

SUMMARY
Hepatocellular carcinoma (HCC) remains one of the deadliest malignancies worldwide. One major obstacle to treatment is a lack of effective molecular-targeted therapies. In this study, we find that EphA2 expression and signaling are enriched in human HCC and associated with poor prognosis. Loss of EphA2 suppresses the initiation and growth of HCC both in vitro and in vivo. Furthermore, CRISPR/CAS9-mediated EphA2 inhibition significantly delays tumor development in a genetically engineered murine model of HCC. Mechanistically, we discover that targeting EphA2 suppresses both AKT and JAK1/STAT3 signaling, two separate oncogenic pathways in HCC. We also identify a small molecule kinase inhibitor of EphA2 that suppresses tumor progression in a murine HCC model. Together, our results suggest EphA2 as a promising therapeutic target for HCC.
Graphical Abstract
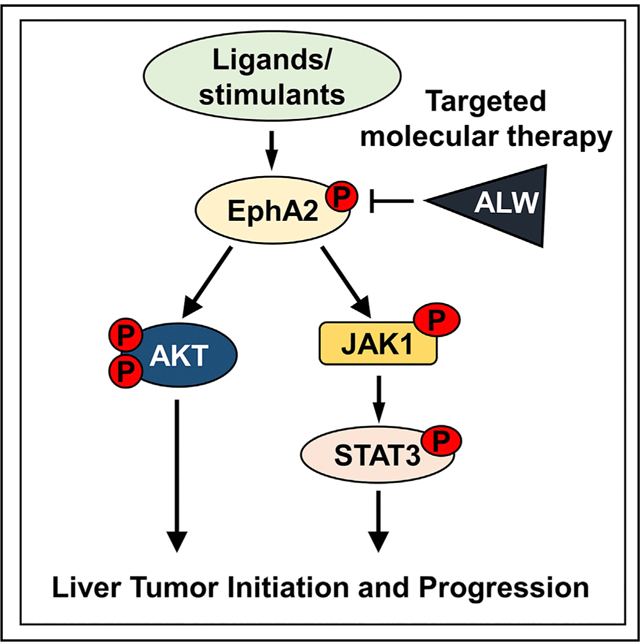
In brief
Wang et al. define the role of receptor tyrosine kinase EphA2 in the development of hepatocellular carcinoma and elucidate its mechanisms of action. Inhibition of EphA2 is a promising strategy for the treatment of advanced hepatocellular carcinoma.
INTRODUCTION
Hepatocellular carcinoma (HCC) accounts for 90% of all primary liver cancers worldwide (Ferlay et al., 2015). Liver cancer is the sixth most common cancer, and the fourth most common cause of cancer death worldwide (Villanueva, 2019). The prognosis for patients with HCC is poor, with a 5-year survival of 18% (Villanueva, 2019). The Food and Drug Administration (FDA) approved the VEGFR inhibitor sorafenib as a first-line treatment for advanced HCC in 2008. Sorafenib provides limited clinical benefit, increasing overall survival (OS) by 2.8 months compared to placebo, and achieving an overall response rate of less than 2% (Llovet et al., 2008). In recent years, additional molecular agents such as the first-line treatment lenvatinib, second-line treatments regorafenib, cabozantinib, and checkpoint inhibitors nivolumab and pembrolizumab have been approved by the FDA. However, all are marked by low response rates and limited overall survival benefit. Recently, atezolizumab, combined with bevacizumab, showed better overall and progression-free survival outcomes than sorafenib in patients with unresectable hepatocellular carcinoma (Finn et al., 2020). The exciting results led to the approval of this combination as the first-line treatment by the FDA for HCC patients. However, 58.6% of patients receiving atezolizumab-bevacizumab still had disease progression or died within 12 months after the treatment (Finn et al., 2020).
A significant challenge in drug development is that HCC has the fewest somatic mutations among solid tumors that can be targeted pharmacologically (Villanueva, 2019). The majority of approved therapies for advanced HCC primarily targets VEGFR, which targets the tumor vasculature but not the crucial oncodrivers directly. Available treatments could benefit patients with amplification of the VEGFA genomic locus, the prevalence of this copy number aberration is low (5%–10%) (Horwitz et al., 2014; Llovet, 2014). Given the lack of effective treatment options for patients with advanced HCC, there is an urgent need to identify new druggable molecular targets.
EphA2 is a receptor tyrosine kinase implicated in many human diseases, including cancer, inflammatory disorders, and neurological disorders (Boyd et al., 2014; Kania and Klein, 2016; Pasquale, 2010). On binding with its ligands, ephrins, EphA2 activates its kinase activity, which leads to the autophosphorylation of its juxtamembrane Tyr residues (Y588 and Y594) (Binns et al., 2000; Fang et al., 2008). These events are essential for activation of ephrins-EphA2-directed cellular responses, including cell growth, migration, differentiation, and more (Kania and Klein, 2016; Pasquale, 2010). EphA2 overexpression can also promote migration in glioma and prostate cancer cells in a ligand-independent manner (Miao et al., 2009).
Because EphA2 was initially discovered in human carcinomas over 30 years ago, many studies have investigated its functional roles in tumorigenesis (Hirai et al., 1987). High EphA2 expression and signaling are frequently detected in many cancers (including brain, breast, lung, etc.) and are associated with poor patient outcomes (Boyd et al., 2014; Pasquale, 2010; Wykosky and Debinski, 2008). For example, overexpression of EphA2 was sufficient to malignantly transform mammary epithelial cells (Hochgräfe et al., 2010; Zelinski et al., 2001). In glioblastoma, EphA2 promotes self-renewal and invasion of tumor-propagating cells (Binda et al., 2012; Miao et al., 2015). Recently, EphA2 expressed on tumor cells was shown to suppress T cell-mediated anti-tumor responses, and inhibition of EphA2 reversed T cell exhaustion and sensitized tumors to immunotherapy (Markosyan et al., 2019).
The role of EphA2 in HCC has not been well explored. Several clinical reports suggested that expression of EphA2 and its ligands correlated to tumor progression, invasion, metastasis, and poor prognosis in patients with HCC (Cui et al., 2010; Patil et al., 2009; Yang et al., 2009). Other studies have also shown that microRNAs impair tumorigenesis and enhance radiation therapy’s efficacy through direct inhibition of EphA2 expression in HCC (Li et al., 2015; Xiang et al., 2019). Interestingly, a recent study showed that EphA2 is highly activated in sorafenib-resistant HCC cells, suggesting EphA2 may promote drug resistance to current therapy in HCC (Leung et al., 2020). However, the role of EphA2 and its mechanism of action in HCC development remain unknown. In this study, we discovered mechanisms underlying how EphA2 regulated tumor initiation and progression in HCC and explored the therapeutic potential of targeting EphA2 in HCC.
RESULTS
High EphA2 signaling and expression is detected in HCC patient samples and is associated with poor prognosis
Previous studies showed that phosphorylation at the juxtamembrane domain tyrosine 588 of EphA2 (Y588) is essential for its signaling activity (Binns et al., 2000; Fang et al., 2008). To explore the role of EphA2 signaling in HCC, we performed Y588 p-EphA2 and total EphA2 immunohistochemistry (IHC) on tissue microarrays from a retrospective cohort of 153 HCC specimens and 63 non-tumor liver tissues (Table S1). Y588 p-EphA2 and total EphA2 protein levels were assessed by a hepatic pathologist blinded to the patient outcome (Figures 1A and S1A). High expression of Y588 p-EphA2 and total EphA2 was detectable in 33.33% and 30.06% of HCC cases, respectively, while in non-tumor liver tissues, p-EphA2 and EphA2 high expression was only detectable in 4.76% and 9.52% of patients, respectively. Our results show that increased expression of p-EphA2 and total EphA2 correlates with HCC pathology (Figures 1B–1D). Furthermore, high mRNA expression of EphA2 in The Cancer Genome Atlas (TCGA) human HCC dataset was associated with poor prognosis (Figure 1E). Because Ephrin A class ligands (EFNAs) are known to activate EphA2 signaling (Boyd et al., 2014; Kania and Klein, 2016; Pasquale, 2010), we also analyzed the expression of EFNAs in the TCGA HCC dataset and found that 4 out of 5 EFNAs were highly expressed in HCC and were associated with poor prognosis (Figures 1F, S1B, and S1C). To further confirm these results, we analyzed another HCC cohort, GSE14520 (Roessler et al., 2010). In this cohort, although EphA2 expression (median cutoff) was not significantly associated with poor prognosis (Figure S1D), HCC patients with the highest (top 25%) expression of EphA2 had worse survival compared to the patients with lowest (bottom 25%) expression of EphA2 (Figure S1E). Among five ligands, high expression of EFNA4, but not others, was significantly associated with poor prognosis (Figures S1F–S1J). Hazard ratios analysis for 3-year survival also confirmed these findings (Table S2). The differences seen from these two cohorts might be due to different background, patient number, and other factors among the patients. Collectively, these data suggest that high EphA2 signaling is correlated with poor prognosis in HCC.
Figure 1. High EphA2 signaling and expression is detected in HCC patient samples and is associated with poor prognosis.
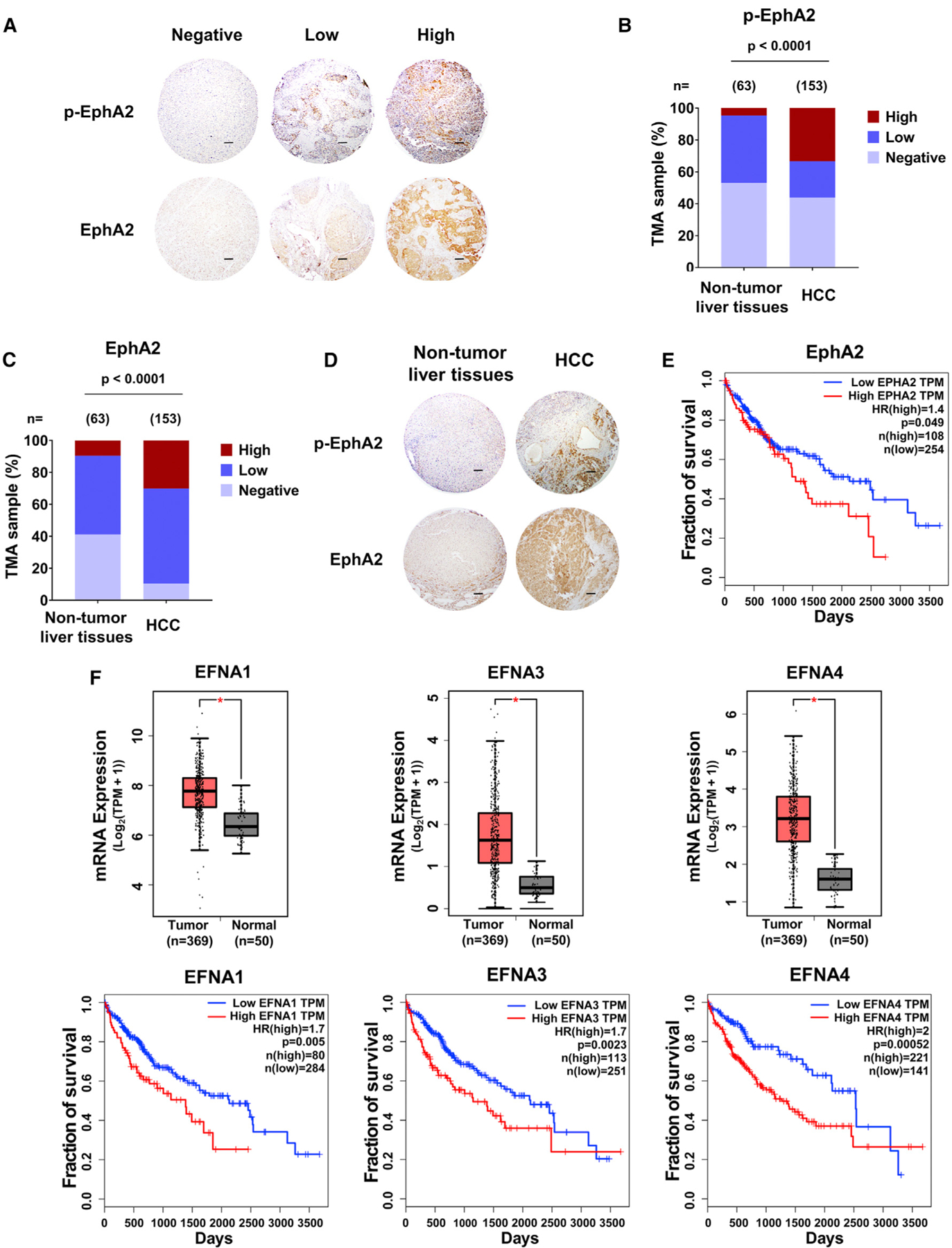
(A) Representative IHC images of Y588 p-EphA2 and total EphA2 in HCC tissue microarray classified as negative, low, or high expression. Scale bars, 100 μm.
(B) Y588 p-EphA2 expression levels in TMAs of normal and HCC liver samples.
(C) Total EphA2 expression levels in TMAs of normal and HCC liver samples.
(D) Representative examples of normal versus HCC TMAs measured in (B) and (C). Scale bars, 100 μm.
(E) Kaplan-Meier plot of overall survival of HCC patients stratified by EphA2 expression levels from the TCGA database (GEPIA).
(F) Top: a boxplot of relative mRNA expression levels of EFNAs comparing normal versus HCC tissue. Bottom: Kaplan-Meier plot of overall survival of HCC patients stratified by EFNA expression levels. Data from TCGA (GEPIA).
Statistical significance was determined by chi-square test (B) and (C), two-tailed Student’s t test *p < 0.0001 boxplots (F), and log-rank test Kaplan-Meier plot (E and F).
EphA2 drives HCC tumor growth
In light of these observations, we sought to test the functional role of EphA2 signaling in HCC. First, we assessed EphA2 expression and activity in seven human HCC cell lines and found all expressed EphA2 and Y588 p-EphA2 (Figure 2A). Next, we selected three human HCC cell lines with high or modest levels of p-EphA2 and EphA2 expression (Huh7, Hep3B, and SNU475) and attenuated the expression of EphA2 by lentiviral-short hairpin RNA (shRNA) knockdown (Figure 2B). We found that knockdown of EphA2 impaired the proliferation of all these HCC cells (Figure 2C). To verify the role of EphA2 in HCC in vivo, we established xenograft models with Huh7 cells (control or shRNA-mediated suppression of EphA2). Following implantation of tumor cells at day 0, controls showed the first sign of tumor at ~18 days and a linear increase in tumor volume during the subsequent week. In contrast, EphA2 knockdown mice showed no tumor sign for up to 28 days (Figure 2D). We observed similar results in the Hep3B xenograft models (Figure 2E). All cells for xenograft studies were screened for viability via Trypan blue before injection; however, we cannot rule out a possible role of EphA2 in maintenance/survival after cell implantation. Overall, our results suggested that EphA2 is crucial for tumor growth in HCC.
Figure 2. EphA2 drives HCC tumor proliferation and initiation.
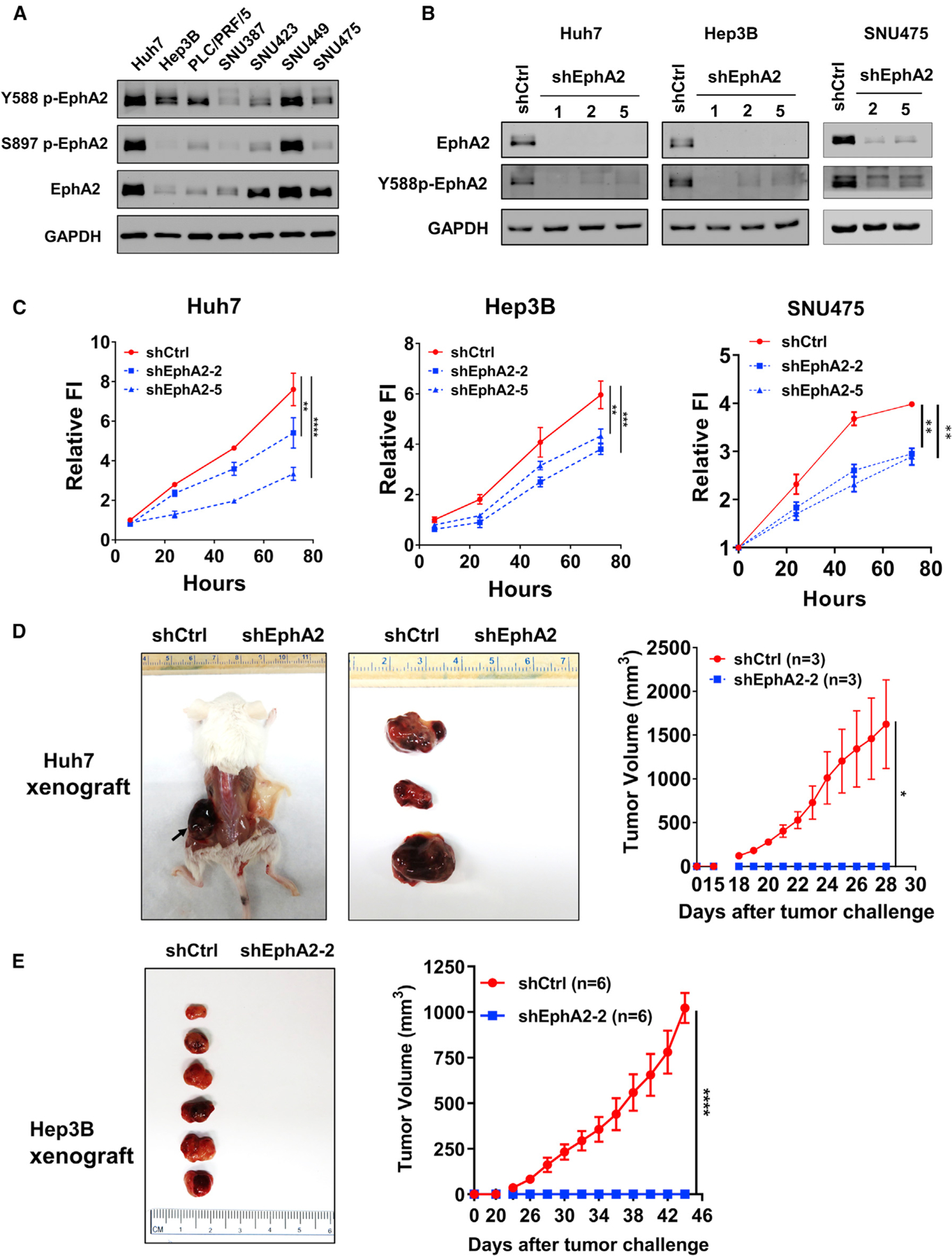
(A) Western blot showed EphA2 activation in 7 independent HCC cell lines. GAPDH as a loading control.
(B) Western blot validated EphA2 knockdown efficiency after lentiviral transduction of 3 independent EphA2 shRNAs 7 days after puromycin selection.
(C) Cell proliferation studies of control versus EphA2 knockdown Huh7, Hep3B, and SNU475 cells at day 7 after puromycin selection. Cell proliferation was evaluated using the Alamar blue assay. The ratio of proliferation is normalized to at 6 h after initial seeding of the cells and measured at 24, 48, and 72 h. Values are mean ± SD (n = 4).
(D) Left: representative picture of NSG-A2 mice 28 days after injection with 3 × 106 Huh7 cells. Left flank: shCtrl. Right flank: shEphA2. N = 3 mice per group. Right, daily measurements of primary tumor size. Values are mean ± SEM.
(E) Left: representative picture of NSG-A2 mice 44 days after injection with 3 × 106 Hep3B cells. Left flank: shCtrl. Right flank: shEphA2. N = 6 mice per group. Right, daily measurements of primary tumor size. Values are mean ± SEM.
Statistical significance was determined by one-ANOVA analysis with Dunnett’s multiple comparisons test (C) or two-tailed Student’s t test (D and E); *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001.
Targeting EphA2 suppresses tumor initiation and progression and enhances overall survival in MET/CAT induced murine model of HCC
We have previously utilized several established genetically engineered mouse models (GEMM) of HCC to study hepatocarcinogenesis (Shang et al., 2015, 2019; Wang et al., 2018). The advantages of the GEMM model are: (1) tumor develops at its site of origin and retains its native vasculature and microenvironment; (2) mice are immunocompetent; and (3) mice exhibit comparable clinical symptoms such as hepatomegaly and ascites. To further determine the role of EphA2 in HCC development, we used the c-MET (MET)/β-catenin (CAT)-driven HCC model, which is useful for studying the functions of genes in HCC development because of its clinical relevance (Cieply et al., 2009; Kaposi-Novak et al., 2006; Tao et al., 2016; Tward et al., 2007). Both p-EphA2 and EphA2 are highly expressed in MET/CAT-induced liver tumors (Figure 3A). To study the role of EphA2 in this model, we combined the MET/CAT model with CRISPR-Cas9-mediated inhibition of EphA2 expression in the mouse liver (Figure 3B) (Cong et al., 2013; Xue et al., 2014). We cloned three pX330 vectors, each co-expressing Cas9 and 1 of 3 independent single guide RNAs (sgRNAs) selected to target EphA2. Of the three sgEphA2 constructs tested, sgEphA2–2, was chosen for further experiments because of its high knockout efficacy (Figure 3C). The effectiveness and specificity of sgEphA2–2 were further validated using immunofluorescence and immunoblot (Figures 3D and 3E). Ten days after the injection of MET/CAT, almost all of the transformed hepatocytes, which expressed high c-MET, also had high EphA2 expression (Figure 3E), while there was very little detectable EphA2 in normal murine liver tissue or non-transformed cells (Figures 3A and 3E). These data suggest that detectable EphA2 was specifically induced in transformed cells. Because hydrodynamic injection equivalently delivers plasmids into the same cells (Sebestyén et al., 2006; Shang et al., 2015), sgEphA2–2 efficiently deleted EphA2 in the transformed cells, which was indicated by almost complete ablation of the EphA2 signals in mouse liver (Figure 3E).
Figure 3. Targeting EphA2 suppresses tumor progression and enhances overall survival in MET/CAT-induced murine model of HCC.
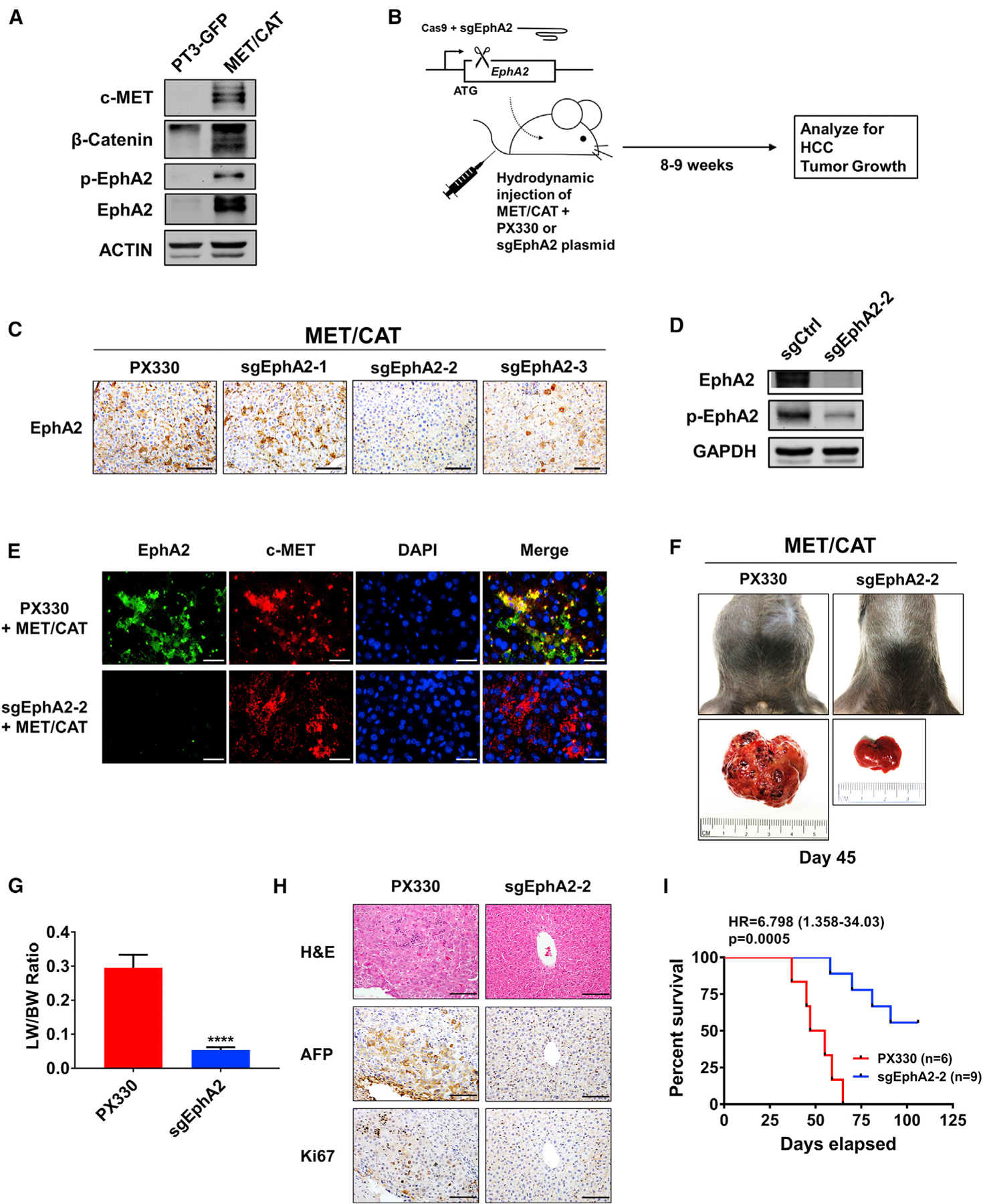
(A) Western blot validating MET/CAT HCC model and analyzing for EphA2 signaling and expression. ACTIN as a loading control.
(B) Schematic of the experiment of targeted inhibition of EphA2 in MET/CAT induced murine HCC model. pX330 plasmids expressing Cas9 and sgRNA targeting EphA2 (sgEphA2) or empty vector (PX330) were hydrodynamically delivered to mice liver in conjunction with MET/CAT; mice were observed for development of HCC for 8–9 weeks. n = 6 mice in the PX330 group, and n = 9 mice in the sgEphA2 group.
(C) IHC validated the efficacy of 3 independent sgEphA2 constructs in MET/CAT model 10 days after injection. Scale bars, 100 μm.
(D) Western blot confirmed the efficacy of sgEphA2–2 in MET/CAT model 10 days after injection. GAPDH as a loading control.
(E) Immunofluorescence analyzing EphA2 knockout in the context of MET overexpression in MET/CAT model 10 days after injection. EphA2, green;, c-MET, red. Nuclei were stained with DAPI (blue). Scale bars, 30 μm.
(F) Top: a representative image of the mouse abdomen in MET/CAT mouse treated with PX330 or sgEphA2 45 days after injection. Bottom: gross pictures of livers extracted from mice from the upper panels.
(G) Liver-to-bodyweight ratios were calculated for MET/CAT mice injected with PX330 or sgEphA2 for 8 weeks (56 days). N = 3 mice per group.
(H) Livers of MET/CAT mice injected with PX330 or sgEphA2 were collected at day 45 for H&E and IHC for HCC and proliferative markers, AFP and Ki67. Scale bars, 100 μm.
(I) Kaplan-Meier plot for experiment illustrated in (B) represented the percent of survival (y axis) at days elapsed after injection (x axis).
Statistical significance was determined by the two-tailed Student’s t test ****p < 0.0001 (G) and log-rank test (I).
Notably, mice receiving sgEphA2–2 had a significant reduction of tumor burden compared to the control on day 45 after the injection, whereas the control mice developed ascites, hepatomegaly, and tumor mass (Figures 3F, 3G, and S2A). Histologic examination of PX330 control livers showed hallmark HCC features, including increased cellular density, nuclear polymorphism, vesicular chromatin, intratumor, and extramedullary hematopoiesis with nucleated red blood cells (Figure 3H). Immunohistochemical staining for alpha-fetoprotein (AFP), an HCC marker, was dramatically lower in the livers of sgEphA2–2 mice compared to PX330 control mice (Figure 3H). Significantly, all of the PX330 control mice died with advanced liver cancer within 37–65 days, whereas sgEphA2-2 mice developed tumors at long latency, as 45% of the mice were symptom-free and were still alive beyond 106 days (Figure 3I). Intriguingly, sgEphA2–2 mice that eventually developed HCC had re-expression of EphA2 in the tumor region (Figures S2B and S2C). These data suggest that deletion of EphA2 was incomplete, resulting in eventual initiation and development of long-latency tumors in these mice. Taken together, our results demonstrate that EphA2 is an important promotor for the initiation and development of MET/CAT induced HCC.
EphA2 promotes the development of HCC partially through activation of the AKT pathway
To investigate the underlying mechanism of EphA2-induced tumor development, we performed RNA sequencing on EphA2 knockdown and scrambled control Huh7 cells. Gene set enrichment analysis (GSEA) revealed that many signaling pathways (Table S3A), including genes induced by AKT signaling (Figure 4A), were affected by EphA2 knockdown. AKT signaling drew our attention because AKT has been shown to play a critical role in promoting HCC development (Galicia et al., 2010; Grabinski et al., 2012; Stauffer et al., 2011). Previous studies have also demonstrated that the modality of EphA2 regulation of AKT is highly variable and tissue-dependent; that is, ligand-induced EphA2 signaling can either activate or inhibit AKT under different conditions (Chang et al., 2008; Miao et al., 2009; Pasquale, 2010). In line with our RNA-sequencing data, we found that phosphorylation of AKT at both Ser473 and Thr308 sites, which are critical for AKT activation (Alessi et al., 1996; Song et al., 2019), was substantially suppressed in both Huh7 and Hep3B EphA2 knockdown cells compared with the scrambled control (Figure 4B). Similarly, knocking out EphA2 decreased AKT activity in our MET/CAT model of HCC (Figures S3A–S3D). Overexpression of EphA2 increased AKT activation in PLC/PRF/5 cells (Figure S3E), which expresses a low level of EphA2 (Figure 2A). Overall, these data suggest that EphA2 promotes AKT signaling in the context of HCC.
Figure 4. EphA2 promotes the initiation and growth of HCC partially through activation of the AKT pathway.
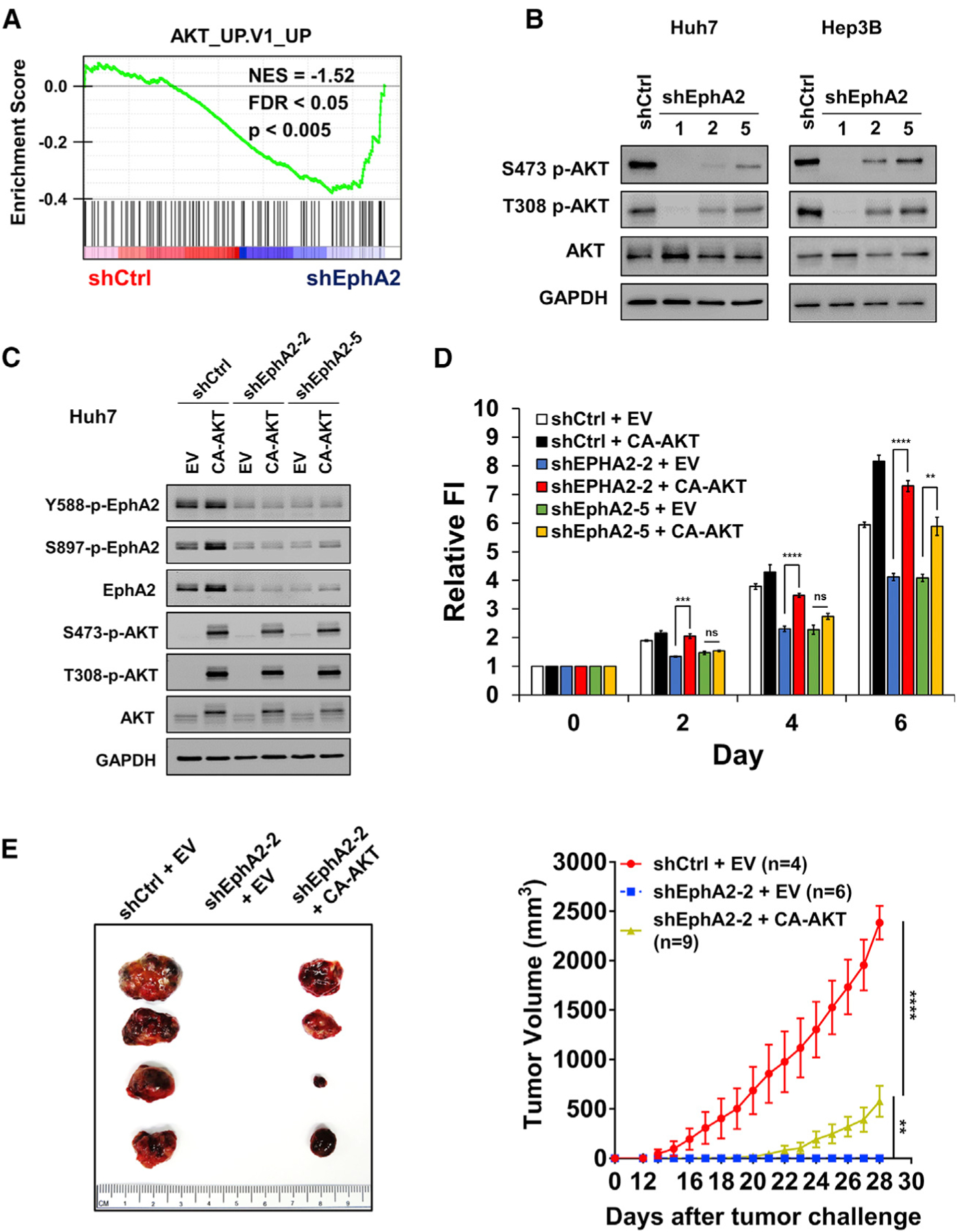
(A) GSEA revealed that genes in the AKT pathway are highly enriched in EphA2 knockdown Huh7 cells.
(B) Cell lysate of EphA2 knockdown Huh7 and Hep3B cells were immunoblotted for p-AKT and total AKT. GAPDH as a loading control.
(C) Western blot validated overexpression of constitutively active AKT (CA-AKT) in the context of two independent shRNA mediated EphA2 knockdown in Huh7 cells.
(D) Cell proliferation study was conducted using cells described in (C) using the Alamar blue assay. Fluorescence intensity was measured at day 0 (6 h after seeding) and subsequently at day 2, 4, and 6. Values are mean ± SD (n = 4).
(E) Left: representative picture of tumors extracted from NSG-A2 mice 28 days after subcutaneous injection with cells described in (C) and (D). n = 4, 6, 9 mice in shCtrl + EV, shEphA2 + EV, and shEphA2 + CA-AKT, respectively. Right: daily measurements of primary tumor size. Values are mean ± SEM.
Statistical significance was determined by two-tailed Student’s t test (D) and one-ANOVA analysis with Dunnett’s multiple comparisons test (E) **p < 0.01, ***p < 0.001 and ****p < 0.0001.
To further validate our hypothesis regarding the relationship between EphA2 and AKT in HCC, we expressed a constitutively active form of AKT (CA-AKT) in the context of EphA2 knockdown Huh7 cells (Figure 4C). Expression of CA-AKT rescued the proliferation of EphA2 knockdown cells in vitro (Figure 4D). Intriguingly, when tumor cells were implanted in our HCC xenograft model, CA-AKT expression only partially rescued tumor growth in EphA2 knockdown cells (Figure 4E). Moreover, MK-2206, a selective inhibitor of pan-AKT, suppressed AKT activity, and HCC cell growth (Figures S4A–S4C). Overall, the results suggest that EphA2 drives the development of HCC partially through AKT signaling.
EphA2 drives HCC tumor development partially through activation of STAT3 signaling
Because expression of CA-AKT only partially rescued tumor development in EphA2 knockdown cells in xenograft models, we suspected that additional signaling pathways might be involved in EphA2 signaling. RNA-sequencing data indicated that the JAK/STAT3 pathway is the second most enriched pathway affected by EphA2 knockdown (Figure 5A; Table S3A), suggesting that STAT3 is a downstream target of EphA2 in HCC. STAT3 is a crucial oncogene that promotes tumor initiation and progression in a majority of human malignancies, including HCC (Calvisi et al., 2006; He and Karin, 2011; He et al., 2010a; Huynh et al., 2019; Johnson et al., 2018; Li et al., 2006; Yu et al., 2014). In addition, STAT3 signaling promotes stem-cell-like properties in cancers by transcription of crucial pluripotent factors such as SOX2 and KLF4, leading to tumor initiation, relapse, and drug resistance (Hall et al., 2009; Huynh et al., 2019; Schroeder et al., 2014; Yu et al., 2014). A previous study showed that EphA2 acts against fungal infection by increasing JAK2/STAT3 signaling (Swidergall et al., 2018); however, no study has reported EphA2 signaling can activate STAT3 in cancer. Although knockdown of EphA2 did not affect STAT3 mRNA expression levels in HCC cells (Figure S5A), EphA2 knockdown in HCC cells suppressed phosphorylation of STAT3 at Tyr705 (Figure 5B), which is critical for STAT3 activation (Darnell et al., 1994). Notably, the level of total STAT3 was also decreased by knockdown of EphA2 in HCC cells (Figure 5B), which might be due to reduced phosphorylation of STAT3, because several studies showed that tyrosine phosphorylation, such as at the Y705 position, stabilizes STAT3 protein (Murase, 2013; Park et al., 2000). Similarly, knocking out EphA2 attenuated STAT3 activity in our MET/CAT HCC model (Figures 5C, S3A, S3C, S3D, and S6A). Additionally, overexpression of EphA2 increased STAT3 signaling in PLC/PRF/5 cells (Figure S3E). These results provide evidence that STAT3 is a downstream effector of EphA2 signaling in HCC. Previous studies showed that STAT3 could be a downstream effector of AKT signaling (Abdelhamed et al., 2016; Yokogami et al., 2000). We found that overexpression of CA-AKT did not affect STAT3 signaling in shEphA2 cells (Figure 5D). Consistently, inhibition of AKT by MK-2206 did not suppress STAT3 activity in parental HCC cells (Figures S4A and S4B). These results suggest that regulation of STAT3 by EphA2 in HCC is not through AKT signaling.
Figure 5. EphA2 drives tumor initiation and development partially through activation of STAT3 signaling.
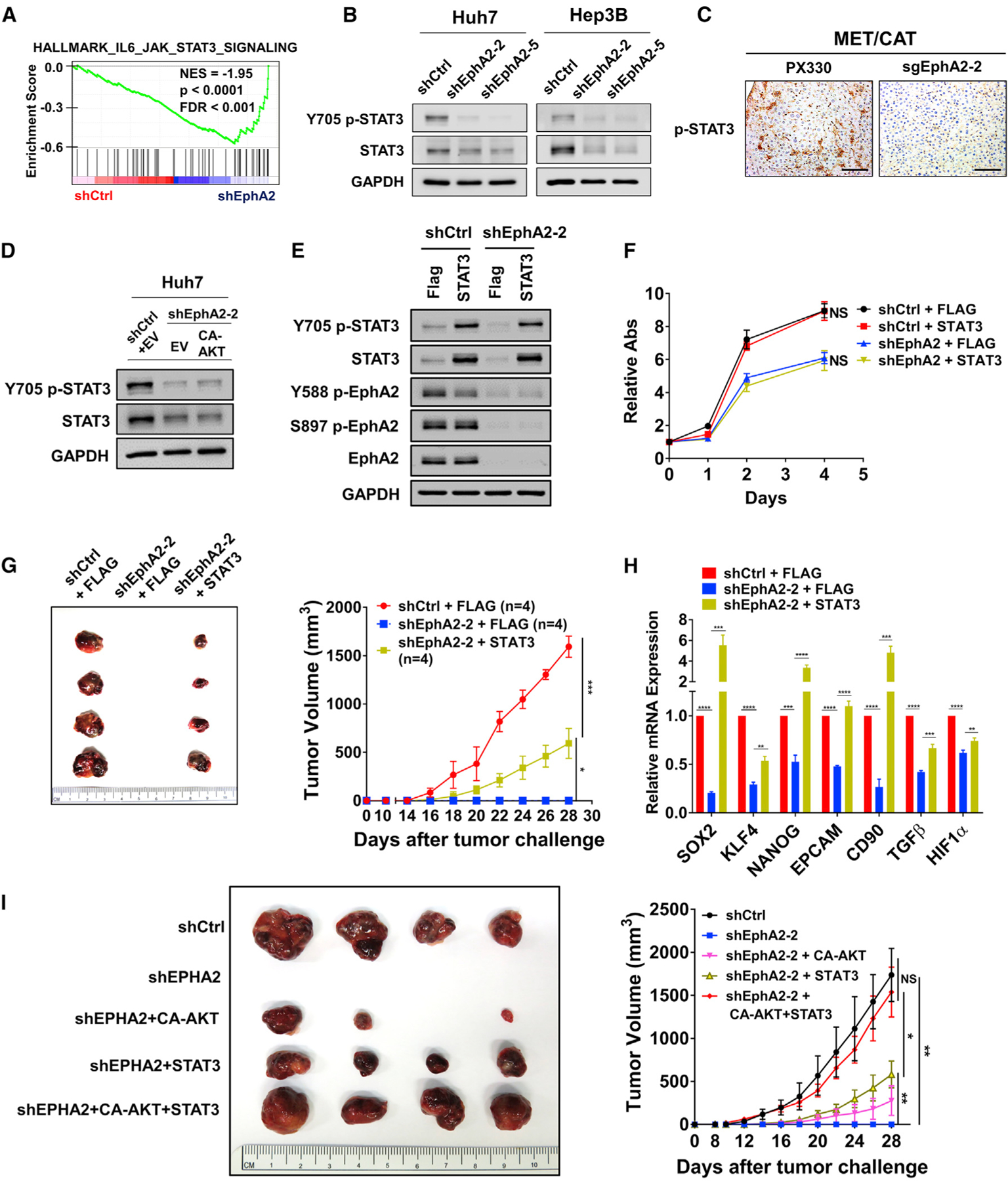
(A) GSEA revealed that genes in the JAK/STAT3 pathway are highly enriched in EphA2 knockdown Huh7 cells.
(B) Cell lysate of EphA2 knockdown Huh7 and Hep3B cells were immunoblotted for p-STAT3 and total STAT3. GAPDH as a loading control.
(C) Liver of EphA2 knockout MET/CAT mice was extracted and immunohistochemically assessed for p-STAT3 expression comparing to control (PX330). Scale bar, 100 μm.
(D) Cell lysate of EphA2 knockdown Huh7 cells with overexpression of CA-AKT was immunoblotted for p-STAT3 and total STAT3.
(E) Western blot validated overexpression of STAT3 in the context of shRNA mediated EphA2 knockdown in Huh7 cells.
(F) Cell proliferation study was conducted using cells described in (C) using the Alamar blue assay. The ratio of proliferation was calculated by normalizing fluorescent intensity to day 0 (6 h after seeding of the cells) and subsequently measured at day 1, 2, and 4 days. Values are mean ± SD (n = 4).
(G) Left: representative picture of tumors extracted from NSG-A2 mice 28 days after subcutaneous injection with cells described in (E) and (F). n = 4 mice in each group. Right: primary tumor size was recorded every 2 days. Values are mean ± SEM.
(H) qPCR analysis of the indicated gene expression in HCC cells described in (F). Values are mean ± SD (n = 3).
(I) Left: representative picture of tumors extracted from NSG-A2 mice 28 days after subcutaneous injection with the indicated cells. n = 4 mice in each group. Right: primary tumor size was recorded every 2 days. Values are mean ± SEM.
Statistical significance was determined by one-ANOVA analysis with Tukey’s multiple comparisons test (F) and (G) and two-tailed Student’s t test (H). NS, not significant; *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001.
Further, we found that STAT3 overexpression had no significant effect on the growth of scrambled Huh7 cells in vitro and in vivo (Figures 5E, 5F, S6B, and S6C). STAT3 overexpression also did not affect the proliferation of EphA2 knockdown Huh7 cells in vitro (Figure 5F). However, BBI608, a STAT3 inhibitor, suppressed STAT3 activity, and HCC cell growth in Huh7 cells without affecting AKT activity (Figures S6D and S6E). The results suggest that overexpression of STAT3 alone is not sufficient to promote HCC cell growth, but STAT3 activity is necessary for HCC cell growth. In the Huh7 xenograft model, overexpression of STAT3 significantly rescued and initiated tumor growth in the EphA2 knockdown condition (Figure 5G), and a repertoire of tumor-initiating and pluripotent factors suppressed in the EphA2 knockdown was dramatically rescued by STAT3 overexpression in HCC cells in vitro (Figure 5H). Overall, these results show that EphA2 drives tumor development also partially through activating STAT3. Furthermore, the combined overexpression of CA-AKT and STAT3 almost completely rescued the reduced tumor development by EphA2 knockdown in a xenograft model (Figures 5I and S6F), and demonstrates that EphA2 drives tumor development mainly through the activation of both AKT and STAT3 signaling.
EphA2 activates STAT3 signaling via JAK1 in HCC
Next, we investigated how EphA2 activates STAT3 in HCC. JAK family non-receptor tyrosine kinases (JAKs) can directly activate STAT3 through canonical activation of STAT3 in many malignancies, including HCC (Johnson et al., 2018; Yu et al., 2014). JAKs can be activated by RTKs such as EGFR, PDGFR, and FGFR (Andl et al., 2004; Huynh et al., 2019), which led us to test whether EphA2 activates STAT3 via JAKs. RNA-sequencing data showed that JAK1 had substantial expression in HCC cells while the expression of JAK2 was modest, and JAK3 expression was very low (Figure S7A). We further confirmed that little JAK2 protein expression was detected in HCC cells by immunoblot (Figure S7B). We, therefore, examined if EphA2 can activate JAK1 in HCC cells. Phosphorylation of JAK1 at Tyr1034/1035, which is critical for activation of JAK1 (Leonard and O’Shea, 1998), was suppressed when EphA2 was knocked down in both Huh7 and Hep3B cells (Figure 6A). Suppression of JAK1 phosphorylation was associated with substantially lower STAT3 activation (Figure 6A). Knocking out EphA2 in the MET/CAT model of HCC also suppressed JAK1 activation (Figures 6B, S3A, S3C, and S3D). Furthermore, we knocked down JAK1 in both Huh7 and Hep3B using small interfering RNA (siRNA) and found that STAT3 signaling was attenuated by JAK1 knockdown (Figure 6C). Moreover, treatment with a JAK inhibitor (pyridine 6) (Thompson et al., 2002) potently inhibited STAT3 signaling in Huh7 cells (Figure S7C).
Figure 6. EphA2 activates STAT3 signaling via JAK1 in HCC, and EphA2 signaling is positively correlated with AKT and JAK1/STAT3 activation in HCC patient samples.
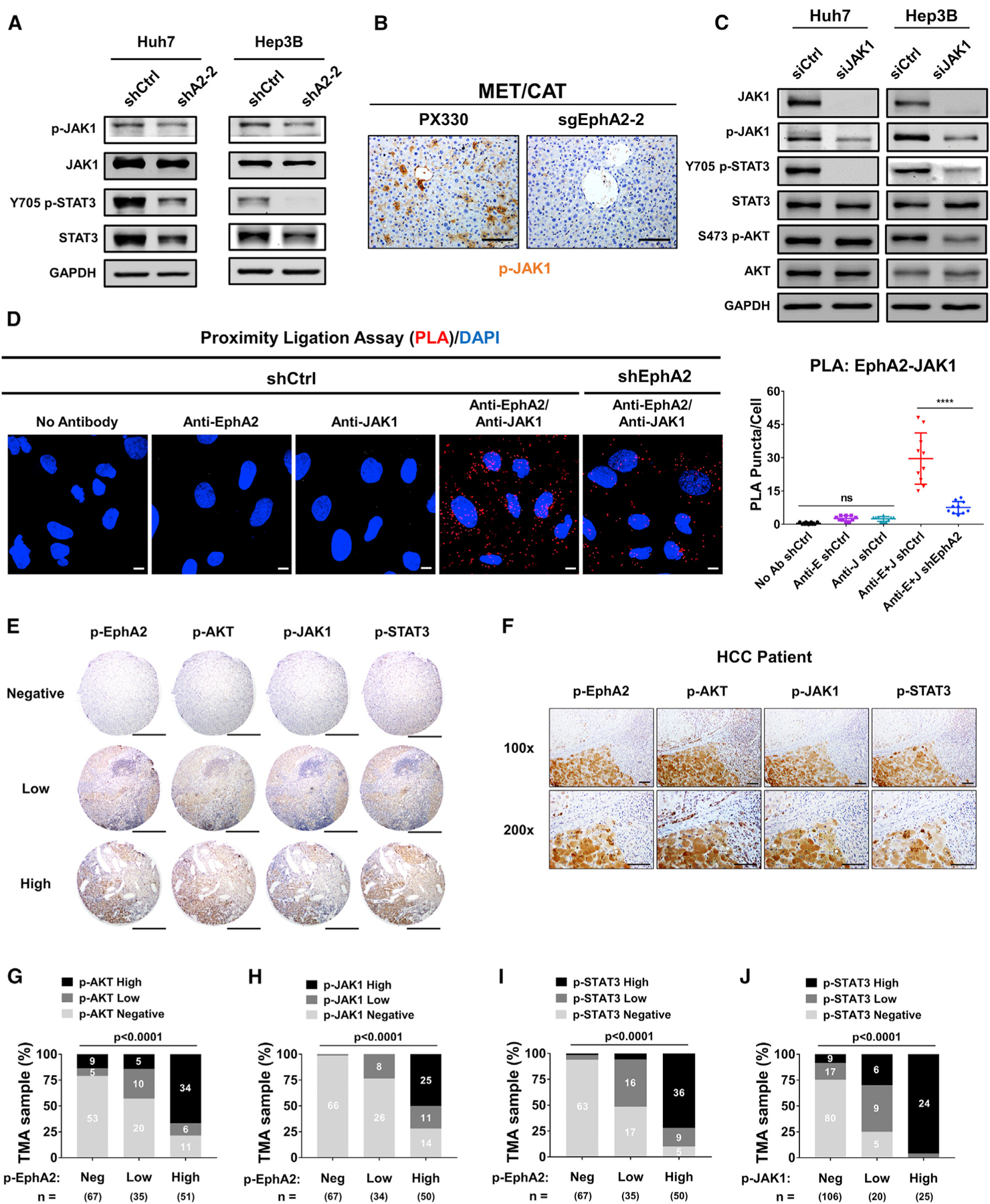
(A) Cell lysate of EphA2 knockdown Huh7 and Hep3B cells were immunoblotted for p-JAK1, JAK1, p-STAT3, and total STAT3. GAPDH as a loading control.
(B) The liver of EphA2 knockout MET/CAT mice was extracted, and IHC was used to assess for p-JAK1 expression comparing to control (PX330). Scale bar, 100 μm.
(C) Huh7 and Hep3B cells were transfected with siRNA targeting JAK1. 48 h after the transfection, the protein was extracted and immunoblotted for JAK1, p-JAK1, p-STAT3, STAT3, p-AKT, and AKT expression.
(D) Left: the interaction of EphA2 and JAK1 was quantified using proximity ligation assay (PLA) in Huh7 scramble (shCtrl) and EphA2 knockdown (shEphA2) cells. Positive PLA interaction (red). Nuclei were stained with DAPI (blue). Scale bar, 10 μm. Right: PLA Puncta per cell was quantified using Imaris Bitplane, n = 10. Statistical significance was determined by one-ANOVA analysis with Tukey’s multiple comparisons test (D). ns, not significant; *p < 0.05, ****p < 0.0001.
(E) Representative immunohistochemistry (IHC) images and correlation of Y588 p-EphA2, p-AKT, p-JAK1, and p-STAT3 in HCC tissue microarray classified as negative, low, or high expression. Scale bars, 500 μm.
(F) Representative high-power magnification of IHC images of HCC showing a correlation between Y588 p-EphA2, p-AKT, p-JAK1, and p-STAT3. Scale bar, 100 μm.
(G–J) Correlation analysis of all HCC TMA tissues between Y588 p-EphA2 and p-AKT (C), p-EphA2 and p-JAK1 (D), Y588 p-EphA2 and p-STAT3 (E), and p-JAK1 and p-STAT3. Statistical significance was determined by a chi-square test (C–F).
Interleukin (IL)-6 can activate JAK1/STAT3 and plays a critical role in HCC development (Bergmann et al., 2017; Lokau et al., 2019). We found no significant decrease in transcript levels of IL-6/GP130 family genes except IL-6ST (a modest reduction) in EphA2 knockdown Huh7 cells compared to scramble control (Figure S5B). We further performed western blot and found the protein levels of GP130 were not significantly changed by EphA2 knockdown (Figure S5C). Besides, secreted IL-6 levels were comparable in cell culture supernatants of EphA2 knockdown and scrambled HCC cells (Figure S5D). These data suggest that the mechanism of EphA2 in promoting STAT3 activity is most likely not due to paracrine or autocrine regulation of IL-6. A previous study showed that EphA4, another member of the Ephrin receptor family, can directly interact with JAK2 and activate STAT3 signaling at the neuromuscular junction (Lai et al., 2004), raising the possibility that EphA2 can activate STAT3 in a similar manner. Notably, using proximity ligation assay (PLA) (Söderberg et al., 2006; Weibrecht et al., 2010), we found notable interaction between EphA2 and JAK1 in the scrambled HCC cells, which was significantly reduced by the EphA2 knockdown (Figure 6D). Taken together, these results indicate that EphA2 promotes STAT3 signaling through activation of JAK1.
EphA2 signaling is positively correlated with AKT and JAK1/STAT3 activation in HCC patient samples
Our data show that EphA2 promotes HCC initiation and development by activating both AKT and JAK1/STAT3 in cell-based and animal models. To determine whether the EphA2/AKT and EphA2/JAK1/STAT3 axes are relevant in human HCC, we examined p-EphA2, p-AKT, p-JAK1, and p-STAT3 by IHC in a tissue microarray including 153 human HCC specimens. Notably, p-EphA2 expression was positively correlated with the activation status of AKT, JAK1, and STAT3 within the same tumor region (Figures 6E–6I). There was also a strong positive correlation between p-JAK1 and p-STAT3 expression, highlighting the importance of JAK1 as an activator of STAT3 in HCC (Figure 6J). Taken together, these results suggest that EphA2 activates both AKT and JAK1/STAT3 signaling in human HCC.
Pharmacologic targeting of EphA2 impairs growth and progression of HCC in vitro and in vivo
So far, we have shown that the receptor tyrosine kinase-EphA2-is a critical oncodriver that promotes the initiation and progression of HCC. Given the success of targeting RTKs with specific small molecule kinase inhibitors (Ferguson and Gray, 2018), we next explored the translational potential of targeting EphA2 with the small molecule inhibitor ALW-II-41–27 (ALW). ALW is a potent and selective EphA2 inhibitor, which was recently shown to exhibit anti-tumor activity in many solid malignancies (Amato et al., 2014, 2016; Choi et al., 2009; Martini et al., 2019). ALW potently impaired the growth of all six HCC cell lines (Figure 7A). In addition, ALW effectively decreased the phosphorylation of EphA2 at Y588 and its downstream effectors (p-AKT, p-JAK1, and p-STAT3) in HCC cells (Figure 7B). To test the effect of ALW on HCC in vivo, we treated mice from a human xenograft model of HCC with ALW and observed the impact on tumor growth. We found that while the vehicle-treated control tumors grew rapidly, the ALW-treated tumors showed complete inhibition of tumor growth for 7 days and notably even showed tumor regression for the first 3 days of treatment (Figure 7C). Furthermore, consistent with in vitro data, tumors of mice treated with ALW showed a dramatic decrease in phosphorylation of EphA2 and its downstream targets p-AKT, p-JAK1, and p-STAT3 (Figure 7D). Overall, these results demonstrated that therapeutic targeting of EphA2 impaired the growth and progression of HCC in vitro and in vivo.
Figure 7. Pharmacologic targeting of EphA2 impairs growth and progression of HCC in vitro and in vivo.
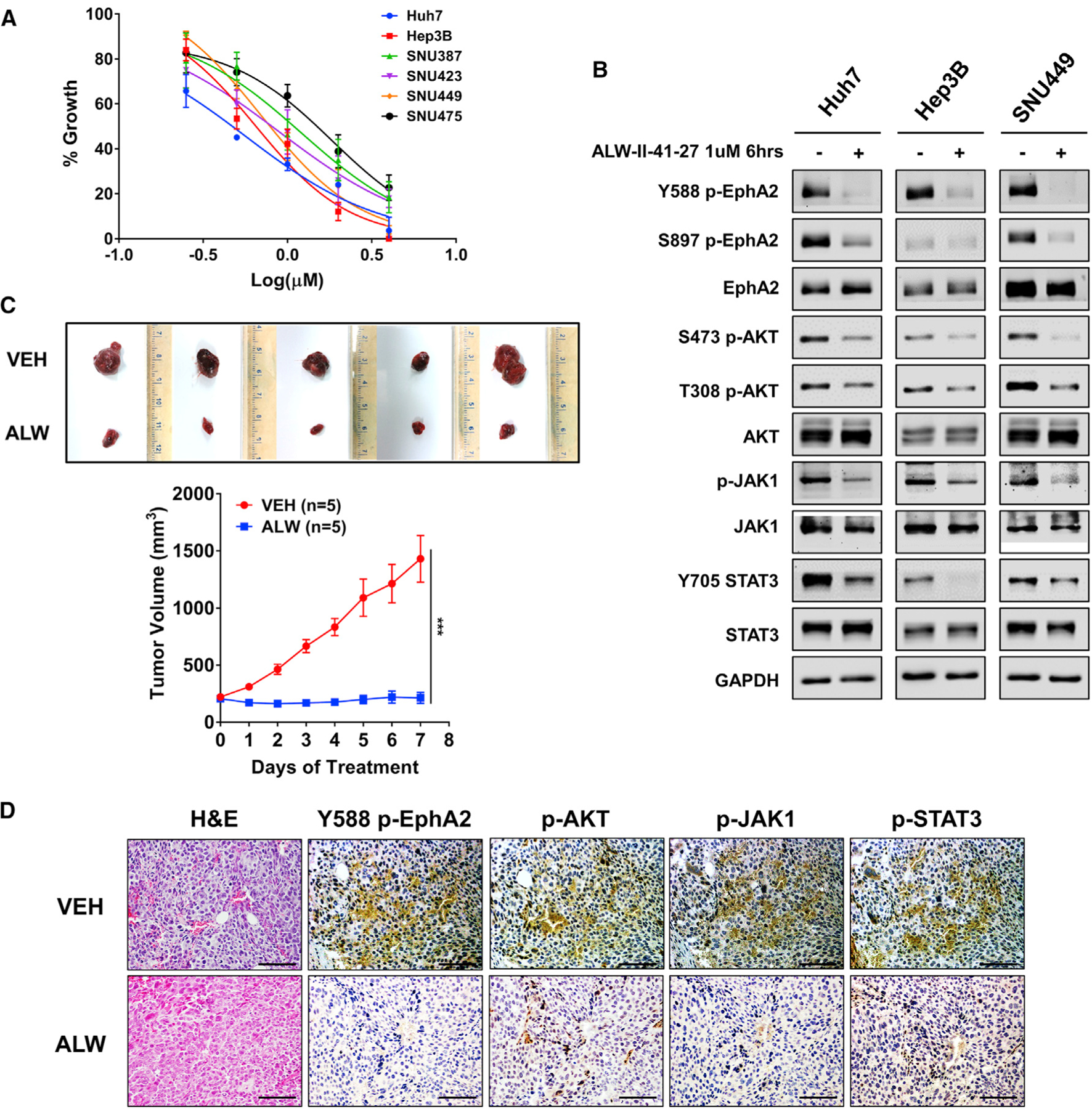
(A) The effect of ALW-II-41–27 on cell proliferation of 6 HCC cell lines was assessed after 48 h of treatment using Alamar blue assay. Data are shown as percent of fluorescence comparing to DMSO control. Values are mean ± SD (n = 4).
(B) Western blot analysis of indicated proteins at the selected time and concentration of ALW-II-41–27 in Huh7, Hep3B, and SNU449 cells. GAPDH as a loading control.
(C) Image of tumors extracted from NSG-A2 mice subcutaneously injected with 5 × 106 Huh7 cells; 7 days after treatment with 15 mg/kg/day ALW-II-41–27 or vehicle. Treatment began after the tumor reached 200 mm3. n = 5 mice per group. Bottom: daily measurements of primary tumor size. Values are mean ± SD. Statistical significance was determined by the two-tailed Student’s t test. ***p < 0.001.
(D) Tumors from mice treated with ALW-II-41–27 or vehicle described in (D) were collected for H&E and assessed for Y588 p-EphA2, p-AKT, p-JAK1, and p-STAT3 expression by IHC. Scale bar 100 μm.
DISCUSSION
HCC remains one of the most lethal malignancies worldwide because of the immense challenges in preventing, diagnosing, and treating the disease (Villanueva, 2019). The currently approved medications for advanced HCC provide patients with limited clinical benefits (Llovet et al., 2015; Villanueva, 2019). A significant barrier to drug development is the lack of understanding of critical drivers of oncogenesis and tumor progression (Llovet et al., 2015). Here, we established EphA2 as an oncogene that promotes tumor initiation and progression in HCC, and inhibition of EphA2 suppressed HCC tumor initiation and growth. We found that high EphA2 signaling and expression is observed in over 33% of HCC patient samples and is associated with worse clinical outcomes. Therefore, many HCC patients may benefit from the inhibition of EphA2 signaling.
The nature of EphA2 signaling is highly diverse and tissue-dependent, and many studies have reported contradictory responses from the same ligand-EphA2 interaction in different cell types (Pasquale, 2010; Wykosky and Debinski, 2008). The reasons for this are still unclear. One possible explanation is that spatial-mechanical properties of EphA2 clustering elicit different downstream signaling based on the chemo-physical properties of the inherent cell type (Himanen et al., 2010; Kania and Klein, 2016; Salaita et al., 2010). Furthermore, tissue-specific responses are driven by the pre-programmed epigenetic signatures across tissue types (Haigis et al., 2019). Thus, differential clustering of EphA2 governed by pre-determined epigenetic landscape allows for a wide array and even contradictory cellular responses across cancers, highlighted by how EphA2 regulates AKT in different cancers. For example, in glioblastoma, ligand-induced EphA2 signaling inactivates AKT by dephosphorylation (Miao et al., 2009). In contrast, the same ligand-induced EphA2 signaling promotes AKT signaling by phosphorylation in pancreatic cancer (Chang et al., 2008). We found that functional inhibition of EphA2 decreases AKT activity in HCC cells, suggesting that EphA2 activates AKT in HCC, further supported by the evidence that EFNA1-Fc (a soluble recombinant EphA2 ligand) activated AKT signaling in HCC cells (Figure S7D). AKT is activated in up to 31.2% of HCC cases and plays a crucial role in the malignant transformation of hepatocytes to HCC (Matter et al., 2014; Villanueva et al., 2008). EphA2 activation is significantly associated with high levels of AKT activity in human HCC (Figure 6G). Therefore, targeting EphA2 could provide an effective therapeutic strategy for AKT-driven HCC. It is notable that shEphA2–2 HCC cell clones expressed less activated AKT (Figure 4B), but proliferated faster than the shEphA2–5 cell clones (Figure 2C). One explanation is that EphA2-mediated HCC cell proliferation is not solely dependent on AKT signaling, and there are other mechanisms involved. It is also possible that alterative or compensatory pathways are activated when knockdown of EphA2 suppresses AKT signaling. We will look into these possibilities in our future studies.
STAT3 is a crucial oncogene in many cancers, including HCC (He and Karin, 2011; Yu et al., 2014). Targeting STAT3 could provide numerous benefits, including inhibition of tumor initiation, growth, metastasis, and resistance to conventional therapy and immunotherapy (Huynh et al., 2019; Johnson et al., 2018; Yu et al., 2014). However, STAT3 is often deemed “undrug-gable” due to the lack of an intrinsic enzymatic site (Wong et al., 2017). Here, we discovered that EphA2 activates STAT3 in HCC, which is further supported by the finding that EFNA1-Fc activated STAT3 in HCC cells (Figure S7D). We found that STAT3 activation by EphA2 in HCC is not through AKT or para-/autocrine regulation of IL-6. Instead, we demonstrated that EphA2 promoted STAT3 through the regulation of JAK1 in HCC cells. Further, we observed a direct interaction between EphA2 and JAK1 in HCC cells. Intriguingly, we found that JAK1 and EphA2 interact in the perinuclear region (Figure 7D), despite JAKs being canonically thought to be exclusively in the cytoplasm associated with transmembrane receptor proteins on the cell membrane (Darnell et al., 1994; Stark et al., 1998). Although the exact mechanism is unclear, EphA2 may exhibit perinuclear endosomal signaling to activate JAK1/STAT3. Many studies have shown that RTKs, on ligand binding and activation, are internalized and can continue to recruit and activate signaling pathways within intracellular vesicles (Kermorgant and Parker, 2008; Sigismund et al., 2008). Furthermore, studies have shown that EphA2 exhibits diverse signaling in endosomes, especially in the perinuclear region (Boissier et al., 2013; Sabet et al., 2015). Reports also demonstrated that EGFR requires clathrin-mediated endocytosis to activate AKT and MAPK (Sigismund et al., 2008). Another RTK, c-MET, has been shown to need trafficking to a perinuclear endosome to activate STAT3 (Kermorgant and Parker, 2008). Thus, we hypothesize that EphA2 activation of JAK1/STAT3 might similarly depend on the perinuclear trafficking of EphA2 and JAK1. Taken together, our results show that EphA2 activates STAT3 by interacting with JAK1 and provides a potential approach to target STAT3 in HCC. Given the critical oncogenic roles of both EphA2 and STAT3 in many cancers, we suspect that the EphA2/JAK1/STAT3 axis is not limited to HCC. For example, previous studies provide independent evidence that either EphA2 or STAT3 is critical for glioma tumor progression and contribute to the glioma stem cell phenotype (Binda et al., 2012; Schaefer et al., 2002; Sherry et al., 2009; Wykosky et al., 2005). The data regarding EphA2 and STAT3 in gliomas suggest that the two pathways might be connected in glioma tumors. Future studies should elucidate the connection between EphA2 and STAT3 in other cancers and explore the therapeutic potential of targeting EphA2, especially in cancers driven by the STAT3 pathway.
Considering the diverse biological functions of EphA2 signaling and the lack of understanding of its functions in HCC, further investigation of its role in HCC is warranted. In addition to the two pathways highlighted in this study, GSEA also revealed oncogenic pathways such as KRAS signaling and angiogenesis (Table S3A), which were regulated by EphA2 signaling in other cancers (Macrae et al., 2005; Sáinz-Jaspeado et al., 2013). Thus, we aim to investigate how EphA2 regulates these oncogenic pathways in HCC in our future studies.
Currently, several EphA2 therapeutics are in clinical trials for cancers in which the oncogenic function of EphA2 is well-established, such as breast cancer, melanoma, and glioblastoma (Boyd et al., 2014). However, a majority of therapies do not target EphA2 functions directly but use EphA2 as bait. For example, MM-310 is a modified EphA2-sensing nanoparticle that delivers docetaxel to EphA2-positive tumors. Although dasatinib has been investigated clinically as an EphA2 inhibitor, it primarily targets SRC and ABL (Gnoni et al., 2011). In this study, we demonstrated the therapeutic potential of targeting EphA2 in HCC by showing that a potent and selective EphA2 inhibitor (ALW-II-41–27) significantly suppresses tumor growth in vitro and in vivo models of HCC. Our findings support the clinical investigation to assess the safety and efficacy of EphA2 inhibitors such as ALWII-41–27 in the treatment of advanced HCC, especially in patients showing activation of EphA2.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
The lead contact for this manuscript is Dr. Wei Qiu (wqiu@luc.edu).
Materials availiability
Further information and requests for resources generated in this study should be directed to and will be fulfilled by the lead contact.
Data and code availiability
The datasets and code utilized in this study are available at GEO: GSE141880.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
HCC Cell lines were acquired from the American Type Culture Collection (ATCC®) or the Japanese Cancer Research Resources Bank (JCRB). Huh7, Hep3B, and PLC/PRF/5 were maintained in Dulbecco’s modified Eagle’s (DMEM) high glucose medium (Thermo Scientific, Waltham, MA) supplemented with 10% fetal bovine serum (Tissue Culture Biologicals), penicillin/streptomycin (Sigma-Aldrich, St. Louis, MO) in a humidified atmosphere of 5% CO2 at 37°C. SNU387, SNU423, SNU449, and SNU475 were cultured in RPMI 1640 supplemented with 10% fetal bovine serum (Tissue Culture Biologicals), 1× penicillin/streptomycin (Sigma-Aldrich, St. Louis, MO) at 37°C and 5% CO2.
Mice Models
All animals received humane care according to the “Guide for the Care and Use of Laboratory Animals” (https://login.archer.luhs.org/login?qurl=http://oacu.od.nih.gov%2fac_cbt%2fguide3.htm). All animal procedures were approved by the Institutional Animal Care and Use Committee at Loyola University Chicago. The mice were housed in micro-isolator cages in a room illuminated from 7:00 AM to 7:00 PM (12:12-hr light-dark cycle) and allowed access to water and chow ad libitum.
For xenograft studies: NSG-A2 mice were purchased from Jackson Laboratory. Mice were all 8 – 10 weeks old, 1:1 males to female, and evenly distributed in the experimental and control groups. For the EphA2 knockdown and AKT and STAT3 rescue studies, 3×106 cells were injected into the right and/or left flank of the mice (depending on the experiment). Mice were monitored for tumor growth by a digital caliper. Growth volume was calculated as volume = length × width2/2. The tumor was extracted for the analysis described in the paper. The ALW-II-41–27 study method was adopted by Dr. Jin Chen Lab (Vanderbilt) (Song et al., 2017). 5×106 cells were injected into the right flank of the mice. When the tumor reached ≥ 200 mm3 at day 10, mice were treated with 15 mg/kg of ALW-II-41–27 dissolved in 10%1-methyl-2-pyrrolidinone, and 90% polyethylene glycol 300 (Sigma) injected intraperitoneally once daily for seven days. Tumors were measured daily, and tumor volume was calculated as described previously.
C57BL/6 mice were purchased from Jackson Laboratory the in vivo CRISPR and MET/CAT HCC models. Mice were all 8 – 10 weeks old, 1:1 male to female, and evenly distributed in experimental and control groups. To examine whether EphA2 is activated in the MET/CAT model (Shang et al., 2015), 50 μg of total plasmids, encoding the Sleeping Beauty transposase (HSB2) and transposons with oncogenes MET/CAT (22.5μg pT3-EF1α-c-MET(human) + 22.5μg pT3-EF1α-ΔN90-β-catenin (human)+ 5μg HSB2) (Shang et al., 2015), were injected hydrodynamically into age- and sex-matched mice. Mice were maintained on the standard diet and euthanized after seven weeks, and livers were collected for analysis. To knockout EphA2 in the MET/CAT-induced HCC model, age-and-sex-matched C57BL/6J mice were divided into two groups: one received MET/CAT + PX330 control, while the other received MET/CAT+ sgEphA2–2. 72.5 μg of total plasmids was dissolved in 2 mL of 0.9% saline and hydrodynamically injected in each mouse. The plasmids encode the single vector PX330-sgEphA2-CRISPR CAS9 (sgEphA2) or pX330-U6-Chimeric_BB-CBhhSpCas9 (PX330, empty vector control), Sleeping Beauty transposase (HSB2) and transposons with oncogenes MET/CAT (22.5μg sgEphA2 or 22.5 μg PX330 + 22.5μg pT3-EF1α-c-MET(human) + 22.5μg pT3-EF1α-ΔN90-β-catenin (human) + 5 μg HSB2). Mice were monitored for approximately 6–9 weeks total as this is the average period for MET/CAT mice to develop end-stage HCC symptoms, including ascites and liver failure. Mice were then euthanized, and livers were collected for analysis.
METHOD DETAILS
Plasmids
The plasmids pT3-internal ribosome entry site (IRES) - green fluorescent protein (GFP), pT3-CAT, and pT3-MET and pT3-EphA2 were generated by Gateway cloning (Shang et al., 2015). sgEphA2–1, sgEphA2–2, and sgEphA3 target sequences were designed based on library and protocol from Feng Zhang Lab (Cong et al., 2013), and the final plasmids pX330-sgEphA2–1, pX330-sgEphA2–2, pX330-sgEphA2–3 were constructed by cloning EphA2-target sequences into pX330-U6-Chimeric_BB-CBh-hSpCas9 backbone following the protocol from Feng Zhang Lab (Cong et al., 2013). The plasmids were purified using the GeneJET Plasmid Maxiprep Kit (Thermo Fisher Scientific) for hydrodynamic tail vein injection.
The following plasmids were purchased: pCMV-dR8.2 dvpr (Addgene, #8455). pCMV-VSV-G (Addgene, #8454), pLKO.1-puroshEphA2–1(Millipore Sigma, TRCN0000231647), pLKO.1-puro-shEphA2–2 (Millipore Sigma, TRCN0000195734), pLKO.1-puroshEphA2–5 (Millipore Sigma, TRCN0000231648), FG12 (Addgene, #14884), pMD2.G (Addgene, #12259), pEXP304-STAT3/V5 (DNASU, HsCD00443857). The pEXP304-FLAG/V5 plasmid was generously donated from Dr. Takashi Shimamura (University of Illinois Chicago).
TMA analysis
IHC was performed as previously described (Shang et al., 2015). Human tissue microarrays (TMAs) were obtained from the Mayo Clinic. 153 cases of HCC and 63 non-tumor liver tissues (free of significant pathologies) were analyzed. The source of patients, demographic information, and their clinical presentations are shown in Table S1. An IRB (#707–03) was approved by the Mayo Clinic for this study. We summarized categorical data as frequency counts and percentages, and continuous measures as means, standard deviations, medians, and ranges. Categorical variables were compared using the chi-square test or Fisher’s exact test. Continuous variables were compared using the one-way ANOVA test or Kruskal–Wallis test.
Western Blot Analysis
Cells/tissue lysate was prepared in RIPA lysis, and extraction buffer (Thermo Scientific) supplemented with proteinase (Thermo-Scientific A32953) and phosphatase (Thermo-scientific A32957). Protein was quantified using the Pierce protein assay (Thermo-Scientific #1861426). SDS–polyacrylamide gel electrophoresis was performed using 10% acrylamide gels, and proteins were transferred to nitrocellulose membranes using the Trans-Blot Turbo transfer system (Bio-Rad). Membranes were blocked in 5% milk for 1 hour and incubated overnight at 4°C with primary antibodies with manufacturer-recommended concentrations in 5% bovine serum albumin. After several washes with TBS-T (20 mM Tris (pH 7.6), 140 mM NaCl, and 0.1% Tween 20), blots were incubated with the appropriate HRP-conjugated secondary antibody (Thermo-Fisher Scientific) and imaged using the iBrightCL1000 (Thermo-Fisher). Signals were quantified using ImageJ.
Immunohistochemistry and immunofluorescence
Mouse liver tissues were dissected and placed in 10% buffered formalin overnight at room temperature and dehydrated in a series of graded alcohol. Human HCC specimens were provided by Loyola University Medical Center Pathology Core, and human and mouse liver tissues were paraffin-embedded by the Loyola Pathology Core. Human HCC tissue microarrays were provided by Dr. Robert Lewis and the Mayo Clinic Hepatobiliary SPORE. Tissue blocks were cut into 4 μm sections, dewaxed, and rehydrated. After heat-mediated-citrate-based antigen retrieval, slides were then washed in TBS and blocked using 5% goat serum for 1 h at room temperature before incubation with primary antibodies for 1 hour: Y588 p-EphA2 (1:100), EphA2 (1:200), s473 p-AKT (1:200), p-JAK1 (1:100), p-STAT3 (1:100), AFP (1:200), Ki67 (1:200). Slides were washed in TBST and incubated with biotin-conjugated goat secondary antibody (Thermo Fisher Scientific, depending on the type of species) for 1 hour. Next, slides were incubated with VECTASTAIN® ABC HRP reagent for 30 min at room temperature. Slides were washed in TBST followed by detection by DAB staining for 5 min (Vector Laboratories, SK-4100) for 5 min and finally counterstained with hematoxylin. The IHC signals were quantified visually. The staining was scored as – (0, negative), + (1, weak signal), + + (2, moderate signal), + + + (3, strong signal) by three independent observers, including a pathologist from Loyola University Chicago, who was masked as to the patient outcome. The average score k was calculated and categorized as negative (k = 0), low (0 < k < 1.5), or high (k > 1.5)
For immunofluorescence, following the incubation with primary antibody and TBST wash, fluorophore-conjugated goat secondary antibody (Thermo Fisher Scientific, depending on the species) was incubated for 1 hour. Slides were then washed in TBST followed by nuclear staining with DAPI mounting media (Vector Laboratories, H-1200)
Transient transfection
PLC/PRF/5 cells were transfected with pT3-EF1α-EphA2 or control pT3-EF1α-GFP plasmids by lipofectamine 3000 (Invitrogen, L3000001) according to the manufacturer’s instructions. Forty-eight hours after transfection, total cell lysates were collected used in western blotting.
Lentivirus particle production and transduction
All lentiviral particles were produced in HEK293 cells. The protocol was based on Addgene’s lentivirus production protocol with modifications. HEK293 packaging cells were plated onto 10 cm dishes at 3×106 cells for overnight. Two packaging plasmids pCMV-dR8.2 dvpr (Addgene, #8455) pCMV-VSV-G (Addgene, #8454) plus pLKO-based transfer plasmids were diluted in Opti-MEM (GIBCO) with 1 mg/ml PEI at the DNA: PEI ratio of 1:4. Plasmid mixtures were transfected into cells, and the media was replaced with complete DMEM 18 hours after transfection. Lentivirus was harvested 72 hours post-transfection. Collected media was centrifuged at 500 × g for 10 min, and the supernatant was extracted and stored at −80°C. Lentivirus transduction was performed following the Addgene PLKO.1 protocol.
Gene knockdown
Knockdown of EphA2 in HCC cell lines was performed using lentiviral mediate shRNA expression. pLKO.1-puro-shEphA2–1(CCGGCCATCAAGATGCAGCAGTATACTCGAGTATACTGCTGCATCTTGATGGTTTTTG), pLKO.1-puro-shEphA2–2 (CCGGGATAAGTTTCTATTCTGTCAGCTCGAGCTGACAGAATAGAAACTTATCTTTTTTG), pLKO.1-puro-shEphA2–5 (CCGGTCGGACAGACATATAGGATATCTCGAGATATCCTATATGTCTGTCCGATTTTTG) and scramble control were purchased from Millipore Sigma. Lentiviral particles were produced in HEK293 cells. Cells were selected with puromycin (1 μg/mL).
Knockdown of JAK1 in HCC cell lines was performed using siRNA. siGENOME SMARTpool siRNA targeting JAK1 (CCACAUAGCUGAUCUGAAA; UGAAAUCACUCACAUUGUA; UAAGGAACCUCUAUCAUGA; GCAGGUGGCUGUUAAAUCU) was purchased from Dharmacon/Horizon and transfected into HCC cells at a concentration of 100 nM using Lipofectamine RNAiMAX reagent (Thermo-Fisher, #13778075) prepared in OptiMEM (Thermo-Fisher, #31985070) according to manufacturer’s protocol. After 48 hours, knockdown efficiency was assessed by western blot.
Cell Viability Assays
HCC cell lines acquired from American Type Culture Collection (ATCC®) or the Japanese Cancer Research Resources Bank (JCRB) were cultured in DMEM or RPMI 1640 supplemented with 10% fetal bovine serum (Tissue Culture Biologicals), 1 × penicillin/streptomycin (Sigma-Aldrich, St. Louis, MO) at 37°C and 5% CO2.
For cell proliferation studies, HCC cells were seeded into 96-well plates (5 × 103 cells/well). At indicated time points, culture media was removed, and alamarBlue (BUF012A; BioRad, Hercules, CA) solution (1:10 dilution in DMEM) was added to the cells. After a 4-hours-incubation at 37°C, fluorescence values were measured with a fluorescent plate reader at 530–560 nm excitation/590 nm emission.
For IC50 of ALW-II-41–27(ALW) studies, HCC cells were seeded into 96-well plates (5 × 103 cells/well). After 24 hours, cells were treated with 0, 0,25, 0.5, 1, 2, or 4 μM of ALW. After 48 hours, culture media was removed, and alamarBlue (BUF012A; BioRad, Hercules, CA) solution (1:10 dilution in DMEM) was added to the cells. After 4 hours of incubation at 37°C, fluorescence values were measured with a fluorescent plate reader at 530–560 nm excitation/590 nm emission.
For IC50 of MK-2206 and BBI608 studies, HCC cells were seeded into 96-well plates (5 × 103 cells/well). After 24 hours, cells were treated with different doses (0–10 μM) of MK-2206 (S1078, Selleck chemicals) or BBI608 (S7977, Selleck chemicals). According to the manufacturer’s instructions, after 48 hours, cell viability was measured with CCK-8 kit (Dojindo, CK04).
High-resolution hepatic ultrasound
Hepatic ultrasound was performed using micro-ultrasound system (Vevo 2100, Visualsonics) with 40 MHz ultrasound transducer (MS550D, Visualsonics) by the Small Animal Core Facility at Loyola University Chicago Health Sciences Division.
RNA isolation, RNA sequencing, dataset analysis, and qPCR
All total RNA was extracted using the RNeasy Plus Mini Kit (QIAGEN) following the manufacturer’s protocol. RNA-seq was performed by Novogene Corporation and analyzed by Dr. Jun Li from the University of Notre Dame. Gene set enrichment analysis was performed using GSEA software (Broad MIT, http://www.gsea-msigdb.org/gsea/index.jsp). A detailed user guide can be found from https://www.gsea-msigdb.org/gsea/doc/GSEAUserGuideFrame.html. In brief, expression database files were prepared, and data files were then loaded into GSEA. The analysis parameters (Hosted MSigDB Gene Sets, H1, and C6) were set, and analysis was run as the user guide indicated.
All RNA expression data from The Cancer Genome Atlas Liver Hepatocellular Carcinoma cohort was analyzed using Gene Expression Profiling Interactive Analysis (GEPIA) (Tang et al., 2017). The data of GSE14520 was retrieved from the Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=gse14520). To create the Kaplan-Meier plots, Python 3.0 was used to analyze the original data. Specifically, censored data was generated by dividing patients into two groups. The censored group consisted of patients who had not experienced death by the end of the study time. Patient data was also separated into two groups based on the expression level indicated by expression values (mean of 25% top versus 25% bottom). Binary censored data, binary expression group data, and survival times were imported into R 3.6.0. Kaplan-Meier plots were created using survival, survminer, dplyr packages, Surv, survfit, and ggsurvplot functions. P-values were generated by including the command “pval = TRUE” in the ggsurvplot function from the survminer library. Hazard ratios for 3 years survival were analyzed based on the median cutoff, and the p value is for the effect of each dichotomized gene in univariable Cox proportional hazards analysis.
For qPCR, 1 μg of RNA was reverse transcribed with iScript cDNA Synthesis Kit (Bio-Rad). qRT-PCR was performed with iTaq Universal SYBR Green Supermix (Bio-Rad) on the Real-Time PCR System. Primer pairs were selected from the Primer Bank (https://pga.mgh.harvard.edu/primerbank/) (Table S3B). Relative expression values for each gene of interest were obtained by normalizing to GAPDH mRNA expression using the ΔΔCt method.
Proximity Ligation Assay
Proximity ligation assay (PLA) was performed using the Duolink® In Situ Red Starter Kit Mouse/Rabbit (Millipore Sigma) according to the manufacturer’s instructions. In brief, cells were seeded on an 8 well-Nunc Lab-Tek II CC2 Chamber Slide System (Thermo Fisher) at 17.5×103/well overnight, then fixed with 4% paraformaldehyde for 30 min at room temperature and washed in PBS, followed by permeabilization with 0.1% Triton X-100 for 10 min. After washing with Wash Buffer A (Millipore Sigma) followed by blocking with Duolink Blocking Buffer (Millipore Sigma) for 30 min at room temperature, cells were incubated with primary antibodies (EphA2, 1:200 Santa Cruz and JAK1, 1:100, Cell Signaling) overnight at 4°C. The next day, cells were washed repeatedly in Wash Buffer A, followed by incubation with appropriate Duolink secondary antibodies (Millipore Sigma) for 1 hour at 37°C according to the manufacturer’s protocol. After washing with Wash Buffer A at room temperature, ligation, and amplification steps of the PLA were performed according to the manufacturer’s protocol. After final washes with Wash Buffer B at room temperature, slides were mounted with Corning® 24×50 mm Rectangular #1 Cover Glass (Corning) using Duolink® In Situ Mounting Medium with DAPI (Millipore Sigma).
EFNA1-FC experiment
The stock solution of Recombinant Human Ephrin-A1 Fc Chimera Protein (R&D Systems; EFNA1-Fc) was prepared in PBS at 100 μg/mL and diluted to 0.1 μg/mL in DMEM based culture medium. After HCC cells were seeded in a 6-well-culture plate 1.7×105/well overnight, the medium was removed, and the cells were treated with 0.1 μg/mL EFNA1-Fc for 0, 2, 4, 8, 12, 16, 24, or 32 hours. Cell lysates were collected at these experimental times and analyzed by western blot.
IL6 ELISA analysis
The culture supernatants from the scrambled and EphA2 knockdown Huh7 cells were collected, and IL-6 protein levels were measured using Human IL-6 ELISA Kit (RAB0306, Sigma-Aldrich), according to the manufacturer’s instructions.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical tests used are indicated in the figure legends. Student’s t test (unpaired, two-tailed) was used to assess significance between experiment and control groups. One-way ANOVA was used to determine if there is statistical significance between multiple experiment groups. The log-rank test was used to assess the significance of differences in survival between the control and EphA2 knockout group. Chi-square test was used to determine the significance of correlations in TMAs. p < 0.05 was considered significant. plots and statistical analyses were done using Prism version 7 software (GraphPad) and Excel.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| EphA2 (C-3) | Santa Cruz | sc-398832 |
| EphA2 (phospho Y588) (D7X2L) | Cell signaling | 12677S; RRID:AB_2797989 |
| EphA2 (phospho S897) (D9A1) | Cell signaling | 6347S; RRID:AB_11220420 |
| Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody | Thermo Fisher Scientific | 31432; RRID:AB_228302 |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody | Thermo Fisher Scientific | 31462; RRID:AB_228338 |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Biotin | Thermo Fisher Scientific | 31822; RRID:AB_228337 |
| Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Biotin | Thermo Fisher Scientific | 31802; RRID:AB_228301 |
| AKT (phospho S473) (D9E) | Cell signaling | 4060S; RRID:AB_2315049 |
| AKT (phospho T308) (244F9) | Cell signaling | 4056S; RRID:AB_331163 |
| AKT | Cell signaling | 9272S; RRID:AB_329827 |
| β-Actin | Sigma-Aldrich | A5441; RRID:AB_476744 |
| GADPH (GA1R) | Thermo Fisher Scientific | MA5–15738; RRID:AB_10977387 |
| GP130 | R&D | MAB2281; RRID:AB_2296041 |
| STAT3 (phospho Y705) (D3A7) | Cell signaling | 9145S; RRID:AB_2491009 |
| STAT3 (124H6) | Cell signaling | 9139; RRID:AB_331757 |
| MET (D1C2) | Cell signaling | 8198; RRID:AB_10858224 |
| β-Catenin | BD Biosciences | 610153; RRID:AB_397554 |
| Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific | A-11001, RRID:AB_2534069 |
| Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | Thermo Fisher Scientific | A-11037; RRID:AB_2534095 |
| Alpha-1-Fetoprotein Pathology Antibody | Agilent | A0008; RRID:AB_2650473 |
| Ki-67 (D3B5) | Cell signaling | 12202; RRID:AB_2620142 |
| JAK1 (phospho Y1034/1035) (D7N4Z) | Cell signaling | 74129; RRID:AB_2799851 |
| Jak1 (6G4) | Cell signaling | 3344, RRID:AB_2265054 |
| Bacterial and virus strains | ||
| MAX Efficiency Stbl2 Competent Cells | Thermo Fisher Scientific | 10268019 |
| MAX Efficiency DH5α Competent Cells | Thermo Fisher Scientific | 18258012 |
| Biological samples | ||
| Normal + HCC human liver samples | Mayo Clinic Rochester; Dr. Lewis R. Roberts | https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga |
| Chemicals, peptides, and recombinant proteins | ||
| ALW-II-41–27 | APExBIO | A3165 |
| Critical commercial assays | ||
| AlamarBlue® | BIO-RAD | BUF012B |
| GeneJET Endo-Free Plasmid Maxiprep Kit | Thermo Fisher Scientific | K0861 |
| RNeasy Mini Kit | QIAGEN | 74106 |
| Duolink® In Situ Red Starter Kit Mouse/Rabbit | Millipore Sigma | DUO92101 |
| Deposited data | ||
| RNaseq EphA2 knockdown | This Paper | Database: GEO, GSE141880 |
| RNaseq expression analysis of HCC and normal liver tissue | Database: TCGA | https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga |
| Experimental models: cell lines | ||
| Huh7 | JCRB | JCRB0403 |
| Hep3B | ATCC | HB-8064 |
| PLC/PRF/5 | ATCC | CRL-8024 |
| SNU387 | ATCC | CRL-2237 |
| SNU423 | ATCC | CRL-2238 |
| SNU449 | ATCC | CRL-2234 |
| SNU475 | ATCC | CRL-2236 |
| Experimental models: organisms/strains | ||
| NSG-A2 | Jackson Laboratory | 009617 |
| C57BL/6 | Jackson Laboratory | 000664 |
| Oligonucleotides | ||
| Primer sequences for qRT-PCR | Table S3B | N/A |
| Human EphA2 shRNAshEphA2–1: CCGGCCATCAAGATGCAGCAGTATACTCGAGTATACTGCTGCATCTTGATGGTTTTTG | Millipore Sigma | TRCN0000231647 |
| Human EphA2 shRNAshEphA2–2: CCGGGATAAGTTTCTATTCTGTCAGCTCGAGCTGACAGAATAGAAACTTATCTTTTTTG | Millipore Sigma | TRCN0000195734 |
| Human EphA2 shRNAshEphA2–5: CCGGTCGGACAGACATATAGGATATCTCGAGATATCCTATATGTCTGTCCGATTTTTG | Millipore Sigma | TRCN0000231648 |
| Human JAK1: siGENOME SMARTpool siRNA | Dharmacon/Horizon | SO-2796483G |
| Human JAK1: CCACAUAGCUGAUCUGAAA | Dharmacon/Horizon | D-003145–05 |
| Human JAK1: UGAAAUCACUCACAUUGUA | Dharmacon/Horizon | D-003145–06 |
| Human JAK1: UAAGGAACCUCUAUCAUGA | Dharmacon/Horizon | D-003145–07 |
| Human JAK1: GCAGGUGGCUGUUAAAUCU | Dharmacon/Horizon | D-003145–08 |
| Mouse EphA2 sgRNAsgEphA2–1: CAACGTGGTATCCGGCGACC | Dr. Feng Zhang; Cong et al., 2013 | MGLibA_16318 |
| Mouse EphA2 sgRNAsgEphA2–2: TTCGCTGTCGAAGCACGCAA | Dr. Feng Zhang; Cong et al., 2013 | MGLibA_16319 |
| Mouse EphA2 sgRNAsgEphA2–3: CTGCTGACCGTGATCTCGTC | Dr. Feng Zhang; Cong et al., 2013 | MGLibA_16320 |
| Recombinant DNA | ||
| pCMV-dR8.2 dvpr | Stewart et al., 2003 | 8455; RRID:Addgene_8455 |
| pCMV-VSV-G | Stewart et al., 2003 | 8454; RRID:Addgene_8454 |
| pLKO.1-puro-shEphA2–1 | Millipore Sigma | TRCN0000231647 |
| pLKO.1-puro-shEphA2–2 | Millipore Sigma | TRCN0000195734 |
| pLKO.1-puro-shEphA2–5 | Millipore Sigma | TRCN0000231648 |
| FG12 | Qin et al., 2003 | 14884; RRID:Addgene_14884 |
| FG12-CMV-CA-AKT | Dr. Mitchell Denning, LUC | Yadav and Denning, 2011 |
| pMD2.G | Dr. Didier Trono | 12259; RRID:Addgene_12259 |
| pLX304 | Yang et al., 2011 | RRID:Addgene_25890 |
| pEXP304-STAT3/V5 | Sinnberg et al., 2016; DNASU | HsCD00443857; PMID: 27428425 |
| pEXP304-FLAG/V5 | Dr. Takashi Shimamura, UIC | N/A |
| pT3-CAT | Khanna et al., 2004; Shang et al., 2015 | N/A |
| pT3-MET | Khanna et al., 2004; Shang et al., 2015 | N/A |
| pT3-GFP | Shang et al., 2015 | N/A |
| pT3-EphA2 | This Paper | N/A |
| pX330-U6-Chimeric_BB-CBh-hSpCas9 (PX330) | Dr. Feng Zhang; Cong et al., 2013 | Addgene plasmid # 42230; RRID:Addgene_42230 |
| pX330-sgEphA2–1 | This Paper | N/A |
| pX330-sgEphA2–2 | This Paper | N/A |
| pX330-sgEphA2–3 | This Paper | N/A |
| Software and algorithms | ||
| PRISM | GraphPad Software | Version 7 |
| GEPIA | Tang et al., 2017 | http://gepia.cancer-pku.cn/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| GSEA | Broad Institute | http://www.gsea-msigdb.org/gsea/index.jsp |
| Bitplane Imaris | Oxford Instruments | https://imaris.oxinst.com/packages |
Highlights.
High EphA2 signaling is associated with worse clinical outcomes in HCC patients
Inhibition of EphA2 suppresses HCC growth both in vitro and in vivo
EphA2 promotes hepatocarcinogenesis by dual activation of AKT and JAK1/STAT3 signaling
EphA2 inhibitor ALW-II-41–27 suppresses HCC growth
ACKNOWLEDGMENTS
We thank Dr. Andrew Dingwall, Dr. Nancy Zeleznik-Le, and Dr. Jiwang Zhang for their helpful advice on this project. This work was supported by the American Cancer Society (RSG-18-107 to W.Q.) and the National Cancer Institute (R01CA197128 to W.Q. and P50CA210964 to Mayo Clinic Hepatobiliary SPORE). The content is solely the authors’ responsibility and does not necessarily represent the official views of the American Cancer Society or the National Cancer Institute.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.108765.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abdelhamed S, Ogura K, Yokoyama S, Saiki I, and Hayakawa Y (2016). AKT-STAT3 Pathway as a Downstream Target of EGFR Signaling to Regulate PD-L1 Expression on NSCLC cells. J. Cancer 7, 1579–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, and Hemmings BA (1996). Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 15, 6541–6551. [PMC free article] [PubMed] [Google Scholar]
- Amato KR, Wang S, Hastings AK, Youngblood VM, Santapuram PR, Chen H, Cates JM, Colvin DC, Ye F, Brantley-Sieders DM, et al. (2014). Genetic and pharmacologic inhibition of EPHA2 promotes apoptosis in NSCLC. J. Clin. Invest 124, 2037–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amato KR, Wang S, Tan L, Hastings AK, Song W, Lovly CM, Meador CB, Ye F, Lu P, Balko JM, et al. (2016). EPHA2 Blockade Overcomes Acquired Resistance to EGFR Kinase Inhibitors in Lung Cancer. Cancer Res. 76, 305–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andl CD, Mizushima T, Oyama K, Bowser M, Nakagawa H, and Rustgi AK (2004). EGFR-induced cell migration is mediated predominantly by the JAK-STAT pathway in primary esophageal keratinocytes. Am. J. Physiol. Gastrointest. Liver Physiol 287, G1227–G1237. [DOI] [PubMed] [Google Scholar]
- Bergmann J, Müller M, Baumann N, Reichert M, Heneweer C, Bolik J, Lücke K, Gruber S, Carambia A, Boretius S, et al. (2017). IL-6 trans-signaling is essential for the development of hepatocellular carcinoma in mice. Hepatology 65, 89–103. [DOI] [PubMed] [Google Scholar]
- Binda E, Visioli A, Giani F, Lamorte G, Copetti M, Pitter KL, Huse JT, Cajola L, Zanetti N, DiMeco F, et al. (2012). The EphA2 receptor drives self-renewal and tumorigenicity in stem-like tumor-propagating cells from human glioblastomas. Cancer Cell 22, 765–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binns KL, Taylor PP, Sicheri F, Pawson T, and Holland SJ (2000). Phosphorylation of Tyrosine Residues in the Kinase Domain and Juxtamembrane Region Regulates the Biological and Catalytic Activities of Eph Receptors. Mol. Cell Biol 20, 4791–4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissier P, Chen J, and Huynh-Do U (2013). EphA2 signaling following endocytosis: role of Tiam1. Traffic 14, 1255–1271.24112471 [Google Scholar]
- Boyd AW, Bartlett PF, and Lackmann M (2014). Therapeutic targeting of EPH receptors and their ligands. Nat. Rev. Drug Discov 13, 39–62. [DOI] [PubMed] [Google Scholar]
- Calvisi DF, Ladu S, Gorden A, Farina M, Conner EA, Lee JS, Factor VM, and Thorgeirsson SS (2006). Ubiquitous activation of Ras and Jak/Stat pathways in human HCC. Gastroenterology 130, 1117–1128. [DOI] [PubMed] [Google Scholar]
- Chang Q, Jorgensen C, Pawson T, and Hedley DW (2008). Effects of dasatinib on EphA2 receptor tyrosine kinase activity and downstream signalling in pancreatic cancer. Br. J. Cancer 99, 1074–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y, Syeda F, Walker JR, Finerty PJ Jr., Cuerrier D, Wojciechowski A, Liu Q, Dhe-Paganon S, and Gray NS (2009). Discovery and structural analysis of Eph receptor tyrosine kinase inhibitors. Bioorg. Med. Chem. Lett 19, 4467–4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cieply B, Zeng G, Proverbs-Singh T, Geller DA, and Monga SPS (2009). Unique phenotype of hepatocellular cancers with exon-3 mutations in beta-catenin gene. Hepatology 49, 821–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, and Zhang F (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui X-D, Lee M-J, Yu G-R, Kim I-H, Yu H-C, Song E-Y, and Kim D-G (2010). EFNA1 ligand and its receptor EphA2: potential biomarkers for hepatocellular carcinoma. Int. J. Cancer 126, 940–949. [DOI] [PubMed] [Google Scholar]
- Darnell JE Jr., Kerr IM, and Stark GR (1994). Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 264, 1415–1421. [DOI] [PubMed] [Google Scholar]
- Fang WB, Brantley-Sieders DM, Hwang Y, Ham A-JL, and Chen J (2008). Identification and Functional Analysis of Phosphorylated Tyrosine Residues within EphA2 Receptor Tyrosine Kinase. J. Biol. Chem 283, 16017–16026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson FM, and Gray NS (2018). Kinase inhibitors: the road ahead. Nat. Rev. Drug Discov 17, 353–377. [DOI] [PubMed] [Google Scholar]
- Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, and Bray F (2015). Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 136, E359–E386. [DOI] [PubMed] [Google Scholar]
- Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, Kudo M, Breder V, Merle P, Kaseb AO, et al. ; IMbrave150 Investigators (2020). Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med 382, 1894–1905. [DOI] [PubMed] [Google Scholar]
- Galicia VA, He L, Dang H, Kanel G, Vendryes C, French BA, Zeng N, Bayan JA, Ding W, Wang KS, et al. (2010). Expansion of hepatic tumor progenitor cells in Pten-null mice requires liver injury and is reversed by loss of AKT2. Gastroenterology 139, 2170–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnoni A, Marech I, Silvestris N, Vacca A, and Lorusso V (2011). Dasatinib: an anti-tumour agent via Src inhibition. Curr. Drug Targets 12, 563–578. [DOI] [PubMed] [Google Scholar]
- Grabinski N, Ewald F, Hofmann BT, Staufer K, Schumacher U, Nashan B, and Jücker M (2012). Combined targeting of AKT and mTOR synergistically inhibits proliferation of hepatocellular carcinoma cells. Mol. Cancer 11, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigis KM, Cichowski K, and Elledge SJ (2019). Tissue-specificity in cancer: The rule, not the exception. Science 363, 1150–1151. [DOI] [PubMed] [Google Scholar]
- Hall J, Guo G, Wray J, Eyres I, Nichols J, Grotewold L, Morfopoulou S, Humphreys P, Mansfield W, Walker R, et al. (2009). Oct4 and LIF/Stat3 additively induce Krüppel factors to sustain embryonic stem cell self-renewal. Cell Stem Cell 5, 597–609. [DOI] [PubMed] [Google Scholar]
- He G, and Karin M (2011). NF-κB and STAT3 - key players in liver inflammation and cancer. Cell Res. 21, 159–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He G, Yu G-Y, Temkin V, Ogata H, Kuntzen C, Sakurai T, Sieghart W, Peck-Radosavljevic M, Leffert HL, and Karin M (2010a). Hepatocyte IKK-beta/NF-kappaB inhibits tumor promotion and progression by preventing oxidative stress-driven STAT3 activation. Cancer Cell 17, 286–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himanen JP, Yermekbayeva L, Janes PW, Walker JR, Xu K, Atapattu L, Rajashankar KR, Mensinga A, Lackmann M, Nikolov DB, and Dhe-Paganon S (2010). Architecture of Eph receptor clusters. Proc. Natl. Acad. Sci. USA 107, 10860–10865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai H, Maru Y, Hagiwara K, Nishida J, and Takaku F (1987). A novel putative tyrosine kinase receptor encoded by the eph gene. Science 238, 1717–1720. [DOI] [PubMed] [Google Scholar]
- Hochgräfe F, Zhang L, O’Toole SA, Browne BC, Pinese M, Porta Cubas A, Lehrbach GM, Croucher DR, Rickwood D, Boulghourjian A, et al. (2010). Tyrosine phosphorylation profiling reveals the signaling network characteristics of Basal breast cancer cells. Cancer Res. 70, 9391–9401. [DOI] [PubMed] [Google Scholar]
- Horwitz E, Stein I, Andreozzi M, Nemeth J, Shoham A, Pappo O, Schweitzer N, Tornillo L, Kanarek N, Quagliata L, et al. (2014). Human and mouse VEGFA-amplified hepatocellular carcinomas are highly sensitive to sorafenib treatment. Cancer Discov. 4, 730–743. [DOI] [PubMed] [Google Scholar]
- Huynh J, Chand A, Gough D, and Ernst M (2019). Therapeutically exploiting STAT3 activity in cancer - using tissue repair as a road map. Nat. Rev. Cancer 19, 82–96. [DOI] [PubMed] [Google Scholar]
- Johnson DE, O’Keefe RA, and Grandis JR (2018). Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol 15, 234–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kania A, and Klein R (2016). Mechanisms of ephrin-Eph signalling in development, physiology and disease. Nat. Rev. Mol. Cell Biol 17, 240–256. [DOI] [PubMed] [Google Scholar]
- Kaposi-Novak P, Lee J-S, Gòmez-Quiroz L, Coulouarn C, Factor VM, and Thorgeirsson SS (2006). Met-regulated expression signature defines a subset of human hepatocellular carcinomas with poor prognosis and aggressive phenotype. J. Clin. Invest 116, 1582–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kermorgant S, and Parker PJ (2008). Receptor trafficking controls weak signal delivery: a strategy used by c-Met for STAT3 nuclear accumulation. J. Cell Biol 182, 855–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna C, Wan X, Bose S, Cassaday R, Olomu O, Mendoza A, Yeung C, Gorlick R, Hewitt SM, and Helman LJ (2004). The membrane-cyto-skeleton linker ezrin is necessary for osteosarcoma metastasis. Nat. Med 10, 182–186. [DOI] [PubMed] [Google Scholar]
- Lai K-O, Chen Y, Po H-M, Lok K-C, Gong K, and Ip NY (2004). Identification of the Jak/Stat proteins as novel downstream targets of EphA4 signaling in muscle: implications in the regulation of acetylcholinesterase expression. J. Biol. Chem 279, 13383–13392. [DOI] [PubMed] [Google Scholar]
- Leonard WJ, and O’Shea JJ (1998). Jaks and STATs: biological implications. Annu. Rev. Immunol 16, 293–322. [DOI] [PubMed] [Google Scholar]
- Leung CON, Tong M, Chung KPS, Zhou L, Che N, Tang KH, Ding J, Lau EYT, Ng IOL, Ma S, and Lee TKW (2020). Overriding Adaptive Resistance to Sorafenib Through Combination Therapy With Src Homology 2 Domain-Containing Phosphatase 2 Blockade in Hepatocellular Carcinoma. Hepatology 72, 155–168. [DOI] [PubMed] [Google Scholar]
- Li WC, Ye SL, Sun RX, Liu YK, Tang ZY, Kim Y, Karras JG, and Zhang H (2006). Inhibition of growth and metastasis of human hepatocellular carcinoma by antisense oligonucleotide targeting signal transducer and activator of transcription 3. Clin. Cancer Res 12, 7140–7148. [DOI] [PubMed] [Google Scholar]
- Li H, Sun Q, Han B, Yu X, Hu B, and Hu S (2015). MiR-26b inhibits hepatocellular carcinoma cell proliferation, migration, and invasion by targeting EphA2. Int. J. Clin. Exp. Pathol 8, 4782–4790. [PMC free article] [PubMed] [Google Scholar]
- Llovet JM (2014). Focal gains of VEGFA: candidate predictors of sorafenib response in hepatocellular carcinoma. Cancer Cell 25, 560–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc J-F, de Oliveira AC, Santoro A, Raoul J-L, Forner A, et al. ; SHARP Investigators Study Group (2008). Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med 359, 378–390. [DOI] [PubMed] [Google Scholar]
- Llovet JM, Villanueva A, Lachenmayer A, and Finn RS (2015). Advances in targeted therapies for hepatocellular carcinoma in the genomic era. Nat. Rev. Clin. Oncol 12, 408–424. [DOI] [PubMed] [Google Scholar]
- Lokau J, Schoeder V, Haybaeck J, and Garbers C (2019). Jak-Stat Signaling Induced by Interleukin-6 Family Cytokines in Hepatocellular Carcinoma. Cancers (Basel) 11, 1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macrae M, Neve RM, Rodriguez-Viciana P, Haqq C, Yeh J, Chen C, Gray JW, and McCormick F (2005). A conditional feedback loop regulates Ras activity through EphA2. Cancer Cell 8, 111–118. [DOI] [PubMed] [Google Scholar]
- Markosyan N, Li J, Sun YH, Richman LP, Lin JH, Yan F, Quinones L, Sela Y, Yamazoe T, Gordon N, et al. (2019). Tumor cell-intrinsic EPHA2 suppresses anti-tumor immunity by regulating PTGS2 (COX-2). J. Clin. Invest 129, 3594–3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini G, Cardone C, Vitiello PP, Belli V, Napolitano S, Troiani T, Ciardiello D, Della Corte CM, Morgillo F, Matrone N, et al. (2019). EPHA2 Is a Predictive Biomarker of Resistance and a Potential Therapeutic Target for Improving Antiepidermal Growth Factor Receptor Therapy in Colorectal Cancer. Mol. Cancer Ther 18, 845–855. [DOI] [PubMed] [Google Scholar]
- Matter MS, Decaens T, Andersen JB, and Thorgeirsson SS (2014). Targeting the mTOR pathway in hepatocellular carcinoma: current state and future trends. J. Hepatol 60, 855–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao H, Li D-Q, Mukherjee A, Guo H, Petty A, Cutter J, Basilion JP, Sedor J, Wu J, Danielpour D, et al. (2009). EphA2 mediates ligand-dependent inhibition and ligand-independent promotion of cell migration and invasion via a reciprocal regulatory loop with Akt. Cancer Cell 16, 9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao H, Gale NW, Guo H, Qian J, Petty A, Kaspar J, Murphy AJ, Valenzuela DM, Yancopoulos G, Hambardzumyan D, et al. (2015). EphA2 promotes infiltrative invasion of glioma stem cells in vivo through cross-talk with Akt and regulates stem cell properties. Oncogene 34, 558–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murase S (2013). Signal transducer and activator of transcription 3 (STAT3) degradation by proteasome controls a developmental switch in neurotrophin dependence. J. Biol. Chem 288, 20151–20161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park OK, Schaefer LK, Wang W, and Schaefer TS (2000). Dimer stability as a determinant of differential DNA binding activity of Stat3 isoforms. J. Biol. Chem 275, 32244–32249. [DOI] [PubMed] [Google Scholar]
- Pasquale EB (2010). Eph receptors and ephrins in cancer: bidirectional signalling and beyond. Nat. Rev. Cancer 10, 165–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil MA, Lee SA, Macias E, Lam ET, Xu C, Jones KD, Ho C, Rodriguez-Puebla M, and Chen X (2009). Role of cyclin D1 as a mediator of c-Met- and beta-catenin-induced hepatocarcinogenesis. Cancer Res. 69, 253–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin X-F, An DS, Chen ISY, and Baltimore D (2003). Inhibiting HIV-1 infection in human T cells by lentiviral-mediated delivery of small interfering RNA against CCR5. Proc. Natl. Acad. Sci. USA 100, 183–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessler S, Jia HL, Budhu A, Forgues M, Ye QH, Lee JS, Thorgeirsson SS, Sun Z, Tang ZY, Qin LX, and Wang XW (2010). A unique metastasis gene signature enables prediction of tumor relapse in early-stage hepatocellular carcinoma patients. Cancer Res. 70, 10202–10212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabet O, Stockert R, Xouri G, Brüggemann Y, Stanoev A, and Bastiaens PIH (2015). Ubiquitination switches EphA2 vesicular traffic from a continuous safeguard to a finite signalling mode. Nat. Commun 6, 8047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sáinz-Jaspeado M, Huertas-Martinez J, Lagares-Tena L, Martin Liberal J, Mateo-Lozano S, de Alava E, de Torres C, Mora J, Del Muro XG, and Tirado OM (2013). EphA2-induced angiogenesis in ewing sarcoma cells works through bFGF production and is dependent on caveolin-1. PLoS ONE 8, e71449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salaita K, Nair PM, Petit RS, Neve RM, Das D, Gray JW, and Groves JT (2010). Restriction of receptor movement alters cellular response: physical force sensing by EphA2. Science 327, 1380–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer LK, Ren Z, Fuller GN, and Schaefer TS (2002). Constitutive activation of Stat3α in brain tumors: localization to tumor endothelial cells and activation by the endothelial tyrosine kinase receptor (VEGFR-2). Oncogene 21, 2058–2065. [DOI] [PubMed] [Google Scholar]
- Schroeder A, Herrmann A, Cherryholmes G, Kowolik C, Buettner R, Pal S, Yu H, Müller-Newen G, and Jove R (2014). Loss of androgen receptor expression promotes a stem-like cell phenotype in prostate cancer through STAT3 signaling. Cancer Res. 74, 1227–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebestyén MG, Budker VG, Budker T, Subbotin VM, Zhang G, Mona-han SD, Lewis DL, Wong SC, Hagstrom JE, and Wolff JA (2006). Mechanism of plasmid delivery by hydrodynamic tail vein injection. I. Hepatocyte uptake of various molecules. J. Gene Med 8, 852–873. [DOI] [PubMed] [Google Scholar]
- Shang N, Arteaga M, Zaidi A, Stauffer J, Cotler SJ, Zeleznik-Le NJ, Zhang J, and Qiu W (2015). FAK is required for c-Met/β-catenin-driven hepatocarcinogenesis. Hepatology 61, 214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang N, Wang H, Bank T, Perera A, Joyce C, Kuffel G, Zilliox MJ, Cotler SJ, Ding X, Dhanarajan A, et al. (2019). Focal Adhesion Kinase and β-Catenin Cooperate to Induce Hepatocellular Carcinoma. Hepatology 70, 1631–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry MM, Reeves A, Wu JK, and Cochran BH (2009). STAT3 is required for proliferation and maintenance of multipotency in glioblastoma stem cells. Stem Cells 27, 2383–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigismund S, Argenzio E, Tosoni D, Cavallaro E, Polo S, and Di Fiore PP (2008). Clathrin-mediated internalization is essential for sustained EGFR signaling but dispensable for degradation. Dev. Cell 15, 209–219. [DOI] [PubMed] [Google Scholar]
- Sinnberg T, Makino E, Krueger MA, Velic A, Macek B, Rothbauer U, Groll N, Pötz O, Czemmel S, Niessner H, et al. (2016). A Nexus Consisting of Beta-Catenin and Stat3 Attenuates BRAF Inhibitor Efficacy and Mediates Acquired Resistance to Vemurafenib. EBioMedicine 8, 132–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söderberg O, Gullberg M, Jarvius M, Ridderstråle K, Leuchowius K-J, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson L-G, and Landegren U (2006). Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 3, 995–1000. [DOI] [PubMed] [Google Scholar]
- Song W, Hwang Y, Youngblood VM, Cook RS, Balko JM, Chen J, and Brantley-Sieders DM (2017). Targeting EphA2 impairs cell cycle progression and growth of basal-like/triple-negative breast cancers. Oncogene 36, 5620–5630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M, Bode AM, Dong Z, and Lee MH (2019). AKT as a Therapeutic Target for Cancer. Cancer Res. 79, 1019–1031. [DOI] [PubMed] [Google Scholar]
- Stark GR, Kerr IM, Williams BR, Silverman RH, and Schreiber RD (1998). How cells respond to interferons. Annu. Rev. Biochem 67, 227–264. [DOI] [PubMed] [Google Scholar]
- Stauffer JK, Scarzello AJ, Andersen JB, De Kluyver RL, Back TC, Weiss JM, Thorgeirsson SS, and Wiltrout RH (2011). Coactivation of AKT and β-catenin in mice rapidly induces formation of lipogenic liver tumors. Cancer Res. 71, 2718–2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen ISY, Hahn WC, Sharp PA, et al. (2003). Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9, 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swidergall M, Solis NV, Lionakis MS, and Filler SG (2018). EphA2 is an epithelial cell pattern recognition receptor for fungal β-glucans. Nat. Microbiol 3, 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Li C, Kang B, Gao G, Li C, and Zhang Z (2017). GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 45 (W1), W98–W102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao J, Xu E, Zhao Y, Singh S, Li X, Couchy G, Chen X, Zucman-Rossi J, Chikina M, and Monga SPS (2016). Modeling a human hepatocellular carcinoma subset in mice through coexpression of met and point-mutant β-catenin. Hepatology 64, 1587–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JE, Cubbon RM, Cummings RT, Wicker LS, Frankshun R, Cunningham BR, Cameron PM, Meinke PT, Liverton N, Weng Y, and DeMartino JA (2002). Photochemical preparation of a pyridone containing tetracycle: a Jak protein kinase inhibitor. Bioorg. Med. Chem. Lett 12, 1219–1223. [DOI] [PubMed] [Google Scholar]
- Tward AD, Jones KD, Yant S, Cheung ST, Fan ST, Chen X, Kay MA, Wang R, and Bishop JM (2007). Distinct pathways of genomic progression to benign and malignant tumors of the liver. Proc. Natl. Acad. Sci. USA 104, 14771–14776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva A (2019). Hepatocellular Carcinoma. N. Engl. J. Med 380, 1450–1462. [DOI] [PubMed] [Google Scholar]
- Villanueva A, Chiang DY, Newell P, Peix J, Thung S, Alsinet C, Tovar V, Roayaie S, Minguez B, Sole M, et al. (2008). Pivotal role of mTOR signaling in hepatocellular carcinoma. Gastroenterology 135, 1972–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Bank T, Malnassy G, Arteaga M, Shang N, Dalheim A, Ding X, Cotler SJ, Denning MF, Nishimura MI, et al. (2018). Inhibition of insulin-like growth factor 1 receptor enhances the efficacy of sorafenib in inhibiting hepatocellular carcinoma cell growth and survival. Hepatol. Commun 2, 732–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weibrecht I, Leuchowius K-J, Clausson C-M, Conze T, Jarvius M, Howell WM, Kamali-Moghaddam M, and Söderberg O (2010). Proximity ligation assays: a recent addition to the proteomics toolbox. Expert Rev. Proteomics 7, 401–409. [DOI] [PubMed] [Google Scholar]
- Wong ALA, Hirpara JL, Pervaiz S, Eu J-Q, Sethi G, and Goh B-C (2017). Do STAT3 inhibitors have potential in the future for cancer therapy? Expert Opin. Investig. Drugs 26, 883–887. [DOI] [PubMed] [Google Scholar]
- Wykosky J, and Debinski W (2008). The EphA2 receptor and ephrinA1 ligand in solid tumors: function and therapeutic targeting. Mol. Cancer Res 6, 1795–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wykosky J, Gibo DM, Stanton C, and Debinski W (2005). EphA2 as a novel molecular marker and target in glioblastoma multiforme. Mol. Cancer Res 3, 541–551. [DOI] [PubMed] [Google Scholar]
- Xiang Y, Huang Y, Sun H, Pan Y, Wu M, and Zhang J (2019). Deregulation of miR-520d-3p promotes hepatocellular carcinoma development via lncRNA MIAT regulation and EPHA2 signaling activation. Biomed. Pharmac-other 109, 1630–1639. [DOI] [PubMed] [Google Scholar]
- Xue W, Chen S, Yin H, Tammela T, Papagiannakopoulos T, Joshi NS, Cai W, Yang G, Bronson R, Crowley DG, et al. (2014). CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature 514, 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav V, and Denning MF (2011). Fyn is induced by Ras/PI3K/Akt signaling and is required for enhanced invasion/migration. Mol. Carcinog 50, 346–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang P, Yuan W, He J, Wang J, Yu L, Jin X, Hu Y, Liao M, Chen Z, and Zhang Y (2009). Overexpression of EphA2, MMP-9, and MVD-CD34 in hepatocellular carcinoma: Implications for tumor progression and prognosis. Hepatol. Res 39, 1169–1177. [DOI] [PubMed] [Google Scholar]
- Yang X, Boehm JS, Yang X, Salehi-Ashtiani K, Hao T, Shen Y, Lubonja R, Thomas SR, Alkan O, Bhimdi T, et al. (2011). A public genome-scale lentiviral expression library of human ORFs. Nat. Methods 8, 659–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokogami K, Wakisaka S, Avruch J, and Reeves SA (2000). Serine phosphorylation and maximal activation of STAT3 during CNTF signaling is mediated by the rapamycin target mTOR. Curr. Biol 10, 47–50. [DOI] [PubMed] [Google Scholar]
- Yu H, Lee H, Herrmann A, Buettner R, and Jove R (2014). Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat. Rev. Cancer 14, 736–746. [DOI] [PubMed] [Google Scholar]
- Zelinski DP, Zantek ND, Stewart JC, Irizarry AR, and Kinch MS (2001). EphA2 overexpression causes tumorigenesis of mammary epithelial cells. Cancer Res. 61, 2301–2306. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.