

Abstract
The mitochondrial permeability transition pore (mPTP) is a channel that, when open, is responsible for a dramatic increase in the permeability of the mitochondrial inner membrane, a process known as the mitochondrial permeability transition (mPT). mPTP activation during Ca2+ dyshomeostasis and oxidative stress disrupts normal mitochondrial function and induces cell death. mPTP opening has been implicated as a critical event in many diseases, including hypoxic injuries, neurodegeneration, and diabetes. Discoveries of recent years indicate that mPTP demonstrates very complicated behavior and regulation, and depending on specific induction or stress conditions, it can function as a high-conductance pore, a small channel, or a non-specific membrane leak. The focus of this review is to summarize the literature on the electrophysiological properties of the mPTP and to evaluate the evidence that it has multiple molecular identities. This review also provides perspective on how an electrophysiological approach can be used to quantitatively investigate the biophysical properties of the mPTP under physiological, pharmacological, pathophysiological, and disease conditions.
Keywords: mitochondrial permeability transition pore, calcium, ion channel, ROS, patch-clamp, mitochondria
Introduction
Mitochondrial permeability transition (mPT) is a phenomenon of the sudden increase of permeability of the inner mitochondrial membrane (IMM). It is believed that mPT occurs through the opening of a big non-selective channel called the mitochondrial permeability transition pore (mPTP). Once the mPTP opens, mitochondria lose the membrane potential and proton gradient across the IMM that serves as a driving force for ATP synthase to produce ATP. A resulting shortage of cellular ATP disrupts the normal metabolism of the cell and eventually leads to cell death.
mPTP opening in vitro is stimulated by elevated Ca2+ and potentiated by reactive oxygen species (ROS), and it results in swelling of mitochondria. In extreme conditions, the swelling of mitochondria can lead to the rupture of the outer mitochondrial membrane (OMM). The mPTP plays a crucial role in necrotic and apoptotic cell death during numerous pathologies, including both acute and chronic stress [2–5]. Numerous studies have shown that infarction is associated with elevated levels of ROS and Ca2+ overload of the cells, followed by the loss of mitochondrial membrane potential and release of cytochrome c. All of these outcomes are consistent with the opening of mPTP, which makes it an ideal therapeutic target to stop tissue damage.
The molecular structure of the mPTP is not well defined. However, it is generally accepted that mPTP opening is strongly regulated by cyclophilin D (CypD), which is a mitochondrial chaperone protein with foldase activity. The mPTP can be inhibited by cyclosporin A (CsA), an immunosuppressive drug that acts through CypD [6, 7]. Thus, the mPTP can be commonly defined as a CypD-dependent large pore.
The bulk of the knowledge about the mPTP comes from experiments using isolated mitochondria or permeabilized cells. mPTP opening usually manifests in a rapid decrease of light scattering in mitochondrial suspensions due to mitochondrial swelling; a sudden loss of mitochondrial ability to uptake Ca2+ from exogenous Ca2+ boluses (Ca2+ retention capacity, CRC), a sudden leakage of fluorescent dyes accumulated in the matrix, or a sudden depolarization of mitochondrial membrane potential (reviewed in [8]).
The most direct and specific method to study mPT pore/channel activity is patch clamping of mitoplasts, the mitochondria with partially removed OMM. This method directly measures the electrical activity of mPTP in the IMM. Recent experimental evidence suggests that mPTP can occur through multiple molecular pathways, underscoring the critical need for electrophysiological approaches to discriminate between different mPTP mechanisms. The primary goal of the present review is to summarize current literature on electrophysiological studies of the mPTP and to discuss these data in the context of the implications of the biophysical properties of mPTP in mitochondrial, cellular, and organismal physiology and pathology. Furthermore, we will provide a perspective on potential future directions for electrophysiological studies of the mPTP.
Electrophysiology approach to study mitochondria
The patch-clamp technique for studies of the mitochondrial ion channels
Patch-clamp is a powerful experimental approach for measuring electrical activity of the biological membranes directly. Patch-clamp was initially developed to measure currents and membrane potential of the cells in cultures or tissues, and it is still widely used for this purpose. More recently, it was adapted to define ionic currents through mitochondrial membranes. Although recordings of channel activity in purified OMM have been reported [9, 10], the patch-clamp technique is usually used to characterize the properties of IMM [11,12].
To provide the access for the patch pipette to the inner membrane, OMM is first disrupted mechanically either with a French press or with osmotic shock [13, 14]. The disrupted mitochondria are then placed in the hypo-osmotic media for swelling in order to form the vesicles composed of the inner membrane with attached parts of OMM, structures referred to as the mitoplasts in early electrophysiological studies [15]. Mitoplasts can also be formed with passive swelling in the absence of mitochondrial substrates without the prior mechanical rupture of OMM. Single-channel activity is usually recorded in excised patch-clamp configuration [16, 17]. With the excised patch, the glass pipette extracts part of the membrane and registers only the currents through the extracted part, resulting in a probability of “catching” the activity of single channels. Meanwhile, whole mitoplast configuration [15, 18] allows measurement of the total conductance of IMM with multiple channels. In this configuration, the glass electrode stays attached to the mitoplast, and voltage pulse or suction is applied to rupture the patch and provide access to the mitochondrial matrix and the rest part of a membrane (Figure 1).
Figure 1. The electrophysiological approach towards investigation of the mitochondrial permeability transition pores.
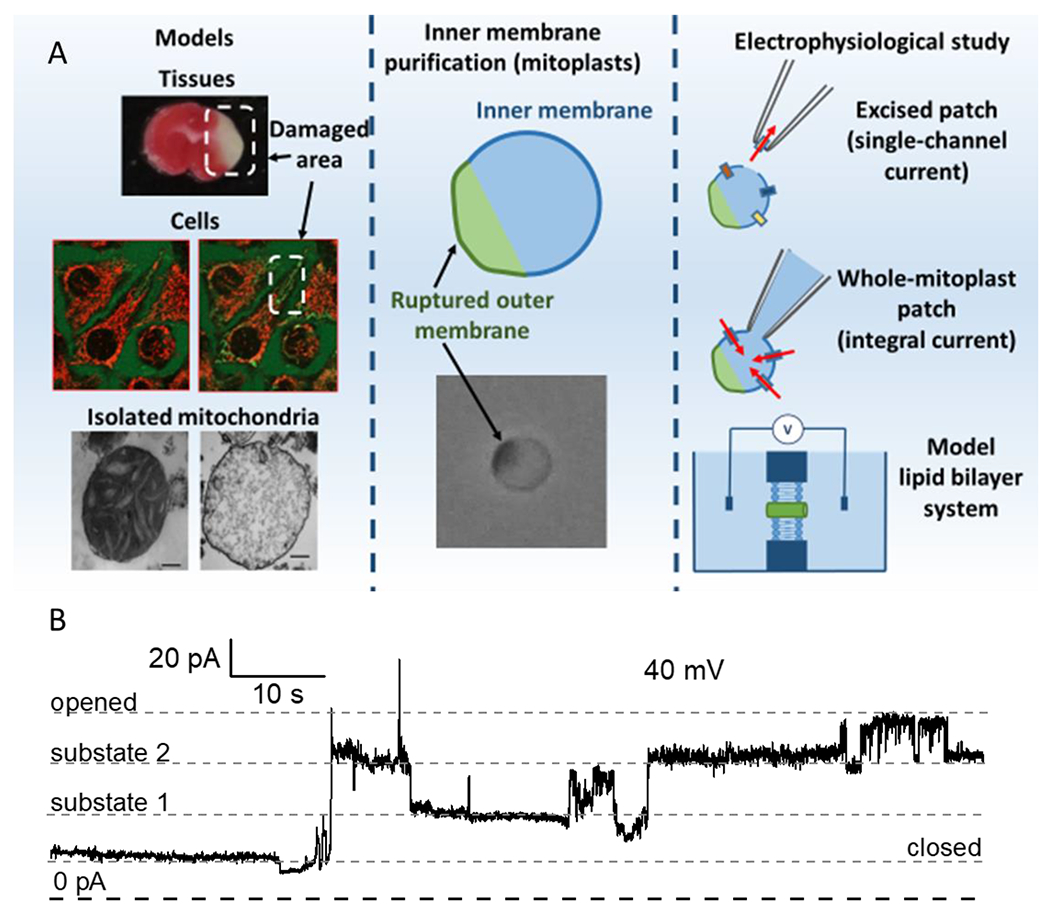
A. Before the assay, mitochondria need to be isolated either from diseased or healthy tissues. Following the isolation, experiments are performed in the mitoplasts (mitochondrial inner membrane vesicles with an aggregated outer mitochondrial membrane in green/dark color attached). Currents can be measured in the excised or whole mitoplast mode. In a complementary approach, channel activity can be measured in the model bilayer membranes (e.g. black lipid membranes) with purified and reconstituted components of the mPTP. B. Representative single channel mPTP current recording obtained using excised patch-clamp configuration of mouse liver mitoplast. Note the presence of the multiple conductance sub-states.
The first attempt to record mitochondrial electrical activity was made by the Tedeschi group in 1969 [19]. By impaling a microelectrode inside the mitochondria, they measured the membrane potential and resistance of Drosophila mitochondria. Due to a rather invasive impalement of the microelectrode, the tiny mitochondria were frequently and rapidly injured so that the recordings were not sustainable and reliable. The first reported successful recording of ion channel activity in the IMM was made by Sorgato et al. in 1987 [15]. The authors presented the voltage-clamp recordings of the currents through the IMM in two patch-clamp configurations—whole mitoplast mode and mitoplast attached mode—and detected an anion permeable channel [15].
Electrophysiology of mPTP from native mitochondrial membranes
The properties of the native channel that was later recognized as mPTP were first described by two independent groups that investigated the ion channel activity of the IMM in the presence of increased concentrations of Ca2+ [16, 17]. To investigate the properties of IMM, Petronilli et al. (1989) used osmotic shock to remove the OMM of the isolated mitochondria and applied an excised patch-clamp to mitoplasts. In these experiments, an excised patch was performed in the “inside-out” configuration in which the bath side of the patch corresponds to the matrix side of the membrane. Channel activity was induced by the addition of Ca2+ to the matrix side of the patch membrane. These experimental conditions led to the detection of the channels with multiple conductance states ranging from 30 pS up to 1.3 nS in symmetrical 150 mM KCl solution. The researchers concluded that channels higher than 300 pS appeared to be the sub-conductance states of a giant 1.3 nS channel [17]. In 1991 the same group reported that this Ca2+-activated channel is inhibited by Mg2+, CsA, and ADP when applied from the matrix side [20]. This “Megachannel” lacked ion selectivity and represented the properties correspondent to permeability transition of mitochondria [21].
An independent experimental study that characterizes the properties of the mPTP channel in the native membrane was then performed by the Kinnally group, which reported the presence of conductance levels of 10-20, 45, 80, 120-150, 300 and 1000 pS within the same patch [16]. Ion selectivity of conductance levels was weak and variable (slightly anionic for 45 pS whereas slightly cationic for 120-150 and 300 pS). However, the appearance of the channel with sub-levels up to 1000 pS depended on the presence of Ca2+ chelator in isolation media [22]. When mitochondria were isolated in the absence of EGTA, channel activity was detected in 96% of patched membranes. In contrast, the presence of Ca2+ chelator in isolation buffer led to a decrease in detection to 8%. Thus, in this case, activation of the “Multiconductance” channel was initiated by Ca2+ applied from the cytoplasmic site. Subsequent elimination of Ca2+ from the recording solution with EGTA did not abolish channel activity [22]. The addition of Ca2+ from the matrix side of the membrane led to activity on the sublevel with lower conductance. The channel activity was also potentiated by voltages above ±60 mV range. The Multiconductance channel was sensitive to CsA and to several other cationic amphiphiles (e.g., amiodarone, quinine, propranolol) [23]. Inhibition by these drugs caused stepwise closure of the channel by the same conductance steps as were observed during voltage activation. The electrophysiological mPTP properties defined by these two research groups are summarized in Table 1. As can be seen, the described channels have some differences in activation patterns and selectivity. Interestingly, the Megachannel was activated in the excised membrane by Ca2+ added to the matrix side and can be inhibited by its elimination, whereas the Multiconductance channel was detected when mitochondria were exposed to Ca2+ during isolation (i.e., it was activated with external Ca2+). These discrepancies in activation could explain different properties of the native channel in IMM, but it should be noted that conditions of activation could change the activity of the channel.
Table 1.
Comparison of two early patch-clamp studies of mPTP
| Megachannel (Petronilli, Szabo) | Multiconductance channel (Kinnally) | |
|---|---|---|
| Activation | Ca2+ from matrix side | Ca2+ from cytoplasmic site |
| Channel size | Up to 1.3 nS | Up to 1 nS* |
| Selectivity | Unselective | Sub-levels slightly selective |
| Inhibition | CsA, washout Ca2+ | CsA, amiodarone, quinine, propranolol |
| Voltage dependence | Yes | Yes |
later the same group reported channel conductance of up to 2.7 nS [25].
Despite of some differences in the Multiconductance channel of the Kinnally group and in the Megachannel of Zoratti group, their electrophysiological properties correspond to mPTP. It is likely that these channels could be opened only in pathological conditions of Ca2+ overload and not in normal physiological conditions. We should note that the values of ion conductance reported by both groups are consistent with the activity in which even the single channel opening would depolarize the membrane completely. Indeed, simple calculations show that respiratory chain activity can compensate only current at the femtoampere range [24]. Thus, the mPTP channel seen in these patch-clamp recordings is most likely associated with the pathological pore opening that causes high amplitude swelling rather than with the low rate ion fluxes through a “leaky” but overall intact membrane.
In search of the molecular structure of the mPTP
Reconstitution of purified proteins
The structure of the mPTP channel is still a matter of debate. In an attempt to identify the possible components of mPTP, several purified mitochondrial proteins have been tested for their ability to form a channel in model lipid bilayers or liposomes.
Moran et al. (1990) performed the first experiments with purified mitochondrial proteins by purifying and reconstituting contact sites isolated from rat brain mitochondria into liposomes. Subsequent patch-clamp study of these liposomes detected channels, which were grouped by conductance ranging from < 100 pS, 475-550 pS to 1000 pS. The last two groups had no voltage dependence [11]. These results suggested that the mPT channel could require the components of both mitochondrial membranes. Although the exact protein composition of the contact sites was not presented, presumably these sites contained VDAC in the OMM and ATP synthase and adenine nucleotide translocator (ANT) protein complexes in the IMM, as demonstrated by Brdiczka’s group [26]. Later experiments with VDAC knockout mitochondria showed that this OMM channel was not required for mPTP, but the IMM proteins of this complex remained as candidates [27].
A further search for putative components mostly focused on the proteins from IMM. Based on the sensitivity of mPTP to the inhibitors of ANT, it was proposed that ANT can be a structural unit of the mPTP channel [6]. Indeed, ANT extracted from bovine heart mitoplasts and reconstituted to liposomes was able to form channels with multiple sublevels of conductance in the range of 300-600 pS [28]. Notably, these experiments were done in 100 mM KCI solution as opposed to 150 mM KCI in patch-clamp of native membranes experiments, making the comparison of the channel conductance somewhat ambiguous. This channel was slightly cationic and had no voltage dependence but switched to a closed state at voltages >150-180 mV of either sign. The ANT formed channel was activated by Ca2+, as elimination of Ca2+ from the media reversibly deactivated activity. Channel activity was also reversibly blocked by low pH. An inhibitor of ANT, bongkrekic acid (BKA), decreased the channel activity and completely blocked it in combination with ADP.
CsA is known to inhibit mPTP via interaction with mitochondrial chaperone CypD. The channel formed by reconstituted ANT was not sensitive to CsA, which can be explained by the lack of CypD in the purified and reconstituted channel. A follow-up study by the same group addressed the functional relationship between ANT and CypD by investigating the properties of recombinant ANT from Neurospora crassa expressed in E. coli [29]. Single-channel patch-clamp showed that recombinant ANT reconstituted in membrane forms a channel with only slightly different properties, including a conductance of 500-700 nS in 100 mM KCI and slight cation selectivity. The channel activity was not symmetrically voltage dependent, as high positive voltage dramatically decreases conductance of the channel while high negative voltage usually does not change conductance. The channel formed by recombinant ANT was also inhibited by BKA and completely blocked by BKA together with ADP, and it was sensitive not only to Ca2+ but also to the pro-oxidant tert-butyl hydroperoxide (t-BOOH). Interestingly, a decrease in Ca2+ concentration did not lead to changes in the properties of the channel but did lead to a decrease in its detection frequency. The addition of recombinant CypD to the recording solution also made this channel sensitive to the CsA [29]. It should be noted that while this preparation was not highly purified leaving the possibility of the presence of other channels. The detected channel activity was sensitive to the specific ANT ligands, which suggests the likelihood of a direct involvement of the ANT. However, one cannot rule out that ANT might serve as a regulatory component for an unidentified channel pore forming protein and the binding of these ligands to ANT inhibits its regulatory function for channel to open.
Overall, experiments with reconstituted protein provided strong support to the idea that the mPTP can be formed from ANT. Interestingly, further studies demonstrated that cells deficient of ANT still can develop mPT albeit required much higher amounts of Ca2+ [30]. This mPTP still was inhibited by CsA and was not sensitive to the ANT ligands (BKA and atractyloside). No electrophysiological measurements were performed in these mitochondria, so it was not clear whether mPT was caused by the same channel in both wild-type and ANT KO mitochondria.
Mitochondrial ATP synthase has also been extensively studied as a candidate for mPTP by using a purification and reconstitution strategy. Reconstitution experiments suggest that ATP synthase can participate in the formation of the large pore by several mechanisms. These possible mechanisms include a direct channel formation by c-subunit peptide monomers [31], or by the “c-ring” of the F0 part of the ATP synthase complex [32] or by the dimers of the ATP synthase complex with the putative pore-forming part located between two monomers [33].
C-subunit of sheep liver and bovine brain ATP synthase was reported to facilitate the oscillation of sodium currents when reconstituted in plant phospholipid [34]. Although these experiments were not done in the context of mPTP, they were the first to demonstrate the ability of the c-subunit to form ion channels. C-subunit channel activity has also been shown to depend on its phosphorylation status [31]. Further, c-subunit was present in chloroform extracts of the mitochondrial polyhydroxybutyrate/Ca2+/polyphosphate complexes. These extracts, when reconstituted into planar lipid bilayers, demonstrated channel activity resembling native mPTP, suggesting the possibility that interactions between all these macromolecules might be important for the mPTP formation [1, 35]. Interestingly, a very recent study demonstrated that a fully synthetic c-subunit is an amyloidogenic peptide capable of forming fibrils and β-sheet oligomers [36]. It is tantalizing to hypothesize that the misfolded protein pathway of mPTP formation proposed by Lemasters’ group [37] could involve the mechanism of membrane permeabilization by c-subunit misfolding that involves spontaneous formation of β-sheet oligomers by the mechanism described for other channel forming amyloids amyloid-β and α-synuclein, in the context of neurodegeneration [38]. Notably, both amyloid-β and α-synuclein have been implicated as participants in mPTP formation [39–41]. With this respect it is also worth mentioning that some ionophores like mastoparan can facilitate CsA sensitive membrane permeabilization when added to the mitochondria in submicromolar concentrations. However, higher concentrations of these ionophores (mastoparan or alamethicin) induce calcium independent and CsA insensitive mPT that makes it intriguing to detect channel activity in these preparations in order to see how it compares to the classical mPTP [42].
Extensive investigation of the link between the mPTP, c-subunit peptide, and ATP synthase complex has been done by the Jonas group (reviewed in [43, 44]). This group showed that a purified mammalian c-subunit of ATP synthase could form a non-selective ion channel when reconstituted into liposomes [45]. In these experiments, patch clamping of liposomes with a purified c-subunit revealed the presence of non-selective channels with a sub-conductance state of ~100 pS and peak conductance up to 1.5-2.0 nS. The c-subunit channel closes at negative voltages and can be blocked by AMP, ADP, or ATP, and antibodies to c-subunit. The channel was not affected by usual mPTP regulators, Ca2+ and CsA, probably because of the absence of regulatory components. However, a monomeric ATP synthase that contained all regulatory sites also formed channel activity that was increased by the addition of recombinant CypD and became CsA sensitive. This could indicate that the regulatory component of the channel is present in the extra membrane space. Moreover, the authors showed that similar channel activity could be stimulated in submitochondrial vesicles (SMVs) by Ca2+, and this activity can be blocked by c-subunit antibodies and CsA. After urea-purification, channels in SMVs lose the sensitivity to CsA and Ca2+, indicating dissociation of the regulatory site from the channel. The interpretation that mPTP can be directly formed by the complete monomers of ATP synthase was later confirmed by the reconstitution of the highly purified intact complexes, as demonstrated by Cryo-EM [46]. It was demonstrated that the complexes that were used in the study maintained all morphological features of the intact complexes. In should be mentioned that the exact mechanism of how ATP synthase complex can be assembled/converted into the large pore remains elusive. Interestingly, very recent Cryo-EM structure of the complete ATP synthase done in the presence of Ca2+ suggests dramatic rearrangement and loss of normal c-ring structure [47]. Although this study did not measure channel activity, it supports the notion that conformational changes in the c-ring might play a critical role in the pore formation [48, 49].
mPTP derived from ATP synthase might also occur through the formation of the pore at the interface between two monomers. Giorgio et al. reported that gel-purified dimers, but not monomers, of ATP synthase form a channel in a model lipid bilayer in the presence of Ca2+ and the addition of benzodiazepine 423 [50]. Benzodiazepine 423 has a common binding site with CypD on oligomycin sensitivity-conferring protein (OSCP) subunit of the lateral stalk of ATP synthase, and therefore it can replace CypD and facilitate channel formation in the lipid bilayer. In the presence of benzodiazepine 423 and Ca2+, ATP synthase dimers formed the multiple sub-conductance states channel with a maximal conductance up to 1.0–1.3 nS in 150 mM KCI. The channel was blocked by Mg2+, ADP, and ATP synthase inhibitor AMP-PNP, but was not sensitive to CsA or ANT ligands BKA and atractyloside. Later the same group provided additional rigorous support for their conclusions by showing the presence of dimers and tetramers in the active fraction using Cryo-EM. It should be noted though, as in the case of monomers studies by Jonas group, that the exact mechanism of dimer (or tetramer) induced pore formation remains elusive and will require further structural studies. Discussion regarding some possible mechanisms of the pore formation by the ATPase complexes can be found in a recent theoretical paper by Gerle [51].
Overall, experiments with reconstituted proteins showed that mitochondria have numerous possibilities to assemble channels with electrophysiological properties that resemble Ca2+-activated, CsA-sensitive mPTP. These experiments do not definitively establish which protein is responsible for the mPTP. However, they raise the possibility of multiple pathways for mPT development.
Patch-clamp experiments with genetically modified mitochondria
In another approach to isolate the molecular identity of the mPTP, researchers have used patch-clamp to study the channel activity of various knockout models. The use of patch clamp is particularly useful. Indeed, one of the limitations associated with KO studies is that they can produce mitochondria with significantly compromised metabolism. The altered metabolic activity might lead to indirect changes in mPTP activity, when assayed in the intact organelles. This makes application of the patch-clamp essential since it directly measures channel activity driven by applied voltage in the purified membranes under well-define experimental conditions, allowing to exclude other parameters that might alter biophysical properties of the channel indirectly.
Genetic manipulations to delete putative mPTP components were first applied to yeast. Lohret et al. reported on a VDAC-less yeast strain that has channel activity very similar to wild-type strain [52]. Later the same group reported that proteoliposomes containing inner membrane from mitochondria of wild-type and ANT-deficient yeast strains contain a channel of similar ion selectivity, voltage dependence, and peak conductance [53]. These studies challenged the idea of VDAC and ANT as essential structural components of the mPTP.
The involvement of ANT and ATP synthase in the mPTP was further studied by patch-clamp experiments of the corresponding knockout constructs. Before patch-clamp experiments, these models were characterized extensively by non-electrophysiological methods. It was demonstrated that liver mitochondria from ANT-deficient mice could still undergo permeability transition (measured by the swelling and CRC assays). Notably, mPT initiation required three times more Ca2+ compared to wild-type mitochondria. Moreover, the deletion of ANT led to the loss of sensitivity of mPT to the ANT ligands [30]. Another set of experiments with human near-haploid HAP1-A12 cells, which lack various components of ATP synthase, also showed the ability to undergo mPT [54–56]. Therefore, genetic deletion of the two most discussed candidates (i.e. ANT and parts of the ATP synthase) did not result in the total blockage of the mPT phenomena, but the properties of mPT seemed to change. These results could point to the idea that multiple molecular pathways could be responsible for the mPT phenomena. It has been proposed previously that misfolding proteins could be a mechanism for multiple molecular pathways for mPTP formation [37]. According to this mechanism any damaged and clustered proteins when overcome chaperon activity could form unregulated pores in the conditions of calcium exposure. The idea that mPT can be caused by distinct channels was recently confirmed electrophysiologically [32]. Using excised patch clamp configuration, ATPase c-subunit deficient HAP1-A12 cells in the presence of Ca2+ were shown to still contain a CsA sensitive channel, but its properties differed from the channel in wild-type HAP1 cells. The channel in HAP1-A12 cells had a smaller conductance of 300 pS on average versus 1.3 nS in wild-type cells, and it was sensitive to the inhibitors of ANT, ADP, and BKA. The channels of both conductance were large enough to increase the permeability of IMM, and thus mPT, but these channels had distinct properties.
Electrophysiological study of ANT triple null Mouse Embryonic Fibroblast (MEF) cells showed complete block mPTP, whereas ANT null mitochondria from mouse liver also required KO of Pfif gene, which encodes CypD [57]. These data again point to the existence of several, perhaps tissue dependent mPTP pathways with a distinct molecular nature.
Taken together, these studies of using genetically modified models indicate that mitochondria can undergo permeability transition by the formation of at least two different channels in a tissue- or cell type-dependent manner.
The diversity of mPTP properties, identified by multiple studies, have challenged the old paradigm of the unique pathway of channel formation during mPT. Electrophysiological and pharmacological properties of channels vary greatly from one model of mPT to another. The new hypothesis of multiple mPT phenotypes has recently become more accepted [58–60]. Figure 2 summarizes possible mPT pathways that can be present in the mitochondria (Fig 2). Specific mPT phenotypes could be tissue- or pathophysiology-dependent and require further investigation.
Figure 2. Possible mechanisms of the formation of the mPTP.
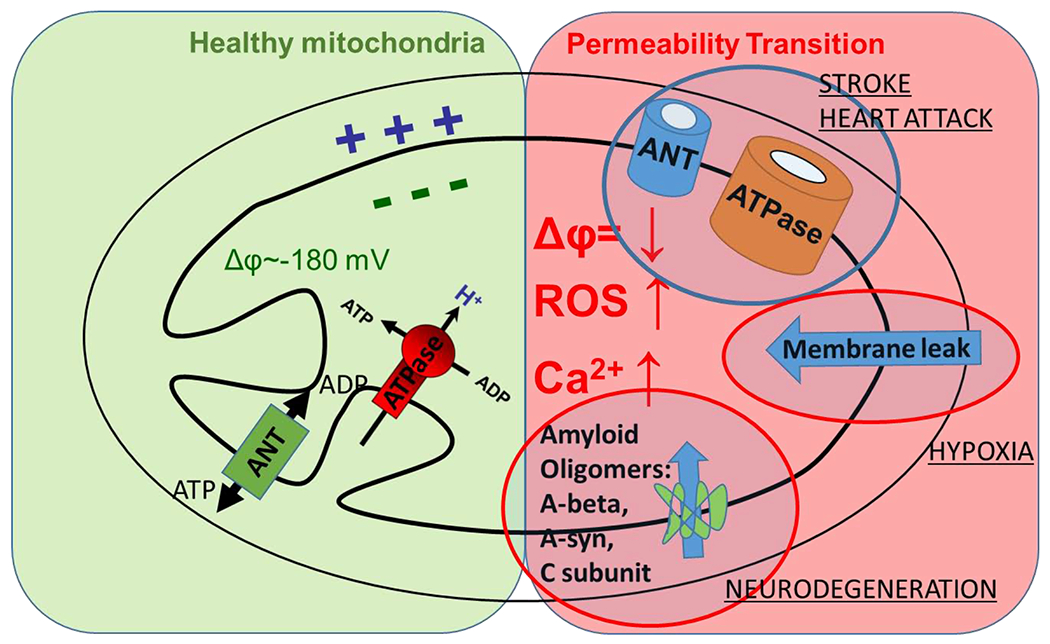
Under normal physiological conditions, the mitochondrial inner membrane is largely impermeable to ions and contains functional ATP synthase and ANT. Upon permeability transition pore development, mPTP can be formed by several putative molecular mechanisms: 1) conversion of the ATP synthase or ANT into the large pores – currently established mechanisms; 2) non-specific membrane leak through the lipid bilayer; 3) direct formation of the mPTP by misfolded amyloid peptides.
Electrophysiology of the mPTP in disease-relevant conditions
mPTP activation has been proposed to play a key role in the development of many pathologies, including several types of acute hypoxic-ischemic injuries, chronic neurodegeneration, metabolic disorders, and cancer [61]. As outlined above, the mPTP likely occurs through several independent pathways. Considering the wide range of conditions that can evoke the mPTP, it is critically important to investigate which particular path of the mPTP is activated under what specific stress conditions. The electrophysiological approach can complement approaches traditionally used to detect and investigate mPTP, which including measurements of membrane depolarization, CRC, or mitochondrial swelling [62, 63]. These indirect methods allow reliable detection of mPTP but cannot provide detailed biophysical information on which specific channel has been activated by which specific stress or on the nature of membrane permeabilization and whether it involves an opening of the pore or non-specific leak across the lipid bilayer. A direct patch-clamp approach towards measurements of the mPTP in diseased membranes might be able to address this issue and help to clarify several questions that currently remain unresolved. These experiments can involve patch-clamp investigations of mitoplasts that have been isolated from human tissues known to experience mPTP-dependent stress or from animals representing genetic models of disease. Similar strategy was used previously to investigate how stress affects the activity of the channels of the mitochondrial outer membrane [10, 64]. While this line of the investigation in the context of mPTP remains largely unexplored, several recent studies have demonstrated the feasibility of this approach and its potential for wider use in the near future.
mPTP activity during hypoxic injury
One example of how electrophysiology is used to uncover the mechanism of mPTP is a recent study in white matter brain injury (WMI), a condition that is caused by brief episodes of hypoxia (intermittent hypoxia) in prematurely born children [65]. Unlike acute ischemia seen during stoke, WMI does not cause extensive neuronal loss but is characterized by the disruption of axon myelination and developmental brain abnormalities. The researchers found that at the animal and cellular levels, this process is dependent on CypD and CsA, suggesting the involvement of mPTP. However, they did not observe extensive swelling of the mitochondria, indicating that mPTP is not activated in its high conductance mode. Patch-clamp of the mitochondrial membranes from the diseased tissue uncovered that WMI is associated with mPTP functioning as a small “leaky” channel with no resolvable transitions (suggesting the sub pS conductance of the single conducting unit) but not as a large conductance pore which has a conductance of 1.5 nS. It will be interesting to investigate whether more severe ischemia-reperfusion mediated brain injury will activate the “canonical” large conductance mPTP, regularly seen in Ca2+ overloaded mitochondria.
In perspective, this electrophysiological approach opens very exciting possibilities for investigating the signaling mechanisms regulating mPTP activity during the progression of diseases. For example, under various disease stresses, through post-translational/transcriptional modification, a specific mPTP can have an up or down regulation of channel expression levels, or it can open at different sub-conductance states or at different frequencies. A recent study performed in vivo and at the level of isolated mitochondria established the link between CypD phosphorylation, mPTP opening, and cell death during ischemia-reperfusion injury [66]. This study demonstrated that CypD phosphorylation at specific residue S191 controls its ability to activate mPTP. Patch-clamp applied directly to these mitochondria will improve understanding of the specific modulations of the mPTP that are caused by CypD phosphorylation at the single channel activity level.
mPTP activity in neurodegeneration
mPTP with complicated conductance patterns can be present in conditions associated with neuronal development abnormalities and neurodegeneration of genetic nature. In these settings, the patch-clamp approach can be instrumental in defining the nature of mPTP and possible candidate proteins involved in the process. This approach was successfully used recently to characterize “non-canonical” mPTP involved in Fragile X syndrome (FXS), a genetic disorder that is the most common inherited cause of intellectual disability [43]. At the neuronal level, FXS is characterized by defects in synaptic plasticity, excitotoxicity, and increased excitability. This study established that FXS conditions are linked to abnormalities in mitochondria. Specifically, mitochondria appeared to be swollen, have an increased number of dense granules, and decreased mitochondrial membrane potential. All of these characteristics suggested possible involvement of mPTP in disrupting the normal mitochondrial function. The patch-clamp approach applied to these mitochondria showed that mitochondrial abnormalities are caused by increased activity of the dexpramipexole-sensitive ATP synthase channel. Activation of this channel includes increased peak conductance that caused uncoupling of oxidative phosphorylation, leading to disruption of energy homeostasis and loss of normal neuronal function.
mPTP activity in diabetes
An electrophysiological approach was successfully applied to investigate functional details underlying the increased mPTP activity in mitochondria isolated from diabetic brain tissues [67]. In these experiments, mPTP was investigated in mice with streptozotocin-induced diabetes. Light scattering assays done with intact isolated mitochondria demonstrated increased rates and degree of swelling in diabetic mitochondria. This swelling was sensitive to the CsA, indicating involvement of the mPTP. The patch-clamp experiments done with the mitoplasts from this preparation showed that increased mPTP activity is caused by the increase in open probability of the channel, while the size and regulation of the channel remained the same in both the control and diseased preparations.
Conclusion
In recent years, it has become evident that the mPTP channel likely has multiple molecular identities. The electrophysiological approach will be able to differentiate these multiple mPTP pathways at the level of single channel and whole mitochondrion conductance. Use of this approach in combination with genetic models will help to clarify molecular and regulatory mechanisms of mPTP under physiological and disease conditions. This quantitative information about the biophysical properties of mPTP will help in the discovery of novel drug targets for the treatment of human diseases.
Highlights.
This review summarizes the studies of using electrophysiological techniques to characterize the properties of mPTP
We discuss that multiple molecular mechanisms of the mPTP can potentially coexist based on the electrophysiological data
We discuss the unique advantages of applying electrophysiological approaches in studying the mPTP under disease relevant conditions
Acknowledgments
This work was supported by an NIH R01GM115570, United States grant and an American Heart Association, United Sates grant (16GRNT27260229) (to E.V.P.), NIH R01HL093671, R01HL137266, R01HL142864 & R01HL122124 (to S-S. S.). We thank Jennifer Wilson for the English language editing on the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Competing Interests
The authors declare that they have no conflict of interest.
References
- [1].Elustondo PA, Nichols M, Negoda A, Thirumaran A, Zakharian E, Robertson GS, Pavlov EV, Mitochondrial permeability transition pore induction is linked to formation of the complex of ATPase C-subunit, polyhydroxybutyrate and inorganic polyphosphate, Cell Death Discov 2 (2016) 16070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Duchen MR, Roles of mitochondria in health and disease, Diabetes 53 Suppl 1 (2004) S96–102. [DOI] [PubMed] [Google Scholar]
- [3].Lemasters JJ, Nieminen AL, Qian T, Trost LC, Elmore SP, Nishimura Y, Crowe RA, Cascio WE, Bradham CA, Brenner DA, Herman B, The mitochondrial permeability transition in cell death: a common mechanism in necrosis, apoptosis and autophagy, Biochim. Biophys. Acta 1366(1-2) (1998) 177–196. [DOI] [PubMed] [Google Scholar]
- [4].Lemasters JJ, Nieminen AL, Qian T, Trost LC, Herman B, The mitochondrial permeability transition in toxic, hypoxic and reperfusion injury, Mol Cell Biochem 174(1-2) (1997) 159–65. [PubMed] [Google Scholar]
- [5].Murphy E, Bernardi P, Cohen M, Di Lisa F, Forte M, Molkentin JD, Ovize M, Fondation Leducq Transatlantic Network of Excellence Targeting Mitochondria to Treat Heart Disease, Circ Res 124(9) (2019) 1294–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Halestrap AP, Davidson AM, Inhibition of Ca2(+)-induced large-amplitude swelling of liver and heart mitochondria by cyclosporin is probably caused by the inhibitor binding to mitochondrial-matrix peptidyl-prolyl cis-trans isomerase and preventing it interacting with the adenine nucleotide translocase, Biochem. J 268(1) (1990) 153–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Halestrap AP, Connern CP, Griffiths EJ, Kerr PM, Cyclosporin A binding to mitochondrial cyclophilin inhibits the permeability transition pore and protects hearts from ischaemia/reperfusion injury, Mol. Cell Biochem 174(1-2) (1997) 167–172. [PubMed] [Google Scholar]
- [8].Zoratti M, Szabo I, The mitochondrial permeability transition, Biochim. Biophys. Acta 1241(2) (1995) 139–176. [DOI] [PubMed] [Google Scholar]
- [9].Guo L, Pietkiewicz D, Pavlov EV, Grigoriev SM, Kasianowicz JJ, Dejean LM, Korsmeyer SJ, Antonsson B, Kinnally KW, Effects of cytochrome c on the mitochondrial apoptosis-induced channel MAC, Am. J. Physiol Cell Physiol 286(5) (2004) C1109–C1117. [DOI] [PubMed] [Google Scholar]
- [10].Pavlov EV, Priault M, Pietkiewicz D, Cheng EH, Antonsson B, Manon S, Korsmeyer SJ, Mannella CA, Kinnally KW, A novel, high conductance channel of mitochondria linked to apoptosis in mammalian cells and Bax expression in yeast, J Cell Biol 155(5) (2001) 725–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Moran O, Sandri G, Panfili E, Stuhmer W, Sorgato MC, Electrophysiological characterization of contact sites in brain mitochondria, J Biol Chem 265(2) (1990) 908–13. [PubMed] [Google Scholar]
- [12].Moran O, Sciancalepore M, Sandri G, Panfili E, Bassi R, Ballarin C, Sorgato MC, Ionic permeability of the mitochondrial outer membrane, Eur Biophys J 20(6) (1992) 311–9. [DOI] [PubMed] [Google Scholar]
- [13].Decker GL, Greenawalt JW, Ultrastructural and biochemical studies of mitoplasts and outer membranes derived from French-pressed mitochondria. Advances in mitochondrial subfractionation, J Ultrastruct Res 59(1) (1977) 44–56. [DOI] [PubMed] [Google Scholar]
- [14].Pallotti F, Lenaz G, Isolation and subfractionation of mitochondria from animal cells and tissue culture lines, Methods Cell Biol 80 (2007) 3–44. [DOI] [PubMed] [Google Scholar]
- [15].Sorgato MC, Keller BU, Stuhmer W, Patch-clamping of the inner mitochondrial membrane reveals a voltage-dependent ion channel, Nature 330(6147) (1987) 498–500. [DOI] [PubMed] [Google Scholar]
- [16].Kinnally KW, Campo ML, Tedeschi H, Mitochondrial channel activity studied by patch-clamping mitoplasts, J. Bioenerg. Biomembr 21(4) (1989) 497–506. [DOI] [PubMed] [Google Scholar]
- [17].Petronilli V, Szabo I, Zoratti M, The inner mitochondrial membrane contains ion-conducting channels similar to those found in bacteria, FEBS Lett 259(1) (1989) 137–143. [DOI] [PubMed] [Google Scholar]
- [18].Kirichok Y, Krapivinsky G, Clapham DE, The mitochondrial calcium uniporter is a highly selective ion channel, Nature 427(6972) (2004) 360–364. [DOI] [PubMed] [Google Scholar]
- [19].Tupper JT, Tedeschi H, Microelectrode studies on the membrane properties of isolated mitochondria, Proc Natl Acad Sci U S A 63(2) (1969) 370–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Szabo I, Zoratti M, The mitochondrial megachannel is the permeability transition pore, J. Bioenerg. Biomembr 24(1) (1992) 111–117. [DOI] [PubMed] [Google Scholar]
- [21].Hunter DR, Haworth RA, Southard JH, Relationship between configuration, function, and permeability in calcium-treated mitochondria, J Biol Chem 251(16) (1976) 5069–77. [PubMed] [Google Scholar]
- [22].Kinnally KW, Zorov D, Antonenko Y, Perini S, Calcium modulation of mitochondrial inner membrane channel activity, Biochem. Biophys. Res. Commun 176(3) (1991) 1183–1188. [DOI] [PubMed] [Google Scholar]
- [23].Kinnally KW, Antonenko YN, Zorov DB, Modulation of inner mitochondrial membrane channel activity, J. Bioenerg. Biomembr 24(1) (1992) 99–110. [DOI] [PubMed] [Google Scholar]
- [24].Kane DA, Pavlov EV, Calculation of ion currents across the inner membrane of functionally intact mitochondria, Channels (Austin) 7(6) (2013) 426–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zorov DB, Kinnally KW, Tedeschi H, Voltage activation of heart inner mitochondrial membrane channels, J Bioenerg Biomembr 24(1) (1992) 119–24. [DOI] [PubMed] [Google Scholar]
- [26].Beutner G, Ruck A, Riede B, Welte W, Brdiczka D, Complexes between kinases, mitochondrial porin and adenylate translocator in rat brain resemble the permeability transition pore, FEBS Lett 396(2-3) (1996) 189–95. [DOI] [PubMed] [Google Scholar]
- [27].Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD, Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death, Nat. Cell Biol 9(5) (2007) 550–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Brustovetsky N, Klingenberg M, Mitochondrial ADP/ATP carrier can be reversibly converted into a large channel by Ca2+, Biochemistry 35(26) (1996) 8483–8488. [DOI] [PubMed] [Google Scholar]
- [29].Brustovetsky N, Tropschug M, Heimpel S, Heidkamper D, Klingenberg M, A large Ca2+-dependent channel formed by recombinant ADP/ATP carrier from Neurospora crassa resembles the mitochondrial permeability transition pore, Biochemistry 41(39) (2002) 11804–11. [DOI] [PubMed] [Google Scholar]
- [30].Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, Wallace DC, The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore, Nature 427(6973) (2004) 461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Azarashvili T, Odinokova I, Bakunts A, Ternovsky V, Krestinina O, Tyynela J, Saris NE, Potential role of subunit c of F0F1-ATPase and subunit c of storage body in the mitochondrial permeability transition. Effect of the phosphorylation status of subunit c on pore opening, Cell Calcium 55(2) (2014) 69–77. [DOI] [PubMed] [Google Scholar]
- [32].Neginskaya MA, Solesio ME, Berezhnaya EV, Amodeo GF, Mnatsakanyan N, Jonas EA, Pavlov EV, ATP Synthase C-Subunit-Deficient Mitochondria Have a Small Cyclosporine A-Sensitive Channel, but Lack the Permeability Transition Pore, Cell Rep 26(1) (2019) 11–17 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Urbani A, Giorgio V, Carrer A, Franchin C, Arrigoni G, Jiko C, Abe K, Maeda S, Shinzawa-Itoh K, Bogers JFM, McMillan DGG, Gerle C, Szabo I, Bernardi P, Purified F-ATP synthase forms a Ca(2+)-dependent high-conductance channel matching the mitochondrial permeability transition pore, Nat Commun 10(1) (2019) 4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].McGeoch JE, Guidotti G, A 0.1-700 Hz current through a voltage-clamped pore: candidate protein for initiator of neural oscillations, Brain Res 766(1–2) (1997) 188–194. [DOI] [PubMed] [Google Scholar]
- [35].Pavlov E, Zakharian E, Bladen C, Diao CT, Grimbly C, Reusch RN, French RJ, A large, voltage-dependent channel, isolated from mitochondria by water-free chloroform extraction, Biophys. J 88(4) (2005) 2614–2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Amodeo GF, Lee BY, Krilyuk N, Filice CT, Valyuk D, Otzen D, Noskov S, Leonenko Z, Pavlov EV, C subunit of the ATP synthase is an amyloidogenic channel-forming peptide: possible implications in mitochondrial pathogenesis, bioRxiv (2020) 2020.01.16.908335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].He L, Lemasters JJ, Regulated and unregulated mitochondrial permeability transition pores: a new paradigm of pore structure and function?, FEBS Lett 512(1–3) (2002) 1–7. [DOI] [PubMed] [Google Scholar]
- [38].Dobson CM, Protein folding and misfolding, Nature 426(6968) (2003) 884–90. [DOI] [PubMed] [Google Scholar]
- [39].Ludtmann MHR, Angelova PR, Horrocks MH, Choi ML, Rodrigues M, Baev AY, Berezhnov AV, Yao Z, Little D, Banushi B, Al-Menhali AS, Ranasinghe RT, Whiten DR, Yapom R, Dolt KS, Devine MJ, Gissen P, Kunath T, Jaganjac M, Pavlov EV, Klenerman D, Abramov AY, Gandhi S, alpha-synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson’s disease, Nat Commun 9(1) (2018) 2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Guo L, Du H, Yan S, Wu X, McKhann GM, Chen JX, Yan SS, Cyclophilin D deficiency rescues axonal mitochondrial transport in Alzheimer’s neurons, PLoS One 8(1) (2013) e54914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Du H, Guo L, Wu X, Sosunov AA, McKhann GM, Chen JX, Yan SS, Cyclophilin D deficiency rescues Abeta-impaired PKA/CREB signaling and alleviates synaptic degeneration, Biochim Biophys Acta 1842(12 Pt A) (2014) 2517–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Pfeiffer DR, Gudz TI, Novgorodov SA, Erdahl WL, The peptide mastoparan is a potent facilitator of the mitochondrial permeability transition, J Biol Chem 270(9) (1995) 4923–32. [DOI] [PubMed] [Google Scholar]
- [43].Licznerski P, Park HA, Rolyan H, Chen R, Mnatsakanyan N, Miranda P, Graham M, Wu J, Cruz-Reyes N, Mehta N, Sohail S, Salcedo J, Song E, Effman C, Effman S, Brandao L, Xu GN, Braker A, Gribkoff VK, Levy RJ, Jonas EA, ATP Synthase c-Subunit Leak Causes Aberrant Cellular Metabolism in Fragile X Syndrome, Cell (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Jonas EA, Porter GA Jr., Beutner G, Mnatsakanyan N, Alavian KN, Cell death disguised: The mitochondrial permeability transition pore as the c-subunit of the FF ATP synthase, Pharmacol. Res (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Alavian KN, Beutner G, Lazrove E, Sacchetti S, Park HA, Licznerski P, Li H, Nabili P, Hockensmith K, Graham M, Porter GA Jr., Jonas EA, An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore, Proc. Natl. Acad. Sci. U. S. A 111(29) (2014) 10580–10585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Mnatsakanyan N, Llaguno MC, Yang Y, Yan Y, Weber J, Sigworth FJ, Jonas EA, A mitochondrial megachannel resides in monomeric F1FO ATP synthase, Nat Commun 10(1) (2019) 5823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Pinke G, Zhou L, Sazanov LA, Cryo-EM structure of the entire mammalian F-type ATP synthase, Nat Struct Mol Biol (2020). [DOI] [PubMed] [Google Scholar]
- [48].Gerle C, Mitochondrial F-ATP synthase as the permeability transition pore, Pharmacol Res 160 (2020) 105081. [DOI] [PubMed] [Google Scholar]
- [49].Mnatsakanyan N, Jonas EA, ATP synthase c-subunit ring as the channel of mitochondrial permeability transition: Regulator of metabolism in development and degeneration, J Mol Cell Cardiol 144(2020)109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Giorgio V, von SS, Antoniel M, Fabbro A, Fogolari F, Forte M, Glick GD, Petronilli V, Zoratti M, Szabo I, Lippe G, Bernardi P, Dimers of mitochondrial ATP synthase form the permeability transition pore, Proc. Natl. Acad. Sci. U. S. A 110(15) (2013) 5887–5892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Gerle C, On the structural possibility of pore-forming mitochondrial FoF1 ATP synthase, Biochim Biophys Acta 1857(8) (2016) 1191–1196. [DOI] [PubMed] [Google Scholar]
- [52].Lohret TA, Kinnally KW, Multiple conductance channel activity of wild-type and voltage-dependent anion-selective channel (VDAC)-less yeast mitochondria, Biophys J 68(6) (1995) 2299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lohret TA, Murphy RC, Drgon T, Kinnally KW, Activity of the mitochondrial multiple conductance channel is independent of the adenine nucleotide translocator, J Biol Chem 271(9) (1996) 4846–9. [DOI] [PubMed] [Google Scholar]
- [54].Carroll J, He J, Ding S, Fearnley IM, Walker JE, Persistence of the permeability transition pore in human mitochondria devoid of an assembled ATP synthase, Proc Natl Acad Sci U S A 116(26) (2019) 12816–12821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].He J, Carroll J, Ding S, Fearnley IM, Walker JE, Permeability transition in human mitochondria persists in the absence of peripheral stalk subunits of ATP synthase, Proc Natl Acad Sci U S A 114(34) (2017) 9086–9091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].He J, Ford HC, Carroll J, Ding S, Fearnley IM, Walker JE, Persistence of the mitochondrial permeability transition in the absence of subunit c of human ATP synthase, Proc Natl Acad Sci U S A 114(13) (2017) 3409–3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Karch J, Bround MJ, Khalil H, Sargent MA, Latchman N, Terada N, Peixoto PM, Molkentin JD, Inhibition of mitochondrial permeability transition by deletion of the ANT family and CypD, Sci Adv 5(8) (2019) eaaw4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Bernardi P, Mechanisms for Ca(2+)-dependent permeability transition in mitochondria, Proc Natl Acad Sci U S A (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Carraro M, Carrer A, Urbani A, Bernardi P, Molecular nature and regulation of the mitochondrial permeability transition pore(s), drug target(s) in cardioprotection, J Mol Cell Cardiol 144 (2020) 76–86. [DOI] [PubMed] [Google Scholar]
- [60].Bround MJ, Bers DM, Molkentin JD, A 20/20 view of ANT function in mitochondrial biology and necrotic cell death, J Mol Cell Cardiol (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Bonora M, Patergnani S, Ramaccini D, Morciano G, Pedriali G, Kahsay AE, Bouhamida E, Giorgi C, Wieckowski MR, Pinton P, Physiopathology of the Permeability Transition Pore: Molecular Mechanisms in Human Pathology, Biomolecules 10(7) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Baev AY, Elustondo PA, Negoda A, Pavlov EV, Osmotic regulation of the mitochondrial permeability transition pore investigated by light scattering, fluorescence and electron microscopy techniques, Anal Biochem (2017). [DOI] [PubMed] [Google Scholar]
- [63].Neginskaya MA, Strubbe JO, Amodeo GF, West BA, Yakar S, Bazil JN, Pavlov EV, The very low number of calcium-induced permeability transition pores in the single mitochondrion, J Gen Physiol 152(10) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Bonanni L, Chachar M, Jover-Mengual T, Li H, Jones A, Yokota H, Ofengeim D, Flannery RJ, Miyawaki T, Cho CH, Polster BM, Pypaert M, Hardwick JM, Sensi SL, Zukin RS, Jonas EA, Zinc-dependent multi-conductance channel activity in mitochondria isolated from ischemic brain, J Neurosci 26(25) (2006) 6851–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Niatsetskaya Z, Sosunov S, Stepanova A, Goldman J, Galkin A, Neginskaya M, Pavlov E, Ten V, Cyclophilin D-dependent oligodendrocyte mitochondrial ion leak contributes to neonatal white matter injury, J Clin Invest (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Hurst S, Gonnot F, Dia M, Crola Da Silva C, Gomez L, Sheu SS, Phosphorylation of cyclophilin D at serine 191 regulates mitochondrial permeability transition pore opening and cell death after ischemia-reperfusion, Cell Death Dis 11(8) (2020) 661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Yan S, Du F, Wu L, Zhang Z, Zhong C, Yu Q, Wang Y, Lue LF, Walker DG, Douglas JT, Yan SS, F1F0 ATP Synthase-Cyclophilin D Interaction Contributes to Diabetes-Induced Synaptic Dysfunction and Cognitive Decline, Diabetes 65(11) (2016) 3482–3494. [DOI] [PMC free article] [PubMed] [Google Scholar]