

ABSTRACT
Irreversible electroporation (IRE) is an effective method for treating pancreatic ductal adenocarcinoma (PDAC). It remains unclear whether IRE can induce a specific immune response by stimulating macrophages. Here, the associated markers of macrophages were analyzed after exposure to tumor culture supernatant (TSN) of tumor cells treated with electroporation. Subcutaneous and orthotopic PDAC models were also used to evaluate the effect of macrophage polarization induced by IRE. Aside from its direct killing effect, IRE could induce the immunogenic cell death of tumor cells by increasing the synthesis and secretion of damage associated molecular patterns. Moreover, IRE could increase the release of HMGB1, which activates the MAPK-p38 pathway and leads to the increased expression of M1 markers in macrophages, through binding to the receptor of the advanced glycation end-product (RAGE) receptor. M1 polarization was inhibited by the inhibitors of HMGB1 release, the RAGE receptor, and the MAPK-p38 signaling pathway, but it was activated by rHMGB1 or the stimulator of MAPK-p38. In addition, the promotion of M1 macrophage polarization was enhanced by the positive-feedback release or expression of HMGB1 and RAGE through the MAPK-ERK pathway in macrophages. The promotion of M1 macrophage polarization induced by IRE provided a specific rationale for the combination of IRE and immune therapy in treating PDAC.
KEYWORDS: Irreversible electroporation, pancreatic cancer, macrophage, polarization, immunogenic cell death
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the fourth leading cause of cancer-specific death in the world.1 The highly immune-suppressive microenvironment of PDAC, which is characterized by the enrichment of fibrotic stroma and the infiltration of suppressive immune cells, further limits the efficacy of treatments, such as immune checkpoint blockade or immunotherapy.2 Tumor-associated macrophages (TAMs) play an important role in the development and progression of PDAC.3 In response to different stimuli, TAMs can be polarized and differentiated into classically activated M1 or alternatively activated M2 status.4 M1 macrophages play a critical role in tumor suppression by upregulating the expression of proinflammatory mediators and inducing an inflammatory state. Conversely, M2 macrophages contribute to the progression of the disease through the secretion of anti-inflammatory cytokines.5 Macrophages polarize to M2-type and exhibit distinct tumor-promoting activities after interacting with tumor cells and other components within the tumor microenvironment.6 The expansion of the M2 macrophage is another mechanism through which a desmoplastic stroma is formed, 7 promoting the immune escape of tumors. Higher levels of M2-macrophages have been clinically correlated with PDAC malignancy.8 Reprogramming the polarization of TAMs from immune-suppressive to immunogenic phenotypes may provide new insights into the field of immune therapy in PDAC.9
As one of the most lethal and challenging malignancies, PDAC has a dismal prognosis with a 5-year survival rate of only 8%.1 Surgical resection remains the only curative modality for patients with PC. However, most patients were diagnosed at advanced stages and the surgical resection rate was only 20%.10 This is because 40% of PDAC patients were diagnosed of diseases major vascular involvement and another 40% were present with distant metastases.11 For those with vascular involvement, which is also known as locally advanced pancreatic cancer (LAPC), chemotherapy is the recommended therapy, although it can only provide limited benefit for these patients.12 Recently, irreversible electroporation (IRE) has emerged as a newly developed non-thermal ablative technology that produces an extremely high electric field across cells, inducing cell membrane disruption and tumor cell death.13 Furthermore, IRE has shown to be an effective method for the treatment of PDAC with promising survival results.13–15 The non-thermal ablative characteristic of IRE ensures that the clinical effect is free of the heat-sink effect and leaves supporting tissue largely unaffected.16 It has become clear that antitumor therapy is more successful when it can kill tumor cells directly and simultaneously induce an immunogenic form of cell death.17,18 Immunogenic cell death (ICD), which is characterized by the regulated secretion of damage-associated molecular patterns (DAMPs), mainly including high mobility group B1 (HMGB1), calreticulin, and heat shock protein (HSP), suggests the activation of an immune response specific for cancer cells.19,20 Previous studies have shown that IRE could prime or boost tumor-specific immunity.21–24 We hypothesize that the molecules released from the dying cells following IRE contribute to the activation of antigen-presenting cells and induce anti-tumor immune responses. However, the alterations of major infiltrated cells, especially macrophages, which constitute the most important component of the tumor microenvironment (TME), have not been evaluated. In the present study, we aim to illustrate that IRE enhances the specific anti-tumor immune response through promoting M1 TAM polarization in PDAC.
Results
The direct killing effect of IRE on pancreatic cancer cells
To assess the efficacy of IRE on pancreatic cancer, tumor cell suspensions were exposed to electric fields with different field strengths, including 500 V/cm, 750 V/cm, 1000 V/cm, 1250 V/cm, and 1500 V/cm. The electric pulse numbers were set to 8, 16, and 24. The parameters for electroporation were as follows: pulse duration = 100 µs, pulse repetition frequency = 1 Hz. To detect cell viability, a CCK8 analysis was applied immediately following electroporation at each field strength level (Figure S1a). As the electric field strengths and pulse numbers increased, tumor cell viability decreased gradually. At 1500 V/cm, cell viability decreased by more than 98% compared to the control group after treatment. It was also demonstrated that the proportions of late apoptosis increased dramatically along with the increasing electric field strengths (Figure S1b). Additionally, although the ratios of irreversible perforation decreased with time under an electric field strength of 750 V/cm, they almost remained unaltered when the electric field strength was as high as 1500 V/cm (Figure S1c).
We further evaluated the antitumor efficacy of IRE in the orthotopic and subcutaneous pancreatic cancer models. The median length of survival was 20 days for mice in the untreated control group and sham-operation group while mice with orthotopic tumors in the IRE group achieved a significantly longer median survival of more than 40 days (p< .05). Furthermore, tumor sizes at one week after treatment in the IRE group were significantly smaller than those in the untreated and sham-operation groups (p< .01). Similar results were also obtained in the subcutaneous pancreatic cancer model (Figure S1d).
IRE induces ICD of tumor cells
To explore whether IRE induces ICD that can be exploited in immunotherapy, we assessed changes in the markers of ICD. IRE resulted in the increased expression of HMGB1, HSP70, and calreticulin, with increasing levels of electric field strength (Figure (1a, b, c)). Additionally, using FC analysis, it was shown that the quantification of surface exposed HMGB1, HSP70, and calreticulin of tumor cells after electroporation was elevated in an electric field strength-dependent manner (Figure 1d). Moreover, the translocation from the nucleus to the cytoplasm and the release of HMGB1 were also observed in vivo. The expression of HMGB1, HSP70, and calreticulin increased significantly after IRE (Figure (1e, f)).
Figure 1.

IRE induces ICD of tumor cells. (a) The expression of intracellular DAMPs of Panc-1 at 24 hours after tumor cells being treated with electric fields with different field strengths. (b) The expression of intracellular DAMPs of Bxpc-3 at 24 hours after tumor cells being treated with electric fields with different field strengths. The expression of HMGB1, HSP70, and calreticulin increased significantly along with the increasing strength of electroporation. (c) The analysis of released HMGB1 in TSN of tumor cells at 24 hours after tumor cells being treated with electric fields with different field strengths by ELISA. The levels of secreted HMGB1 in TSN of Panc-1, Bxpc-3, and Panc02 increased significantly along with the increasing strength of electroporation. (d) The quantification of surface exposed HMGB1, HSP70, and calreticulin of tumor cells after electroporation using flow cytometry. It was shown that the quantification of surface exposed HMGB1, HSP70, and calreticulin of tumor cells after electroporation was elevated in an electric field strength-dependent manner. (e) The increased expression of intracellular DAMPs of tumor cells treated with IRE in orthotopic and subcutaneous pancreatic cancer models (IHC). (f) The increased expression of intracellular DAMPs of tumor cells treated with IRE in orthotopic and subcutaneous pancreatic cancer models (IHF). Orthotopic or subcutaneous tumors from the sham operation group were used as control. IRE increased the expression of HMGB1, HSP70, and calreticulin in orthotopic or subcutaneous tumors. DAPI was used to indicate the nuclear of cells. One-way analysis of variance (ANOVA) with Bonferroni comparison test was performed. * p < .05, **, p < .01, ***, p < .001, NS. not significant
The effect of TSN on macrophage polarization
Additionally, tumor culture supernatant (TSN) of tumor cells treated with electric fields with different field strengths was added to the culture of PMA-stimulated THP-1 or RAW264.7 cells. The mRNA analysis demonstrated that TNF-α, CCL2, and IL-1ß mRNA levels were significantly up-regulated in an electric field strength-dependent manner (Figure 2a). Furthermore, expressions of the markers of M1 polarization of macrophages, including HLA-DR or CD16/3225,26 on the cell surface of THP-1 or RAW264.7, showed an electric field strength-dependent increase, while CD163 or CD206 were expressed to a lesser degree than control cells with TSN stimulation (Figure 2b, Figure S2a and b). Apart from the changes to the phenotypic characteristics of macrophages, the TSN of tumor cells treated with electroporation also induced dramatic changes in cell morphology. The exposure to TSN for 3 days also caused cells to spread and flatten into a round, pancake-like shape, 27 suggesting the M1 polarization of macrophages (Figure S2c).
Figure 2.

The effect of TSN on macrophage polarization. (a) The comparisons of mRNA levels of cytokines from THP-1 stimulated by TSN of Panc-1-treated with electric fields. The mRNA levels of M1 markers increased along with the increasing electric fields, while the mRNA levels of M2 markers did not show a continuous increase along with the increasing electric fields. (b) The expression of surface markers of THP-1 stimulated by TSN of Bxpc-3 treated with electric fields. Increased expression of HLA-DR on THP-1 along with the increasing electric fields was observed. (c) IRE could increase infiltration of M1 macrophage in pancreatic tumor tissue (IHC). Orthotopic or subcutaneous tumor tissue was used as control. (d) IRE could increase the expression of CD16/32 in infiltrated macrophages in pancreatic tumor tissue. Orthotopic or subcutaneous tumor tissue was used as control. F4/80 was used to indicate macrophage; CD16/32 was the marker of M1 macrophage; CD206 was the marker of M2 macrophage; DAPI was used to indicate the nuclear of cells. (e) The expression of CD16/32 in macrophage from tumor tissue, spleen, peripheral blood, and lymph node in an orthotopic model of mouse pancreatic cancer after IRE treatment. IRE increased the expression of CD16/32 on the surface of macrophages from these tissues in the orthotopic model of mouse pancreatic cancer. (f) The expression of CD16/32 in macrophages from tumor tissue, spleen, peripheral blood, and lymph node in the subcutaneous model of mouse pancreatic cancer after IRE treatment. IRE increased the expression of CD16/32 on the surface of macrophages from these tissues in the subcutaneous model of mouse pancreatic cancer. Orthotopic or subcutaneous tumors from the sham operation group were used as control. One-way analysis of variance (ANOVA) with Bonferroni comparison test was performed. * p < .05, **, p < .01, ***, p < .001, NS. not significant
We then explored immune cells in the orthotopic and subcutaneous pancreatic cancer models 7 days after IRE treatments. The number of representative macrophages per gram of tumor tissue was presented in Figure (2c, 2d). There were significantly higher frequencies of total infiltrated M1 macrophages within the tumor and regional lymph node in the IRE groups as compared to the control group (Figure (2(e, f)) and Figure S2e). There were no significant differences in the frequencies of M2 macrophages within the regional lymph node between the IRE and control groups. Additionally, a decreased infiltration of M2 macrophages after IRE was observed in the tumor and spleen tissue (Figure 2f and Figure S2f).
The effect of HMGB1 on macrophage polarization
Recombinant HMGB1 (rHMGB1) was added to the culture of PMA-stimulated THP-1 and RAW264.7 cells. It was shown that the levels of HLA-DR on macrophage surface were increased in an rHMGB1’s dose-dependent manner (Figure 3a). Most significantly, HLA-DR expression increased by over 10-fold following exposure to rHMGB1 with a concentration of 0.6ug/ml, compared to that of the control group. Furthermore, rHMGB1-induced morphology changed from a rod-like to a round shape, which also suggested the maturation of M1-polarized macrophages (Figure 3b).
Figure 3.
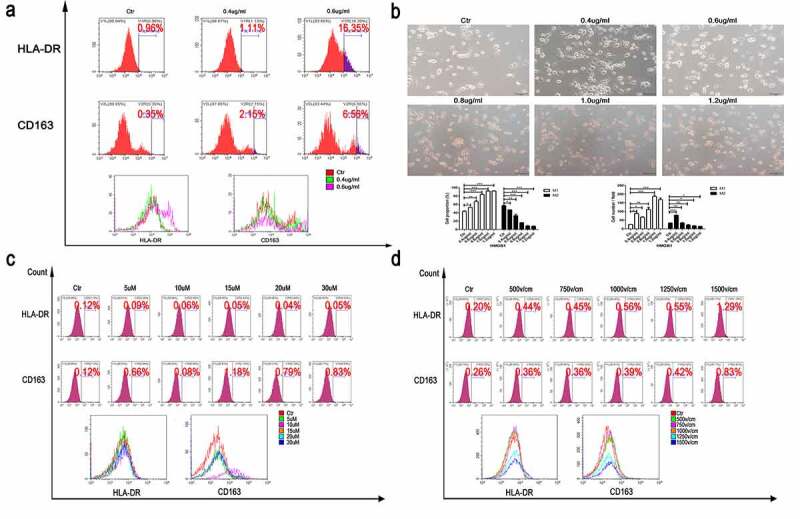
The effect of rHMGB1 on macrophage polarization. (a) The expression of surface markers of THP-1 stimulated by rHMGB1 with different concentrations for three days. Increased expression of HLA-DR of macrophage under the stimulation of higher doses of rHMGB1, compared with those of lower doses. (b) The morphologic changes of THP-1 stimulated by rHMGB1 with different concentrations for three days. The proportions of round and pancake-like shaped macrophages increased along with the elevated doses of rHMGB1, indicating that HMGB1 could promote the polarization of macrophages.(c) The expression of surface markers of macrophages stimulated by TSN of Panc-1 treated with electric fields after pretreatment with Glycyrrhizic Acid of different concentrations. (d) The expression of surface markers of macrophages stimulated by TSN of Panc-1 treated with electric fields after pretreatment with Glycyrrhizic Acid with a concentration of 10uM. After being treated with Glycyrrhizic Acid with a concentration of 10uM, no significant changes were observed in the expression levels of HLA-DR of macrophages stimulated by TSN of tumor cells treated with electric fields. The inhibitor was added to the cell suspension with the specific concentration 30 minutes prior to electroporation. * p < .05, **, p < .01, ***, p < .001, NS. not significant
To further substantiate the effect of HMGB1 on the polarization of macrophages, an inhibitor of HMGB1, Glycyrrhizic Acid (Gly; T2741, TargetMol, USA), was used in the subsequent experiments. Gly significantly inhibited the synthesis and secretion of HMGB1 (Figure S3a and b) and significantly prevented the formation of round-shaped macrophages stimulated with the TSN from tumor cells for 3 days after IRE treatments (Figure S3c). Similar results were also obtained in the FC assay. After being pretreated with Gly of varying doses, the elevation of cell surface M1 markers in THP-1 or RAW264.7 following IRE treatment was not observed (Figure 3c). We also found that 10uM Gly induced significant HMGB1 inhibition in the TSN and this concentration was selected in the following experiments. As detected by FC, the inhibition of M1 polarization of THP-1 and RAW264.7 was reproduced in the TSN of Panc-1, Bxpc-3 and Pan02, which were pretreated with Gly (Figure 3d, Figure S3d). Similar results were also obtained as those detected by RT-qPCR (Figure S3e). The gradual rise in mRNA levels of M1 markers along with the increasing electric field strength in macrophages, including TNF-α, CCL2, and IL-1ß, was inhibited or even reversed after the exposure of Gly.
HMGB1 upregulated RAGE expression and levels of autocrine HMGB1 in macrophage
To understand the mechanisms underlying TSN-mediated HMGB1 release, the culture of macrophages was replaced with fresh RPMI 1640 medium supplemented with 10% FBS after a brief (6 hours) exposure, and HMGB1 levels in the new culture medium were determined 24 hours after the onset of TSN stimulation. WB analysis revealed that the up-regulation of HMGB1 and receptor of advanced glycation end-product (RAGE) levels in macrophages occurred in an electric field strength level-dependent manner (Figure S4a). Additionally, the ELISA assay also showed that the TSN from cells after being treated with high strength of electric fields triggered a more pronounced and robust HMGB1 release from macrophages (Figure S4b). The expression of RAGE on macrophages was also explored in orthotopic and subcutaneous pancreatic cancer models at 7 days after IRE treatments. Compared with tumors without any treatment, IRE significantly elevated the expression of RAGE on macrophages in pancreatic cancer models (Figure S1c and d).
MAPK-p38 activation is indispensable for the M1 polarization of macrophages
Previous studies indicated that MAPK and NF-κB could play key roles in the differentiation of macrophages.28–30 We observed that the phosphorylation of MAPK-p38 (Figure 4a) and MAPK-ERK (Figure (4b, c)) was elevated after exposure to TSN of tumor cells treated with IRE compared with the control TSN. Compared with the control group, phosphorylated MAPK-ERK did not show an increase in an electric field strength level-dependent manner after tumor cells were pretreated with Gly. Although in Panc-1 group, there seemed to be an increase in the protein level of phosphorylated MAPK-ERK, the ascensional range was markedly decreased compared with that of the control group. In the Gly group, the protein level of phosphorylated MAPK-ERK was almost similar among different subgroups of varied electric field strength. In addition, the pretreatment of macrophage cell cultures with inhibitors for MAPK-p38 (SB203580, S1076, Selleck, Shanghai, China), but not MAPK-ERK (PD98059, S1177, Selleck) inhibited the promotion of M1 polarization of macrophages (Figure 4d). We observed that the M1 polarization of macrophages was linked to the activation of MAPK-p38 signaling pathways in an HMGB1 dose-dependent manner (Figure S5). Consistently, pretreatment with an activator of MAPK-p38 [chromium picolinate (100 nM), T4575, TargetMol] dramatically enhanced M1 polarization of macrophages (Figure 4e), suggesting that the activation of MAPK-p38 was involved in the M1 polarization of macrophages induced by HMGB1.
Figure 4.

MAPK-p38 activation is indispensable for the M1 polarization of macrophages. (a) The changes of some signaling pathways of THP-1 stimulated by TSN from Bxpc-3 and Panc-1 treated with electric fields. The western blot analysis demonstrated that expression of MAPK-p38 was increased in THP-1 stimulated by TSN from tumor cells treated with IRE. (b) The changes of MAPK-ERK signaling pathway of THP-1 stimulated by TSN of Panc-1 after the pretreatment with Glycyrrhizic Acid (10uM) or not. The western blot analysis demonstrated that increased expression of MAPK-p38 in THP-1 stimulated by TSN from Panc-1 treated with IRE was inhibited by Glycyrrhizic Acid. (c) The changes of MAPK-ERK signaling pathway of THP-1 stimulated by TSN of Bxpc-3 after the pretreatment with Glycyrrhizic Acid (10uM) or not. The western blot analysis demonstrated that increased expression of MAPK-p38 in THP-1 stimulated by TSN from Bxpc-3 treated with IRE was inhibited by Glycyrrhizic Acid. (d) The expression of surface markers of macrophages stimulated by TSN treated with electric fields after pretreatment with different inhibitors. The increased expression of M1 marker (CD16/32 and HLA-DR) in THP-1 stimulated by rHMGB1 or TSN from tumor cells treated with IRE was inhibited by the inhibitor of MAPK-p38 (SB203580, 10uM), not the inhibitor of MAPK ERK (PD98059, 10uM). (e) The expression of surface markers of macrophages stimulated by chromium picolinate (100 nM). The expression of the M1 marker (CD16/32 and HLA-DR) was stimulated by the activator of MAPK-p38, similar to that stimulated with rHMGB1 or TSN from tumor cells treated with IRE. The inhibitor was added to the cell suspension with the specific concentration 30 minutes prior to electroporation
HMGB1-activated MAPK-ERK, inducing an increased RAGE expression and the release of autocrine HMGB1 in macrophage
Our results showed that under the stimulation of rHMGB1 or TSN of tumor cells treated with IRE, the expression of RAGE increased significantly. Additionally, pretreatment of macrophage cell cultures with inhibitors for RAGE (FPS-ZM1, T3259, TargetMol) inhibited the activation of the MAPK-p38 signaling pathway (Figure 5a) and the promotion of M1 polarization of macrophages (Figure 5b), This illustrated that HMGB1 could induce M1 macrophage polarization via RAGE. Moreover, a positive-feedback mechanism is developed between the binding of HMGB1 and RAGE and the increasing release of HMGB1 and the expression of RAGE, leading to the enhancement of M1 macrophage polarization. Considering MAPK plays a key role in the induction of HMGB1, we analyzed the secretion of HMGB1 and the expression of RAGE on the cell surface of macrophages after the inhibition of MAPK-p38 and MAPK-ERK. We observed that the inhibition of MAPK-p38 had little impact on the expression of RAGE (Figure 5c) and the secretion of HMGB1 (Figure 5d). In addition, RAGE on the cell surface of macrophages increased significantly after macrophages were stimulated with rHMGB1, while the effect was converted by pretreatment with FPS-ZM1, not SB203580 (Figure 5e). Additionally, we observed that the phosphorylation of MAPK-ERK was inhibited after exposure to TSN of IRE-treated tumor cells which were pretreated with Gly (Figure (4b, c)) or FPS-ZM1. As shown in Figure S6a, without FPS-ZM1, there was a significant increase in p-ERK in an electric field strength level-dependent manner. With FPS-ZM1, it was shown that the ratios of p-ERK and t-ERK were similar among different subgroups of varied electric field strength. Therefore, the ascensional range of subgroups with FPS-ZM1 was markedly decreased compared with that of the control group (without FPS-ZM1). The pretreatment of macrophage cell cultures with PD98059 or FPS-ZM1 also significantly decreased the secretion of HMGB1 (Figure S6b) and expression of RAGE, as detected by FC analyses (Figure S6c). This suggested that the MAPK-ERK signaling pathway, which was activated after the binding of HMGB1 to RAGE, played an important role in the RAGE expression and release of autocrine HMGB1 of macrophages.
Figure 5.
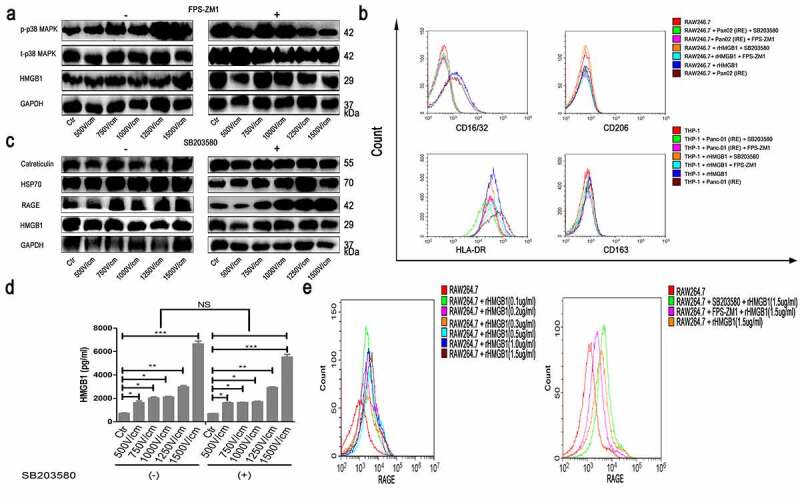
HMGB1 activated MAPK-ERK, inducing increased RAGE expression and release of autocrine HMGB1 in macrophages. (a) The changes of MAPK-p38 signaling pathway of RAW264.7 stimulated by TSN of Pan02 treated with electric fields after pretreatment with FPS-ZM1 (100 nM) or not. The western blot analysis demonstrated that increased expression of MAPK-p38 in THP-1 stimulated by TSN from tumor cells treated with IRE was inhibited by the inhibitor of RAGE (FPS-ZM1, 100 nM). (b) The expression of surface markers of macrophages stimulated by TSN of tumor cells treated with IRE or rHMGB1 after pretreatment with SB203580 or FPS-ZM1. The increased expression of M1 marker (CD16/32 and HLA-DR) in THP-1 stimulated by rHMGB1 or TSN from tumor cells treated with IRE was inhibited by the inhibitor of MAPK-p38 (SB203580, 10uM) and the inhibitor of RAGE (FPS-ZM1, 100 nM). (c) The expression of DAMPs of RAW264.7 stimulated by TSN of Pan02 treated with electric fields after pretreatment with SB203580 or not. The western blot analysis demonstrated that increased expression of RAGE in THP-1 stimulated by TSN from tumor cells treated with IRE was inhibited by the inhibitor of MAPK-p38 (SB203580). (d) The concentration measurement of HMGB1 of THP-1 stimulated by TSN of Panc-1 treated with electric fields after pretreatment with SB203580. The increased self-secretion of HMGB1 of THP-1 stimulated by TSN from tumor cells treated with IRE was not inhibited by the inhibitor of MAPK-p38 (SB203580, 10uM). (e) The expression of RAGE on the cellular surface of RAW264.7 stimulated by rHMGB1 with different concentrations. The increased self-expression of RAGE in THP-1 stimulated by rHMGB1 was inhibited by the inhibitor of RAGE (FPS-ZM1, 100 nM), not the inhibitor of MAPK-p38 (SB203580, 10uM). The inhibitor was added to the cell suspension with the specific concentration 30 minutes prior to electroporation
The enhancement of phagocytosis in macrophages stimulated by HMGB1 or TSN of tumor cells treated with IRE
The capacity of phagocytosis of fluorescent particles or dying tumor cells by macrophages was also analyzed by FC and IHF. Compared with macrophages without stimulation after exposure to HMGB1 or TSN of tumor cells treated with IRE, significantly larger proportions of stimulated macrophages were defined as cells that were double-positive for CFSE and eFluor 670 (Figure S7a and Figure 6a). These results were confirmed by IHF, demonstrating that both HMGB1 and TSN of IRE-treated tumors enhanced the phagocytosis in dying cells treated with IRE by macrophages and that this effect could be inhibited by the inhibitors of HMGB1, RAGE, and MAPK-p38 (Figure S7b and Figure 6b).
Figure 6.

The enhancement of phagocytosis in macrophages stimulated by rHMGB1 or TSN of tumor cells treated with IRE. (a) The detection of the ability of phagocytosis of tumor cells after IRE treatment in macrophages stimulated by rHMGB1 and TSN of tumor cells treated with electric fields. Upon the stimulation of rHMGB1 and TSN of tumor cells treated with electric fields, the proportions of macrophages that were double-positive for CFSE and eFluor 670 significantly elevated in an electric field strength-dependent manner, indicating that the proportions of activated macrophages were elevated Upon the stimulation of rHMGB1 and TSN of tumor cells treated with IRE. (b) The immunofluorescence co-localization analysis of RAW264.7 stimulated by TSN of Pan02 treated with IRE or rHMGB1 and fluorescent particles or Pan02 cells after IRE treatment. The increased ratios of stimulated macrophages which were defined as cells that were double-positive for CFSE and eFluor 670 stimulated by rHMGB1 or TSN of tumor cells treated with IRE were inhibited by the inhibitor of the release of HMGB1 (Glycyrrhizic Acid, 10uM), the inhibitor of RAGE (FPS-ZM1, 100 nM) and the inhibitor of MAPK-p38 (SB203580, 10uM). The inhibitor was added to the cell suspension with the specific concentration 30 minutes prior to electroporation
Discussion
Several existing studies indicated that IRE induces immune responses that provide local or systemic protection against tumor recurrence.18,23,31 This effect has been accompanied by an increase in CD4+ and CD8+ T cells, not only in TME but also in peripheral blood, which could be regarded as T cell activation.22,23 The activation of the function or increase in the number of T cells can be partly dependent on the increase of co-stimulatory signals, indicating the activation of prime immune responses. Additionally, unlike with surgical resection, the treated tumors are not removed from the body, but the dying cell remnants induced by IRE remain available to be taken up by phagocytic leukocytes. In keeping with this notion, this study explored the immune changes of apoptotic cells after IRE treatment and their effects on macrophages. We found that the apoptosis of tumor cells induced by IRE was accompanied by the release of DAMPs, which activated the infiltrating macrophages and promoted M1 polarization. The increase of phagocytosis in dying tumor cells by M1-polarized macrophages acted as tumor antigens, which in turn activated specific immune responses against the tumor.
Previous studies have indicated that IRE could mediate immune activation by increasing the number of infiltrating immune cells and by activating their function, 17,32 and served as an in situ tumor vaccination.33 In clinical practice, 1500 V/cm was adopted in the IRE procedure, which had the most powerful killing and perforation effect. Additionally, in this study, we showed that IRE induced the ICD of tumor cells by inducing them to release DAMP patterns. As the main component of DAMPs, the release of HMGB1 is a key event in the initiation of ICD.34 HMGB1 was released into the extracellular space by tumor cells after IRE. Previous studies have shown that HMGB1 influenced the differentiation of macrophages in systemic lupus erythematosus.35 Therefore, the effects of HMGB1 on macrophages might provide an avenue through which to explain the immune response induced by IRE.
Interestingly, macrophages with paradoxical characteristics were identified in TME, including M1 macrophages aimed at suppressing tumor progression and M2 macrophages aimed at driving tumor progression.4,36 In the original immune-suppressive TME of pancreatic cancer, M2 occupied the majority of TAMs and diminished the killing of tumor cells by cytotoxic T cells and NK cells.37 We found that under the stimulation of HMGB1, the expression of M1 markers, increased significantly in TAM. As shown by our results, HMGB1 released from dying tumor cells could reprogram the polarization of TAMs from immune-suppressive (M2) to immune-promoting (M1) phenotypes, illustrating that HMGB1 may provide new insights into the field of immune changes in PDAC after IRE.
Furthermore, through binding to the RAGE receptor on the cell surface of macrophages, HMGB1 induced an increase in RAGE expression and the release of autocrine HMGB1 in macrophages. This positive-feedback mechanism eventually led to the enhancement of M1 macrophage polarization. Additionally, the ability of macrophage phagocytosis was also significantly enhanced after exposure to TSN. The protection of not only larger vessels, but also micro-vessels by IRE, 21 was helpful for the increased infiltration of M1 macrophages and other immune cells. In line with our results, previous studies showed that HMGB1 induced changes in the formation of macrophages and promoted adhesive ability and migration in vivo and in vitro, 38 which was extremely important for the function of antigen presentation in macrophages. Studies also consistently showed that IRE induces more robust antigen release and T-cell activation as compared to other forms of cryo- or heat-ablation.39 The promotion of M1 polarization and the enhancement of macrophage phagocytosis will eventually induce the activation of T cells and special immune response. Therefore, local ablation through IRE provided pancreatic cancer, a poorly immunogenic tumor, with more opportunities for the combination with immune therapy, such as immune checkpoint inhibition.
As the main receptor of HMGB1, RAGE has a cytoplasmic domain that lacks endogenous tyrosine kinase activity, suggesting that RAGE might interact with cytoplasmic binding partners to trigger the recruitment of downstream effector pathways.40 Although studies suggested that several signaling cascades, including MAPK-ERK, -p38, and NF-κB, could be activated by RAGE ligand, 41 there remained a lack of clarity around which signaling pathway was triggered by the combination of HMGB1 and RAGE after IRE. We observed that MAPK-ERK and -p38 were significantly activated in macrophages after exposure to the TSN of tumor cells treated with IRE. Additionally, specific inhibitors of MAPK-p38 significantly abrogated HMGB1-induced M1 macrophage polarization. Moreover, the promotion of M1 macrophage polarization could be recovered by the specific activator for MAPK-p38, while the increased release or expression of HMGB1 and RAGE was effectively abrogated by the inhibitor of MAPK-ERK. Taken together, our results supported the potential role for MAPK-p38 in the regulation of M1 macrophage polarization induced by HMGB1. Simultaneously, a positive-feedback mechanism for the release or expression of HMGB1 and RAGE was finalized by activating the MAPK-ERK signaling pathway.42,43 As an important inflammatory factor, HMGB1 can be released or function through different pathways in response to different stimuli. IRE was illustrated to induce reactive oxygen stress (ROS), which contributed to the translocation of HMGB1 from the nucleus.44–46 Additionally, HMGB1 also facilitated ROS function, which acted as a signaling mediator in various pathways, including phosphorylation of MAPK-ERK and -p38 pathways.47 Therefore, through binding HMGB1 to RAGE, our study implicated RAGE signaling for the first time in macrophage responses. We evidenced that the accumulation or activation of M1 macrophages was stimulated by activating MAPK-p38 and was enhanced by a positive-feedback mechanism for the release or expression of HMGB1 and RAGE through the MAPK-ERK pathway in macrophages (Figure S8). The results of this study were limited by the lack of validation with the clinical biopsy tissue before and after IRE. More evidences based on the human biopsy tissue were needed to consolidate the results of our study.
In conclusion, we revealed that IRE could induce the ICD of tumor cells by releasing DAMPs in the current study. The released HMGB1 from tumor cells after IRE had promoted M1 macrophage polarization by activating MAPK-p38, which could be positively enhanced by the activation of the MAPK-ERK signaling pathway. Therefore, our finding of the promotion of M1 macrophage polarization induced by ICD provides a specific rationale for the combination of IRE and immune therapy in treating pancreatic cancer.
Methods
Cell lines and animal models
Human pancreatic adenocarcinoma cell lines Panc-1 and Bxpc-3, human monocyte cell line THP-1, murine pancreatic adenocarcinoma cell line Pan02, murine macrophage cell line RAW264.7 were purchased from Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Cells were maintained at 37°C in a humidified incubator and 5% CO2 atmosphere in DMEM or RPMI 1640 medium supplemented with 10% heat inactivated fetal bovine serum (FBS) (Gibco, California, USA), 1% of Penicillin-Streptomycin (10000 U/ml; Life Technology, USA). Similar with other studies, 48 tumor supernatant (TSN) was collected after specific cells were cultured for 24 h. THP-1 cells were culture and treated with 100 ng/ml phorbol 12-myristate 13-acetate (PMA) (P1585, Sigma-Aldrich, USA) and cultured for 72 h to generate a macrophage phenotype as previously described.49
Animals were maintained and studies were carried out in accordance with institutional guidelines. In accordance with the method described in previous study, 50 the subcutaneous and orthotopic pancreatic cancer model were established by subcutaneous incubation of 6 × 106 Pan02 cells into the left back and the parenchyma of the pancreas of 6-week-old C57BL/6 mice, respectively. After 3 weeks, palpable tumors had developed whose diameters researched 7–8 mm. This study was approved by the Institutional Review Board of Sun Yat-sen University Cancer Center. All animal studies complied with relevant ethical regulations for animal testing and research, and were approved by the Institutional Animal Care and Use Committee of Sun Yat-sen University Cancer Center.
Electroporation
The electroporation of percutaneous or orthotopic tumor in vivo and was performed according to the methods reported in our previous study.18 For the electroporation experiments of cells in vitro, tumor cells were trypsinized, resuspended in phosphate-buffered saline (PBS) at 2 × 106 cells mL−1, and added to an electroporation cuvette (1652088; BTX, Holliston, Massachusetts, 01746, USA) embedded with two aluminum plate electrodes 4 mm apart. The cell suspension was in direct contact with the plate electrodes, and subjected to electroporation at room temperature with the following parameters: voltage: 200–600 V; pulse duration: 100 µs; pulse repetition frequency: 1 Hz; number of repetition pulses: 8, 16, and 24. After electroporation, the cell suspension was kept on ice and analyzed or used within 30 minutes. Similar with other studies, 48 TSN was collected after specific cells were cultured for 3 days after electroporation with different strength levels and the cell debris was filtered. The parameters were in consistent with clinically used values in other studies.17
Electroporation experiments in vivo were performed using an ECM 830 square wave pulse electroporator (BTX Harvard Apparatus, Holliston, MA) when tumors researched 7–8 mm. Through puncturing the skin above the tumor or a small abdominal incision, the subcutaneous or orthotopic tumor was bracketed along the long axis using the two-needle probe with a 5 mm gap followed by the delivery of electric pulses. The electric array fully penetrated the whole tumor to maximize the effect of electroporation, with the following parameters: voltage: 1000 V; pulse duration: 100 ms; pulse frequency: 1 Hz; pulse number: 80. For control, surgical procedures and needles placement were performed to tumors without electric pulses using the same anesthetic conditions. Five animals were used for each experimental setting in vivo experiments and three independent repetitions were performed for each experiment.
Cell viability assay
Tumor cell viability after IRE treatment was detected as previously described using Cell Counting Kit 8 (CCK-8; CK04, Dojindo Laboratories, Japan).51 Briefly, triturated cells were seeded in 96-well flat-bottomed plates at a density of 2 × 103 in 100 μL of conditioned medium per well, and 10 μL of CCK-8 solution was added to each well. After 2 h of incubation, the absorbance of each well at a wavelength of 450 nm was quantified. To explore the death types, AO-EB (Invitrogen) double staining was performed. After 15 min of incubation, pictures were captured by confocal laser scanning microscopy (Nikon).
Analysis of tumor-infiltrated immune cells
Mice bearing subcutaneous and orthotopic tumors were euthanized 7 days after IRE, and tumors were harvested and dissociated using a mouse tumor dissociation kit according to manufacturer’s recommendations (Miltenyi Biotec, Germany). For the analysis of tissue-derived lymphocytes, tumor tissues, LN, and spleens harvested from mice were chopped into small pieces and mashed through a 70 um strainer. The red blood cells were lysed by red cell lysis buffer (TIANGEN). Then, cells were washed with PBS containing 1% FBS and stained with FITC-conjugated anti-mouse CD206 (141705, Biolegend, San Diego, USA), PE-conjugated anti-mouse CD16/32 (101307, Biolegend), APC-conjugated anti-mouse F4/80 (123115, Biolegend), respectively, on ice for 15 min (3 × 106 cells/sample). The sample were washed three times and resuspended in 200 uL of cold PBS containing 2% FBS and 1 mM EDTA for analysis using flow cytometry (FC; CytoFLEX, Beckman Coulter, USA).
Analysis of tumor cells and macrophages in vitro
For in vitro electroporation detection, we utilized PI (Invitrogen) staining. PI (640905, BioLegend) was added to the cell suspension simultaneously or after electroporation at 10 μg/mL. After incubation for 15 minutes, the transfection efficiencies of the samples were measured. To determine apoptosis, after electroporation, the resuspended tumor cells were stained with Annexin V-FITC/PI (640905, BioLegend), and analyzed by FC. To analyze the polarization of macrophages, the macrophages were stained with FITC-conjugated anti-human HLA-DR (11–9956-42, eBioscience, San Diego, USA), PE-conjugated anti-human CD206 (12–2069-42, eBioscience), PE-conjugated anti-human CD163 (333606, Biolegend), PE-conjugated anti-mouse CD16/32 (101307, Biolegend), and APC-conjugated anti-mouse F4/80 (123115, Biolegend) .
Western blot (WB) analysis
WB analysis was performed as previously described.51 Cells were lysed using the M-PER Mammalian Protein Extraction Reagent (ThERMO Scientific), and sodium dodecyl sulfate-polyacrylamide gel electrophoresis was performed. The following antibodies were used: HMGB1 (1:1000, ab79823, Abcam), HSP70 (1:1000, ab181606, Abcam), Calreticulin (1:1000, ab92516, Abcam), RAGE (1:1000, ab216329, Abcam), phosphorylated NF-κB [1:1000, 3033, Cell Signaling Technology (CST)], NF-κB (1:1000, 8242, CST), phosphorylated Akt (1:1000, 4060S, CST), Akt (1:1000, 4691S, CST), phospho-MAPK Family Antibody Kit (1:1000, 9910, CST), a MAPK Family Antibody Kit (1:1000, 9926, CST), β-Actin (1:1000, 4970S, CST) and GAPDH (1:1000, 60004-1-Ig; Proteintech). Proteins were visualized by using an ECL kit (4AW011; purchased from 4A Biotech Co., Ltd).Images were prepared using ChemiDoc XRS + system (BioRad, China) and quantification analyses were performed by Bio-Rad Image Lab software (BioRad, China).
Enzyme-linked immunosorbent assay (ELISA)
The supernatant of treated cells was collected at 24 hours after tumor cells being treated with electroporation and centrifuged at 12000 × g for 1 minute. The HMGB1 levels in the supernatant of treated cells was analyzed using ELISA (JYM0485Mo and JYM0485Hu, Jiyinmei, Wuhan, China) as described by the manufacturer.
Reverse transcription-quantitative PCR (RT-qPCR)
Total RNA was extracted using TRIzol reagent (15596026, Invitrogen, Carlsbad, USA). The reverse procedure was in according to our previous study.52 GAPDH was used as the internal control. RT-qPCR was performed using (FSQ-301, TOYOBO Co., Osaka, Japan) and following primer pairs:
GAPDH: CTCCTCCTGTTCGACAGTCAGC (forward), CCCAATA- CGACCAAATCCGTT (reverse);
HMGB1: CGCTTTTGTGATGGAGTGCT (forward); AGGGAAAA- ACTTTGCCATCCC (reverse);
HSP70: TTTTACCACTGAGCAAGTGACTG (forward); ACAAGG- AACCGAAACAACACA (reverse);
CD163: TTTGTCAACTTGAGTCCCTTCAC (forward); TCCCGCT- ACACTTGTTTTCAC (reverse);
TGF-ß: CAATTCCTGGCGATACCTCAG (forward); GCACAACTC- CGGTGACATCAA (reverse);
CD206: GGGTTGCTATCACTCTCTATGC (forward); TTTCTTGTC- TGTTGCCGTAGTT (reverse);
TNF-α: GAGGCCAAGCCCTGGTAT (forward); CGGGCCGATTG- ATCTCAGC (reverse);
CCL2: AAGATCTCAGTGCAGAGGCTCG (forward); CACAGATC- TCCTTGGCCACAA (reverse);
IL-1ß: TTCGACACATGGGATAACGAGG (forward); TTTTTGCTG- TGAGTCCCGGAG (reverse);
Immunohistochemistry (IHC) and immunohistofluorescence (IHF)
IHC and IHF were performed as previously reported.18 Briefly, tumor section (4 μm) were dewaxed in xylene, hydrated in decreasing concentrations for 30 min, washed with phosphate-buffered saline, and probed with monoclonal antibodies or isotype controls at 4°C overnight. The primary antibodies used were the same as the ones mentioned above. After being washed, the sections were incubated with biotinylated goat anti-rabbit or anti-mouse IgG at room temperature for 2 h. Immunostaining was visualized with streptavidin/peroxidase complex and diaminobenzidine, and sections were counterstained with hematoxylin. Slides were visualized under a bright-field microscope at ×40 and ×400 magnification. Immunofluorescence staining images were taken by ZEISS microscope (LSM880, Germany). Positive cells were quantified using ImagePro Plus software (Media Cybernetics) and expressed as mean ± SEM in high-powered fields detected by confocal microscopy.
Phagocytosis assay measurement
FC and IHF were used to analyze the phagocytosis capacity of macrophages. Fluorescent particles or apoptotic tumor cells after IRE were labeled with carboxyfluorescein succinimidylester (CFSE, 21888–25 mg-F, Sigma-Aldrich) and added to polarized macrophages which were stained with eFluor 670 (65–0840-85, eBioscience), for 2 h at 37°C in air enriched with 5% CO2. Macrophages that had phagocytosed apoptotic tumor cells were defined as cells which were double-positive for CFSE and eFluor 670.
Statistical methods
Statistical values were expressed as mean ± standard error of the mean. Statistical differences between groups were calculated either using the student t test or One-way analysis of variance (ANOVA) with post-hoc multiple comparisons depending on the data, using GraphPad Prism 8.0 software. The log-rank test was used in Kaplan-Meier survival analysis. A p value of less than 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
The authors would like to thank Dr. Xin Huang and Dr. Juncheng Wang for excellent technical assistance.
Funding Statement
This work was supported by grants from Guangdong Basic and Applied Basic Research Foundation (2020A1515110954), National Natural Science Funds (No.81972299, No. 81672390) and the National Key Research and Development Plan (No.2017YFC0910002).
Data availability statement
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request. The authenticity of this article has been validated by uploading the key raw data onto the Research Data Deposit public platform (www.researchdata.org.cn), with the approval RDD number as RDDB2020000877.
Disclosure of Competing Interest
No potential conflicts of interest were disclosed.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.Siegel RL, Miller KD, Jemal A.. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–12. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 2.Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research. 2012;18:4266–4276. doi: 10.1158/1078-0432.Ccr-11-3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heusinkveld M, Van Der Burg SH. Identification and manipulation of tumor associated macrophages in human cancers. J Transl Med. 2011;9:216. doi: 10.1186/1479-5876-9-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol. 2011;11:750–761. doi: 10.1038/nri3088. [DOI] [PubMed] [Google Scholar]
- 5.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nature Reviews Immunology. 2011;11(11):723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sica A, Schioppa T, Mantovani A, Allavena P. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: potential targets of anti-cancer therapy. European Journal of Cancer (Oxford, England: 1990). 2006;42:717–727. doi: 10.1016/j.ejca.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 7.Parisi L, Gini E, Baci D, Tremolati M, Fanuli M, Bassani B, Farronato G, Bruno A, Mortara L. Macrophage polarization in chronic inflammatory diseases: killers or builders? Journal of Immunology Research. 2018;2018:8917804. doi: 10.1155/2018/8917804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kurahara H, Shinchi H, Mataki Y, Maemura K, Noma H, Kubo F, Sakoda M, Ueno S, Natsugoe S, Takao S. Significance of M2-polarized tumor-associated macrophage in pancreatic cancer. J Surg Res. 2011;167(2):e211–219. doi: 10.1016/j.jss.2009.05.026. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Q, Green MD, Lang X, Lazarus J, Parsels JD, Wei S, Parsels LA, Shi J, Ramnath N, Wahl DR, et al. Inhibition of ATM increases interferon signaling and sensitizes pancreatic cancer to immune checkpoint blockade therapy. Cancer Res. 2019;79:3940–3951. doi: 10.1158/0008-5472.Can-19-0761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hidalgo M. Pancreatic cancer. The New England Journal of Medicine. 1605-1617;2010(362). doi: 10.1056/NEJMra0901557. [DOI] [PubMed] [Google Scholar]
- 11.Lafranceschina S, Brunetti O, Delvecchio A, Conticchio M, Ammendola M, Currò G, Piardi T, De’angelis N, Silvestris N, Memeo R. Systematic review of irreversible electroporation role in management of locally advanced pancreatic cancer. Cancers. 2019:11. doi: 10.3390/cancers11111718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Porcelli L, Quatrale AE, Mantuano P, Leo MG, Silvestris N, Rolland JF, Carioggia E, Lioce M, Paradiso A, Azzariti A. Optimize radiochemotherapy in pancreatic cancer: PARP inhibitors a new therapeutic opportunity. Mol Oncol. 2013;7:308–322. doi: 10.1016/j.molonc.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martin RC 2nd, McFarland K, Ellis S, Velanovich V. Irreversible electroporation in locally advanced pancreatic cancer: potential improved overall survival. Ann Surg Oncol. 2013;20(Suppl 3):S443–449. doi: 10.1245/s10434-012-2736-1. [DOI] [PubMed] [Google Scholar]
- 14.He C, Huang X, Zhang Y, Cai Z, Lin X, Li S. Comparison of survival between irreversible electroporation followed by chemotherapy and chemotherapy alone for locally advanced pancreatic cancer. Front Oncol. 2020;10:6. doi: 10.3389/fonc.2020.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martin RC 2nd, Kwon D, Chalikonda S, Sellers M, Kotz E, Scoggins C, McMasters KM, Watkins K. Treatment of 200 locally advanced (stage III) pancreatic adenocarcinoma patients with irreversible electroporation: safety and efficacy. Ann Surg. 2015;262:486–494. discussion 492-484. doi: 10.1097/sla.0000000000001441. [DOI] [PubMed] [Google Scholar]
- 16.Mansson C, Brahmstaedt R, Nilsson A, Nygren P, Karlson BM. Percutaneous irreversible electroporation for treatment of locally advanced pancreatic cancer following chemotherapy or radiochemotherapy. European Journal of Surgical Oncology: The Journal of the European Society of Surgical Oncology and the British Association of Surgical Oncology. 2016;42:1401–1406. doi: 10.1016/j.ejso.2016.01.024. [DOI] [PubMed] [Google Scholar]
- 17.Zhao J, Wen X, Tian L, Li T, Xu C, Wen X, Melancon MP, Gupta S, Shen B, Peng W, et al. Irreversible electroporation reverses resistance to immune checkpoint blockade in pancreatic cancer. Nat Commun. 2019;10:899. doi: 10.1038/s41467-019-08782-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.He C, Huang X, Zhang Y, Lin X, Li S. T-cell activation and immune memory enhancement induced by irreversible electroporation in pancreatic cancer. Clin Transl Med. 2020. doi: 10.1002/ctm2.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17:97–111. doi: 10.1038/nri.2016.107. [DOI] [PubMed] [Google Scholar]
- 20.Kepp O, Senovilla L, Vitale I, Vacchelli E, Adjemian S, Agostinis P, Apetoh L, Aranda F, Barnaba V, Bloy N, et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology. 2014;3:e955691. doi: 10.4161/21624011.2014.955691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bulvik BE, Rozenblum N, Gourevich S, Ahmed M, Andriyanov AV, Galun E, Goldberg SN. Irreversible electroporation versus radiofrequency ablation: a comparison of local and systemic effects in a small-animal model. Radiology. 2016;280:413–424. doi: 10.1148/radiol.2015151166. [DOI] [PubMed] [Google Scholar]
- 22.He C, Wang J, Sun S, Zhang Y, Li S. Immunomodulatory effect after irreversible electroporation in patients with locally advanced pancreatic cancer. J Oncol. 2019;2019:9346017. doi: 10.1155/2019/9346017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scheffer HJ, Stam AGM, Geboers B, Vroomen L, Ruarus A, De Bruijn B, Van Den Tol MP, Kazemier G, Meijerink MR, De Gruijl TD. Irreversible electroporation of locally advanced pancreatic cancer transiently alleviates immune suppression and creates a window for antitumor T cell activation. Oncoimmunology. 2019;8:1652532. doi: 10.1080/2162402x.2019.1652532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pandit H, Hong YK, Li Y, Rostas J, Pulliam Z, Li SP, Martin RCG. Evaluating the Regulatory Immunomodulation Effect of Irreversible Electroporation (IRE) in pancreatic adenocarcinoma. Ann Surg Oncol. 2019;26:800–806. doi: 10.1245/s10434-018-07144-3. [DOI] [PubMed] [Google Scholar]
- 25.Wu K, Yuan Y, Yu H, Dai X, Wang S, Sun Z, Wang F, Fei H, Lin Q, Jiang H, et al. The gut microbial metabolite trimethylamine N-oxide aggravates GVHD by inducing M1 macrophage polarization in mice. Blood. 2020;136:501–515. doi: 10.1182/blood.2019003990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Viceconte N, Burguillos MA, Herrera AJ, De Pablos RM, Joseph B, Venero JL. Neuromelanin activates proinflammatory microglia through a caspase-8-dependent mechanism. J Neuroinflammation. 2015;12:5. doi: 10.1186/s12974-014-0228-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buchacher T, Ohradanova-Repic A, Stockinger H, Fischer MB, Weber V.. Polarization of human macrophages favors survival of the intracellular pathogen chlamydia pneumoniae. PloS One. 2015;10:e0143593. doi: 10.1371/journal.pone.0143593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sagar V, Vatapalli R, Lysy B, Pamarthy S, Anker JF, Rodriguez Y, Han H, Unno K, Stadler WM, Catalona WJ, et al. EPHB4 inhibition activates ER stress to promote immunogenic cell death of prostate cancer cells. Cell Death Dis. 2019;10:801. doi: 10.1038/s41419-019-2042-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou H, Jin C, Cui L, Xing H, Liu J, Liao W, Liao H, Yu Y. HMGB1 contributes to the irradiation-induced endothelial barrier injury through receptor for advanced glycation endproducts (RAGE). J Cell Physiol. 2018;233:6714–6721. doi: 10.1002/jcp.26341. [DOI] [PubMed] [Google Scholar]
- 30.Xu Y, Toure F, Qu W, Lin L, Song F, Shen X, Rosario R, Garcia J, Schmidt AM, Yan SF. Advanced glycation end product (AGE)-receptor for AGE (RAGE) signaling and up-regulation of Egr-1 in hypoxic macrophages. J Biol Chem. 2010;285:23233–23240. doi: 10.1074/jbc.M110.117457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Narayanan JSS, Ray P, Hayashi T, Whisenant TC, Vicente D, Carson DA, Miller AM, Schoenberger SP, White RR. Irreversible electroporation combined with checkpoint blockade and TLR7 stimulation induces antitumor immunity in a murine pancreatic cancer model. Cancer Immunology Research. 1714-1726;2019(7). doi: 10.1158/2326-6066.Cir-19-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Neal RE 2nd, Rossmeisl JH Jr., Robertson JL, Arena CB, Davis EM, Singh RN, Stallings J, Davalos RV. Improved local and systemic anti-tumor efficacy for irreversible electroporation in immunocompetent versus immunodeficient mice. PloS One. 2013;8:e64559. doi: 10.1371/journal.pone.0064559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O’Brien MA, Power DG, Clover AJ, Bird B, Soden DM, Forde PF. Local tumour ablative therapies: opportunities for maximising immune engagement and activation. Biochim Biophys Acta. 2014;1846:510–523. doi: 10.1016/j.bbcan.2014.09.005. [DOI] [PubMed] [Google Scholar]
- 34.Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2013;31:51–72. doi: 10.1146/annurev-immunol-032712-100008. [DOI] [PubMed] [Google Scholar]
- 35.Schaper F, De Leeuw K, Horst G, Bootsma H, Limburg PC, Heeringa P, Bijl M, Westra J. High mobility group box 1 skews macrophage polarization and negatively influences phagocytosis of apoptotic cells. Rheumatology (Oxford, England). 2016;55:2260–2270. doi: 10.1093/rheumatology/kew324. [DOI] [PubMed] [Google Scholar]
- 36.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang H, Mooney DJ. Biomaterial-assisted targeted modulation of immune cells in cancer treatment. Nat Mater. 2018;17:761–772. doi: 10.1038/s41563-018-0147-9. [DOI] [PubMed] [Google Scholar]
- 38.Zhou J, Bai W, Liu Q, Cui J, Zhang W. Intermittent hypoxia enhances thp-1 monocyte adhesion and chemotaxis and promotes m1 macrophage polarization via RAGE. Biomed Res Int. 2018;2018:1650456. doi: 10.1155/2018/1650456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shao Q, O’Flanagan S, Lam T, Roy P, Pelaez F, Burbach BJ, Azarin SM, Shimizu Y, Bischof JC. Engineering T cell response to cancer antigens by choice of focal therapeutic conditions. International Journal of Hyperthermia: The Official Journal of European Society for Hyperthermic Oncology, North American Hyperthermia Group. 2019;36:130–138. doi: 10.1080/02656736.2018.1539253. [DOI] [PubMed] [Google Scholar]
- 40.Hudson BI, Kalea AZ, Del Mar Arriero M, Harja E, Boulanger E, D’Agati V, Schmidt AM. Interaction of the RAGE cytoplasmic domain with diaphanous-1 is required for ligand-stimulated cellular migration through activation of Rac1 and Cdc42. J Biol Chem. 2015;12(49):34457–34468. doi: 10.1186/s12974-014-0228-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Taguchi A, Blood DC, Del Toro G, Canet A, Lee DC, Qu W, Tanji N, Lu Y, Lalla E, Fu C, et al. Blockade of RAGE-amphoterin signalling suppresses tumour growth and metastases. Nature. 2000;405:354–360. doi: 10.1038/35012626. [DOI] [PubMed] [Google Scholar]
- 42.Tang D, Shi Y, Kang R, Li T, Xiao W, Wang H, Xiao X. Hydrogen peroxide stimulates macrophages and monocytes to actively release HMGB1. J Leukoc Biol. 2007;81:741–747. doi: 10.1189/jlb.0806540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen G, Li J, Ochani M, Rendon-Mitchell B, Qiang X, Susarla S, Ulloa L, Yang H, Fan S, Goyert SM, et al. Bacterial endotoxin stimulates macrophages to release HMGB1 partly through CD14- and TNF-dependent mechanisms. J Leukoc Biol. 2004;76:994–1001. doi: 10.1189/jlb.0404242. [DOI] [PubMed] [Google Scholar]
- 44.Yadav DK, Kumar S, Choi EH, Kim MH. Electric-field-induced electroporation and permeation of reactive oxygen species across a skin membrane. J Biomol Struct Dyn. 2020:1–20. doi: 10.1080/07391102.2020.1730972. [DOI] [PubMed] [Google Scholar]
- 45.Rossi A, Pakhomova ON, Mollica PA, Casciola M, Mangalanathan U, Pakhomov AG, Muratori C. Nanosecond pulsed electric fields induce endoplasmic reticulum stress accompanied by immunogenic cell death in murine models of lymphoma and colorectal cancer. Cancers. 2019:11. doi: 10.3390/cancers11122034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tang D, Kang R, Livesey KM, Cheh C-W, Farkas A, Loughran P, Hoppe G, Bianchi ME, Tracey KJ, Zeh HJ 3rd, et al. Endogenous HMGB1 regulates autophagy. J Cell Biol. 2019;11(5):881–892. doi: 10.3390/cancers11122034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saunders JA, Rogers LC, Klomsiri C, Poole LB, Daniel LW. Reactive oxygen species mediate lysophosphatidic acid induced signaling in ovarian cancer cells. Free Radic Biol Med. 2010;49:2058–2067. doi: 10.1016/j.freeradbiomed.2010.10.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen D-P, Ning W-R, Li X-F, Wei Y, Lao X-M, Wang J-C, Wu Y, Zheng L. Peritumoral monocytes induce cancer cell autophagy to facilitate the progression of human hepatocellular carcinoma. Autophagy. 2018;14(8):1335–1346. doi: 10.1080/15548627.2018.1474994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ye H, Zhou Q, Zheng S, Li G, Lin Q, Wei L, Fu Z, Zhang B, Liu Y, Li Z, et al. Tumor-associated macrophages promote progression and the Warburg effect via CCL18/NF-kB/VCAM-1 pathway in pancreatic ductal adenocarcinoma. Cell Death Dis. 2018;14(5):453. doi: 10.1080/15548627.2018.1474994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jiang Y-J, Lee C-L, Wang Q, Zhou Z-W, Yang F, Jin C, Fu D-L. Establishment of an orthotopic pancreatic cancer mouse model: cells suspended and injected in Matrigel. World Journal of Gastroenterology. 2014;20(28):9476–9485. doi: 10.3748/wjg.v20.i28.9476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sun S, Xue D, Chen Z, Ou-Yang Y, Zhang J, Mai J, Gu J, Lu W, Liu X, Liu W, et al. R406 elicits anti-Warburg effect via Syk-dependent and -independent mechanisms to trigger apoptosis in glioma stem cells. Cell Death Dis. 2019;10(5):358. doi: 10.1038/s41419-019-1587-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang X, He C, Lin G, Lu L, Xing K, Hua X, Sun S, Mao Y, Song Y, Wang J, et al. Induced CD10 expression during monocyte-to-macrophage differentiation identifies a unique subset of macrophages in pancreatic ductal adenocarcinoma. Biochem Biophys Res Commun. 2020. doi: 10.1016/j.bbrc.2020.02.042. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request. The authenticity of this article has been validated by uploading the key raw data onto the Research Data Deposit public platform (www.researchdata.org.cn), with the approval RDD number as RDDB2020000877.