

Abstract
Eruca sativa Mill. (Brassicaceae) is an important edible vegetable and a potential medicinal plant due to the antibacterial activity of its seed oil. Here, the complete chloroplast (cp) genome of E. sativa was de novo assembled with a combination of long PacBio reads and short Illumina reads. The E. sativa cp genome had a quadripartite structure that was 153,522 bp in size, consisting of one large single-copy region of 83,320 bp and one small single-copy region of 17,786 bp which were separated by two inverted repeat (IRa and IRb) regions of 26,208 bp. This complete cp genome harbored 113 unique genes: 79 protein-coding genes, 30 tRNA genes, and four rRNA genes. Forty-nine long repetitive sequences and 69 simple sequence repeats were identified in the E. sativa cp genome. A codon usage analysis of the E. sativa cp genome showed a bias toward codons ending in A/T. The E. sativa cp genome was similar in size, gene composition, and linearity of the structural region when compared with other Brassicaceae cp genomes. Moreover, the analysis of the synonymous (Ks) and non-synonymous (Ka) substitution rates demonstrated that protein-coding genes generally underwent purifying selection pressure, expect ycf1, ycf2, and rps12. A phylogenetic analysis determined that E. sativa is evolutionarily close to important Brassica species, indicating that it may be possible to transfer favorable E. sativa alleles into other Brassica species. Our results will be helpful to advance genetic improvement and breeding of E. sativa, and will provide valuable information for utilizing E. sativa as an important resource to improve other Brassica species.
Introduction
Eruca sativa Mill. is an annual or perennial species of Eruca (Brassicaceae), mainly distributed in Europe and Western Asia. E. sativa is believed to have originated from the Mediterranean region and has been widely used as an oil crop and edible vegetable [1]. Due to the fragrance of its leaves, E. sativa is also a popular salad and spice in Middle Eastern and European countries [2]. Moreover, recent studies have demonstrated that E. sativa has many medicinal and therapeutic properties, including antioxidant and antimicrobial activities [3] as well as he ability to reduce neuroinflammation and testicular silver toxicity [4]. Additionally, E. sativa is resistant to white rust, drought, and aphids [5], traits that are urgently needed for cultivated Brassica species. Therefore, many attempts have been made to use somatic fusion and sexual hybridization to transfer such desirable agronomic traits from E. sativa to other Brassica species [6]. A phylogenetic analysis between E. sativa and other species in Brassicaceae family will undoubtedly improve the ability to transfer desirable agronomic traits to related species.
Chloroplasts (cp) have an independent circular genome and play essential roles in photosynthesis, development, and the physiology of green plants [7, 8]. Cp genomes generally have a quadripartite cyclic structure (120–160 kb in size), and harbor 110–130 unique genes [9]. The typical quadripartite cyclic structure of most angiosperms is comprised of a large single copy (LSC) region and a small single copy (SSC) region, which are divided by a pair of inverted repeats (IRa and IRb) [10, 11]. The evolutionary rate of the cp genome is much lower than that of the nuclear genome [12], due to fewer recombination incidents, lower nucleotide replacement rates, and the typical maternal inheritance of the cp genome. Therefore, the cp genome has been widely employed to decipher the genealogical relationships among species [13–16].
Many cp genomes have recently been decoded due to the advancements in next-generation sequencing technology, particularly third-generation sequencing technology which yields reads longer than 10 kb [16–21]. Thus, phylogenetic analyses based on cp genome data have become increasingly popular, even in small taxonomic groups [22–24]. Although a large number of cp genomes of Brassicaceae species have been sequenced, the plastid genome of E. sativa is not available.
In the present study, the complete cp genome of E. sativa was de novo assembled with a combination of long PacBio reads and short Illumina reads, and the features of this cp genome were fully elucidated. Next, 59 cp genomes of other species in the Brassicaceae family from GenBank were used to determine the genealogical relationships between E. sativa and other species. Our results will enable further genetic improvements and breeding of E. sativa and provide valuable information for utilizing E. sativa as a resource to improve of important Brassica species.
Materials and methods
Plant materials and cp DNA extraction
E. sativa seeds were provided by Professor Zaiyun Li (Huazhong Agriculture University), and cultivated in a glasshouse at Guizhou Normal University (Guiyang, China). A total of 5 g of fresh young E. sativa leaves of were collected to isolate cp DNA using the Plant DNA Extraction Mini Kit C (Onrew, Beijing) according to the manufacturer’s instructions. After determining the integrity of the DNA, 1 μg of DNA was fragmented, and a short-insert library (with the insertion of 450 bp) was constructed for Illumina sequencing (HiSeq X Ten), according to the manufacturer’s instructions (Illumina, USA). Then, 5 μg DNA was used to prepare the DNA libraries with insert sizes of 20 kb for PacBio sequencing, according to the manufacturer’s instructions (Pacific Bioscience Inc., Menlo Park, CA, USA). All the raw data, including short Illumina reads and long PacBio reads were submitted to the figshare (https://figshare.com) with the DOI: 10.6084/m9.figshare.13653515.
Cp genome assembly and genes annotation
The 150 bp paired-end reads were produced by the Illumina sequencing platform. After removing the sequencing adapters and low-quality reads, the clean reads were obtained by Trimmomatic [25], according to the default options. To remove the nuclear reads, the clean reads were aligned to the published Arabidopsis thaliana (NC_000932) cp genome using BLASR (Basic Local Alignment with Successive Refinement) [26] (E-value: 10–6). Then, these selected short reads were used to assemble scaffolds using SOAPdenovo v2.04 according to the default parameters [27]. The low-quality PacBio reads (minimum read length of 500 bp and minimum read quality of 0.80) were removed from the raw data. The long selected PacBio reads were employed to fill the gaps within the scaffolds with PBJelly [28]. To correct possible mis-assemblies and errors, the Illumina reads were aligned to the assembled cp genome using BWA (version 0.5.9) with the default settings [29]. Frame-shift errors were manually corrected during gene prediction.
The Dual Organellar Genome Annotator was used to annotate the E. sativa cp genome with default settings [30]. The start and stop codons of each gene were verified by homology searches using BLAST (Basic Local Alignment Search Tool). Then, the E. sativa circular gene map was drawn in OGDraw software version 1.2 [31]. The well-annotated cp genome of E. sativa is available in the public GenBank database (https://www.ncbi.nlm.nih.gov/) under the accession number of MT013255.
Repeat sequence analyses
The long repeat sequences of the E. sativa cp genome, including the palindrome, reverse, forward, and complement types, were detected by the web-service REPuter (https://bibiserv.cebitec.uni-bielefeld) [32] with the following settings: minimal repeat size, 30; sequence consistency, >90%; and maximum computed repeats to 50. Additionally, simple sequence repeat (SSR) loci were detected using MISA (https://webblast.ipk-gatersleben.de/misa/) [33] with the following settings: 10 repeats for mono-types, five repeats for di-types, four repeats for tri-, and three repeats for tetra-, penta- and hexa-types, respectively.
Codon bias usage analysis
To understand the translation dynamics of the E. sativa cp genome, the CodonW1.4.2 program [34] (http://downloads.fyxm.net/CodonW-76666.html) was employed to calculate the synonymous codon usage of the protein-coding genes with default settings. The relative synonymous codon usage (RSCU) of all coding genes was also analyzed.
Comparison of related cp genomes
The mVISTA program (http://genome.lbl.gov/vista/mvista/submit.shtml) [35] was used to analyze sequence divergence between the E. sativa cp genome and those of six related species. The related cp sequences were downloaded from the National Center for Biotechnology Information (NCBI), including B. rapa (NC_040849), B. oleracea (NC_O41167), B. juncea (NC_0282720), B. nigra (NC_030450), B. napus (NC_016734) and A. thaliana (NC_000932). IRscope (https://irscope.shinyapps.io/irapp/) was used to compare LSC/IRb/SSC/IRa junction regions among the seven selected cp genomes according to the annotated information.
Analysis of the molecular evolution of coding genes
Pairwise comparisons of 79 protein-coding genes shared between E. sativa and six related Brassicaceae species were employed to calculate non-synonymous (Ka) and synonymous (Ks) substitution rates. Pairwise alignments of these genes were carried out by with MAFFT, and the Ka/Ks value was determined with KaKs calculator (version 2.0) according to the default parameters.
Phylogenetic relationship analysis
To analyze the phylogenetic relationships between E. sativa and related Brassicaceae species, 59 Brassicaceae species cp genomes (S1 Table) were downloaded from GenBank to construct phylogenetic trees. In total, 61 homologous protein-coding sequences: atpA, atpB, atpE, atpF, atpH, atpI, clpP, ndhA, ndhB, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, petA, petB, petD, petG, psaA, psaB, psaC, psaI, psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbK, psbL, psbM, psbN, psbT, rbcL, rpl2, rpl16, rpl20, rpl22, rpl23, rpl32, rpl33, rpl36, rpoA, rpoB, rpoC1, rpoC2, rps2, rps3, rps4, rps7, rps8, rps11, rps14, rps18, and ycf4, among the selected Brassicaceae species cp genomes were used to determine the phylogenetic relationships according to the maximum likelihood (ML) method with 1000 replicates using MEGA7 [36].
Results and discussion
Cp genome assembly and features
The Illumina sequencing platform generated 8,078 Mb of raw data, resulting in an average coverage of more than 50,000 over the cp genome. After removing the adapters and low-quality reads, 7,722 Mb of clean data were obtained with an average Q20 of 97.62%. The PacBio platform generated 20,921 subreads with an N50 length of 4,698 bp and an average length of 4,002 bp (S2 Table). Both the Illumina reads and the PacBio subreads were used together to assemble the E. sativa cp genome (see Materials and Methods section). The complete E. sativa cp genome had a quadripartite structure comprised of 153,522 bp, including an SSC region of 17,786 bp and an LSC region of 83,320 bp, which were separated by a pair of inverted repeats (IRa and IRb) of 26,208 bp (Table 1, Fig 1). The average GC content of the cp genome was 36.38%, and the IR regions had the highest GC content (42.25%), followed by the LSC (34.15%), and SSC regions (29.23%). The E. sativa cp genome encoded 113 unique genes: 79 protein-coding genes, 30 tRNAs, and four rRNAs. This result is similar to previous findings on the whole cp genomes of B. juncea and B. oleracea [37]. The average gene length was 867 bp, and protein-coding gene regions accounted for 65.65% of the total sequence. The total length of the genic regions was 74,547 bp, representing 48.46% of the whole cp genome. A total of 82 genes, including 59 protein-coding genes and 23 tRNAs, were observed in the LSC regions. A total of 28 genes: five protein-coding genes, five tRNAs, and four rRNAs, were repeated in the IR regions, while only 11 genes were found in the SSC regions. Among the 113 genes, 14 genes (eight protein-coding genes and five tRNAs) harbored a single intron, whereas three genes (rps12, clpP, and ycf3) possessed two introns (Table 2). Moreover, rps12 was a trans-spliced gene, as reported previously [38]. Detailed information about the gene copy number, the number of introns, and the gene functions are listed in Table 2.
Table 1. The detail characteristics of the complete cp genome of E. sativa.
| Category | Items | Descriptions |
|---|---|---|
| Construction of cp genome | LSC region (bp) | 83,320 |
| IRA region (bp) | 26,208 | |
| SSC region (bp) | 17,786 | |
| IRB region (bp) | 26,208 | |
| Genome Size (bp) | 153,522 | |
| Gene content | Total genes (unique) | 113 |
| Protein-coding genes | 79 | |
| tRNAs | 30 | |
| rRNAs | 4 | |
| Two copy genes | 17 | |
| Genes on LSC region (total) | 82 | |
| Genes on IRA region (total) | 18 | |
| Genes on SSC region (total) | 11 | |
| Genes on IRB region (total) | 19 | |
| Gene total length (bp) | 74,547 | |
| Average of genes length (bp) | 867 | |
| Gene length / Genome (%) | 48.56 | |
| GC content | GC content of LSC region (%) | 34.15 |
| GC content of IRA region (%) | 42.35 | |
| GC content of SSC region (%) | 29.23 | |
| GC content of IRB region (%) | 42.35 | |
| Overall GC content (%) | 36.38 |
Fig 1. The circle gene map of the E. sativa cp genome.

Genes drawn outside and inside of the circle are transcribed clockwise and counterclockwise, respectively. Genes belonging to different functional groups are color coded. The darker gray in the inner circle corresponds to GC content. SSC region, LSC region, and inverted repeats (IRA and IRB) are indicated.
Table 2. Summary of assembled gene functions of E. sativa cp genome.
| Category for genes | Groups of genes | Name of genes |
|---|---|---|
| Genes involvingin photosynthesis | Subunits of photosystem | ndhI, ndhJ, ndhK, psaA, psaB, psaC, psaI, psaJ, psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI,psbJ, psbK, psbL, psbM, psbN, psbT, psbZ |
| Subunits of cytochrome b/f complex | petA, petBb, petD, petG, petL, petN | |
| Large subunit of Rubisco | rbcL | |
| Subunits of ATP synthase | atpA, atpB, atpE, atpFb, atpH, atpI | |
| Subunits of NADH-dehydrogenase | ndhAb, ndhBa,b, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH | |
| Self-replication | Ribosomal RNA genes | rrn16a,rrn23a,rrn4.5a,rrn5a |
| Transfer RNA genes | trnA-UGCa,b, trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnfM-CAU, trnG-GCCb, trnG-UCC, trnH-GUG, trnI-CAUa, trnI-GAUa,b, trnK-UUUb, trnL-CAAa, trnL-UAAb, trnL-UAG, trnM-CAU, trnN-GUUa, trnP-UGG, trnQ-UUG, trnR-ACGa, trnR-UCU, trnS-GCU, trnS-GGA, trnS-UGA, trnT-GGU, trnT-UGU, trnV-GAC, trnV-UACa,b, trnW-CCA, trnY-GUA | |
| Small subunit of ribosome | rps11, rps12a,c, rps14, rps15, rps16b, rps18, rps19, rps2 | |
| Large subunit of ribosome | rps3, rps4, rps7a, rps8, rpl14, rpl16b, rpl2a,b, rpl20, rpl22, rpl23a,rpl32, rpl33 | |
| DNA-dependent RNA polymerase | rpl36, rpoA, rpoB, rpoC1b, rpoC2 | |
| Other genes | Maturase | matK |
| Envelope membrane protein | cemA | |
| Subunit of acetyl-CoA | accD | |
| C-type cytochrome synthesis gene | ccsA | |
| Protease | clpPc | |
| Functionally unknown genes | Conserved Open reading frames | ycf1, ycf2a, ycf3c, ycf4, ycf15 |
a, b, c The letters indicate the gene with two copes, harboring one intron and two introns, respectively.
Long repeat sequence and SSR analysis
Long repeat sequences exist widely throughout the genome, and play an essential role in gene expression and regulation. Furthermore, due to the high polymorphism present in these regions, long repeat sequences are ideal for generating genetic and physical maps [39, 40]. In the present study, 49 pairs of long repeat sequences were identified, including 19 forward repeats, 28 palindromic repeats, one reverse repeat (44 bp), and one complementary repetition (49 bp) (Table 3; Fig 2). The longest repeat (85 bp) was a forward type located in the LSC region. Among these long repeats, 30 (61.2%) repeats were found in the LSC region. A majority of the repeat pairs (37 of 49, 75.5%) were found in the same regions, including 30 repeats in the LSC region, five repeats in the IR regions, and two repeats in the SSC region. However, 12 repeats, comprising nine palindromic types and three forward types, were detected in different regions. Complementary repeats are infrequent in other cp genomes in Brassicaceae [41, 42].
Table 3. The long repeat sequences detected in E. sativa cp genome.
| No. | Repeat | Type | Repeat 1 Start (bp) | Repeat 2 Start (bp) | Region |
|---|---|---|---|---|---|
| 1 | 30 | P | 61768 | 61768 | LSC |
| 2 | 30 | P | 7627 | 44148 | LSC |
| 3 | 30 | F | 106858 | 106890 | IRB |
| 4 | 30 | P | 106858 | 129922 | IRB; SSC |
| 5 | 30 | P | 106890 | 129954 | IRB; SSC |
| 6 | 32 | P | 6262 | 6262 | LSC |
| 7 | 34 | F | 106854 | 106886 | IRB |
| 8 | 34 | P | 106854 | 129922 | IRB; SSC |
| 9 | 34 | P | 106886 | 129954 | IRB; SSC |
| 10 | 34 | F | 129922 | 129954 | SSC |
| 11 | 35 | P | 75818 | 75818 | LSC |
| 12 | 35 | P | 64224 | 64224 | LSC |
| 13 | 36 | P | 9229 | 9229 | LSC |
| 14 | 36 | P | 35536 | 35536 | LSC |
| 15 | 37 | F | 97938 | 119515 | IRB; SSC |
| 16 | 37 | P | 119515 | 138867 | SSC; IRA |
| 17 | 39 | F | 43041 | 97935 | LSC; IRB |
| 18 | 39 | P | 43041 | 138868 | LSC; IRA |
| 19 | 40 | P | 28188 | 28188 | LSC |
| 20 | 40 | P | 75813 | 75818 | LSC |
| 21 | 40 | F | 148542 | 148563 | IRA |
| 22 | 41 | P | 35531 | 35536 | LSC |
| 23 | 42 | F | 97933 | 119510 | IRB; SSC |
| 24 | 42 | P | 119510 | 138867 | SSC; IRA |
| 25 | 44 | P | 73307 | 73307 | LSC |
| 26 | 44 | R | 35505 | 35505 | LSC |
| 27 | 44 | P | 76826 | 76826 | LSC |
| 28 | 44 | P | 62009 | 62009 | LSC |
| 29 | 45 | P | 112907 | 112907 | SSC |
| 30 | 47 | F | 88239 | 88260 | IRB |
| 31 | 47 | P | 88239 | 148535 | IRB; IRA |
| 32 | 47 | P | 88260 | 148556 | IRB; IRA |
| 33 | 47 | F | 148535 | 148556 | IRA |
| 34 | 49 | P | 29704 | 29704 | LSC |
| 35 | 49 | C | 35518 | 35519 | LSC |
| 36 | 49 | P | 55354 | 55354 | LSC |
| 37 | 50 | P | 185 | 185 | LSC |
| 38 | 52 | F | 37938 | 40162 | LSC |
| 39 | 55 | F | 37935 | 40159 | LSC |
| 40 | 55 | P | 66074 | 66074 | LSC |
| 41 | 56 | P | 179 | 185 | LSC |
| 42 | 58 | F | 37932 | 40156 | LSC |
| 43 | 73 | F | 37917 | 40141 | LSC |
| 44 | 74 | F | 37899 | 40123 | LSC |
| 45 | 76 | F | 37914 | 40138 | LSC |
| 46 | 77 | F | 37896 | 40120 | LSC |
| 47 | 81 | F | 37909 | 40133 | LSC |
| 48 | 82 | F | 37902 | 40126 | LSC |
| 49 | 85 | F | 37905 | 40129 | LSC |
Note: P represents for palindrome, R for reverse, F for forward, and C for complement types.
Fig 2. The long repetitive sequences and simple sequence repeats of E. sativa.

(A) The numbers of long repetitive sequences detected in E. sativa chloroplast genomes, contains four types. F: Forward repeats; R: Reverse repeats; P: Palindrome repeats; C: Complementary. (B) The numbers of SSRs detected in E. sativa chloroplast genomes, contains six types. P1: mono-; P2: di-; P3: tri-; P4: tetra-; P5: penta-; P6: hexanucleotides.
SSR loci are widely found in various species and are useful for studies of molecular evolution and genetic diversity as well as the development of molecular markers essential for plant breeding [43, 44]. Sixty-nine SSRs were identified in the E. sativa cp genome (Table 4; Fig 2), including 44 mononucleotides, 15 dinucleotides, four trinucleotides, two tetranucleotides, two pentanucleotides, and two hexanucleotides with a length of at least 10 bp. Fewer SSR loci were detected than the number of SSR loci reported in other cp genomes of Brassicaceae [16, 18, 41, 45]. Among the 44 mononucleotide repeats (including 15 A type, 27 T type, one G type, and one C type), the longest SSR was one T type of 17 bp, which was found in the SSC regions. Similar distributions of mononucleotide repeats were observed in the cp genomes of B. napus [45], Raphanus sativus [18], Nasturium officinale [41], and Sinapis alba [16]; however, the mononucleotide repeats of the G type was only observed in this cp genome. The AT/TA type contributed to all 14 dinucleotides, and the longest type of dinucleotides was AT type of 20 bp. Four trinucleotide repeats were detected, including two AAT (12 bp) types and two ATT (12 bp), which were located in the LSC and IR regions respectively. Two tetranucleotides repeats, CAAA (12 bp) and ATAG (12 bp), were found in the LSC and SSC regions, respectively. Two pentanucleotides (TGTTG and CAACA) and two hexanucleotides (GAAAGT and GTTAGA) were also detected in this cp genome. The number and types of SSR loci varied extensively compared to other cp genomes in Brassicacea using the same identified software and criteria, which supports the idea that these SSRs can be made into lineage-specific markers for genetic diversity analysis. Furthermore, SSRs have been used as markers to understand the evolutionary history [46, 47].
Table 4. Distribution of SSRs in the E. sativa cp genome.
| SSR Type | Unit | Length | Number | Position on Genome (bp) |
|---|---|---|---|---|
| P1 | A | 10 | 9 | 4258–4267,12905–12914,26968–26998,34531–34540,55461–55470,70959–70968,96292–96301,107217–107226,109509–109518 |
| 11 | 3 | 8250–8260,67289–67299,137563 | ||
| 12 | 2 | 65299–65310,113869–113880 | ||
| 14 | 1 | 64397–64310 | ||
| T | 10 | 12 | 25276–25285,42704–42713,44425–44434,55900–55909,59298–59307,70238–70247,80704–80713,81340–81349,123978–123987,127325–127334,129617–129626,140542–140551 | |
| 11 | 5 | 17535–17545,63532–63542,99270–99280,125451–125461,126154–126164 | ||
| 12 | 2 | 69411–69422,111972–111983 | ||
| 13 | 4 | 41304–41316,77182–77194,81603–81615,81736–81748 | ||
| 15 | 2 | 49040–49054,78519–78533 | ||
| 16 | 1 | 124947–124962 | ||
| 17 | 1 | 114766–114782 | ||
| C | 10 | 1 | 47447–47456 | |
| G | 10 | 1 | 66124–66133 | |
| P2 | AT | 10 | 6 | 13357–13366,833116–83125,107641–107650,120486–120495,129193–129202,143196–143205 |
| 16 | 1 | 30693–30708 | ||
| 18 | 1 | 35541–35558 | ||
| 20 | 1 | 3718–3737 | ||
| TA | 10 | 5 | 6274–6283,18907–18916,93637–93646,111792–111801,122991–123000 | |
| 12 | 1 | 7831–7842 | ||
| P3 | AAT | 12 | 2 | 12649–12660,89584–89595 |
| ATT | 12 | 2 | 45840–45851,147248–147259 | |
| P4 | CAAA | 12 | 1 | 28035–28046 |
| ATAG | 12 | 1 | 111554–111565 | |
| P5 | TGTTG | 15 | 1 | 98769–98783 |
| CAACA | 15 | 1 | 138060–138074 | |
| P6 | GAAAGT | 18 | 1 | 56545–56562 |
| GTTAGA | 18 | 1 | 80995–81012 |
Codon biased usage analysis
Codon usage bias exists widely in plastoms and is believed to play a key role in reshaping the cp genome [48]. Moreover, the codon usage bias of some genes in plastoms likely responds to outside pressure [49]. In this study, the codon usage bias and RSCU were analyzed based on 85 CDs (coding sequences) sequences in the E. sativa cp genome. These CDs generated a sequence of 74,547 bp in length, which encoded 24,849 amino acids (Table 5). Of these acids, 2,658 (10.70%) were leucine, representing the most popular type, followed by isoleucine (2,134 codons, 8.59%) and serine (1915 codons, 7.71%), whereas only 300 (1.21%) cysteines were detected (Fig 3). The detailed information of codon usage of the 85 CDs in the E. sativa cp genome is listed in Fig 4. The RSCU values of 29 codons were >1, indicating that these codons were preferentially used. Among these biased codons, the codon for leucine (UUA), was the most preferred codon with an RSCU value of 2.03. The UGG (tryptophan) and AUG (methionine) codons showed no biased usage (RSCU = 1). All of the biased codons ended with A/U, except UUG, which agrees with the results for the N. officinale [41] and S. alba cp genomes [16], suggesting that codon usage of the cp genome in Brassicaceae is conserved.
Table 5. Summary of codon usage and amino acids patterns of E. sativa cp genome.
| Codon | Number | Amino acids | Ratio of Codon | RSCU | Number of amino acid | Ratio of amino acid |
|---|---|---|---|---|---|---|
| GCA | 369 | Ala | 1.48% | 1.09 | 1348 | 5.42% |
| GCC | 207 | 0.83% | 0.61 | |||
| GCG | 146 | 0.59% | 0.43 | |||
| GCU | 626 | 2.52% | 1.86 | |||
| AGA | 425 | Arg | 1.71% | 1.72 | 1482 | 5.96% |
| AGG | 155 | 0.62% | 0.63 | |||
| CGA | 345 | 1.39% | 1.40 | |||
| CGC | 106 | 0.43% | 0.29 | |||
| CGG | 118 | 0.47% | 0.32 | |||
| CGU | 333 | 1.34% | 0.90 | |||
| AAC | 271 | Asn | 1.09% | 0.46 | 1180 | 4.75% |
| AAU | 909 | 3.66% | 1.54 | |||
| GAC | 190 | Asp | 0.76% | 0.39 | 987 | 3.97% |
| GAU | 797 | 3.21% | 1.61 | |||
| UGC | 73 | Cys | 0.29% | 0.49 | 300 | 1.21% |
| UGU | 227 | 0.91% | 1.51 | |||
| CAA | 680 | Gln | 2.74% | 1.54 | 881 | 3.55% |
| CAG | 201 | 0.81% | 0.46 | |||
| GAA | 960 | Glu | 3.86% | 1.51 | 1271 | 5.11% |
| GAG | 311 | 1.25% | 0.49 | |||
| GGA | 709 | Gly | 2.85% | 1.66 | 1712 | 6.89% |
| GGC | 158 | 0.64% | 0.37 | |||
| GGG | 285 | 1.15% | 0.67 | |||
| GGU | 560 | 2.25% | 1.31 | |||
| CAC | 145 | His | 0.58% | 0.50 | 577 | 2.32% |
| CAU | 432 | 1.74% | 1.50 | |||
| AUA | 663 | Ile | 2.67% | 0.93 | 2134 | 8.59% |
| AUC | 407 | 1.64% | 0.57 | |||
| AUU | 1064 | 4.28% | 1.50 | |||
| CUA | 356 | Leu | 1.43% | 0.80 | 2658 | 10.70% |
| CUC | 173 | 0.70% | 0.39 | |||
| CUG | 166 | 0.67% | 0.37 | |||
| CUU | 554 | 2.23% | 1.25 | |||
| UUA | 900 | 3.62% | 2.03 | |||
| UUG | 509 | 2.05% | 1.15 | |||
| AAA | 962 | Lys | 3.87% | 1.49 | 1291 | 5.20% |
| AAG | 329 | 1.32% | 0.51 | |||
| AUG | 561 | Met | 2.26% | 1.00 | 561 | 2.26% |
| UUC | 482 | Phe | 1.94% | 0.66 | 1466 | 5.90% |
| UUU | 984 | 3.96% | 1.34 | |||
| CCA | 291 | Pro | 1.17% | 1.11 | 1017 | 4.09% |
| CCC | 189 | 0.76% | 0.72 | |||
| CCG | 135 | 0.54% | 0.52 | |||
| CCU | 402 | 1.62% | 1.54 | |||
| AGC | 123 | Ser | 0.49% | 0.39 | 1915 | 7.71% |
| AGU | 388 | 1.56% | 1.22 | |||
| UCA | 374 | 1.51% | 1.17 | |||
| UCC | 284 | 1.14% | 0.89 | |||
| UCG | 191 | 0.77% | 0.60 | |||
| UCU | 555 | 2.23% | 1.74 | |||
| UAA | 51 | TER | 0.21% | 1.78 | 86 | 0.35% |
| UAG | 22 | 0.09% | 0.77 | |||
| UGA | 13 | 0.05% | 0.45 | |||
| ACA | 392 | Thr | 1.58% | 1.22 | 1282 | 5.16% |
| ACC | 225 | 0.91% | 0.70 | |||
| ACG | 143 | 0.58% | 0.45 | |||
| ACU | 522 | 2.10% | 1.63 | |||
| UGG | 419 | Trp | 1.69% | 1.00 | 419 | 1.69% |
| UAC | 172 | Tyr | 0.69% | 0.37 | 919 | 3.70% |
| UAU | 747 | 3.01% | 1.63 | |||
| GUA | 498 | Val | 2.00% | 1.46 | 1363 | 5.49% |
| GUC | 168 | 0.68% | 0.49 | |||
| GUG | 195 | 0.78% | 0.57 | |||
| GUU | 502 | 2.02% | 1.47 |
Fig 3. Codon content of 20 amino acids and stop codons in all protein-coding genes of the E. sativa chloroplast genome.

Those whose RSCU value is greater than 1 are bold by the font.
Fig 4. Comparison of the borders of the LSC, SSC, and IR regions in cp genomes of six species of Brassicaceae.

Comparative analysis of cp genomes of six related species
To detect divergence between the E. sativa cp genome and its related species, six reported cp genome sequences in Brassicaceae were downloaded from the NCBI, including five important Brassica species (B. rapa, B. oleracea, B. juncea, B. nigra, and B. napus), and the model species: A. thaliana. As shown in Table 6, these cp genomes were generally highly conserved. Briefly, the sequences ranged from 152,860 bp (B. napus) to 154,478 bp (A. thaliana) in length, and each component of the quadripartite cycle was comparable among the selected cp genomes. The overall GC content was also quite similar (36.29–36.39%) among these cp genomes. The gene content in these cp genomes was consistent, except that the ycf15 gene was missing in the A. thaliana cp genome. The ycf15 gene was also missing in the S. alba cp genome [16], indicating that this gene was varied widely in Brassicaceae. The adjacent genes and boundaries of LSC/IRb/SSC/IRa among the seven related cp genomes were compared (Fig 4), because the variable boundary regions that are believed to be adriving force for the variation in the angiosperm cp genomes [50]. In this study, the IRb/LSC boundary was located within the coding region of the rps19 gene in all seven selected species. Furthermore, the rps19 gene had an expansion of 113/114 bp in the IRb region in all selected cp genomes. The trnH-GUG gene and rpl2 gene resided at the LSC/IRa border, respectively, in all seven cp genomes, and the trnH gene was 2–30 bp from the border. Additionally, the boundary of IRb/SSC was located in the repetitive regions of the ycf1 and ndhF genes in all seven species, with only 1 bp of ycf1 and 36 bp of ndhF located in the SSC region. The IRa/SSC junction extended into the ycf1 gene in the other five species and the length of the ycf1 genes ranged from 1,027 bp to 1,030 bp in the IRa region, but the ycf1 gene was missing from the IRb/SSC regions in the E. sativa and B. juncea cp genomes. Similar results were observed in mustard species, such as S. alba [16] and S. arvensis.
Table 6. Comparison analyses of cp genomes among seven Brassicaceae species.
| Genome Features | E. sativa | A. thaliana | B. juncea | B. napus | B. nigra | B. rapa | B. oleracea |
|---|---|---|---|---|---|---|---|
| Genome Size (bp) | 153522 | 154478 | 153483 | 152860 | 153633 | 153482 | 153364 |
| LSC length (bp) | 83320 | 84170 | 83268 | 83030 | 83552 | 83282 | 83136 |
| SSC length (bp) | 17786 | 17780 | 17775 | 17760 | 17695 | 17775 | 17834 |
| IR length (bp) | 26208 | 26264 | 26211 | 26035 | 26193 | 26213 | 26197 |
| GC content (%) | 36.38 | 36.29 | 36.36 | 36.32 | 36.39 | 36.36 | 36.36 |
| Genome number | 113 | 113 | 113 | 113 | 114 | 113 | 114 |
| Protein-coding gene | 79 | 79 | 79 | 79 | 80 | 79 | 80 |
| rRNA | 4 | 4 | 4 | 4 | 4 | 4 | 4 |
| tRNA | 30 | 30 | 30 | 30 | 30 | 30 | 30 |
To further detect divergence of the cp genomes among related species, and to verify whether genome rearrangement had taken place in the E. sativa cp genome, we used mVISTA to compare the homology of the whole cp sequence among the seven selected cp genomes of Brassicaceae, using the E. sativa cp genome as a reference (Fig 5). The results showed that no genome structural rearrangement had occurred in any of the selected cp genomes, and the selected cp genomes were highly conserved with a genome similarity of > 90%. However, the non-coding regions were more divergent than the coding regions, and the LSC and SSC regions were also more divergent than the IR regions. Furthermore, the matk, atpA, rpoC2, accD, rpoA, rps19, ycf2, ycf1 and ccsA genes were quite mutable. Synonymous (Ks) and non-synonymous (Ka) nucleotide substitution patterns of genes are important indicators of gene evolution [18]. The Ka/Ks ratio is used to assess whether there are selective pressures on protein-coding genes or to evaluate the rate of gene divergence. Ka/Ks ratios > 1, close to 1, or < 1 indicate that the gene has undergone positive selection, neutral selection, or purifying selection, respectively [51]. In this study, we calculated the Ka/Ks ratios of the E. sativa cp genome compared to six closely related species, including B. rapa, B. oleracea, B. juncea, B. nigra, B. napus and A. thaliana (Fig 6). A total of 79 homologous CDs were selected to calculate the Ka/Ks among the selected cp genomes. The results showed that the average Ka/Ks ratio was 0.17, after removing the genes with Ka or Ks of 0, indicating that the genes in the E. sativa cp genome were subject to strong purifying selection pressures. The majority of genes had a Ka/Ks ratio < 0.5 in all comparisons, and the Ka/Ks ratios of most of the genes were comparable among each of the comparisons, except the rps3, accD, ndhB, rpl22, ycf1, ycf2, and rps12 genes of which the Ka/Ks ratio was elusive. For example, the Ka/Ks ratio of ycf1 was 1.144 in the comparison of B. oleracea vs. E. sativa, but the ratio was < 1 in the other five comparisons. Similar results were observed for ycf2 and rpl22. The ycf1 and ycf2 genes are considered to be pseudogenes and have been believed to have no function in plants for a long time [52, 53]. However, knockout studies have shown that the ycf1 gene is indispensable for plant cell survival [45]. The various Ka/Ks ratios of the ycf2 gene observed in the S. alba cp genome compared to other related species, indicate that the ycf2 gene was reshaped in response to outside selection stress. It is the largest plastid gene reported in angiosperms, but the function of ycf2 is largely unknown [54].
Fig 5. Sequence alignment of seven cp genomes of Brassicaceae by mVISTA, with the annotation of E. sativa as the reference.

The vertical scale indicates the percentage of identity, ranging from 50% to 100%. The horizontal axis indicates the coordinates within the chloroplast genome. Genome regions are color coded as protein-coding, rRNA, tRNA, intron, and conserved noncoding sequences.
Fig 6. The Ka/Ks ratios of 79 protein-coding genes of the E. sativa cp genome versus six closely related species of Brassicaceae.

Phylogenetic analysis of the Brassicaceae
Cp genomes containing a large amount of genetic information have been more accessible with the development of high-throughput sequencing technology. Accordingly, several studies have employed cp genomes to detect the phylogenetic relationships in the Brassicaceae family [15, 16, 18, 41]. Although several studies have systematically elucidated the phylogenetic relationships of Brassicaceae species based on nuclear gene [55–57] and cp gene information [16, 41, 58], the systematic position of E. sativa remains unclear. Based on the AG (cis-regulatory sequences of the floral homeotic gene) gene, E. sativa is closely related to S. alba [59]. Another study employing several cp genes to construct the phylogenetic relationship demonstrated that the genus Eruca had the closest relationship with Diplotaxis harra [58]. However, both studies indicated that the genus Eruca is closely related to the genus Brassica. In this study, the cp genomes of 59 Brassicaceae species were used for the phylogenetic analysis based on 62 homologous CDs. The phylogenetic tree generated 58 branches with node values > 48% (Fig 7). Among these branches, 50 branches had the node values > 90%. Based on the node values, the branches were divided into 14 subclades, and E. sativa, R. sativus, Cakile arabica, S. arvensis, and Brassica species were classified into the same subclade. Intriguingly, E. sativa and B. juncea constructed a single branch supported by a node value of 100%, indicating that E. sativa s was most closely related to B. juncea. Another study demonstrated that E. sativa is closed related to the genus Brassica based on homology of the mitochondrial genome [60]. Furthermore, B. nigra was most closely related to S. arvensis, in line with the previous result that B. nigra is closer to the genus Sinapsis than other Brassica species at the cp genomic level [16]. These studies support that the genera Brasscia, Eruca, Sinapsis, and Raphanus share a similar ancestor, or exchanges/captures of maternal genetic information occurred among these species during speciation.
Fig 7. Phylogenetic relationships of the 59 species of Brassicaceae constructed from the shared protein-coding gene sequences using Maximum Likelihood (ML).
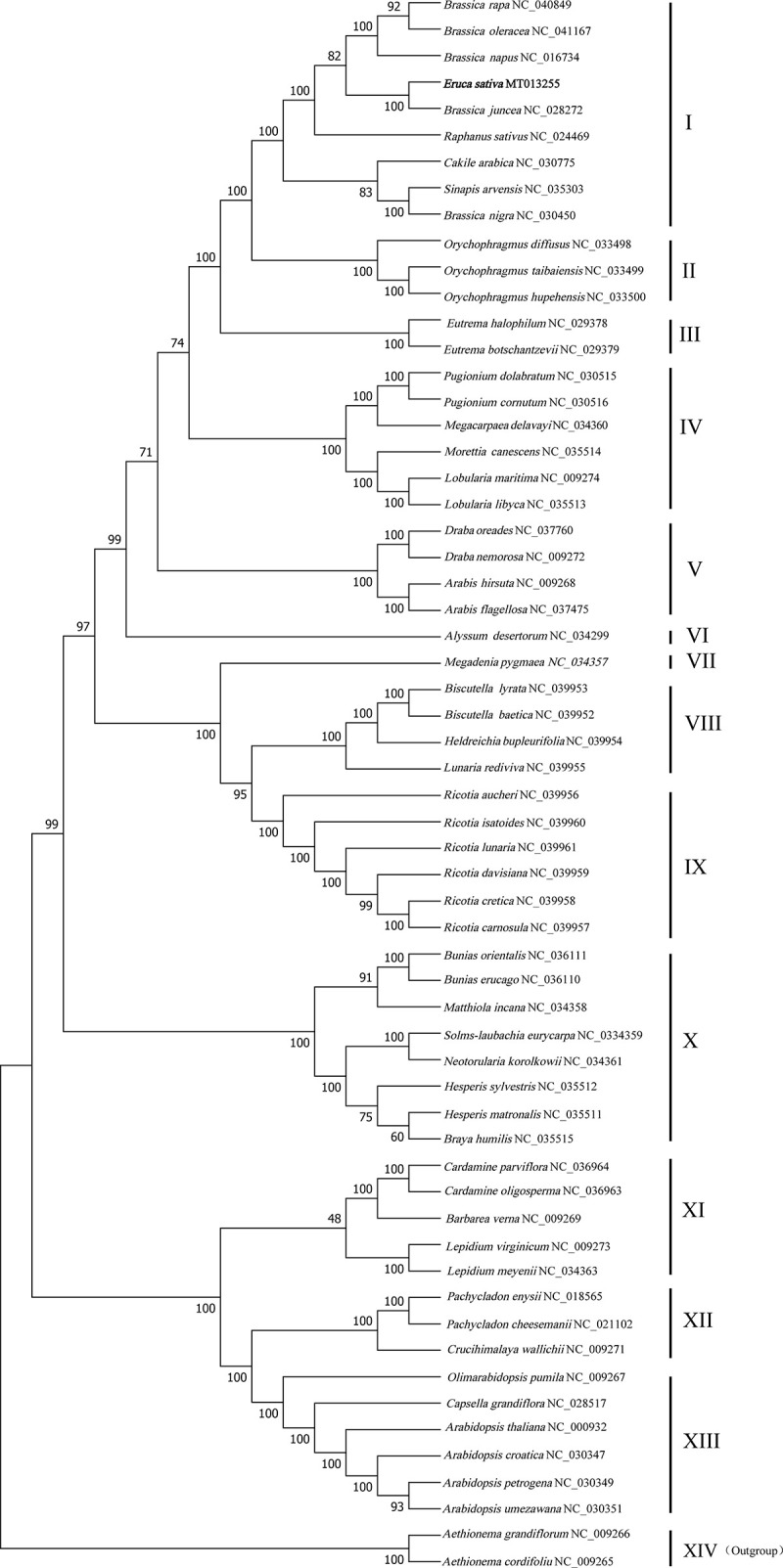
The Aethionema genus formed the outgroup.
Conclusions
In the present study, we obtained the complete cp genome of E. sativa using the combination of PacBio Sequel and Illumina HiSeq reads. The results showed that the cp genome had a typical quadripartite structure of 153,522 bp, consisting of two copies of inverted repeat (IRa and IRb) regions of 26,208 bp separated by one LSC region of 83,320 bp and one SSC region of 17,786 bp. The cp genome harbored 112 unique genes, including 79 protein-coding genes, 29 tRNA genes, and four rRNA genes. The synonymous (Ks) and non-synonymous (Ks) substitution rate analysis showed that protein-coding genes generally underwent purifying selection pressure, except ycf1 and ycf2. A phylogenetic analysis revealed that E. sativa is closely related to agriculturally important Brassica species, and most closely related to B. juncea, indicating that it may be possible to transfer favorable E. sativa alleles into other Brassica species. These results are helpful to further genetic improvement and breeding of E. sativa, and also provide valuable information for understanding the evolutionary history of E. sativa.
Supporting information
(DOCX)
(DOCX)
Abbreviations
- Cp
chloroplast
- IR
inverted repeat
- LSC
large single copy
- SSC
small single copy
- rRNA
ribosomal RNA
- tRNA
transfer RNA
- SSR
simple sequence repeat
- RSCU
the relative synonymous codon usage
- ML
maximum likelihood
- Ka
non-synonymous
- Ks
synonymous
Data Availability
The data is available in the public GenBank database (accession number MT013255). All the raw data, including short Illumina reads and long PacBio reads are available at figshare (DOI: 10.6084/m9.figshare.13653515).
Funding Statement
This research was funded by Open Project of Key Laboratory of Biology and Genetic Improvement of Oil Crops, Ministry of Agriculture and Rural Affairs, P.R. China (KF2019005) by ZB, the Guizhou Provincial Science and Technology Foundation under Grant ([2020]1Y102) by ZB, and the Key Programs for Science, Technology and Social Development of Guizhou (QKHZC[2017]2811) by YWC.
References
- 1.Sharma V, Garg G, Alam A. Extraction and characterization of agronomically and industrially important oil from Eruca sativa (L.) Mill. Am J Biol Chem. 2015; 2: 23–28. [Google Scholar]
- 2.Jansen RK, Cai Z, Raubeson LA, Daniell H, dePamphilis CW, Leebens-Mack J, et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc Natl Acad Sci USA. 2007; 104: 19369–19374. 10.1073/pnas.0709121104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koubaa M, Driss D, Bouaziz F, Ghorbel RE, Chaabouni SE. Antioxidant and antimicrobial activities of solvent extract obtained from rocket (Eruca sativa L.) flowers. Free Radicals Antioxidants. 2015; 5: 29–34. 10.5530/fra.2015.1.5. [DOI] [Google Scholar]
- 4.Gugliandolo A, Giacoppo S, Ficicchia M, Aliquò A, Bramanti P, Mazzon E. Eruca sativa seed extract: A novel natural product able to counteract neuroinflammation. Mol Med Rep. 2018; 17: 6235–6244. 10.3892/mmr.2018.8695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Agnihotri A, Lakshmikumaran M, Shivanna K, Jagannathan V. Embryo rescue of interspecific hybrids of Brassica spinescens × Brassica campestris and DNA analysis. Cell Mol Immunol. 1990; 9: 270–274. 10.1007/978-94-009-2103-0_41. [DOI] [Google Scholar]
- 6.Agnihotri A, Gupta V, Lakshmikumaran MS, Shivanna K, Prakash S, Jagannathan V. Production of Eruca-Brassica hybrids by embryo rescue. Plant Breed. 1998; 104: 281–289. 10.1111/j.1439-0523.1990.tb00437.x. [DOI] [Google Scholar]
- 7.Neuhaus HE, Emes MJ. Nonphotosynthetic metabolism in plastids. Annu Rev Plant Physiol Plant Mol Biol. 2000; 51: 111. 10.1146/annurev.arplant.51.1.111 [DOI] [PubMed] [Google Scholar]
- 8.Howe CJ, Barbrook AC, Koumandou VL, Nisbet RER, Symington HA, Wightman TF. Evolution of the chloroplast genome. Philos Trans R Soc B-Biol Sci. 2003; 358: 99–107. 10.1098/rstb.2002.1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Downie SR, Palmer JD. A chloroplast DNA phylogeny of the Caryophyllales based on structural and inverted repeat restriction site variation. Syst Bot. 1994; 19: 236–252. 10.2307/2419769. [DOI] [Google Scholar]
- 10.Wicke S, Schneeweiss GM, Depamphilis CW, Müller KF, Quandt D. The evolution of the plastid chromosome in land plants: gene content gene order gene function. Plant Mol Biol. 2011; 76: 273–297. 10.1007/s11103-011-9762-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shetty S, Md-Shah M, Makale K, Mohd-Yusuf Y, Khalid N, Othman R. Complete chloroplast genome sequence of corroborates structural heterogeneity of inverted repeats in wild progenitors of cultivated Bananas and Plantains. Plant Genome. 2016; 9: 1–14. 10.3835/plantgenome2015.09.0089 [DOI] [PubMed] [Google Scholar]
- 12.Wolfe KH, Li WH, Sharp PM. Rates of nucleotide substitution vary greatly among plant mitochondrial chloroplast and nuclear DNAs. Proc Natl Acad Sci USA. 1987; 84: 9054–9058. 10.1073/pnas.84.24.9054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moore MJ, Soltis PS, Bell CD, Burleigh JG, Soltis DE. Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. Proc Natl Acad Sci USA. 2014; 107: 4623–4628. 10.1073/pnas.0907801107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shaw J, Shafer HL, Leonard OR, Kovach MJ, Schorr M, Morris AB. Chloroplast DNA sequence utility for the lowest phylogenetic and phylogeographic inferences in angiosperms: the tortoise and the hare IV. Am J Bot. 2014; 101: 1987–2004. 10.3732/ajb.1400398 [DOI] [PubMed] [Google Scholar]
- 15.Zhu B, Gao Z, Luo X, Feng Q, Du X, Weng Q, et al. The complete chloroplast genome sequence of garden cress (Lepidium sativum L.) and its phylogenetic analysis in Brassicaceae family. Mitochondrial DNA B. 2019; 4: 3601–3602. 10.1080/23802359.2019.1677527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Du X, Zeng T, Feng Q, Hu L, Luo X, Weng Q, et al. The complete chloroplast genome sequence of yellow mustard (Sinapis alba L.) and its phylogenetic relationship to other Brassicaceae species. Gene. 2020; 731: 144340. 10.1016/j.gene.2020.144340 [DOI] [PubMed] [Google Scholar]
- 17.Hebert PD, Braukmann TW, Prosser SW, Ratnasingham S, deWaard JR, Ivanova NV, et al. A sequel to sanger: amplicon sequencing that scales. BMC Genomics. 2018; 19: 1–14. 10.1186/s12864-017-4368-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jeong YM, Chung WH, Mun JH, Kim N, Yu HJ. De novo assembly and characterization of the complete chloroplast genome of radish (Raphanus sativus L.). Gene; 2014: 551 39–48. 10.1016/j.gene.2014.08.038. [DOI] [PubMed] [Google Scholar]
- 19.Wang LL, Zhang Y, Yang YC, Du XM, Ren XL, Liu WZ. The complete chloroplast genome of Sinojackia xylocarpa (Ericales: Styracaceae) an endangered plant species endemic to China. Conserv Genet Resour. 2018; 10: 51–54. 10.1007/s12686-017-0763-8. [DOI] [Google Scholar]
- 20.Ren T, Yang Y, Wang J, Zhang R, Liu ZL. Characterization of the complete chloroplast genome sequence of Cardamine macrophylla (Brassicaceae). Conserv Genet Resour. 2018; 10: 627–630. 10.1007/s12686-017-0880-4. [DOI] [Google Scholar]
- 21.Zhang X, Rong C, Qin L, Mo C, Fan L, Yan J, et al. Complete chloroplast genome sequence of Malus hupehensis: genome structure comparative analysis and phylogenetic relationships. Molecules. 2018; 23: 2917. 10.3390/molecules23112917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barrett CF, Davis JI, Leebens-Mack J, Conran JG, Stevenson DW. Plastid genomes and deep relationships among the commelinid monocot angiosperms. Cladistics. 2013; 29: 65–87. 10.1111/j.1096-0031.2012.00418.x. [DOI] [PubMed] [Google Scholar]
- 23.Ma PF, Zhang YX, Zeng CX, Guo ZH, Li DZ. Chloroplast phylogenomic analyses resolve deep-level relationships of an intractable bamboo tribe Arundinarieae (Poaceae). Syst Biol. 2014; 63: 933–950. 10.1093/sysbio/syu054 [DOI] [PubMed] [Google Scholar]
- 24.Carbonell-Caballero J, Alonso R, Ibañez V, Terol J, Talon M, Dopazo JA. Phylogenetic analysis of 34 chloroplast genomes elucidates the relationships between wild and domestic species within the genus Citrus. Mol Biol Evol. 2015; 32: 2015–2035. 10.1093/molbev/msv082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 2014; 30: 2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chaisson M, Tesler G. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC Bioinformatics. 2012; 13: 238–238. 10.1186/1471-2105-13-238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience. 2012; 1: 30. 10.1186/2047-217X-1-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.English AC, Richards S, Han Y, Wang M, Vee V, Qu J, et al. Mind the gap: Upgrading genomes with pacific biosciences RS long-read sequencing technology. PLoS One. 2012; 7: e47768. 10.1371/journal.pone.0047768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009; 25: 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wyman SK, Jansen RK, Boore JL. Automatic annotation of organellar genomes with dogma. Bioinformatics. 2004; 20: 3252–3255. 10.1093/bioinformatics/bth352 [DOI] [PubMed] [Google Scholar]
- 31.Lohse M, Drechsel O, Bock R. Organellar GenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr Genet. 2007; 52: 267–274. 10.1007/s00294-007-0161-y [DOI] [PubMed] [Google Scholar]
- 32.Kurtz S, Choudhuri JV, Ohlebusch E, Schleiermacher C, Stoye J, Giegerich R. REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001; 29: 4633–4642. 10.1093/nar/29.22.4633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beier S, Thiel T, Münch T, Scholz U, Mascher M. MISA-web: a web server for microsatellite prediction. Bioinformatics. 2017; 33: 2583–2585. 10.1093/bioinformatics/btx198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peden J. CodonW. Trinity College, 1997. [Google Scholar]
- 35.Frazer KA, Pachter L, Poliakov A, Rubin EM, Dubchak I. VISTA: computational tools for comparative genomics. Nucleic Acids Res. 2004; 32: 273–279. 10.1093/nar/gkh458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016, 33: 1870–1874. 10.1093/molbev/msw054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seol YJ, Kim K, Kang SH, Perumal S, Lee J, Kim CK. The complete chloroplast genome of two Brassica species, Brassica nigra and B. oleracea. Mitochondrial DNA. 2017; 28: 1–2. 10.3109/19401736.2015.1053063 [DOI] [PubMed] [Google Scholar]
- 38.Lew KA, Manhart JR. The rps 12 gene in Spirogyra maxima (Chlorophyta): its evolutionary significance. J Phycol. 1993; 29: 500–505. 10.1111/j.1529-8817.1993.tb00151.x. [DOI] [Google Scholar]
- 39.Cavaliersmith T. Chloroplast evolution: secondary symbiogenesis and multiple losses. Curr Biol. 2002; 12: R62–4. 10.1016/s0960-9822(01)00675-3 [DOI] [PubMed] [Google Scholar]
- 40.Nie XJ, Lv SZ, Zhang YX, Du XH, Wang L, Biradar SS, et al. Complete chloroplast genome sequence of a major invasive species Crofton weed (Ageratina adenophora). PLoS One. 2012; 7: e36869–. 10.1371/journal.pone.0036869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yan C, Du J, Gao L, Li Y, Hou X. The complete chloroplast genome sequence of watercress (Nasturtium officinale R. Br.): Genome organization adaptive evolution and phylogenetic relationships in Cardamineae. Gene. 2019; 699: 24–36. 10.1016/j.gene.2019.02.075 [DOI] [PubMed] [Google Scholar]
- 42.Gao K, Li J, Khan WU, Zhao T, Yang X, Yang X, et al. Comparative genomic and phylogenetic analyses of Populus section Leuce using complete chloroplast genome sequences. Tree Genet Genomes. 2019; 15: 1–12. 10.1007/s11295-018-1309-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hamada H, Kakunaga T. Potential Z-DNA forming sequences are highly dispersed in the human genome. Nature. 1982; 298: 396–398. 10.1038/298396a0 [DOI] [PubMed] [Google Scholar]
- 44.Doorduin L, Gravendeel B, Lammers Y, Ariyurek Y, Chin-A-Woeng T, Vrieling K. The complete chloroplast genome of 17 individuals of pest species Jacobaea vulgaris: SNPs microsatellites and barcoding markers for population and phylogenetic studies. DNA Res. 2011; 18: 93–105. 10.1093/dnares/dsr002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hu ZY, Hua W, Huang SM, Wang HZ. Complete chloroplast genome sequence of rapeseed (Brassica napus L.) and its evolutionary implications. Genet Resour Crop Evol. 2011; 58: 875–887. 10.1007/s10722-010-9626-9. [DOI] [Google Scholar]
- 46.Powell W, Morgante M, McDevitt R, Vendramin G, Rafalski J. Polymorphic simple sequence repeat regions in chloroplast genomes: applications to the population genetics of pines. Proc Natl Acad Sci USA. 1995; 92: 7759–7763. 10.1073/pnas.92.17.7759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Torokeldiev N, Ziehe M, Gailing O, Finkeldey R. Genetic diversity and structure of natural Juglans regia L. populations in the southern Kyrgyz Republic revealed by nuclear SSR and EST-SSR markers. Tree Genet Genomes. 2019; 15: 5.1–5.12. 10.1007/s11295-018-1311-8. [DOI] [Google Scholar]
- 48.Asaf S, Waqas M, Khan AL, Khan MA, Kang SM, Imran QM, et al. The complete chloroplast genome of wild rice (Oryza minuta) and its comparison to related species. Front Plant Sci. 2017; 8: 304–304. 10.3389/fpls.2017.00304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morton BR. Strand asymmetry and codon usage bias in the chloroplast genome of Euglena gracilis. Proc Natl Acad Sci USA. 1999; 96: 5123–5128. 10.1073/pnas.96.9.5123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim KJ, Lee HL. Complete chloroplast genome sequences from Korean ginseng(Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 2004; 11: 247–261. 10.1093/dnares/11.4.247 [DOI] [PubMed] [Google Scholar]
- 51.Yang Z, Nielsen R. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol Biol Evol. 2000; 17: 32–43. 10.1093/oxfordjournals.molbev.a026236 [DOI] [PubMed] [Google Scholar]
- 52.Douglas SE, Penny SL. The plastid genome of the Cryptophyte alga, Guillardia theta: Complete sequence and conserved synteny groups confirm its common ancestry with red algae. J Mol Evol. 1999; 48: 236–244. 10.1007/pl00006462 [DOI] [PubMed] [Google Scholar]
- 53.De Las Rivas J, Lozano JJ, Ortiz AR. Comparative analysis of chloroplast genomes: functional annotation genome-based phylogeny and deduced evolutionary patterns. Genome Res. 2002; 12: 567–583. 10.1101/gr.209402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Drescher A, Ruf S, Calsa T Jr, Carrer H, Bock R. The two largest chloroplast genome- encoded open reading frames of higher plants are essential genes. The Plant Journal. 2000; 22: 97–104. 10.1046/j.1365-313x.2000.00722.x [DOI] [PubMed] [Google Scholar]
- 55.Zhao B, Liu L, Tan D, Wang J. Analysis of phylogenetic relationships of Brassicaceae species based on Chs sequences. Biochem Syst Ecol. 2010; 38: 731–739. 10.1016/j.bse.2010.06.003. [DOI] [Google Scholar]
- 56.Huang CH, Sun R, Hu Y, Zeng L, Zhang N, Cai L, et al. Resolution of Brassicaceae phylogeny using nuclear genes uncovers nested radiations and supports convergent morphological evolution. Mol Biol Evol. 2016; 33: 394–412. 10.1093/molbev/msv226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cai L, Ma H. Using nuclear genes to reconstruct angiosperm phylogeny at the species level: A case study with Brassicaceae species. J Syst Evol. 2016; 54: 438–452. 10.1111/jse.12204. [DOI] [Google Scholar]
- 58.Arias T, Pires JC. A fully resolved chloroplast phylogeny of the brassica crops and wild relatives (Brassicaceae: Brassiceae): Novel clades and potential taxonomic implications. Taxon. 2012; 61: 980–988. 10.2307/41679344. [DOI] [Google Scholar]
- 59.Hong RL, Hamaguchi L, Busch MA, Weigel D. Regulatory elements of the floral homeotic gene AGAMOUS identified by phylogenetic footprinting and shadowing. Plant Cell. 2003; 15: 1296–1309. 10.1105/tpc.009548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang Y, Chu P, Yang Q, Chang S, Chen J, Hu M, et al. Complete mitochondrial genome of Eruca sativa Mill. (Garden Rocket). PLoS One. 2014; 9: e105748. 10.1371/journal.pone.0105748 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX)
(DOCX)
Data Availability Statement
The data is available in the public GenBank database (accession number MT013255). All the raw data, including short Illumina reads and long PacBio reads are available at figshare (DOI: 10.6084/m9.figshare.13653515).