate response in SIV-infected sooty mangabeys. J. Clin. Invest. 119:3556–3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouladoux, N., Harrison O.J., and Belkaid Y.. 2017. The Mouse Model of Infection with Citrobacter rodentium. Curr. Protoc. Immunol. 119:1–: 25. 10.1002/cpim.34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenchley, J.M., Price D.A., Schacker T.W., Asher T.E., Silvestri G., Rao S., Kazzaz Z., Bornstein E., Lambotte O., Altmann D., et al. 2006. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat. Med. 12:1365–1371. 10.1038/nm1511 [DOI] [PubMed] [Google Scholar]
- Buckley, A., and Turner J.R.. 2018. Cell Biology of Tight Junction Barrier Regulation and Mucosal Disease. Cold Spring Harb. Perspect. Biol. 10. a029314. 10.1101/cshperspect.a029314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canesso, M.C.C., Lemos L., Neves T.C., Marim F.M., Castro T.B.R., Veloso É.S., Queiroz C.P., Ahn J., Santiago H.C., Martins F.S., et al. 2018. The cytosolic sensor STING is required for intestinal homeostasis and control of inflammation. Mucosal Immunol. 11:820–834. 10.1038/mi.2017.88 [DOI] [PubMed] [Google Scholar]
- Caporaso, J.G., Kuczynski J., Stombaugh J., Bittinger K., Bushman F.D., Costello E.K., Fierer N., Peña A.G., Goodrich J.K., Gordon J.I., et al. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods. 7:335–336. 10.1038/nmeth.f.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso, J.G., Lauber C.L., Walters W.A., Berg-Lyons D., Huntley J., Fierer N., Owens S.M., Betley J., Fraser L., Bauer M., et al. 2012. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6:1621–1624. 10.1038/ismej.2012.8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherradi, S., Martineau P., Gongora C., and Del Rio M.. 2019. Claudin gene expression profiles and clinical value in colorectal tumors classified according to their molecular subtype. Cancer Manag. Res. 11:1337–1348. 10.2147/CMAR.S188192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlin, V.S., Wu X., Nguyen C., Dai C., Vallance B.A., Buchan A.M., Boyer L., and Jacobson K.. 2009. Vasoactive intestinal peptide ameliorates intestinal barrier disruption associated with Citrobacter rodentium-induced colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 297:G735–G750. 10.1152/ajpgi.90551.2008 [DOI] [PubMed] [Google Scholar]
- Coulombe, P.A., and Wong P.. 2004. Cytoplasmic intermediate filaments revealed as dynamic and multipurpose scaffolds. Nat. Cell Biol. 6:699–706. 10.1038/ncb0804-699 [DOI] [PubMed] [Google Scholar]
- D’Incà, R., Di Leo V., Corrao G., Martines D., D’Odorico A., Mestriner C., Venturi C., Longo G., and Sturniolo G.C.. 1999. Intestinal permeability test as a predictor of clinical course in Crohn’s disease. Am. J. Gastroenterol. 94:2956–2960. 10.1016/S0002-9270(99)00500-6 [DOI] [PubMed] [Google Scholar]
- Desai, M.S., Seekatz A.M., Koropatkin N.M., Kamada N., Hickey C.A., Wolter M., Pudlo N.A., Kitamoto S., Terrapon N., Muller A., et al. 2016. A Dietary Fiber-Deprived Gut Microbiota Degrades the Colonic Mucus Barrier and Enhances Pathogen Susceptibility. Cell. 167:1339–1353.e21. 10.1016/j.cell.2016.10.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis, T.Z., Hugenholtz P., Larsen N., Rojas M., Brodie E.L., Keller K., Huber T., Dalevi D., Hu P., and Andersen G.L.. 2006. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72:5069–5072. 10.1128/AEM.03006-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didion, J.P., Martin M., and Collins F.S.. 2017. Atropos: specific, sensitive, and speedy trimming of sequencing reads. PeerJ. 5. e3720. 10.7717/peerj.3720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin, A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., and Gingeras T.R.. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 29:15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorhoi, A., Yeremeev V., Nouailles G., J. Weiner, III, Jörg S., Heinemann E., Oberbeck-Müller D., Knaul J.K., Vogelzang A., Reece S.T., et al. 2014. Type I IFN signaling triggers immunopathology in tuberculosis-susceptible mice by modulating lung phagocyte dynamics. Eur. J. Immunol. 44:2380–2393. 10.1002/eji.201344219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichele, D.D., and Kharbanda K.K.. 2017. Dextran sodium sulfate colitis murine model: An indispensable tool for advancing our understanding of inflammatory bowel diseases pathogenesis. World J. Gastroenterol. 23:6016–6029. 10.3748/wjg.v23.i33.6016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estes, J.D., Harris L.D., Klatt N.R., Tabb B., Pittaluga S., Paiardini M., Barclay G.R., Smedley J., Pung R., Oliveira K.M., et al. 2010. Damaged intestinal epithelial integrity linked to microbial translocation in pathogenic simian immunodeficiency virus infections. PLoS Pathog. 6. e1001052. 10.1371/journal.ppat.1001052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabregat, A., Jupe S., Matthews L., Sidiropoulos K., Gillespie M., Garapati P., Haw R., Jassal B., Korninger F., May B., et al. 2018. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 46(D1):D649–D655. 10.1093/nar/gkx1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallimore, A., Glithero A., Godkin A., Tissot A.C., Plückthun A., Elliott T., Hengartner H., and Zinkernagel R.. 1998. Induction and exhaustion of lymphocytic choriomeningitis virus-specific cytotoxic T lymphocytes visualized using soluble tetrameric major histocompatibility complex class I-peptide complexes. J. Exp. Med. 187:1383–1393. 10.1084/jem.187.9.1383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert, J.A., Jansson J.K., and Knight R.. 2014. The Earth Microbiome project: successes and aspirations. BMC Biol. 12:69. 10.1186/s12915-014-0069-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez, A., Navas-Molina J.A., Kosciolek T., McDonald D., Vázquez-Baeza Y., Ackermann G., DeReus J., Janssen S., Swafford A.D., Orchanian S.B., et al. 2018. Qiita: rapid, web-enabled microbiome meta-analysis. Nat. Methods. 15:796–798. 10.1038/s41592-018-0141-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotelli, N.J., and Colwell R.K.. 2001. Quantifying biodiversity: procedures and pitfalls in the measurement and comparison of species richness. Ecol. Lett. 4:379–391. 10.1046/j.1461-0248.2001.00230.x [DOI] [Google Scholar]
- Graham, W.V., He W., Marchiando A.M., Zha J., Singh G., Li H.S., Biswas A., Ong M.L.D.M., Jiang Z.H., Choi W., et al. 2019. Intracellular MLCK1 diversion reverses barrier loss to restore mucosal homeostasis. Nat. Med. 25:690–700. 10.1038/s41591-019-0393-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groom, J.R., and Luster A.D.. 2011. CXCR3 in T cell function. Exp. Cell Res. 317:620–631. 10.1016/j.yexcr.2010.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu, S., Chen D., Zhang J.N., Lv X., Wang K., Duan L.P., Nie Y., and Wu X.L.. 2013. Bacterial community mapping of the mouse gastrointestinal tract. PLoS One. 8. e74957. 10.1371/journal.pone.0074957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbison, J.E., Roth-Schulze A.J., Giles L.C., Tran C.D., Ngui K.M., Penno M.A., Thomson R.L., Wentworth J.M., Colman P.G., Craig M.E., et al. 2019. Gut microbiome dysbiosis and increased intestinal permeability in children with islet autoimmunity and type 1 diabetes: A prospective cohort study. Pediatr. Diabetes. 20:574–583. [DOI] [PubMed] [Google Scholar]
- Harris, L.D., Tabb B., Sodora D.L., Paiardini M., Klatt N.R., Douek D.C., Silvestri G., Müller-Trutwin M., Vasile-Pandrea I., Apetrei C., et al. 2010. Downregulation of robust acute type I interferon responses distinguishes nonpathogenic simian immunodeficiency virus (SIV) infection of natural hosts from pathogenic SIV infection of rhesus macaques. J. Virol. 84:7886–7891. 10.1128/JVI.02612-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto, M., Kamphorst A.O., Im S.J., Kissick H.T., Pillai R.N., Ramalingam S.S., Araki K., and Ahmed R.. 2018. CD8 T Cell Exhaustion in Chronic Infection and Cancer: Opportunities for Interventions. Annu. Rev. Med. 69:301–318. 10.1146/annurev-med-012017-043208 [DOI] [PubMed] [Google Scholar]
- Hedin, C.R., McCarthy N.E., Louis P., Farquharson F.M., McCartney S., Taylor K., Prescott N.J., Murrells T., Stagg A.J., Whelan K., et al. 2014. Altered intestinal microbiota and blood T cell phenotype are shared by patients with Crohn’s disease and their unaffected siblings. Gut. 63:1578–1586. 10.1136/gutjnl-2013-306226 [DOI] [PubMed] [Google Scholar]
- Hillmann, B., Al-Ghalith G.A., Shields-Cutler R.R., Zhu Q., Gohl D.M., Beckman K.B., Knight R., and Knights D.. 2018. Evaluating the Information Content of Shallow Shotgun Metagenomics. mSystems. 3. e00069-18. 10.1128/mSystems.00069-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homann, D., Lewicki H., Brooks D., Eberlein J., Mallet-Designé V., Teyton L., and Oldstone M.B.. 2007. Mapping and restriction of a dominant viral CD4+ T cell core epitope by both MHC class I and MHC class II. Virology. 363:113–123. 10.1016/j.virol.2006.12.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, W.H., Gensheimer J., Hashimoto M., Wieland A., Valanparambil R.M., Li P., Lin J.X., Konieczny B.T., Im S.J., Freeman G.J., et al. 2019. Proliferating Transitory T Cells with an Effector-like Transcriptional Signature Emerge from PD-1+ Stem-like CD8+ T Cells during Chronic Infection. Immunity. 51:1043–1058.e4. 10.1016/j.immuni.2019.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im, S.J., Hashimoto M., Gerner M.Y., Lee J., Kissick H.T., Burger M.C., Shan Q., Hale J.S., Lee J., Nasti T.H., et al. 2016. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature. 537:417–421. 10.1038/nature19330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jostins, L., Ripke S., Weersma R.K., Duerr R.H., McGovern D.P., Hui K.Y., Lee J.C., Schumm L.P., Sharma Y., Anderson C.A., et al. ; International IBD Genetics Consortium (IIBDGC) . 2012. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 491:119–124. 10.1038/nature11582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa, M., and Goto S.. 2000. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30. 10.1093/nar/28.1.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolumam, G.A., Thomas S., Thompson L.J., Sprent J., and Murali-Krishna K.. 2005. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J. Exp. Med. 202:637–650. 10.1084/jem.20050821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead, B., and Salzberg S.L.. 2012. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 9:357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei, Z., Maeda T., Tamura A., Nakamura T., Yamazaki Y., Shiratori H., Yashiro K., Tsukita S., and Hamada H.. 2012. EpCAM contributes to formation of functional tight junction in the intestinal epithelium by recruiting claudin proteins. Dev. Biol. 371:136–145. 10.1016/j.ydbio.2012.07.005 [DOI] [PubMed] [Google Scholar]
- Li, Q., Skinner P.J., Ha S.J., Duan L., Mattila T.L., Hage A., White C., Barber D.L., O’Mara L., Southern P.J., et al. 2009. Visualizing antigen-specific and infected cells in situ predicts outcomes in early viral infection. Science. 323:1726–1729. 10.1126/science.1168676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, R., Fei Y.J., Prasad P.D., Ramamoorthy S., Han H., Yang-Feng T.L., Hediger M.A., Ganapathy V., and Leibach F.H.. 1995. Human intestinal H+/peptide cotransporter. Cloning, functional expression, and chromosomal localization. J. Biol. Chem. 270:6456–6463. 10.1074/jbc.270.12.6456 [DOI] [PubMed] [Google Scholar]
- Liberzon, A., Subramanian A., Pinchback R., Thorvaldsdóttir H., Tamayo P., and Mesirov J.P.. 2011. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 27:1739–1740. 10.1093/bioinformatics/btr260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love, M.I., Huber W., and Anders S.. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunt, S.Y., and Vander Heiden M.G.. 2011. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 27:441–464. 10.1146/annurev-cellbio-092910-154237 [DOI] [PubMed] [Google Scholar]
- Macal, M., Lewis G.M., Kunz S., Flavell R., Harker J.A., and Zúñiga E.I.. 2012. Plasmacytoid dendritic cells are productively infected and activated through TLR-7 early after arenavirus infection. Cell Host Microbe. 11:617–630. 10.1016/j.chom.2012.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machiels, K., Joossens M., Sabino J., De Preter V., Arijs I., Eeckhaut V., Ballet V., Claes K., Van Immerseel F., Verbeke K., et al. 2014. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut. 63:1275–1283. 10.1136/gutjnl-2013-304833 [DOI] [PubMed] [Google Scholar]
- Mahanty, S., Hutchinson K., Agarwal S., McRae M., Rollin P.E., and Pulendran B.. 2003. Cutting edge: impairment of dendritic cells and adaptive immunity by Ebola and Lassa viruses. J. Immunol. 170:2797–2801. 10.4049/jimmunol.170.6.2797 [DOI] [PubMed] [Google Scholar]
- Mancabelli, L., Milani C., Lugli G.A., Turroni F., Cocconi D., van Sinderen D., and Ventura M.. 2017. Identification of universal gut microbial biomarkers of common human intestinal diseases by meta-analysis. FEMS Microbiol. Ecol. 93:93. 10.1093/femsec/fix153 [DOI] [PubMed] [Google Scholar]
- Marchetti, G., Bellistrì G.M., Borghi E., Tincati C., Ferramosca S., La Francesca M., Morace G., Gori A., and Monforte A.D.. 2008. Microbial translocation is associated with sustained failure in CD4+ T-cell reconstitution in HIV-infected patients on long-term highly active antiretroviral therapy. AIDS. 22:2035–2038. 10.1097/QAD.0b013e3283112d29 [DOI] [PubMed] [Google Scholar]
- Marotz, C., Amir A., Humphrey G., Gaffney J., Gogul G., and Knight R.. 2017. DNA extraction for streamlined metagenomics of diverse environmental samples. Biotechniques. 62:290–293. 10.2144/000114559 [DOI] [PubMed] [Google Scholar]
- Martinez-Medina, M., Denizot J., Dreux N., Robin F., Billard E., Bonnet R., Darfeuille-Michaud A., and Barnich N.. 2014. Western diet induces dysbiosis with increased E coli in CEABAC10 mice, alters host barrier function favouring AIEC colonisation. Gut. 63:116–124. 10.1136/gutjnl-2012-304119 [DOI] [PubMed] [Google Scholar]
- Matloubian, M., Kolhekar S.R., Somasundaram T., and Ahmed R.. 1993. Molecular determinants of macrophage tropism and viral persistence: importance of single amino acid changes in the polymerase and glycoprotein of lymphocytic choriomeningitis virus. J. Virol. 67:7340–7349. 10.1128/JVI.67.12.7340-7349.1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki, T., Holst O., and Hardt W.D.. 2012. The bactericidal activity of the C-type lectin RegIIIβ against Gram-negative bacteria involves binding to lipid A. J. Biol. Chem. 287:34844–34855. 10.1074/jbc.M112.399998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molofsky, A.B., Savage A.K., and Locksley R.M.. 2015. Interleukin-33 in Tissue Homeostasis, Injury, and Inflammation. Immunity. 42:1005–1019. 10.1016/j.immuni.2015.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton, J.T., Marotz C., Washburne A., Silverman J., Zaramela L.S., Edlund A., Zengler K., and Knight R.. 2019. Establishing microbial composition measurement standards with reference frames. Nat. Commun. 10:2719. 10.1038/s41467-019-10656-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller, S.N., Matloubian M., Clemens D.M., Sharpe A.H., Freeman G.J., Gangappa S., Larsen C.P., and Ahmed R.. 2007. Viral targeting of fibroblastic reticular cells contributes to immunosuppression and persistence during chronic infection. Proc. Natl. Acad. Sci. USA. 104:15430–15435. 10.1073/pnas.0702579104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee, S., Zheng H., Derebe M.G., Callenberg K.M., Partch C.L., Rollins D., Propheter D.C., Rizo J., Grabe M., Jiang Q.X., et al. 2014. Antibacterial membrane attack by a pore-forming intestinal C-type lectin. Nature. 505:103–107. 10.1038/nature12729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullineaux-Sanders, C., Sanchez-Garrido J., Hopkins E.G.D., Shenoy A.R., Barry R., and Frankel G.. 2019. Citrobacter rodentium-host-microbiota interactions: immunity, bioenergetics and metabolism. Nat. Rev. Microbiol. 17:701–715. 10.1038/s41579-019-0252-z [DOI] [PubMed] [Google Scholar]
- Neil, J.A., Matsuzawa-Ishimoto Y., Kernbauer-Hölzl E., Schuster S.L., Sota S., Venzon M., Dallari S., Galvao Neto A., Hine A., Hudesman D., et al. 2019. IFN-I and IL-22 mediate protective effects of intestinal viral infection. Nat. Microbiol. 4:1737–1749. 10.1038/s41564-019-0470-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, K.B., Watford W.T., Salomon R., Hofmann S.R., Pien G.C., Morinobu A., Gadina M., O’Shea J.J., and Biron C.A.. 2002. Critical role for STAT4 activation by type 1 interferons in the interferon-gamma response to viral infection. Science. 297:2063–2066. 10.1126/science.1074900 [DOI] [PubMed] [Google Scholar]
- Odenwald, M.A., and Turner J.R.. 2013. Intestinal permeability defects: is it time to treat? Clin. Gastroenterol. Hepatol. 11:1075–1083. 10.1016/j.cgh.2013.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odenwald, M.A., and Turner J.R.. 2017. The intestinal epithelial barrier: a therapeutic target? Nat. Rev. Gastroenterol. Hepatol. 14:9–21. 10.1038/nrgastro.2016.169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okayasu, I., Hatakeyama S., Yamada M., Ohkusa T., Inagaki Y., and Nakaya R.. 1990. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology. 98:694–702. 10.1016/0016-5085(90)90290-H [DOI] [PubMed] [Google Scholar]
- Oxenius, A., Bachmann M.F., Ashton-Rickardt P.G., Tonegawa S., Zinkernagel R.M., and Hengartner H.. 1995. Presentation of endogenous viral proteins in association with major histocompatibility complex class II: on the role of intracellular compartmentalization, invariant chain and the TAP transporter system. Eur. J. Immunol. 25:3402–3411. 10.1002/eji.1830251230 [DOI] [PubMed] [Google Scholar]
- Patel, R.M., Myers L.S., Kurundkar A.R., Maheshwari A., Nusrat A., and Lin P.W.. 2012. Probiotic bacteria induce maturation of intestinal claudin 3 expression and barrier function. Am. J. Pathol. 180:626–635. 10.1016/j.ajpath.2011.10.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak, B.G., Shull G.E., Jenkins N.A., and Copeland N.G.. 1996. Mouse chromosomal location of four Na/H exchanger isoform genes. Genomics. 31:261–263. 10.1006/geno.1996.0045 [DOI] [PubMed] [Google Scholar]
- Pertea, M., Pertea G.M., Antonescu C.M., Chang T.C., Mendell J.T., and Salzberg S.L.. 2015. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33:290–295. 10.1038/nbt.3122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson, L.W., and Artis D.. 2014. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 14:141–153. 10.1038/nri3608 [DOI] [PubMed] [Google Scholar]
- Pinchuk, I.V., Mifflin R.C., Saada J.I., and Powell D.W.. 2010. Intestinal mesenchymal cells. Curr. Gastroenterol. Rep. 12:310–318. 10.1007/s11894-010-0135-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponte, R., Mehraj V., Ghali P., Couëdel-Courteille A., Cheynier R., and Routy J.P.. 2016. Reversing Gut Damage in HIV Infection: Using Non-Human Primate Models to Instruct Clinical Research. EBioMedicine. 4:40–49. 10.1016/j.ebiom.2016.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puppa, M.J., White J.P., Sato S., Cairns M., Baynes J.W., and Carson J.A.. 2011. Gut barrier dysfunction in the Apc(Min/+) mouse model of colon cancer cachexia. Biochim. Biophys. Acta. 1812:1601–1606. 10.1016/j.bbadis.2011.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu, A., Min S.H., Jansen M., Malhotra U., Tsai E., Cabelof D.C., Matherly L.H., Zhao R., Akabas M.H., and Goldman I.D.. 2007. Rodent intestinal folate transporters (SLC46A1): secondary structure, functional properties, and response to dietary folate restriction. Am. J. Physiol. Cell Physiol. 293:C1669–C1678. 10.1152/ajpcell.00202.2007 [DOI] [PubMed] [Google Scholar]
- Rooks, M.G., and Garrett W.S.. 2016. Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 16:341–352. 10.1038/nri.2016.42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagné, C., Agulhon C., Ravassard P., Darmon M., Hamon M., El Mestikawy S., Gasnier B., and Giros B.. 2001. Identification and characterization of a lysosomal transporter for small neutral amino acids. Proc. Natl. Acad. Sci. USA. 98:7206–7211. 10.1073/pnas.121183498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauer, D.B., Zabel B.A., Pedraza I.F., O’Hara C.M., Steigerwalt A.G., and Brenner D.J.. 1995. Genetic and biochemical characterization of Citrobacter rodentium sp. nov. J. Clin. Microbiol. 33:2064–2068. 10.1128/JCM.33.8.2064-2068.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz, J.E., Veazey R.S., Kuroda M.J., Levy D.B., Seth A., Mansfield K.G., Nickerson C.E., Lifton M.A., Alvarez X., Lackner A.A., et al. 2001. Simian immunodeficiency virus (SIV)-specific cytotoxic T lymphocytes in gastrointestinal tissues of chronically SIV-infected rhesus monkeys. Blood. 98:3757–3761. 10.1182/blood.V98.13.3757 [DOI] [PubMed] [Google Scholar]
- Sevilla, N., Kunz S., Holz A., Lewicki H., Homann D., Yamada H., Campbell K.P., de La Torre J.C., and Oldstone M.B.. 2000. Immunosuppression and resultant viral persistence by specific viral targeting of dendritic cells. J. Exp. Med. 192:1249–1260. 10.1084/jem.192.9.1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shacklett, B.L., Cox C.A., Sandberg J.K., Stollman N.H., Jacobson M.A., and Nixon D.F.. 2003. Trafficking of human immunodeficiency virus type 1-specific CD8+ T cells to gut-associated lymphoid tissue during chronic infection. J. Virol. 77:5621–5631. 10.1128/JVI.77.10.5621-5631.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahangian, A., Chow E.K., Tian X., Kang J.R., Ghaffari A., Liu S.Y., Belperio J.A., Cheng G., and Deng J.C.. 2009. Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J. Clin. Invest. 119:1910–1920. 10.1172/JCI35412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon, C.E.. 1997. The mathematical theory of communication. 1963. MD Comput. 14:306–317. [PubMed] [Google Scholar]
- Shen, L., Weber C.R., Raleigh D.R., Yu D., and Turner J.R.. 2011. Tight junction pore and leak pathways: a dynamic duo. Annu. Rev. Physiol. 73:283–309. 10.1146/annurev-physiol-012110-142150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin, K., Straight S., and Margolis B.. 2005. PATJ regulates tight junction formation and polarity in mammalian epithelial cells. J. Cell Biol. 168:705–711. 10.1083/jcb.200408064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soler, A.P., Miller R.D., Laughlin K.V., Carp N.Z., Klurfeld D.M., and Mullin J.M.. 1999. Increased tight junctional permeability is associated with the development of colon cancer. Carcinogenesis. 20:1425–1431. 10.1093/carcin/20.8.1425 [DOI] [PubMed] [Google Scholar]
- Stanley, E.R., Chen D.M., and Lin H.S.. 1978. Induction of macrophage production and proliferation by a purified colony stimulating factor. Nature. 274:168–170. 10.1038/274168a0 [DOI] [PubMed] [Google Scholar]
- Stelekati, E., and Wherry E.J.. 2012. Chronic bystander infections and immunity to unrelated antigens. Cell Host Microbe. 12:458–469. 10.1016/j.chom.2012.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturgeon, C., Lan J., and Fasano A.. 2017. Zonulin transgenic mice show altered gut permeability and increased morbidity/mortality in the DSS colitis model. Ann. N. Y. Acad. Sci. 1397:130–142. 10.1111/nyas.13343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su, L., Nalle S.C., Shen L., Turner E.S., Singh G., Breskin L.A., Khramtsova E.A., Khramtsova G., Tsai P.Y., Fu Y.X., et al. 2013. TNFR2 activates MLCK-dependent tight junction dysregulation to cause apoptosis-mediated barrier loss and experimental colitis. Gastroenterology. 145:407–415. 10.1053/j.gastro.2013.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian, A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., et al. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 102:15545–15550. 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumagin, R., Robin A.Z., Nusrat A., and Parkos C.A.. 2014. Transmigrated neutrophils in the intestinal lumen engage ICAM-1 to regulate the epithelial barrier and neutrophil recruitment. Mucosal Immunol. 7:905–915. 10.1038/mi.2013.106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, L., Miyoshi H., Origanti S., Nice T.J., Barger A.C., Manieri N.A., Fogel L.A., French A.R., Piwnica-Worms D., Piwnica-Worms H., et al. 2015. Type I interferons link viral infection to enhanced epithelial turnover and repair. Cell Host Microbe. 17:85–97. 10.1016/j.chom.2014.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundström, G.M., Wahlin A., Nordin-Andersson I., and Suhr O.B.. 1998. Intestinal permeability in patients with acute myeloid leukemia. Eur. J. Haematol. 61:250–254. 10.1111/j.1600-0609.1998.tb01710.x [DOI] [PubMed] [Google Scholar]
- Swiecki, M., and Colonna M.. 2011. Type I interferons: diversity of sources, production pathways and effects on immune responses. Curr. Opin. Virol. 1:463–475. 10.1016/j.coviro.2011.10.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanca, A., Manghina V., Fraumene C., Palomba A., Abbondio M., Deligios M., Silverman M., and Uzzau S.. 2017. Metaproteogenomics Reveals Taxonomic and Functional Changes between Cecal and Fecal Microbiota in Mouse. Front. Microbiol. 8:391. 10.3389/fmicb.2017.00391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teijaro, J.R., Ng C., Lee A.M., Sullivan B.M., Sheehan K.C., Welch M., Schreiber R.D., de la Torre J.C., and Oldstone M.B.. 2013. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science. 340:207–211. 10.1126/science.1235214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaiss, C.A., Levy M., Grosheva I., Zheng D., Soffer E., Blacher E., Braverman S., Tengeler A.C., Barak O., Elazar M., et al. 2018. Hyperglycemia drives intestinal barrier dysfunction and risk for enteric infection. Science. 359:1376–1383. 10.1126/science.aar3318 [DOI] [PubMed] [Google Scholar]
- Tibble, J.A., Sigthorsson G., Bridger S., Fagerhol M.K., and Bjarnason I.. 2000. Surrogate markers of intestinal inflammation are predictive of relapse in patients with inflammatory bowel disease. Gastroenterology. 119:15–22. 10.1053/gast.2000.8523 [DOI] [PubMed] [Google Scholar]
- Tincati, C., Douek D.C., and Marchetti G.. 2016. Gut barrier structure, mucosal immunity and intestinal microbiota in the pathogenesis and treatment of HIV infection. AIDS Res. Ther. 13:19. 10.1186/s12981-016-0103-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinoco, R., Alcalde V., Yang Y., Sauer K., and Zuniga E.I.. 2009. Cell-intrinsic transforming growth factor-beta signaling mediates virus-specific CD8+ T cell deletion and viral persistence in vivo. Immunity. 31:145–157. 10.1016/j.immuni.2009.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trzpis, M., McLaughlin P.M., de Leij L.M., and Harmsen M.C.. 2007. Epithelial cell adhesion molecule: more than a carcinoma marker and adhesion molecule. Am. J. Pathol. 171:386–395. 10.2353/ajpath.2007.070152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, P.Y., Zhang B., He W.Q., Zha J.M., Odenwald M.A., Singh G., Tamura A., Shen L., Sailer A., Yeruva S., et al. 2017. IL-22 Upregulates Epithelial Claudin-2 to Drive Diarrhea and Enteric Pathogen Clearance. Cell Host Microbe. 21:671–681.e4. 10.1016/j.chom.2017.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner, J.R.. 2009. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 9:799–809. 10.1038/nri2653 [DOI] [PubMed] [Google Scholar]
- Utzschneider, D.T., Charmoy M., Chennupati V., Pousse L., Ferreira D.P., Calderon-Copete S., Danilo M., Alfei F., Hofmann M., Wieland D., et al. 2016. T Cell Factor 1-Expressing Memory-like CD8(+) T Cells Sustain the Immune Response to Chronic Viral Infections. Immunity. 45:415–427. 10.1016/j.immuni.2016.07.021 [DOI] [PubMed] [Google Scholar]
- Veazey, R.S., DeMaria M., Chalifoux L.V., Shvetz D.E., Pauley D.R., Knight H.L., Rosenzweig M., Johnson R.P., Desrosiers R.C., and Lackner A.A.. 1998. Gastrointestinal tract as a major site of CD4+ T cell depletion and viral replication in SIV infection. Science. 280:427–431. 10.1126/science.280.5362.427 [DOI] [PubMed] [Google Scholar]
- Venkatesh, M., Mukherjee S., Wang H., Li H., Sun K., Benechet A.P., Qiu Z., Maher L., Redinbo M.R., Phillips R.S., et al. 2014. Symbiotic bacterial metabolites regulate gastrointestinal barrier function via the xenobiotic sensor PXR and Toll-like receptor 4. Immunity. 41:296–310. 10.1016/j.immuni.2014.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virgin, H.W., Wherry E.J., and Ahmed R.. 2009. Redefining chronic viral infection. Cell. 138:30–50. 10.1016/j.cell.2009.06.036 [DOI] [PubMed] [Google Scholar]
- Volynets, V., Reichold A., Bárdos G., Rings A., Bleich A., and Bischoff S.C.. 2016. Assessment of the Intestinal Barrier with Five Different Permeability Tests in Healthy C57BL/6J and BALB/cJ Mice. Dig. Dis. Sci. 61:737–746. 10.1007/s10620-015-3935-y [DOI] [PubMed] [Google Scholar]
- Warner, N.L., Jokinen J.D., Beier J.I., Sokoloski K.J., and Lukashevich I.S.. 2018. Mammarenaviral Infection Is Dependent on Directional Exposure to and Release from Polarized Intestinal Epithelia. Viruses. 10:75. 10.3390/v10020075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber, C.R., Raleigh D.R., Su L., Shen L., Sullivan E.A., Wang Y., and Turner J.R.. 2010. Epithelial myosin light chain kinase activation induces mucosal interleukin-13 expression to alter tight junction ion selectivity. J. Biol. Chem. 285:12037–12046. 10.1074/jbc.M109.064808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler, T.J., and Hinkle P.C.. 1985. The glucose transporter of mammalian cells. Annu. Rev. Physiol. 47:503–517. 10.1146/annurev.ph.47.030185.002443 [DOI] [PubMed] [Google Scholar]
- Wherry, E.J., Blattman J.N., Murali-Krishna K., van der Most R., and Ahmed R.. 2003. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J. Virol. 77:4911–4927. 10.1128/JVI.77.8.4911-4927.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry, E.J., Ha S.J., Kaech S.M., Haining W.N., Sarkar S., Kalia V., Subramaniam S., Blattman J.N., Barber D.L., and Ahmed R.. 2007. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 27:670–684. 10.1016/j.immuni.2007.09.006 [DOI] [PubMed] [Google Scholar]
- Wilairatana, P., Meddings J.B., Ho M., Vannaphan S., and Looareesuwan S.. 1997. Increased gastrointestinal permeability in patients with Plasmodium falciparum malaria. Clin. Infect. Dis. 24:430–435. 10.1093/clinids/24.3.430 [DOI] [PubMed] [Google Scholar]
- Wilson, E.B., Yamada D.H., Elsaesser H., Herskovitz J., Deng J., Cheng G., Aronow B.J., Karp C.L., and Brooks D.G.. 2013. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science. 340:202–207. 10.1126/science.1235208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, T., Ji Y., Moseman E.A., Xu H.C., Manglani M., Kirby M., Anderson S.M., Handon R., Kenyon E., Elkahloun A., et al. 2016. The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci. Immunol. 1. eaai8593. 10.1126/sciimmunol.aai8593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt, J., Vogelsang H., Hübl W., Waldhöer T., and Lochs H.. 1993. Intestinal permeability and the prediction of relapse in Crohn’s disease. Lancet. 341:1437–1439. 10.1016/0140-6736(93)90882-H [DOI] [PubMed] [Google Scholar]
- Yu, A.S., Enck A.H., Lencer W.I., and Schneeberger E.E.. 2003. Claudin-8 expression in Madin-Darby canine kidney cells augments the paracellular barrier to cation permeation. J. Biol. Chem. 278:17350–17359. 10.1074/jbc.M213286200 [DOI] [PubMed] [Google Scholar]
- Zajac, A.J., Blattman J.N., Murali-Krishna K., Sourdive D.J., Suresh M., Altman J.D., and Ahmed R.. 1998. Viral immune evasion due to persistence of activated T cells without effector function. J. Exp. Med. 188:2205–2213. 10.1084/jem.188.12.2205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zander, R., Schauder D., Xin G., Nguyen C., Wu X., Zajac A., and Cui W.. 2019. CD4. Immunity. 51:1028–1042.e4. 10.1016/j.immuni.2019.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuniga, E.I., Macal M., Lewis G.M., and Harker J.A.. 2015. Innate and Adaptive Immune Regulation During Chronic Viral Infections. Annu. Rev. Virol. 2:573–597. 10.1146/annurev-virology-100114-055226 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
lists DEGs in IECs between ARM-infected and uninfected mice on day 9 p.i. (see also Fig. 2).
shows DEGs in IECs between Cl13-infected and uninfected mice on day 9 p.i. (see also Fig. 2).
outlines tight junction–related genes in IECs from Cl13- or ARM-infected and uninfected mice on day 9 p.i. (see also Fig. 2).
shows pathways enriched in IEC transcriptomes from Cl13- versus ARM-infected mice on day 9 p.i. (see also Fig. 2).
lists differentially expressed genes in IECs between Cl13- versus ARM-infected mice on day 9 p.i. (see also Fig. 2).
displays pathways enriched in IEC transcriptomes from isotype-treated versus anti-IFNAR-1–treated Cl13-infected mice on day 9 p.i. (see also Fig. 3).
lists DEGs in IECs between isotype-treated versus anti-IFNAR-1–treated Cl13-infected mice on day 9 p.i. (see also Fig. 3).
shows differentially enriched pathways regulated by type I IFNs from IECs obtained from Cl13- versus ARM-infected mice on day 9 p.i. (see also Fig. 3).
displays DEGs regulated by type I IFNs among DEGs by IECs from Cl13- versus ARM-infected mice on day 9 p.i. (see also Fig. 3).
shows songbird multinomial regression ranks by comparing microbiomes from Cl13-infected mice treated with isotype controls (IgG1) versus IFNAR-1 blocking Abs (anti-IFNAR-1; see also Fig. 5).
Data Availability Statement
All RNA-sequencing data have been deposited to the Gene Expression Omnibus (accession no. GSE155120) and the Sequence Read Archive (accession no. PRJNA648708). All 16S rRNA gene amplicon and shotgun metagenomics sequencing data are publicly available on the European Nucleotide Archive (https://ebi.ac.uk/ena; accession no. ERP123227) and Qiita (https://qiita.ucsd.edu, study ID 11043). On Qiita, processed 16S .fastq and metagenomics .fastq files can be found under preparation ID 31428 and preparation ID 92095, respectively. Detailed metadata for all samples can be found under the “Sample Information” tab.