

ABSTRACT
Long intergenic non-coding RNAs (lincRNAs) are transcripts longer than 200 nucleotides that are transcribed from non-coding loci yet undergo biosynthesis similar to coding mRNAs. The disproportional number of lincRNAs expressed in testes suggests that lincRNAs are important during gametogenesis, but experimental evidence has implicated very few lincRNAs in this process. We took advantage of the relatively limited number of lincRNAs in the genome of the nematode Caenorhabditis elegans to systematically analyse the functions of lincRNAs during meiosis. We deleted six lincRNA genes that are highly and dynamically expressed in the C. elegans gonad and tested the effects on central meiotic processes. Surprisingly, whereas the lincRNA deletions did not strongly impact fertility, germline apoptosis, crossovers, or synapsis, linc-4 was required for somatic growth. Slower growth was observed in linc-4-deletion mutants and in worms depleted of linc-4 using RNAi, indicating that linc-4 transcripts are required for this post-embryonic process. Unexpectedly, analysis of worms depleted of linc-4 in soma versus germline showed that the somatic role stems from linc-4 expression in germline cells. This unique feature suggests that some lincRNAs, like some small non-coding RNAs, are required for germ-soma interactions.
KEYWORDS: Meiosis, lincRNA, non-coding RNA, germline
Background
Long intergenic non-coding RNAs (lincRNAs) are a group of untranslated transcripts, longer than 200 nucleotides, that are transcribed from loci that do not code for proteins. Their biogenesis is similar to that of coding mRNAs, and they are often capped, polyadenylated, and spliced (reviewed in [1–9]). lincRNAs are generally expressed at lower levels than mRNAs, and the expression of lincRNA is usually more restricted to specific tissues and developmental stages [10]. In line with this, several lincRNAs have critical roles during development (reviewed in [6,9–11]). LincRNAs are highly expressed during gametogenesis [12–14], and there is a disproportionally larger number of expressed lincRNAs compared to mRNAs in mammalian testes than in other tissues [15–17]. A recent report estimated that testes have higher amounts of lincRNA than any other vertebrate tissue [18]. Nevertheless, only a few lincRNAs have confirmed roles in gametogenesis in these organisms [19], whereas in Drosophila knockout of 31% of long non-coding RNAs resulted in reduced fertility [19]. A recent careful analysis in Danio rerio found no overt function in fertility for the tested lincRNAs [20]. The authors reasoned that lincRNAs act redundantly and therefore removal of only one lincRNA does not lead to dramatic effects on fertility. If indeed lincRNAs have redundant roles in gametogenesis, this will considerably complicate their study in vertebrates which have relatively high number of lincRNA genes [21]. The number of long non-coding RNA in human is estimated to be in the range of tens of thousands, with about half being intergenic (reviewed in [22]). Therefore, a simple model organism with few lincRNAs is necessary to determine functions of individual lincRNAs in gametogenesis.
The heart of gametogenesis lies in meiosis, the division that reduces chromosome number by half to create haploid gametes. During meiosis, homologous chromosomes pair and undergo crossover recombination events. Meiotic crossovers facilitate the segregation of homologous chromosomes and increase genetic diversity (reviewed in [23–33]). Caenorhabditis elegans offers many advantages in studying the roles of lincRNAs in meiosis. Cell progression throughout meiosis can be followed in the adult hermaphrodite gonad arm since the nuclei are arranged in a spatiotemporal manner from the proliferative region through prophase I to the mature diakinetic oocyte. The C. elegans genome encodes a similar number of coding genes to humans but only hundreds of poly-exonic and a few thousand mono-exonic lincRNAs [34,35]. The lower number of lincRNAs allows more thorough and focused screens to study their meiotic roles. In addition, the expression at each gonadal stage of many of lincRNAs has been characterized [36–38].
Here we report a systematic analysis of the meiotic roles of lincRNAs with high and dynamic expression in the C. elegans gonad. Deletion of the tested lincRNAs had no major effects on fertility or central meiotic events including oogenesis progression, synapsis, chiasmata, or apoptosis. However, germline expression of linc-4 was required for post-embryonic recovery after the release from induced growth arrest. We propose that lincRNAs with high expression in the gonad have redundant functions in fertility but that germline expression of some is required for somatic post-fertilization processes.
Results
Analysis of lincRNAs expression in the C. elegans gonad
To study lincRNA roles in fertility and oogenesis we focused our work on the lincRNAs with relatively high and dynamic expression in the hermaphrodite gonad. Using the oogonial transcriptomic map we published previously [36], we analysed the expression of the 170 lincRNAs identified by the Nam and Bartel [35]. In our previous publication, we used laser capture microdissection to cut the gonad into 10 equally sized pieces, and quantified their transcriptome using CEL-seq (see methods and [36]). Using these databases, we found that among the 170 lincRNAs, two lincRNAs stand out based on their level of expression, linc-168 and linc-7 (Fig. 1). Both are among the top 500 expressed genes in the gonad [36]. The maximum RNA levels of these transcripts in the gonad are more than double than many genes required for meiosis including syp-1, rad-51, and htp-3 (Supplementary Fig. S1). The levels of linc-168 and linc-7 are also considerably higher than levels of other lincRNA genes, and the maximum expression of linc-6, the next highly expressed lincRNA, is less than one-sixth that of linc-7 (Fig. 1). The gonad transcriptome map revealed that about 92% of the genes transcribed from the X chromosome are expressed at low levels in the first half of the gonad corresponding to proliferative, leptotene/zygotene, and pachytene regions (sections 1–5 in Fig. 1), but X chromosome genes are expressed in the late pachytene, diplotene, and diakinesis regions (sections 6–10 in Fig. 1) [36,39,40]. We were therefore surprised to find that three lincRNAs, linc-7, linc-6, and linc-20, which are transcribed from the X chromosome, have higher levels of expression in the first half of the gonad than the second (Fig. 1), suggesting specific roles during early oogenesis. We therefore included these three lincRNAs in our analysis. linc-4 was also of interest, as it is highly expressed in the gonad, close to that of linc-6, but in contrast to linc-6, the expression of linc-4 drops during early diakinesis and rises again during late diakinesis (Fig. 1). linc-9 was included since it is highly expressed, has an expression profile similar to that of linc-4, and is also highly paralogous to linc-20 (Supplementary Fig. S2). As was reported before [35] the sequences of these six lincRNAs are poorly conserved (BLAST bit-score with C. briggsae range 33–45).
Figure 1.
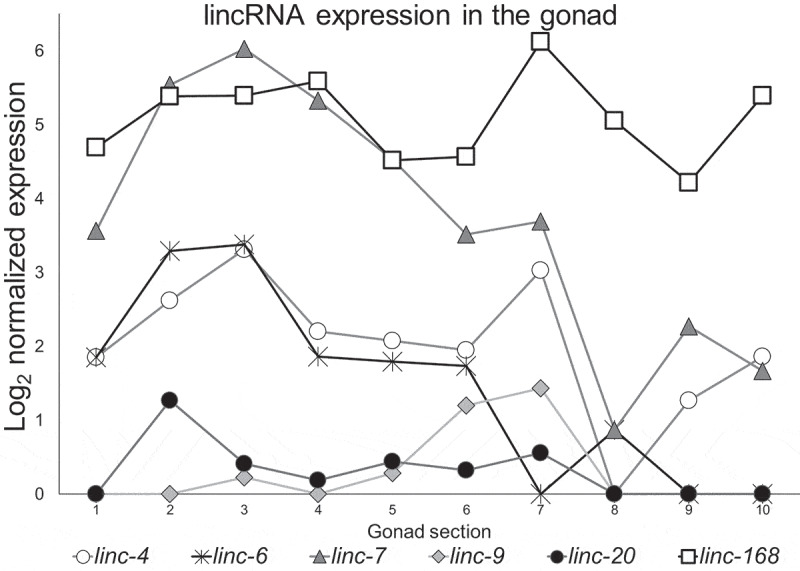
Expression patterns of highly expressed lincRNAs. Log2 of normalized expression values of the six lincRNAs with the high levels of expression along the gonad from proliferative (1) to mature oocyte (10) stage. Adapted from.[36]
We created full genomic deletions in the six lincRNA genes, linc-168, linc-7, linc-6, linc-20, linc-4, and linc-9 in C. elegans (Supplementary Fig. S3, Supplementary Table S1) to ensure loss-of-function, as was previously done [41,42]. After isolating homozygous strains, we outcrossed the strains at least five times, and verified that all the bases of the genes were deleted in both alleles using genotyping of the progeny of isolated L4 larvie and Sanger sequencing (Supplementary Fig. S4-S5). All further experiments were conducted using the outcrossed strains.
lincRNAs with high levels of gonadal expression are not required for fertility
To test the effects of lincRNA genes on fertility we quantified the brood sizes of the six lincRNA mutant strains. Wild-type worms laid an average of 304 ± 47 embryos, and most of the lincRNA deletion strains laid comparable numbers of embryos (Fig. 2A). Deletion of linc-4 and linc-6 led to a minor, yet significant decrease in brood size compared with wild-type worms (282 ± 50, 283 ± 37, vs 304 ± 47, respectively, p value < 0.01, by the two-tailed Mann–Whitney test). These results indicate that the highly expressed lincRNA genes we tested are not critical for regulation of brood size.
Figure 2.
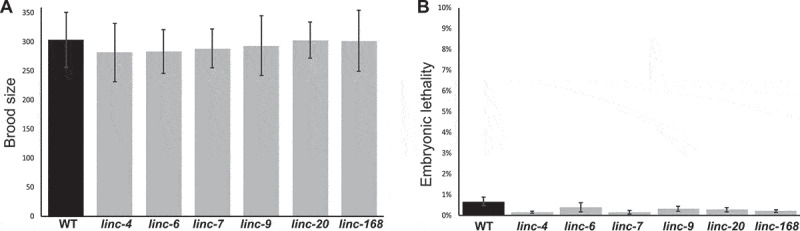
Deletion of highly expressed lincRNA genes does not lead to loss of fertility or embryonic lethality. A. Average progeny brood sizes for indicated lines. For each strain n > 18. B. Percentages of embryonic lethality per worm of wild type and lincRNA deletion worms of the indicated lines. For each strain n > 5200 from > 18 worms
Egg laying occurs over several days once worms reach adulthood, and mutant strains may lay eggs with different dynamics than the wild-type worms. For example, some mutants with longer lifespans than typical lay eggs with slower dynamics than wild-type worms [43]. The lincRNA-deletions strains had egg-laying dynamics similar to wild type. For example, most of the progeny were laid on the second day by wild-type worms (190 ± 33) and by the lincRNA-mutant strains (range: 191–208). These data indicate that lack of expression of any of the individual lincRNAs tested did not change the number of eggs laid or the dynamics of egg laying.
Defects in oogenesis can lead to embryonic lethality. We therefore compared the percentage of embryos that did not hatch between the wild-type worms and the lincRNA-deleted lines. We found that in wildtype as well as in the mutant worms levels of embryonic lethality were low (<1%, Fig. 2B), suggesting that in the mutant lines there are no major meiotic or embryonic developmental failures. Taken together these results indicate that lincRNAs with high or dynamic expression in the C. elegans gonad are not required for fertility or embryonic survival.
Deletion of highly expressed lincRNAs does not change oogenesis progression, apoptosis rates, or the numbers of bivalent chromosomes or chiasma
To gain insight into the reason for the reduction in the brood size of linc-4 and linc-6 mutants and find whether these and other mutants have meiotic defects, we analysed the effects of the lincRNA mutations on several meiotic processes. The order of nuclei in the adult C. elegans gonad allows examination of oogenesis progression. We tested whether lincRNAs are required for normal staging and correct formation of mature oocytes by DAPI staining of dissected gonads. In wild-type gonads the distal region contains proliferative nuclei undergoing mitosis, and then proceed to meiosis at the leptotene/zygotene region where homologous chromosomes pair. The homologs undergo close association known as synapsis in the pachytene region and in diakinesis six individual bivalent chromosomes are observed. The gonads of the lincRNA deletion strains did not differ from those of the wild-type gonads indicating that lack of expression of the six tested lincRNAs does not change oogenesis progression (data not shown).
During the transition from diplotene to diakinesis the chromosomes remodel and individual bivalent chromosomes can be observed by DAPI staining. These bivalent chromosomes are visible as rods or cruciforms around the single chiasma, the cytological outcome of the single crossover that occurs in each homolog pair. Lack of at least one crossover prevents the stable connection between the homologs at diakinesis and more than six DAPI stained bodies are observed as described previously [44–53]. Conversely, when double-strand break repair pathways are perturbed, chromosome clusters form, and less than six bodies are detected [54]. We therefore quantified the number of DAPI-stained bodies in mature oocytes to evaluate crossover. In wild-type gonads, we almost always detected six DAPI-stained bodies (average 6.0 ± 0.3). In all the lincRNA mutant strains we found similar numbers of DAPI-stained bodies (range: 5.8–6.0, Fig. 3A). These results indicate that normal homologous recombination occurs in the absence of the lincRNAs evaluated.
Figure 3.
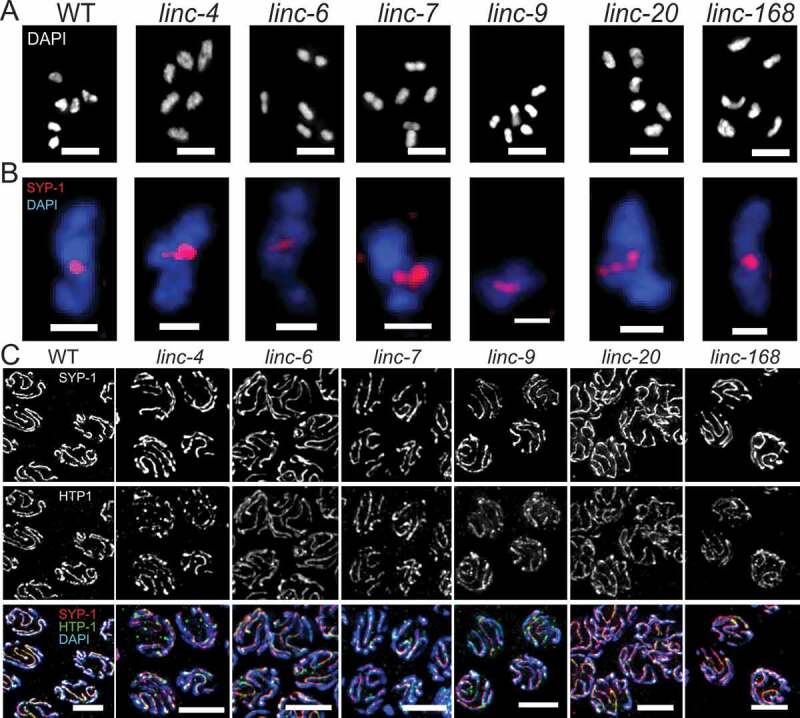
Deletion of the lincRNA genes does not alter, chiasma number and formation or disassembly of the synaptonemal complex. A. Representative images of DAPI-stained mature oocytes of wild-type and lincRNA-deletion worms. Scale bars, 4 μM. B. Representative images of single bivalent chromosomes of the indicated genotypes stained with SYP-1 (red) and DAPI (blue). Scale bars, 1 μM. C. Representative mid-pachytene nuclei of wild-type and lincRNA-deletion worms stained with SYP-1 (red), HTP (green), and DAPI (blue). Scale bars, 4 μM
In many eukaryote germline cells, apoptosis is induced upon failure to correctly execute critical prophase I processes (reviewed in [55,56]). In C. elegans hermaphrodite gonads there is a physiological level of apoptosis rate, which is exacerbated by failure of synapsis or failure to repair DNA double-strand breaks [57–59]. Apoptosis levels are also influenced by other pathways [60–62]. We evaluated germline apoptosis by staining worms with acridine orange as previously described [57,63]. In wild-type worms, we observed an average of 3 ± 2 apoptotic nuclei in the band region (Fig. 4), which is in line with a previous report [64]. The number of apoptotic cells in the lincRNA-deleted worms did not differ significantly from the number observed in wild-type worms (Fig. 4).
Figure 4.
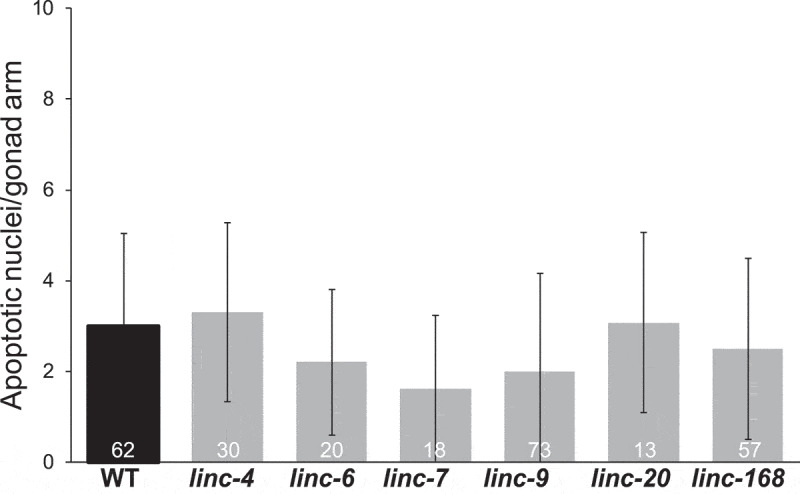
Numbers of apoptotic nuclei are similar in wild-type and lincRNA-deleted worms. Average number of apoptotic nuclei per gonad bend as detected by acridine orange staining of wild-type and lincRNA-deletion strains. The n values are listed on each bar
Over 98% of bivalent chromosomes in wild-type C. elegans gonads undergo a single inter-homolog crossover [65–70], which results in the formation of a single chiasma during late diakinesis. To test whether deletions of any of the six lincRNAs affected the crossover interference that maintains this ratio, we quantified chiasmata numbers. In wild-type worms and in the lincRNA mutants, we detected only bivalent chromosomes with cruciform- and rod-shaped bodies with one chiasma (Fig. 3B). Taken together these results suggest that the lincRNAs evaluated here do not influence crossover number or formation of bivalent chromosomes.
The synaptonemal complex is properly formed and disassembled in lincRNA-deletion strains
Inter-homolog crossovers mature within the synaptonemal complex, a tripartite proteinaceous structure that binds the homologs during pachytene. Assembly of the synaptonemal complex starts with loading of an axial elements on each of the axes of the homologs, which are then linked by the central element (reviewed in [32,71]). We tested whether the synaptonemal complex is properly formed and disassembled in our lincRNA mutant strains by co-staining for HTP-1, an axial element, and SYP-1, a central element of the synaptonemal complex. In wild-type worms, SYP-1 is associated with the chromosomes as foci in leptotene/zygotene, and in pachytene it is fully colocalized with HTP-1 in elongated tracks between DAPI-stained axes (reviewed in [72]). We detected this pattern in gonads of wild-type and all mutant strains. To test if the loss of lincRNA expression led to partial synapsis, we focused on mid-pachytene nuclei and followed HTP-1 tracks. In all nuclei we examined of both wild-type and lincRNA-mutant worms there was no HTP-1 staining that was not accompanied by SYP-1 staining (Fig. 3C).
Disassembly of the synaptonemal complex follows a non-symmetrical pattern. At the end of pachytene, proteins of the central element are removed from regions of the chromosomes that will become the long arms but remain in the regions of the bivalent axes that will be remodelled into the short arms; central element proteins, including SYP-1, are not present in mature oocytes [73–75]. In both wild-type and the lincRNA-mutant strains, SYP-1 staining was restricted to the short arms during diplotene and early diakinesis (Fig. 3B). The last oocytes to contain SYP-1 staining were mostly those at positions −3 or −4 from the spermatheca, and SYP-1 was not observed in the mature oocytes of any of the tested strains. Taken together, these results suggest that lincRNAs have no role in SC formation and disassembly.
Analysis of the levels of endogenous siRNAs mapping antisense to the lincRNAs
The data presented above indicated that the lincRNAs we tested have no major role in fertility, yet they may still have cellular roles. Previous analysis indicated that many lincRNAs are mapped antisense to clusters of endogenous siRNAs [35]. This publication did not provide specific information regarding the lincRNAs we examine here. Therefore, to find if these lincRNA sites could also lead to high levels of siRNAs, we used our previously published small RNA sequencing databases [76] to evaluate the contribution of lincRNA genes to endogenous siRNAs. We quantified the small RNA reads aligned in antisense orientation to all genes (Table S2), and found that two of the six lincRNAs discussed here stand out: linc-6 (average of 410 RPM) and linc-7 (average of 942 RPM). The rest of the lincRNAs have much lower small RNA values (range 0–15, Table S2 and Fig. S6). Taken together, this observation suggests that lincRNAs which map to a wide range of values of endogenous siRNAs have no overt fertility roles.
The lincRNA linc-4 is uniquely expressed as the first gene in an operon and is required for recovery following growth arrest
linc-4 is the first gene of the CEOP2754 operon, which also includes the protein-coding F13H8.2 gene. To determine whether this feature is exceptional among lincRNAs, we manually determined which of the lincRNAs identified by Nam and Bartel [35] are annotated as being transcribed from within an operon. We found that only five lincRNAs, linc-4, linc-30, linc-81, linc-90, and linc-126, just 2.9% of the lincRNAs in C. elegans, are transcribed from operons. This is a significantly lower percentage than coding genes, of which at least 15% are expressed from operons [77] (p< 10−7 by hypergeometric distribution analysis). Moreover, linc-4 is the only lincRNA that is transcribed from the first gene in an operon. Reinke et al. reported that almost all operon genes are expressed during oogenesis [78], and thus linc-4 expression from an operon suggests a germline role.
Previous work showed that operons are enriched with genes involved in growth [79]. Many genes on operons are required for viability, and several viable mutations in operon genes lead to reduced growth rate following L1 arrest [79]. To test if the loss of linc-4 expression impairs the recovery from L1 arrest, we isolated embryos, allowed them to enter arrest in a nutrient-free medium and measured their sizes following the release from the arrest. Worms with the linc-4 mutation grew more slowly than wild-type worms and extent of growth reduction depended on the length of arrest (Fig. 5A-C). These results suggest that linc-4 is involved in recovery from growth arrest.
Figure 5.
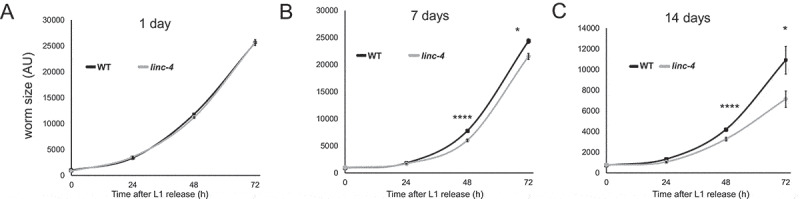
Deletion oflinc-4leads to slower growth following starvation. Size (in AU) of wild-type and linc-4-mutant worms were measured over 72 hours after the release from A) 1 day, B) 7 days, and C) 14 days of L1 starvation arrest. ****< 0.00001, *< 0.05 p value by the two-tailed Mann-Whitney test. For each data point n > 21
It is possible that the genomic deletion reduces the transcription of F13H8.2, the only other gene in the CEOP2754 operon, and thus leads to the growth delays. We therefore quantified the level of F13H8.2 mRNA by RT-qPCR in wild-type and linc-4 mutant worms. F13H8.2 mRNA levels were not significantly different in the two strains (Supplementary Fig. S7). The smaller size we measured in linc-4, could either be due to smaller cell size, or due to slower development. To find if the later contributes to the smaller size, we evaluated the developmental stage of the worms following the release from growth arrest. We found that after seven days of arrest, linc-4 worm populations included more worms with delayed development compared to wild type. For example, after 48 hours 93% of the wild-type worms reached the L4 stage vs 70% of linc-4 worms (n ≥ 122, Fisher’s exact test, p< 0.00001). Similarly, after 72 hours 97% of wild-type worms were gravid adults, compared to 77% of linc-4 (n ≥ 34, Fisher’s exact test, p< 0.01). This observation suggests that the slower growth of linc-4 worms is, at least in part, due to slower development.
It remained possible that the effect on growth we observed is due to the genomic deletion and not the lack of linc-4 transcripts as has been reported when other lincRNAs were deleted [80]. To verify that linc-4 is indeed required for growth, we depleted linc-4 using the RNA interference (RNAi) method and measured worm sizes following release from L1 starvation arrest. We found a considerable growth delay in worms depleted of linc-4 compared with control worms (Fig. 6A). Taken together these results indicate that linc-4 is important for growth following L1 arrest.
Figure 6.
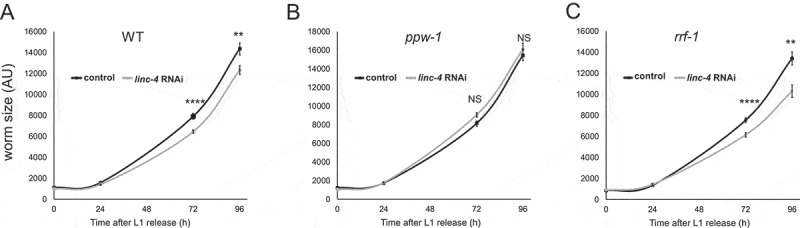
Germ-linelinc-4transcripts are required for normal growth following starvation. Size (in AU) of A) wild-type, B) ppw-1-mutant, or C) rrf-1-mutant backgrounds were measured over 96 hours after the release from 4 days of L1 starvation arrest.****< 0.00001, **< 0.01 p value by the two-tailed Mann-Whitney test. NS indicates not significant. For each data point n > 13
Depletion of linc-4 in the germline, but not in the soma, leads to reduced growth rate following arrest
The post-embryonic growth effect we detected in worms lacking linc-4 suggested that the effect stems from somatic expression of linc-4 rather than from expression in the gonad. To test this hypothesis, we utilized a strain with a mutation in the ppw-1 gene. This mutation blocks germline RNAi but does not prevent RNAi activity in the soma [81]. Surprisingly, there was not a significant difference in the growth rates of linc-4 depleted and control ppw-1-mutant worms (Fig. 6B). To test if the effect of linc-4 on growth rate results from its expression in the germline, we used a strain with the rrf-1 mutation. This mutation dramatically reduces RNAi activity in most somatic tissues but not in the germline [82] (see discussion and [83]). In this strain, linc-4 RNAi depletion led to significantly slower growth after L1 arrest compared to growth of the control rrf-1 worms (Fig. 6C). Taken together these results suggest that the expression of linc-4 in the germline plays a role in the normal recovery from L1 growth arrest.
Deletion of linc-4 leads to changes in gene expression
How could linc-4 affect worms’ growth through the germline? To gain more insight into the molecular mechanism of linc-4, we performed RNA-seq analyses and compared genes’ expression between WT and linc-4 worms. We found 1059 genes that were significantly differentially expressed (DEGs) in linc-4 compared to WT, 364 were downregulated and 695 were upregulated (see supplemental data). Gene Ontology analysis indicated that 19 terms were significantly enriched (Bonferroni-corrected p-value < 0.05) within the list of DEGs, some of which could be attributed to fertility (e.g., ‘reproduction’ – Bonferroni-corrected p-value – 1.04E-08, ‘gamete generation’ Bonferroni-corrected p-value – 1.41E-02, and ‘oogenesis’ Bonferroni-corrected p-value – 9.81E-07, Table S3). Interestingly, cuticle-related terms were also found to be enriched (e.g., ‘structural constituent of cuticle’ Bonferroni-corrected p-value – 4.83E-12, and ‘collagen trimer’ Bonferroni-corrected p-value – 1.45E-11, Table S3). The cuticle is critical for moulting and growth [84], and so changes in the expression of these genes can explain the effect on the recovery after L1 growth arrest we found in linc-4. Together these data suggest that linc-4 expression can affect normal gene expression connected to oogenesis, embryogenesis, and components of the cuticle.
Discussion
The study of the developmental and cellular roles of lincRNAs is challenging (reviewed in [10]). lincRNAs are not translated, and small indels that lead to gene disruption in protein-coding genes, are not predicted to lead to null alleles in lincRNA genes, and so complete deletions are needed when engineering loss-of-function mutants. Similarly, since lincRNAs are not translated, tagging the products of lincRNA genes is also harder. Moreover, it is difficult to study the functions of specific lincRNAs. One reason for this is the low evolutionary conservation that limits cross-species extrapolation of results. Furthermore, even when full genomic deletion of a lincRNA is achieved, there is often no gross phenotype [20,41]. Wei et al. assessed brood sizes (among other gross phenotypes) of many lincRNA mutants and found that very few had brood sizes that differed from wild-type worms [41] (see below).
In the current work, we analysed the effects of deletions of lincRNAs with high levels of germline expression. As done previously [41], we made sure our mutants were null in the lincRNA of interest by completely deleting the gene locus and outcrossing the strains. Instead of screening for gross phenotypes, we tested different aspects related to meiosis including synapsis, germ cell apoptosis, chiasma numbers, brood size, and embryo viability. For the six lincRNAs deleted, there were no major perturbations compared to wild-type worms.
The evolutionary advantage of producing these transcripts in the gonads, in some cases at very high levels, is unclear since elimination of most of them does not change fertility. It is possible that several lincRNAs perform the same function in fertility, and thus phenotypes will manifest only when two or more lincRNA genes are deleted. Testing this possibility without prior association between the lincRNAs examined would be challenging in C. elegans. It is also possible that these lincRNAs have roles that cannot be detected in laboratory conditions but are required at other environments or conditions as previously suggested [85]. The low level of evolutionary conservation of lincRNA sequence [2] raises another option. It is possible that these transcripts do not contribute to the survival of the organism but were newly introduced into the genome. This possibility is supported by the findings that many lincRNA genes were introduced into the genome by transposable elements [86]. It should be noted, however, that often times the structures and syntenic positions of lincRNAs are conserved even when the sequence is not [87,88].
Our results suggest that the level of expression of a lincRNA in the gonad is not a good predictor of a role in oogenesis. One caveat for that conclusion is that in this work we only analysed lincRNAs for which gonad expression has been quantified, namely the 170 lincRNAs identified by Nam and Bartel [35]. Approximately 130 multi-exonic and 3000 mono-exonic lincRNAs were recently identified by Akay et al. [34]. Although it is possible that some of the latter lincRNAs may have higher levels of expression than those analysed here, there are other indications that supports the lack of correlation between fertility roles and gonad expression. A recent report analysed phenotypes in 155 lincRNA deletion lines; only linc-10 and linc-155 mutant worms had significantly smaller brood sizes than wild-type worms, yet these lincRNAs were not in the top 30% of lincRNAs most highly expressed in young adult worms [41]. Moreover, in the previously published gonad transcriptomic map, linc-155 was not detected at all [36]. This observation, and our result indicating that linc-4 is highly expressed in the gonad but that the effect of its deletion can be observed only after the release from L1 growth arrest, raise another possibility. We propose that in some cases lincRNAs affect processes in different tissues or at a different developmental stage from those in which the transcript is expressed.
linc-4 is the third most highly expressed lincRNA in the gonad; however, its deletion resulted in no meiotic phenotype, except for minor reduction in brood size. Interestingly, the growth of the strain lacking linc-4 was considerably slower than that of the wild-type strain after L1 arrest. Effects of reductions in levels of the linc-4 RNA in rrf-1 and ppw-1 backgrounds suggest that the role of linc-4 in this post-embryonic process stems from the germline. One caveat is that a previous publication indicated that some somatic tissues are RNAi proficient in the rrf-1 strain [83]. Nevertheless, the lack of an effect of linc-4 depletion in the ppw-1 background indicates that the germline expression of linc-4 is important for post-embryonic growth. Consistent with this possibility, our transcriptomic data show that the expression of many genes associated with fertility and oogenesis, as well as with the cuticle is altered when linc-4 is deleted. These data further support the possibility that linc-4 acts in the germline to attenuate gene expression which later affects expression of collagen genes required for growth. It will be interesting in the future to find how linc-4 control gene expression in these tissues.
Our results suggest two non-mutually exclusive possibilities: 1) It is possible that factors created during oogenesis and early embryogenesis assist in growth after the arrest, or 2) linc-4 acts before, during, or after the release in the primordial germline cells of the L1 larva to allow rapid recovery after nutrition is provided. Finding the specific cellular localization of linc-4 in embryonic and larval stages is technically challenging since the mature transcript is only 216 nucleotides long, thus limiting the use of smFISH [89].
Whatever the molecular mechanism of linc-4 action, our work demonstrates that to study the roles of lincRNAs in development requires a holistic and broad view and different strategies than those established for functional analysis of coding genes. In the last few years, it has become apparent that small non-coding RNAs take part in the communications between soma and germ cells (reviewed in [90]). At least in C. elegans, small non-coding RNAs act to transmit silencing and epigenetic marks (reviewed in [91]). Our study suggests that lincRNAs may also be involved in soma and germ communication.
Materials and methods
Strains and alleles
All strains were cultured under standard conditions at 20°C [92]. The N2 Bristol strain was utilized as the wild-type background. Worms were grown on NGM plates with Escherichia coli OP50 [92]. Unless otherwise stated, all experiments were conducted using adult hermaphrodites 20–24 h post-L4 stage. The following strains with mutations and chromosomal rearrangements were used: NL3511: ppw-1(pk1425) I and RB798: rrf-1(ok589) I. Strains engineered in this work are listed in Supplementary Table S1.
Generation of engineered strains by CRISPR-Cas9 genome editing
To generate the linc-6(huj4) deletion, we used the procedure described by Friedland et al. [93] with the modifications described previously [42]. The sequences of the sgRNAs are listed in Supplementary Table S1. The complete deletion of the huj4 allele was verified by sequencing of the linc-6 loci. Homozygous strains were outcrossed six times to establish the strain YBT49.
All other deletion strains were generated by CRISPR- Cas9/crRNA-tracrRNA ribonucleoprotein injection using the protocol described by Paix et al. [94] with the modifications detailed by Achache et al. [42]. The sequences of the crRNAs are listed in Supplementary Table S1. All mutations were outcrossed at least five times to establish the homozygous lines used in the study (Supplementary Table S1).
Cytological analysis and immunostaining
DAPI and immunostaining of dissected gonads was carried out as described previously [44,49]. Worms were permeabilized on Superfrost+ slides for 2 min with methanol at −20°C and fixed for 30 min in 4% paraformaldehyde in phosphate‐buffered saline (PBS). Staining with 500 ng/ml DAPI was carried out for 10 minutes followed by destaining in PBS containing 0.1% Tween 20. Slides were mounted with Vectashield anti-fading medium (Vector Laboratories, Burlingame, CA, USA). Primary antibodies used were rabbit α-HTP-1 (1:200 dilution, a kind gift from E. Martinez-Perez, Imperial College London) and goat α-SYP-1 (1:200 dilution, a kind gift from S. Smolikove, The University of Iowa). The secondary antibodies used were Cy2-donkey anti-rabbit, and Cy3-donkey anti-goat (Jackson ImmunoResearch Laboratories, West Grove, PA, USA). A minimum of 20 gonads were examined for each strain.
qRT-PCR
For quantitative real-time PCR (qRT-PCR), total RNA was isolated from whole worms using Direct-zol Miniprep Plus (Zymo Research, Irvine, CA, USA), according to the manufacturer’s instructions. Worms were subjected to nine rapid freeze-thaw cycles in Trizol (Invitrogen, Waltham, MA, USA) prior to RNA isolation. Next, 2 μg of total RNA were reverse-transcribed using SuperScript® III Reverse Transcriptase (Invitrogen). qPCR was performed using Power SYBR Green Mastermix (Applied Biosystems, Waltham, MA, USA) using an Applied Biosystems Step One Plus apparatus. Experiments were performed in triplicate, and all data were normalized to gpd-1. The primers used were as follows:
F13H8.2 Forward: CAA AGC TGC GCG GAA GA
F13H8.2 Reverse: TGT TGT GTC AGA TGT CGC TTT TC
gpd-1 Forward: ACT CGT CCA TTT TCG ATG CT
gpd-1 Reverse: TCG ACA ACA CGG TTC GAG TA
Imaging and microscopy
Images were acquired using the Olympus IX83 fluorescence microscope system. Optical Z-sections were collected at 0.30/0.60-µm increments with the Hamamatsu Orca Flash 4.0 v3 and CellSens Dimension imaging software (Olympus, Tokyo, Japan). Pictures were deconvolved using AutoQuant X3 (Media Cybernetics, Silver Spring, MD, United States).
For counting of bivalent chromosomes and chiasma, at least 20 DAPI-stained oocytes were examined per genotype. The number of oocytes and chiasma were manually quantified using ImageJ.
For analysis of synaptonemal complex assembly, at least 35 SYP-1- and HTP-1-stained nuclei were examined per genotype. Colocalization of the axial and central axes was manually assessed using ImageJ. Disassembly of the synaptonemal complex was analysed in SYP-1-stained diplotene and early diakinesis nuclei.
Quantitative analysis of germ-cell apoptosis
Germ-cell corpses were scored in adult hermaphrodites at 20 h post-L4 using acridine orange as described previously [95]. A minimum of 13 gonads were scored for each genotype.
Progeny quantification
The brood sizes were determined by placing at least 18 individual L4 worms on seeded NGM plates, transferring each worm to a new plate every 24 h, and counting their embryos and hatched progeny over 3 days.
Quantification of lincRNA expression in the gonad
The lincRNA quantification data were extracted from Table S1 of Tzur et al. [36]. All lincRNA data were evaluated, and genes with at least average counts of six were examined. lincRNAs with dynamic expression, as well lincRNAs transcribed from the X chromosome during early oogenesis and their paralogs were further analysed. Fig. 1 shows the log2 normalized expression as extracted from Table S3 of Tzur et al. [36].
Recovery from L1 arrest and size measurements
L1 arrest and recovery experiments were done essentially as described by Zaslaver et al. [79] with the following modifications: Embryos were isolated by bleach and suspended in S-basal without cholesterol. Cholesterol was omitted since it is dissolved in ethanol which was shown to be used by worms as a carbon source [96]. Following 1, 7, and 14 days in S-basal medium, larvae were washed three times in M9 medium and placed on seeded NGM or RNAi plates.
Worm sizes were determined by washing the worms off the plates with M9 and pipetting the suspended worms to unseeded NGM plates. Imaging was done using an Olympus MVX10 at X4 magnification with a QImaing QIClick-equipped camera. Images were analysed using semi-automatic custom-made Matlab scripts. Worms were assayed at the time of release and at 24-h intervals following the release. Only worms that exited the arrested state were evaluated. Under RNAi conditions many worms did not exit the arrest, and so only data obtained after 4 days of arrest were used.
RNAi
To create the linc-4 feeding vector we used a site-directed mutagenesis PCR method [97] to clone the entire linc-4 cDNA into the L4440 vector between the T7 promotors sites. Feeding RNAi experiments were performed at 20°C as described previously [98,99]. Control worms were fed HT115 bacteria carrying the empty pL4440 vector.
Small RNA-Seq Analysis
We analysed three small RNA samples from N2 wild-type worms [76]. The raw FASTQ files are available in NCBI (GSE124049). Adaptor sequences were removed using CutAdapt [100]. Trimmed reads were mapped to the ce11 assembly of the C. elegans genome using ShortStack [101], with specification of mismatches = 0. We next filtered for small RNA reads of 20–23 nucleotide in length, and counted reads that aligned in the antisense orientation to genes as defined by the corresponding Ensembl gff file, using HTSeq count [102]. Protein coding and lincRNA genes values were averaged for comparison with the individual genes described here.
RNA-seq
Young adult N2 and linc-4 worms were collected by 60% sucrose bed. Worms were washed with M9 buffer, and 100 μl packed were lysed with TRIzol reagent (Invitrogen). After 10 freeze-crack cycles in liquid nitrogen, total RNA was extracted using Zymo Research Direct-zol RNA miniprep plus kit. mRNA was enriched using Dynabeads mRNA DIRECT Micro Kit (Invitrogen) according to the manufacturer’s protocol. mRNA was fragmented using Ambion RNA Fragmentation Reagents (AM8740). Fragments were purified using AMPure XP beads (1.5X bead-based cleanup). First strand synthesis was carried out using ThermoFisher SuperScript III Reverse Transcriptase and oligo(dT) primers (Promega) followed by second-strand cDNA synthesis using New England Biolabs NEBNext Ultra II Non-Directional RNA Second Strand Synthesis Module. Double strand cDNA (dscDNA) was purified using AMPure XP beads (1.5X bead-based cleanup). End-It DNA End-Repair Kit (Lucigen) was used to repair and convert the dscDNA to 5′-phosphorylated, blunt-end DNA before 3ʹ-end A-Tailing by DNA Pol I, Large (Klenow) fragment (New England Biolab). Adapters were ligated to 3ʹ-dATP library fragments using Quick Ligase (New England Biolab). Libraries were amplified using Kapa Biosystems HiFi HotStart ReadyMix with 2p fixed primers (2p Fixed, 5ʹ-
AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT-3ʹ and 2p Fixed +barcode, 5ʹ-
CAAGCAGAAGACGGCATACGAGATNNNNNNNNGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT-3ʹ). The final product was purified using AMPure XP beads (0.7X bead-based cleanup). Deep sequencing was carried out on an Illumina NextSeq following the manufacturer’s protocols. Experiments were done in triplicates, and >80 million reads were generated for each genotype.
Differential expression and GO terms analyses
Raw reads were trimmed off low quality and technical bases, as well as trailing poly-A tails, using cutadapt, version 2.10, with parameters -O 1, -m 15 and – use-reads-wildcards. Reads with overall low quality were removed using fastq_quality_filter, FASTX version 0.0.14, with parameters -q 20 and -p 90. Processed reads were aligned to the C. elegans genome version WBcel235 using TopHat2, version 2.1.1. Alignment allowed 5 mismatches and 5-base gaps, and used gene annotations from EnsemblMetazoa release 46. Raw counts per gene were calculated with htseq-count, version 0.12.4, setting – s parameter to ‘no’.
Normalization and differential expression were calculated with the R package DESeq2, version 1.22.2. Calculations were done for genes with at least 10 raw counts using default parameters, without applying independent filtering. Genes were taken as differentially expressed if their adjusted p-value was less than 0.1, their baseMean was above 5 and if the absolute maximum likelihood estimate of the fold change was greater than 5/baseMean^0.5 + 0.4.
Supplementary Material
Acknowledgments
We thank the Caenorhabditis Genetics Center for kindly providing strains. We thank Enrique Martinez-Perez for the HTP-1 antibody and Sarit Smolikove for the SYP-1 antibody. We thank Adam Baer for technical assistance. We thank Alon Zaslaver for guidance, suggestions, and help in writing the manuscript. We thank Yuval Nevo and the team at the Info-CORE, Bioinformatics Unit of the I-CORE Computation Center, The Hebrew University and Hadassah Medical Center, Jerusalem, Israel, for the differential expression analysis. This work was supported by Israel Science Foundation [grant numbers 1283/15, 2090/15] to Y.B.T.
Funding Statement
This work was supported by the Israel Science Foundation [1283/15, 2090/15].
Authors’ contributions
HI and HA performed experiments and analysed data. EK, YR and CB provided technical assistance. HG and YR performed data analysis. EI created new MATLAB script. OR designed the endogenous siRNA analysis. YBT designed experiments and wrote the manuscript.
Availability of data and materials
Strains and plasmids are available upon request. Supplementary Table File S1 contains detailed descriptions of all primers used for genome engineering and genotyping. Transcriptomic data are available at NCBI’s Gene Expression Omnibus, under accession number GSE154322.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Kopp F, Mendell JT.. Functional Classification and Experimental Dissection of Long Noncoding RNAs. Cell. 2018;172:393–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ulitsky I, Bartel DP.. lincRNAs: genomics, evolution, and mechanisms. Cell. 2013;154:26–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Deniz E, Erman B. Long noncoding RNA (lincRNA), a new paradigm in gene expression control. Funct Integr Genomics. 2017;17:135–143. [DOI] [PubMed] [Google Scholar]
- [4].Fico A, Fiorenzano A, Pascale E, et al. Long non-coding RNA in stem cell pluripotency and lineage commitment: functions and evolutionary conservation. Cell Mol Life Sci. 2019;76:1459–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Shields EJ, Petracovici AF, Bonasio R. lncRedibly versatile: biochemical and biological functions of long noncoding RNAs. Biochem J. 2019;476:1083–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Fatica A, Bozzoni I. Long non-coding RNAs: new players in cell differentiation and development. Nat Rev Genet. 2014;15:7–21. [DOI] [PubMed] [Google Scholar]
- [7].Blythe AJ, Fox AH, Bond CS. The ins and outs of lncRNA structure: how, why and what comes next? Biochim Biophys Acta. 2016;1859:46–58. [DOI] [PubMed] [Google Scholar]
- [8].Marques AC, Ponting CP. Intergenic lncRNAs and the evolution of gene expression. Curr Opin Genet Dev. 2014;27:48–53. [DOI] [PubMed] [Google Scholar]
- [9].Melissari MT, Grote P. Roles for long non-coding RNAs in physiology and disease. Pflugers Arch. 2016;468:945–958. [DOI] [PubMed] [Google Scholar]
- [10].Perry RB, Ulitsky I. The functions of long noncoding RNAs in development and stem cells. Development. 2016;143:3882–3894. [DOI] [PubMed] [Google Scholar]
- [11].Taylor DH, Chu ET, Spektor R, et al. Long non-coding RNA regulation of reproduction and development. Mol Reprod Dev. 2015;82:932–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bie B, Wang Y, Li L, et al. Noncoding RNAs: potential players in the self-renewal of mammalian spermatogonial stem cells. Mol Reprod Dev. 2018;85:720–728. [DOI] [PubMed] [Google Scholar]
- [13].Cuomo D, Porreca I, Ceccarelli M, et al. Transcriptional landscape of mouse-aged ovaries reveals a unique set of non-coding RNAs associated with physiological and environmental ovarian dysfunctions. Cell Death Discov. 2018;4:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Luk AC, Chan WY, Rennert OM, et al. Long noncoding RNAs in spermatogenesis: insights from recent high-throughput transcriptome studies. Reproduction. 2014;147:R131–41. [DOI] [PubMed] [Google Scholar]
- [15].Cabili MN, Trapnell C, Goff L, et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011;25:1915–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Necsulea A, Soumillon M, Warnefors M, et al. The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature. 2014;505:635–640. [DOI] [PubMed] [Google Scholar]
- [17].Soumillon M, Necsulea A, Weier M, et al. Cellular source and mechanisms of high transcriptome complexity in the mammalian testis. Cell Rep. 2013;3:2179–2190. [DOI] [PubMed] [Google Scholar]
- [18].Trovero MF, Rodríguez-Casuriaga R, Romeo C, et al . Revealing stage-specific expression patterns of long noncoding RNAs along mouse spermatogenesis. RNA biol. 2020; 17:350–65:e40815. DOI: 10.7554/eLife.40815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wen K, Yang L, Xiong T, et al. Critical roles of long noncoding RNAs in Drosophila spermatogenesis. Genome Res. 2016;26:1233–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Goudarzi M, Berg K, Pieper LM, et al. Individual long non-coding RNAs have no overt functions in zebrafish embryogenesis, viability and fertility. eLife. 2019;8:e40815. doi: 10.7554/eLife.40815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Grote P, Herrmann BG. Long noncoding RNAs in organogenesis: making the difference. Trends Genet. 2015;31:329–335. [DOI] [PubMed] [Google Scholar]
- [22].Uszczynska-Ratajczak B, Lagarde J, Frankish A, et al. Towards a complete map of the human long non-coding RNA transcriptome. Nat Rev Genet. 2018;19:535–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Couteau F, Goodyer W, Zetka M. Finding and keeping your partner during meiosis. Cell Cycle. 2004;3:1014–1016. [PubMed] [Google Scholar]
- [24].Gerton JL, Hawley RS. Homologous chromosome interactions in meiosis: diversity amidst conservation. Nat Rev Genet. 2005;6:477–487. [DOI] [PubMed] [Google Scholar]
- [25].Gray S, Cohen PE. Control of Meiotic Crossovers: from Double-Strand Break Formation to Designation. Annu Rev Genet. 2016;50:175–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Jasin M, Rothstein R. Repair of strand breaks by homologous recombination. Cold Spring Harb Perspect Biol. 2013;5:a012740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Mezard C, Jahns MT, Grelon M. Where to cross? New insights into the location of meiotic crossovers. Trends Genet. 2015;31:393–401. [DOI] [PubMed] [Google Scholar]
- [28].Page SL, Hawley RS. The genetics and molecular biology of the synaptonemal complex. Annu Rev Cell Dev Biol. 2004;20:525–558. [DOI] [PubMed] [Google Scholar]
- [29].Rog O, Dernburg AF. Chromosome pairing and synapsis during Caenorhabditis elegans meiosis. Curr Opin Cell Biol. 2013;25:349–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Woglar A, Jantsch V. Chromosome movement in meiosis I prophase of Caenorhabditis elegans. Chromosoma. 2014;123:15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Yu Z, Kim Y, Dernburg AF. Meiotic recombination and the crossover assurance checkpoint in Caenorhabditis elegans. Semin Cell Dev Biol. 2016;54:106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zetka M. Homologue pairing, recombination and segregation in Caenorhabditis elegans. Genome Dyn. 2009;5:43–55. [DOI] [PubMed] [Google Scholar]
- [33].Zetka M, Rose A. The genetics of meiosis in Caenorhabditis elegans. Trends Genet. 1995;11:27–31. [DOI] [PubMed] [Google Scholar]
- [34].Akay A, Jordan D, Navarro IC, et al. Identification of functional long non-coding RNAs in C. elegans. BMC Biol. 2019;17:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Nam JW, Bartel DP. Long noncoding RNAs in C. elegans. Genome Res. 2012;22:2529–2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Tzur YB, Winter E, Gao J, et al. Spatiotemporal Gene Expression Analysis of the Caenorhabditis elegans Germline Uncovers a Syncytial Expression Switch. Genetics. 2018;210:587–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Diag A, Schilling M, Klironomos F, et al. Spatiotemporal m(i)RNA Architecture and 3ʹ UTR Regulation in the C. elegans Germline. Dev Cell. 2018;47(785–800):e8. [DOI] [PubMed] [Google Scholar]
- [38].Ebbing A, Vertesy A, Betist MC, et al. Spatial Transcriptomics of C. elegans Males and Hermaphrodites Identifies Sex-Specific Differences in Gene Expression Patterns. Dev Cell. 2018;47:801–13 e6. [DOI] [PubMed] [Google Scholar]
- [39].Bean CJ, Schaner CE, Kelly WG. Meiotic pairing and imprinted X chromatin assembly in Caenorhabditis elegans. Nat Genet. 2004;36:100–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kelly WG, Schaner CE, Dernburg AF, et al. X-chromosome silencing in the germline of C. elegans. Development. 2002;129:479–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wei S, Chen H, Dzakah EE, et al. Systematic evaluation of C. elegans lincRNAs with CRISPR knockout mutants. Genome Biol. 2019;20:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Achache H, Laurent L, Hecker-Mimoun Y, et al. Progression of Meiosis Is Coordinated by the Level and Location of MAPK Activation Via OGR-2 in Caenorhabditis elegans. Genetics. 2019;212:213–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Crawford D, Libina N, Kenyon C. Caenorhabditis elegans integrates food and reproductive signals in lifespan determination. Aging Cell. 2007;6:715–721. [DOI] [PubMed] [Google Scholar]
- [44].Colaiacovo MP, MacQueen AJ, Martinez-Perez E, et al. Synaptonemal complex assembly in C. elegans is dispensable for loading strand-exchange proteins but critical for proper completion of recombination. Dev Cell. 2003;5:463–474. [DOI] [PubMed] [Google Scholar]
- [45].Lightfoot J, Testori S, Barroso C, et al. Loading of meiotic cohesin by SCC-2 is required for early processing of DSBs and for the DNA damage checkpoint. Curr Biol. 2011;21:1421–1430. [DOI] [PubMed] [Google Scholar]
- [46].Goodyer W, Kaitna S, Couteau F, et al. HTP-3 links DSB formation with homolog pairing and crossing over during C. elegans meiosis. Dev Cell. 2008;14:263–274. [DOI] [PubMed] [Google Scholar]
- [47].Martinez-Perez E, Villeneuve AM. HTP-1-dependent constraints coordinate homolog pairing and synapsis and promote chiasma formation during C. elegans meiosis. Genes Dev. 2005;19:2727–2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Reddy KC, Villeneuve AM. C. elegans HIM-17 links chromatin modification and competence for initiation of meiotic recombination. Cell. 2004;118:439–452. [DOI] [PubMed] [Google Scholar]
- [49].Saito TT, Youds JL, Boulton SJ, et al. Caenorhabditis elegans HIM-18/SLX-4 interacts with SLX-1 and XPF-1 and maintains genomic integrity in the germline by processing recombination intermediates. PLoS Genet. 2009;5:e1000735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Severson AF. Analysis of Meiotic Sister Chromatid Cohesion in Caenorhabditis elegans. Methods Mol Biol. 2017;1515:65–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Severson AF, Meyer BJ. Divergent kleisin subunits of cohesin specify mechanisms to tether and release meiotic chromosomes. eLife. 2014;3:e03467. doi: 10.7554/eLife.03467.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Yokoo R, Zawadzki KA, Nabeshima K, et al. COSA-1 reveals robust homeostasis and separable licensing and reinforcement steps governing meiotic crossovers. Cell. 2012;149:75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Bhalla N, Wynne DJ, Jantsch V, et al. ZHP-3 acts at crossovers to couple meiotic recombination with synaptonemal complex disassembly and bivalent formation in C. elegans. PLoS Genet. 2008;4:e1000235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Adamo A, Collis SJ, Adelman CA, et al. Preventing nonhomologous end joining suppresses DNA repair defects of Fanconi anemia. Mol Cell. 2010;39:25–35. [DOI] [PubMed] [Google Scholar]
- [55].Cohen PE, Pollack SE, Pollard JW. Genetic analysis of chromosome pairing, recombination, and cell cycle control during first meiotic prophase in mammals. Endocr Rev. 2006;27:398–426. [DOI] [PubMed] [Google Scholar]
- [56].Baum JS, St George JP, McCall K. Programmed cell death in the germline. Semin Cell Dev Biol. 2005;16:245–259. [DOI] [PubMed] [Google Scholar]
- [57].Gartner A, Milstein S, Ahmed S, et al. A conserved checkpoint pathway mediates DNA damage–induced apoptosis and cell cycle arrest in C. elegans. Mol Cell. 2000;5:435–443. [DOI] [PubMed] [Google Scholar]
- [58].Bhalla N, Dernburg AF. A conserved checkpoint monitors meiotic chromosome synapsis in Caenorhabditis elegans. Science. 2005;310:1683–1686. [DOI] [PubMed] [Google Scholar]
- [59].Ye AL, Ragle JM, Conradt B, et al. Differential regulation of germline apoptosis in response to meiotic checkpoint activation. Genetics. 2014;198:995–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Lascarez-Lagunas LI, Silva-Garcia CG, Dinkova TD, et al. LIN-35/Rb causes starvation-induced germ cell apoptosis via CED-9/Bcl2 downregulation in Caenorhabditis elegans. Mol Cell Biol. 2014;34:2499–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Gartner A, Boag PR, Blackwell TK. Germline survival and apoptosis. WormBook. 2008. doi: 10.1895/wormbook.1.145.1. http://www.wormbook.org/chapters/www_germlinesurvival/germlinesurvival.html [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Villanueva-Chimal E, Salinas LS, Fernandez-Cardenas LP, et al. DPFF-1 transcription factor deficiency causes the aberrant activation of MPK-1 and meiotic defects in the Caenorhabditis elegans germline. Genesis. 2017;55:e23072. [DOI] [PubMed] [Google Scholar]
- [63].Mpoke SS, Wolfe J. Differential staining of apoptotic nuclei in living cells: application to macronuclear elimination in Tetrahymena. J Histochem Cytochem. 1997;45:675–683. [DOI] [PubMed] [Google Scholar]
- [64].Lettre G, Kritikou EA, Jaeggi M, et al. Genome-wide RNAi identifies p53-dependent and -independent regulators of germ cell apoptosis in C. elegans. Cell Death Differ. 2004;11:1198–1203. [DOI] [PubMed] [Google Scholar]
- [65].Carlton PM, Farruggio AP, Dernburg AF. A link between meiotic prophase progression and crossover control. PLoS Genet. 2006;2:e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Gabdank I, Fire AZ. Gamete-type dependent crossover interference levels in a defined region of Caenorhabditis elegans chromosome V. G3. 2014;4:117–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Hodgkin J, Horvitz HR, Brenner S. Nondisjunction Mutants of the Nematode CAENORHABDITIS ELEGANS. Genetics. 1979;91:67–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Lim JG, Stine RR, Yanowitz JL. Domain-specific regulation of recombination in Caenorhabditis elegans in response to temperature, age and sex. Genetics. 2008;180:715–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Meneely PM, Farago AF, Kauffman TM. Crossover distribution and high interference for both the X chromosome and an autosome during oogenesis and spermatogenesis in Caenorhabditis elegans. Genetics. 2002;162:1169–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Nabeshima K, Villeneuve AM, Hillers KJ. Chromosome-wide regulation of meiotic crossover formation in Caenorhabditis elegans requires properly assembled chromosome axes. Genetics. 2004;168:1275–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Colaiacovo M. The many facets of SC function during C. elegans meiosis. Chromosoma. 2006;115:195–211. [DOI] [PubMed] [Google Scholar]
- [72].Hillers KJ, Jantsch V, Martinez-Perez E, et al. Meiosis. WormBook. 2017;1–43. DOI: 10.1895/wormbook.1.178.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].de Carvalho CE, Zaaijer S, Smolikov S, et al. LAB-1 antagonizes the Aurora B kinase in C. elegans. Genes Dev. 2008;22:2869–2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Martinez-Perez E, Schvarzstein M, Barroso C, et al. Crossovers trigger a remodeling of meiotic chromosome axis composition that is linked to two-step loss of sister chromatid cohesion. Genes Dev. 2008;22:2886–2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Severson AF, Ling L, van Zuylen V, et al. The axial element protein HTP-3 promotes cohesin loading and meiotic axis assembly in C. elegans to implement the meiotic program of chromosome segregation. Genes Dev. 2009;23:1763–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Posner R, Toker IA, Antonova O, et al. Neuronal Small RNAs Control Behavior Transgenerationally. Cell. 2019;177:1814–26 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Allen MA, Hillier LW, Waterston RH, et al. A global analysis of C. elegans trans-splicing. Genome Res. 2011;21:255–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Reinke V, Cutter AD. Germline expression influences operon organization in the Caenorhabditis elegans genome. Genetics. 2009;181:1219–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Zaslaver A, Baugh LR, Sternberg PW. Metazoan operons accelerate recovery from growth-arrested states. Cell. 2011;145:981–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Bassett AR, Akhtar A, Barlow DP, et al. Considerations when investigating lncRNA function in vivo. eLife. 2014;3:e03058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Tijsterman M, Okihara KL, Thijssen K, et al. PPW-1, a PAZ/PIWI protein required for efficient germline RNAi, is defective in a natural isolate of C. elegans. Curr Biol. 2002;12:1535–1540. [DOI] [PubMed] [Google Scholar]
- [82].Sijen T, Fleenor J, Simmer F, et al. On the role of RNA amplification in dsRNA-triggered gene silencing. Cell. 2001;107:465–476. [DOI] [PubMed] [Google Scholar]
- [83].Kumsta C, Hansen M. C. elegans rrf-1 mutations maintain RNAi efficiency in the soma in addition to the germline. PLoS One. 2012;7:e35428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Page AP, Johnstone IL. The cuticle. WormBook. 2007. doi: 10.1895/wormbook.1.138.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Schreiner WP, Pagliuso DC, Garrigues JM, et al. Remodeling of the Caenorhabditis elegans non-coding RNA transcriptome by heat shock. Nucleic Acids Res. 2019;47:9829–9841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Johnson R, Guigo R. The RIDL hypothesis: transposable elements as functional domains of long noncoding RNAs. Rna. 2014;20:959–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Chen J, Shishkin AA, Zhu X, et al. Evolutionary analysis across mammals reveals distinct classes of long non-coding RNAs. Genome Biol. 2016;17:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Hezroni H, Koppstein D, Schwartz MG, et al. Principles of long noncoding RNA evolution derived from direct comparison of transcriptomes in 17 species. Cell Rep. 2015;11:1110–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Bolkova J, Lanctot C. Quantitative gene expression analysis in Caenorhabditis elegans using single molecule RNA FISH. Methods. 2016;98:42–49. [DOI] [PubMed] [Google Scholar]
- [90].Gao S, Liu Y. Intercepting noncoding messages between germline and soma. Genes Dev. 2012;26:1774–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Kelly WG. Transgenerational epigenetics in the germline cycle of Caenorhabditis elegans. Epigenetics Chromatin. 2014;7:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Friedland AE, Tzur YB, Esvelt KM, et al. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nat Methods. 2013. DOI: 10.1038/nmeth.2532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Paix A, Folkmann A, Rasoloson D, et al. High Efficiency, Homology-Directed Genome Editing in Caenorhabditis elegans Using CRISPR/Cas9 Ribonucleoprotein Complexes. Genetics. 2015. DOI: 10.1534/genetics.115.179382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Kelly KO, Dernburg AF, Stanfield GM, et al. Caenorhabditis elegans msh-5 is required for both normal and radiation-induced meiotic crossing over but not for completion of meiosis. Genetics. 2000;156:617–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Castro PV, Khare S, Young BD, et al. Caenorhabditis elegans battling starvation stress: low levels of ethanol prolong lifespan in L1 larvae. PLoS One. 2012;7:e29984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Weiner MP, Costa GL, Schoettlin W, et al. Site-directed mutagenesis of double-stranded DNA by the polymerase chain reaction. Gene. 1994;151:119–123. [DOI] [PubMed] [Google Scholar]
- [98].Govindan JA, Cheng H, Harris JE, et al. Galphao/i and Galphas signaling function in parallel with the MSP/Eph receptor to control meiotic diapause in C. elegans. Curr Biol. 2006;16:1257–1268. [DOI] [PubMed] [Google Scholar]
- [99].Govindan JA, Nadarajan S, Kim S, et al. Somatic cAMP signaling regulates MSP-dependent oocyte growth and meiotic maturation in C. elegans. Development. 2009;136:2211–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Martin M. CUTADAPT removes adapter sequences from high-throughput sequencing reads. EMBnetjournal. 2011;17(1):10–12. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- [101].Axtell MJ. ShortStack: comprehensive annotation and quantification of small RNA genes. Rna. 2013;19:740–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Anders S, Pyl PT, Huber W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Strains and plasmids are available upon request. Supplementary Table File S1 contains detailed descriptions of all primers used for genome engineering and genotyping. Transcriptomic data are available at NCBI’s Gene Expression Omnibus, under accession number GSE154322.