

Diet-induced obesity, fatty liver, and type 2 diabetes are prevented by lowering apoCIII.
Abstract
Increased levels of apolipoprotein CIII (apoCIII), a key regulator of lipid metabolism, result in obesity-related metabolic derangements. We investigated mechanistically whether lowering or preventing high-fat diet (HFD)–induced increase in apoCIII protects against the detrimental metabolic consequences. Mice, first fed HFD for 10 weeks and thereafter also given an antisense (ASO) to lower apoCIII, already showed reduced levels of apoCIII and metabolic improvements after 4 weeks, despite maintained obesity. Prolonged ASO treatment reversed the metabolic phenotype due to increased lipase activity and receptor-mediated hepatic uptake of lipids. Fatty acids were transferred to the ketogenic pathway, and ketones were used in brown adipose tissue (BAT). This resulted in no fat accumulation and preserved morphology and function of liver and BAT. If ASO treatment started simultaneously with the HFD, mice remained lean and metabolically healthy. Thus, lowering apoCIII protects against and reverses the HFD-induced metabolic phenotype by promoting physiological insulin sensitivity.
INTRODUCTION
Apolipoprotein CIII (apoCIII) is an 8.8-kDa polypeptide and is predominantly produced in liver (1, 2). ApoCIII plays an important role in lipid metabolism by affecting triglyceride-rich lipoprotein (TRL) lipolysis and hepatic uptake of TRL (3–6). It is well known that increased circulating levels of apoCIII are associated with hypertriglyceridemia and cardiovascular diseases (CVDs) (4, 7, 8). Large-scale epidemiological studies have shown that loss-of-function mutations in the apoCIII gene are related to protection against CVD (9), whereas apoCIII gene variants resulting in increased levels of the apolipoprotein are associated with nonalcoholic fatty liver disease (NAFLD), hepatic insulin resistance, and type 2 diabetes mellitus (T2DM) (10, 11). Insulin suppresses the expression of the apoCIII gene, and under T2DM conditions, i.e. hyperglycemia, hyperinsulinemia, and insulin resistance, there is an increased expression of the gene (12–14).
Obesity is a severe risk factor for metabolic derangements, including diabetes mellitus and NAFLD, and it has been shown that serum levels of apoCIII increase upon consumption of high-fat diet (HFD) (1). Previous reports have also shown that global apoCIII deficiency aggravates diet-induced obesity (DIO) and insulin resistance in mice (15). Hence, it seems that either high apoCIII levels or complete deficiency of the apolipoprotein is equally deleterious from the metabolic point of view. Therefore, we hypothesized that lowering of apoCIII protects against HFD-induced metabolic impairments. We show that interference with HFD-induced increase in apoCIII in mice prevents the development of obesity and T2DM. Moreover, we found that reduction of high apoCIII levels in obese mice on HFD reversed their detrimental metabolic phenotype.
RESULTS
Lowering apoCIII in mice on HFD improves their metabolic status
To evaluate the effects of lowering apoCIII in DIO, we designed a prevention and reversibility study where 8-week-old male C57BL6/j (B6) mice were fed an HFD and treated twice per week with intraperitoneal injections with either an active antisense oligonucleotide (ASO) targeting apoCIII mRNA or an inactive/scrambled (Scr) ASO (16), as specified in fig. S1A.
We have performed experiments in three cohorts of mice. Two cohorts were used in the prevention and reversibility studies, and data on body weight (BW) of these two cohorts are provided in Fig. 1A. The third cohort was used for studies of energy homeostasis in metabolic cages, and data on their BWs, which were similar to the other two cohorts, are shown in fig. S1B. The Scr and active ASOs used in our studies were developed and tested for specificity and toxicity by the IONIS Pharmaceutical Company. They showed that the Scr ASO, as well as saline, had no effect on the apoCIII levels, while the active ASO lowered apoCIII in C57BL/6 mice on chow or Western diet (16).
Fig. 1. Lowering apoCIII in mice on HFD improves the metabolic status.
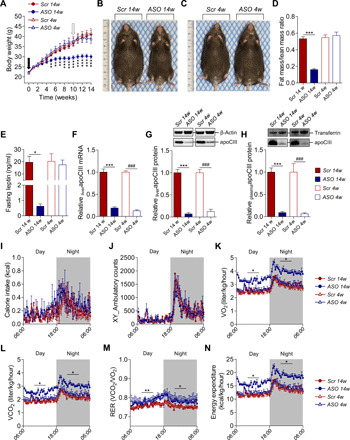
(A) Body weight (BW). Black arrow indicates start of ASO treatment in the prevention study (week 0), and white arrow in the reversibility study (week 10, N = 10 to 11). (B and C) Representative pictures of mice treated for (B) 14 weeks with Scr (left) and ASO (right) and (C) 4 weeks with Scr (left) and ASO (right). (D) Fat–to–lean mass ratio analyzed with NMR (N = 6). (E) Twelve-hour fasting plasma leptin levels (N = 4). (F) Liver mRNA levels of apoCIII analyzed by qRT-PCR (N = 10). (G and H) Representative immunoblots and densitometry analysis of apoCIII in (G) liver and (H) plasma (N = 7 and 6, respectively). (I to N) Parameters evaluated in metabolic cages (CLAMS) at the end of the studies (N = 7 to 8). (I) Calorie intake. (J) Locomotion (x and y axes). (K) Oxygen consumption (VO2). (L) Carbon dioxide production (VCO2). (M) Respiratory exchange ratio (RER). (N) Calculated energy expenditure (EE). Data are presented as mean ± SEM. *ASO 14w versus Scr 14w; #ASO 4w versus Scr 4w. Single symbol, P < 0.05; double symbol, P < 0.01; and triple symbol, P < 0.001. Photo credit: Ismael Valladolid-Acebes, The Rolf Luft Research Center for Diabetes and Endocrinology, Karolinska Institutet.
As absence of apoCIII is known to be deleterious (15), our aim with the ASO treatment was to lower, but not totally suppress, the presence of the apolipoprotein.
Mice on HFD simultaneously given ASO (ASO 14w) had a normal age-related weight gain during the first 4 weeks of the study but thereafter remained lean (P < 0.01 to P < 0.001; Fig. 1A). Scr-treated mice (Scr 14w and Scr 4w) and those first being on HFD for 10 weeks and thereafter treated with the active ASO during the last 4 weeks (ASO 4w) remained obese until the end of the studies (Fig. 1, A to C). Nuclear magnetic resonance (NMR) confirmed these results and showed that fat mass was only reduced in ASO 14w (P < 0.001), while lean mass remained unchanged among the groups (Fig. 1D and fig. S1, C and D). In agreement with these findings, plasma leptin levels were only reduced in the nonobese mice (ASO 14w, P < 0.05; Fig. 1E). ApoCIII mRNA and protein levels were decreased in liver and plasma in both ASO-treated groups, as compared to Scr-treated mice (P < 0.001; Fig. 1, F to H). Furthermore, because the apoCIII gene is located within the apoAI and apoAIV gene cluster (17), we have confirmed that there is no effect of the ASO on liver apoAI or apoAIV mRNA levels (fig. S1, E and F).
To assess energy homeostasis, mice were placed in metabolic cages and evaluated for indirect calorimetry, food intake, and locomotion at the end of the studies. Before placing the animals in metabolic cages, they were acclimatized in single cages for 24 hours in accordance with our ethical permit. Although this may seem like a relatively short period of time, there is little evidence that single housing induces stress, because fecal corticosterone levels of singly housed mice do not differ from those of group-housed individuals (18).
There were no differences in calorie intake or locomotion among the groups (Fig. 1, I and J, and fig. S1, G and H). However, ASO 14w showed an increase in oxygen consumption (VO2, P < 0.05), carbon dioxide production (VCO2, P < 0.05), and, consequently, a higher respiratory exchange ratio (RER; P < 0.05 to P < 0.01), indicating higher energy expenditure (P < 0.05; Fig. 1, K to N, and fig. S1, I to L). Similar tendencies were observed in the obese mice after 4 weeks on ASO, mostly affecting RER (Fig. 1M and fig. S1K).
Lowering apoCIII affects brown adipose tissue
Brown adipose tissue (BAT) thermogenesis is under the control of the sympathetic nervous system via activation of the β3-adrenergic receptor (ADRB3) (19, 20). The expression levels of ADRB3 were increased in BAT from ASO-treated mice (P < 0.05 to P < 0.01; Fig. 2A). In accordance with an enhanced metabolic activity, the body temperature was higher in ASO 14w (P < 0.05) and, after 4 weeks on ASO treatment, it began to increase in the still obese ASO 4w (Fig. 2B). There were distinct differences in BAT morphology with massive fat accumulation in Src-treated mice, a clear improvement already after 4 weeks on ASO, and normal appearance when HFD-induced increase in apoCIII was prevented (Fig. 2C and fig. S2A). It has been shown that BAT is important for clearance of triglycerides (Tgs) in rodents by a process dependent on lipoprotein lipase (LPL) activity (21, 22). Because apoCIII inhibits LPL, we wanted to clarify whether lowering apoCIII could stimulate the activity of LPL in BAT. Support for a more metabolically active BAT in our ASO-treated animals was suggested by the increased LPL activity, which reached statistical significance in ASO 4w (P < 0.01), together with the up-regulation of the thermogenic genes, namely, uncoupling protein 1 (UCP-1), peroxisome proliferator–activated receptor-γ coactivator1α (PGC-1α), cell death–inducing DNA fragmentation factor α–like effector A (CIDEA), and positive regulatory domain–containing 16 (PRDM-16) (P < 0.05 to P < 0.001; Fig. 2, D and E).
Fig. 2. Lowering apoCIII affects BAT and improves glucose homeostasis and insulin sensitivity.
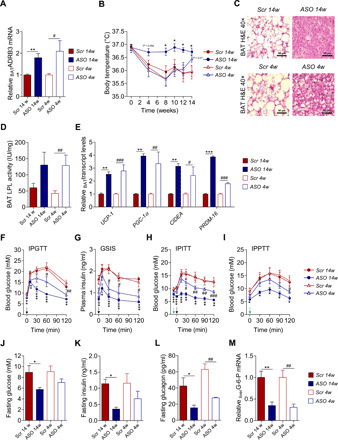
(A) BAT mRNA levels of ADRB3 analyzed by qRT-PCR (N = 6). (B) Body temperature (N = 6). (C) Representative images of hematoxylin and eosin–stained cryosections from BAT. Scale bar, 50 μm (N = 4). (D) LPL activity in BAT (N = 6). (E) Expression of thermogenic genes in BAT by qRT-PCR (N = 6). (F to I) In vivo metabolic tests performed at the end of the studies. Injection of bolus doses is indicated with arrows (black, glucose; blue, insulin; and green, pyruvate). (F) Glucose tolerance test (IPGTT; N = 6), (G) glucose-stimulated insulin secretion (GSIS) during IPGTT (N = 4), (H) insulin tolerance test (IPITT; N = 6), and (I) pyruvate tolerance test (IPPTT; N = 4 to 5. (J to L) Twelve-hour fasting levels of (J) blood glucose (N = 4 to 5), (K) plasma insulin (N = 4), and (L) plasma glucagon (N = 4). (M) Liver mRNA levels of G-6-P analyzed by qRT-PCR (N = 6). Data are presented as mean ± SEM. *ASO 14w versus Scr 14w; #ASO 4w versus Scr 4w. Single symbol, P < 0.05; double symbol, P < 0.01; and triple symbol, P < 0.001.
Improved glucose homeostasis and insulin sensitivity upon apoCIII reduction
Preventing HFD-induced increase in apoCIII preserved insulin sensitivity, and already after 4 weeks of ASO treatment, there was an improvement in glucose and insulin tolerance [intraperitoneal glucose tolerance test (IPGTT) and intraperitoneal insulin tolerance test (IPITT), respectively], as well as glucose-stimulated insulin secretion (GSIS) (P < 0.05 to P < 0.001), despite maintained BW and continuation on HFD (Fig. 2, F to H, and fig. S2, C to E). As a marker of insulin sensitivity, we measured plasma adiponectin (23) and found that all ASO-treated mice had an increase of this adipokine (P < 0.05 to P < 0.01; fig. S2B).
To evaluate hepatic gluconeogenesis, a pyruvate tolerance test [intraperitoneal pyruvate tolerance test (IPPTT)] was performed. The bolus dose of pyruvate elicited a reduced glycemic excursion in ASO 14w (P < 0.05). In addition, in mice treated for 4 weeks, there were signs of improvement reflected by a decrease in hepatic gluconeogenesis (Fig. 2I and fig. S2F). Additional signs of decreased gluconeogenesis in the ASO-treated mice were lower levels of fasting blood glucose (P < 0.05) and plasma insulin (P < 0.05) in ASO 14w and of circulating glucagon in both ASO 4 and 14w (P < 0.05 to P < 0.01; Fig. 2, J to L). Furthermore, the gene encoding glucose-6-phosphatase (G-6-P), the key enzyme regulating liver gluconeogenesis, was down-regulated in liver from all ASO-treated mice (P < 0.01; Fig. 2M).
Liver function in ASO-treated mice on HFD
ApoCIII regulates lipid metabolism by inhibiting lipases (24, 25). Because LPL is not expressed in the liver of adult humans and animals (26), we instead determined hepatic lipase (HL) activity. Our results show that ASO-treated mice exhibited higher liver HL activity, reaching statistical significance in ASO 14w (P < 0.05; Fig. 3A).
Fig. 3. Liver function in ASO-treated mice on HFD.
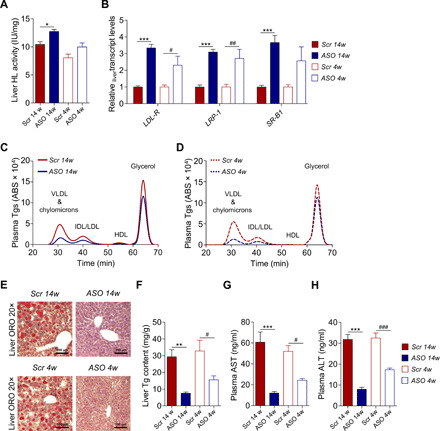
(A) Hepatic lipase (HL) activity in liver samples (N = 6). (B) Gene expression levels of receptors involved in hepatic lipid clearance by qRT-PCR (N = 5 to 6). (C and D) Representative chromatograms for Tg lipid profiles measured in VLDL and chylomicron–, IDL/LDL-, HDL-, and glycerol-eluted plasma fractions at the end of (C) prevention and (D) reversibility study (N = 6). (E) Representative images of ORO-stained cryosections from liver. Nuclei counterstained with hematoxylin. Scale bar, 100 μm (N = 4). (F) Spectrophotometric measurements of liver Tg content (N = 6). (G and H) Nonfasting plasma (G) AST and (H) ALT (N = 6). Data are presented as mean ± SEM. *ASO 14w versus Scr 14w; #ASO 4w versus Scr 4w. Single symbol, P < 0.05; double symbol, P < 0.01; and triple symbol, P < 0.001.
A previous work has demonstrated that decreasing apoCIII enhances low-density lipoprotein (LDL) family receptor–mediated hepatic clearance of TRL particles (5). We found increased expression of LDL receptors (LDLR) and LDLR-related protein 1 (LRP1) already after 4 weeks of ASO treatment (P < 0.05 to P < 0.001), whereas the scavenger receptor class B type 1 (SR-B1) was up-regulated only in ASO 14w (P < 0.001; Fig. 3B). An enhanced hepatic lipid clearance from the systemic circulation together with increased lipase activity in our ASO-treated mice was supported by the improved plasma lipid profiles in these animals, the major differences being in very LDL (VLDL)–Tgs and intermediate-density lipoprotein (IDL)/LDL–Tgs (P < 0.05 to P < 0.001; Fig. 3, C and D, and fig. S3, A to H).
Despite a daily intake of large amounts of fat, the morphology of the liver was preserved when the increase in apoCIII was prevented, while there was steatosis and higher liver Tgs in the Scr-treated mice (P < 0.05 to P < 0.01; Fig. 3, E and F, and fig. S3I). Somewhat unexpected, the still obese mice had, after 4 weeks of ASO treatment, a clear reduction in fat and Tgs in the liver (P < 0.05; Fig. 3, E and F, and fig. S3I). Hence, an important finding in this study is that a lowering in apoCIII prevents/reverses liver fat accumulation.
As a measure of liver function, aspartate aminotransferase (AST) and alanine aminotransferase (ALT) were analyzed in plasma. These enzymes were increased, as a sign of liver damage, in Scr-treated compared to ASO-treated mice (P < 0.05 to P < 0.001; Fig. 3, G and H).
Lipid catabolism and ketogenesis
In liver, expression of the major intracellular lipases that regulate Tg turnover, adipose triglyceride lipase (ATGL) and hormone-sensitive lipase (HSL), was up-regulated in ASO-treated mice (P < 0.05 to P < 0.001; Fig. 4A). The hydrolyzed fatty acids are known to be channeled to fatty acid oxidation (FAO) and to activate peroxisome proliferator–activated receptor-α (PPAR-α) and its target gene carnitine palmitoyltransferase 1a (CPT-1a) (27–29), both of them being up-regulated in animals with reduced apoCIII (P < 0.05 to P < 0.01; Fig. 4A). Estimation of whole-body FAO in vivo, according to the previously described method (30–32), revealed that although the rate of FAO seems to be higher in ASO-treated mice from the prevention study, it was not statistically significant (Fig. 4B and fig. S4A). As obesity and insulin resistance are known activators of de novo lipogenesis (DNL) (33), we analyzed important regulators of this pathway in the liver. Sterol regulatory element–binding protein-1 (SREBP-1), acetyl–coenzyme A (CoA)–carboxylase (ACC), and fatty acid synthase (FAS) were down-regulated in both groups of ASO-treated mice (P < 0.01 to P < 0.001), whereas the expression of cytosolic 3-hydroxy-3methylglutaryl-CoA synthase (cytHMGCoAS) was decreased only in ASO 14w (P < 0.001; fig. S4B).
Fig. 4. Lipid catabolism and ketogenesis.
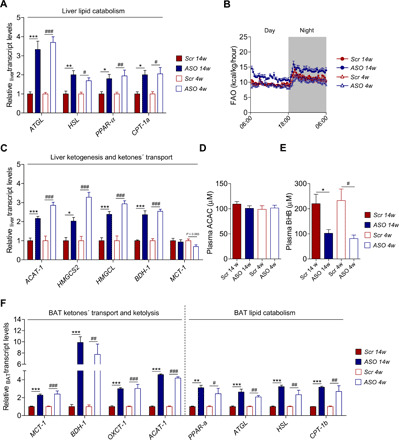
(A) Expression levels of genes involved in liver lipid catabolism by qRT-PCR (N = 6). (B) In vivo FAO (N = 7 to 8). (C) Expression levels of genes involved in liver ketogenesis and ketone transport by qRT-PCR (N = 6). (D and E) Nonfasting plasma levels of (D) ACAC (N = 5) and (E) BHB (N = 5). (F) Expression levels of genes involved in BAT ketone transport and ketolysis, as well as BAT lipid catabolism by qRT-PCR (N = 6). Data are presented as mean ± SEM. *ASO 14w versus Scr 14w; #ASO 4w versus Scr 4w. Single symbol, P < 0.05; double symbol, P < 0.01; and triple symbol, P < 0.001.
Decreasing apoCIII also led to a rise in the hepatic ketogenic pathways with augmented expression of the enzymes acetyl-CoA acetyltransferase (ACAT-1), 3-hydroxy-3-methylglutaryl-CoA synthase 2 (HMGCS2), 3-hydroxy-3-methylglutaryl-CoA lyase (HMGCL), and 3-hydroxybutyrate dehydrogenase 1 (BDH-1) (P < 0.05 to P < 0.001; Fig. 4C). The latter encodes the protein that catalyzes the interconversion of acetoacetate (ACAC) and β-hydroxybutyrate (BHB), the two major ketone bodies produced during fatty acid catabolism (34), and we confirmed that the protein levels and activity of liver BDH-1 were increased in ASO-treated mice (fig. S4, C and D). Plasma levels of ACAC were unaltered, while there was a decrease in BHB in ASO-treated mice (P < 0.05; Fig. 4, D and E). The transcription levels of monocarboxylate transporter-1 (MCT-1), which transports ketone bodies (34), remained unchanged in liver and was up-regulated in BAT (P < 0.001; Fig. 4, C and F). These findings indicate that lowering apoCIII shifts liver lipid metabolism toward ketogenesis and drives extrahepatic uptake of ketones by BAT. Analysis of markers for ketolysis, BDH-1, 3-oxoacid CoA-transferase (OXCT-1), and ACAT-1, showed an increase in BAT from ASO-treated mice (P < 0.01 to P < 0.001; Fig. 4F). Lowering apoCIII also affected lipid catabolism by transcriptional activation of PPAR-α, the intracellular lipases ATGL and HSL, as well as the main transcription factor for FAO in BAT, CPT-1b (P < 0.05 to P < 0.001; Fig. 4F).
ApoCIII reduction reverses the metabolic phenotype induced by HFD
To clarify whether prolongation of ASO treatment is able to reverse DIO and related metabolic derangements, we designed a long-lasting reversibility study where mice were first fed an HFD for 10 weeks and thereafter also treated with either active ASO (HFD + ASO) or Scr (HFD + Scr) for 14 weeks (fig. S5A). After 5 weeks, HFD + ASO mice had a lower BW (P < 0.05 to P < 0.001; Fig. 5A), and at the end of the study, these mice had a similar weight reduction as those in the prevention study (Fig. 5, A and B). In line with the decreased BW, there was a reduction in perigonadal adipose tissue (PgAT) (P < 0.01), lower leptin levels (P < 0.05), and an increase in circulating adiponectin in HFD + ASO mice (P < 0.01; Fig. 5, C to E). Hepatic mRNA and protein levels, as well as plasma apoCIII, remained low in the ASO-treated mice (P < 0.05; Fig. 5, F to H).
Fig. 5. ApoCIII reduction reverses the metabolic phenotype induced by HFD.
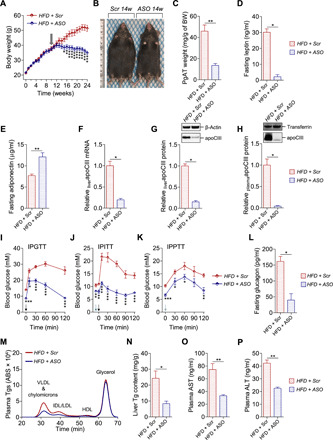
(A) Body weight (BW). Black arrow indicates start of ASO treatment (week 10, N = 12). (B) Representative pictures of mice from the long-lasting reversibility study treated for 14 weeks with Scr (left) and ASO (right). (C) PgAT weight in relation to BW (N = 7). (D and E) Twelve-hour fasting plasma levels of (D) leptin (N = 4) and (E) adiponectin (N = 6). (F) Liver mRNA levels of apoCIII analyzed by qRT-PCR (N = 4). (G and H) Representative immunoblots and densitometry analysis of apoCIII in (G) liver and (H) plasma (N = 4). (I to K) In vivo metabolic tests performed at the end of the study. Injection of bolus doses is indicated with arrows (black, glucose; blue, insulin; and green, pyruvate). (I) IPGTT (N = 5), (J) IPITT (N = 5), and (K) IPPTT (N = 5). (L) Twelve-hour fasting plasma glucagon levels (N = 4). (M) Representative chromatograms for Tg lipid profiles measured in VLDL and chylomicron–, IDL/LDL-, HDL-, and glycerol-eluted plasma fractions at the end of the study (N = 7). (N) Spectrophotometric measurements of liver Tg content (N = 7). (O and P) Nonfasting plasma (O) AST (N = 5) and (P) ALT (N = 5). Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001, HFD + ASO versus HFD + Scr. Photo credit: Ismael Valladolid-Acebes, The Rolf Luft Research Center for Diabetes and Endocrinology, Karolinska Institutet.
Moreover, glucose, insulin, and pyruvate tolerance tests, plasma glucagon, lipid profiles, liver Tgs, and plasma AST and ALT underscore that prolongation of ASO treatment was able to reverse the metabolic phenotype (P < 0.05 to P < 0.001; Fig. 5, I to P, and fig. S5, B to L).
DISCUSSION
High blood glucose levels and insulin resistance in DIO are known to increase apoCIII (13, 14). Several studies in mice and humans indicate that apoCIII regulates multiple steps in lipid metabolism, including hepatic uptake of lipids from the circulation (5, 35, 36), conversion of TRL to LDL, and activity of lipases (24, 2). Receptor-mediated hepatic clearance of lipoprotein particles has been demonstrated to be enhanced by decreasing apoCIII (5, 35, 36). The primary effect of apoCIII reduction is an increased breakdown of circulating lipids due to enhanced lipolysis (Fig. 6). A decrease in circulating Tgs and remnant cholesterol particles, less accumulation of atherogenic lipoproteins in the artery wall, and reduced endothelial inflammation result in a diminished risk for CVD (37).
Fig. 6. Schematic overview of the beneficial metabolic effects of lowering apoCIII in vivo under HFD conditions.
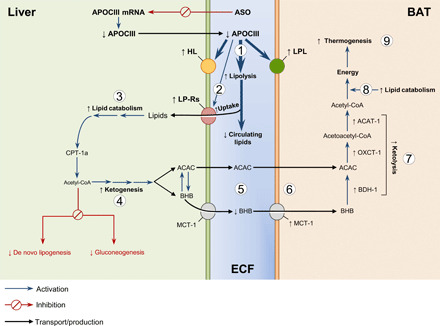
Targeting apoCIII protects against HFD-induced metabolic derangements by increasing lipolysis (1), hepatic clearance of circulating lipids (2), liver lipid catabolism (3), and shifting liver metabolism toward ketogenesis (4). The ketones are taken up (5 and 6) and used for energy production in BAT (7 to 9). LP-Rs, lipoprotein receptors; ECF, extracellular fluid.
We now show that interference with apoCIII during HFD intake activates lipases and improves hepatic clearance and catabolism of lipids from the circulation. Our work also reveals that lowering apoCIII shifts liver lipid metabolism toward ketogenesis and uptake of ketones by BAT. Hence, as a result of decreased levels of the apolipoprotein, there is a normalization in insulin sensitivity and an increase in FAO and ketogenesis, while DNL and gluconeogenesis are inhibited. The ketones are taken up and used for energy production in BAT. The metabolic activity of BAT is inversely correlated with the fraction of fat in the tissue, while the lipid-burning capacity is directly related to insulin sensitivity (38, 39), which was confirmed by our results showing an improved glucose homeostasis and insulin sensitivity in the ASO-treated mice.
The overall positive effect on the metabolic phenotype of the ASO-treated mice on HFD is caused by an efficient use of fat for fuel instead of storage and a maintained/restored insulin sensitivity, preventing deleterious metabolic consequences (Fig. 6). We could not only prevent the detrimental effects of a diet rich in fat but also reverse its metabolic derangements by lowering apoCIII despite a continued intake of HFD.
There are several limitations in our study. One is that the temperature in our animal rooms and in the metabolic cages is 22°C. Thermoneutral temperature for most experimental animals (and naked humans) lies close to 30°C. Hence, animals kept at room temperature have an increased metabolism to maintain body temperature. Chronic exposure to room temperature will thus result in nonshivering thermogenesis, with BAT being the main site. In animals that live at 20°C, cold stress is not induced until around 5°C (40, 41). Because all our mice have been exposed to 22°C for their entire life, it is unlikely that lower than thermoneutral ambient temperature will affect the interpretation of our data.
We are also aware of the fact that there are challenges in analyzing rodent energy metabolism (18, 42). Hence, changes in BW may partly drive the observed effects shown in Fig. 1M and fig. S1K. However, still obese mice on HFD, treated for only 4 weeks with ASO, show a tendency to an increase in RER. Because this limitation is known to us, we do not base our conclusions on the effects of decreasing apoCIII on data solely from the metabolic cages. These results are used together with data on glucose homeostasis, insulin sensitivity, body temperature, morphology of liver and BAT, as well as phenotype of the animals.
Another limitation is that we did not perform tracer studies to support our findings that there is an increased use of ketone bodies in BAT in ASO-treated mice. The reason for not conducting tracer studies is that we did not want to disturb the animals and thereby jeopardize the outcome of the metabolic and mechanistic investigations, which were the focus of the present study. Tracer experiments would have had to be done as separate experiments, and for this study, we think that the results we have with increased expression of the hepatic enzymes ACAT-1, HMGCS2, HMGCL, and BDH-1, the latter also demonstrated at the protein and activity level, which are involved in the ketone body metabolism, the increased expression in BAT of MCT-1, which transports ketone bodies, and the increased expression of the markers for ketolysis BDH-1, OXCT-1, and ACAT-1 support our suggestion for an increased use of ketone bodies in BAT in the ASO-treated mice. Although tracer studies would have made it possible to reach a higher level of evidence, the differences in phenotype, morphology of liver and BAT, and prevention/reversion of obesity and of the in vivo results on glucose homeostasis and insulin sensitivity are clear findings of this study.
In summary, our compiled data suggest that mice consuming HFD and simultaneously being subjected to a lowering in apoCIII have a more efficient metabolism resulting in normal glucose homeostasis, insulin sensitivity, and lack of obesity despite similar food intake and physical activity compared to obese mice solely on HFD.
The fact that increased levels of apoCIII are associated with NAFLD highlights the clinical relevance of our studies (10, 11). NAFLD represents a spectrum of liver damages, ranging from steatosis to nonalcoholic steatohepatitis to cirrhosis (43). NAFLD affects 40 to 50% of the total population in Europe and the United States, and the prevalence is even higher in risk groups with obesity and T2DM. This disease can be considered the hepatic component of the metabolic syndrome and is closely associated with obesity, insulin resistance, and T2DM. Our findings thus identify apoCIII as a central multifaceted drug target in the metabolic syndrome.
MATERIALS AND METHODS
Mice and diets
Male C57BL6/j (B6) mice were purchased from Charles River Laboratories at the age of 6 weeks. Mice, housed three to five animals per cage, were acclimated to our animal facilities for 2 weeks from arrival in a temperature- and humidity-controlled room with 12-hour light/12-hour dark cycles and ad libitum access to water and food (R70, Lantmännen, Sweden). At the age of 8 weeks, all mice were fed an HFD (60% kcal from fat, Open Source Diets D12492, Research Diets). The duration of the diet intervention varied depending on the experimental design, from 14 to 24 weeks. Animal care and experiments were carried out according to the Animal Experiment Ethics Committee at Karolinska Institutet.
Antisense oligonucleotides
Chimeric 20-mer phosphorothioate ASOs containing 2′-O-methoxyethyl groups at positions 1 to 5 and 16 to 20 were provided by IONIS Pharmaceutical Company. The sequence of the active antisense (ASO) against apoCIII gene (ION-353982) was 5′-GAGAATATACTTTCCCCTTA-3′ and of the inactive or scrambled (Scr) (ION-141923) was 5′-CCTTCCCTGAAGGTTCCTCC-3′. Scr and active ASO were developed and tested for specificity and toxicity by the IONIS Pharmaceutical Company, as previously described (16).
Experimental design
We performed the following studies in B6 mice: (i) a prevention study, where ASO treatment started simultaneously with the HFD (in the prevention study, mice were fed an HFD for 14 weeks, starting at week 8 of age, and intraperitoneally administered with either ASO or Scr twice per week at a dose of 25 mg/kg); (ii) an early reversibility study, where mice were fed an HFD for 10 weeks, starting at 8 weeks of age, and thereafter treated intraperitoneally with either ASO or Scr twice per week (25 mg/kg) for four additional weeks, still being fed an HFD (the total duration of the early reversibility study was 14 weeks); and (iii) a long-lasting reversibility study, where mice were fed an HFD for 10 weeks, starting at week 8 of age, and thereafter treated intraperitoneally with either ASO or Scr twice per week (25 mg/kg) for 14 additional weeks, still being fed an HFD (the total duration of the long-lasting reversibility study was 24 weeks). In all studies, BW was determined.
Energy metabolism characterization
To evaluate systemic energy metabolism, mice treated according to the prevention and early reversibility protocols were acclimated to single cages during a 24-hour period. Thereafter, Scr- and ASO-treated mice from both experimental designs were monitored for four consecutive days in the Oxymax Lab Animal Monitoring System [Comprehensive Laboratory Animal Monitoring System (CLAMS), Columbus Instruments] with ad libitum access to food except for a 12-hour fasting period during the last night. The following parameters were continuously monitored: calorie intake, locomotion, VO2, VCO2, and RER. Locomotion was reported as total beam breaks for the x and y axes. Energy expenditure (EE) was calculated as previously described (30–32) using the following formula: EE (kcal/kg per hour ) = (3.815 + 1.232 × RER) × VO2. In vivo FAO was calculated as previously described (30–32) using the following formula: FAO (kcal/kg per hour) = EE × (1 – RER/0.3).
Body composition
Total fat and lean mass were determined in mice from the prevention and early reversibility studies using an EchoMRI-100 system (Echo Medical Systems). The measurement is based on NMR, taking advantage of the different density of the hydrogen nuclei in the adipose tissue, water, and bone, as previously described (44). Fat mass–to–lean mass ratio was calculated by normalizing the total fat mass per the lean mass of every individual mouse.
In mice from the long-lasting reversibility study, the PgAT weight was used as an estimation of fat mass accumulation. Tissue weight was normalized to whole BW and expressed in milligrams of PgAT per gram of BW.
Body temperature
Body temperature was monitored in all studies with a FLUKE 5 1 K/J thermometer with a rectal probe (Fluke Corporation).
Intraperitoneal glucose tolerance test
Glucose tolerance was monitored in 6-hour fasted mice from all studies during daytime. Blood glucose concentrations were measured at basal state (0 min) and 10, 30, 60, and 120 min after an intraperitoneal glucose injection (2 g/kg BW, intraperitoneally). Glucose concentrations were measured with an Accu-Chek Aviva monitoring system (F. Hoffmann–La Roche).
Insulin secretion during IPGTTs
Blood samples were collected in Microvette CB 300 K2 EDTA tubes (SARSTEDT AG & Co. KG) at all time points of the IPGTTs and kept on ice. Thereafter, blood samples were centrifuged at 2500g for 15 min at 4°C; plasma was collected and preserved at –80°C until use. Insulin was analyzed using ultrasensitive mouse insulin enzyme-linked immunosorbent assay (ELISA) kits (Crystal Chem Inc.).
Intraperitoneal insulin tolerance test
To monitor whole-body insulin sensitivity, IPITTs were performed in mice from all studies after 6-hour fasting during daytime. Blood glucose concentrations were measured at basal state. Thereafter, mice were given an intraperitoneal injection of insulin [0.25 IU/kg of BW, diluted in phosphate-buffered saline (PBS), Novo Nordisk]; blood glucose was measured followed by intraperitoneal glucose administration (1 g/kg of BW), as previously described (45). Blood glucose concentrations were measured at 15, 30, 60, 90, and 120 min after glucose injection with an Accu-Chek Aviva monitoring system (F. Hoffmann–La Roche).
Intraperitoneal pyruvate tolerance test
To determine liver gluconeogenesis and hepatic insulin sensitivity in vivo, 12-hour overnight fasted mice received an intraperitoneal injection of pyruvate (2 g/kg of BW, dissolved in PBS). Blood glucose concentrations were measured at basal state (0 min) and 30, 60, 90, and 120 min after the injection with the Accu-Chek Aviva monitoring system (F. Hoffmann–La Roche). IPPTTs were performed in all studies at the end of the experimental designs.
Tissue fixation for histological studies
For tissue fixation, mice were anesthetized with isoflurane and transcardially perfused with PBS followed by freshly prepared 4% paraformaldehyde in PBS (pH 7.4). Liver and BAT were dissected out and postfixed overnight. After fixation, tissue samples were processed with a sucrose gradient [10 to 30% (w/v) sucrose solution in PBS containing 0.01% (w/v) sodium azide and 0.02% (w/v) bacitracin], frozen in dry ice, and preserved at −80°C until use.
Because tissue fixation by alcohol-based fixatives damages tissue lipids, we used 4% neutral-buffered paraformaldehyde solution for fixation. Tissue processing after fixation (i.e., postfixation and cryopreservation by sucrose gradient) never included rinsing in alcohol.
Hematoxylin and eosin staining in BAT
Paraformaldehyde-fixed BAT was sectioned into 20-μm-thick sections with a cryostat (Microm HM500M/Cryostar NX70, Thermo Fisher Scientific) and collected onto SuperFrost Plus microscope slides (VWR International). The sections were rinsed in hematoxylin [Sigma-Aldrich; 50% (v/v) diluted in distilled water] for 6 min, washed with distilled water, and stained with eosin (Histolab) for 30 s. After washing, sections were dried and mounted with VectaMount permanent mounting medium (Vector Laboratories). All procedures were performed in a cold room at 4°C. Once the mounting medium was solidified, the preparations were brought up to room temperature and imaged under 10×, 20×, and 40× magnification objectives using an optical microscope (Leica). The morphological evaluations were performed in three nonconsecutive sections separated by 100 μm. For each section, three fields of view were collected in four individual animals per experimental group.
Liver lipid visualization by Oil Red O staining
Twenty-micrometer-thick cryosections from paraformaldehyde-fixed liver tissue were collected onto SuperFrost Plus microscope slides. After 2-hour equilibration at room temperature, slides were rinsed in 60% (v/v) isopropyl alcohol, stained in freshly prepared 0.1% (v/v) Oil Red O (ORO) (Sigma-Aldrich) in 60% (v/v) isopropyl alcohol solution for 30 min, and washed in distilled water. The ORO-stained sections were rinsed in hematoxylin [Sigma-Aldrich; 50% (v/v) diluted in distilled water] for 2 min, washed in distilled water, and mounted in aqueous media. The sections were immediately imaged under 10×, 20×, and 40× magnification objectives using an optical microscope (Leica). Lipid visualization by ORO staining was performed in three nonconsecutive liver sections separated by 100 μm. For each section, three fields of view were collected in four individual animals per experimental group.
Spectrophotometric analysis of liver lipids
Liver tissues were dissected out, weighed, snap-frozen in liquid nitrogen, and stored at −80°C until use. Hepatic Tg content was determined, as previously described (46), by carcass saponification in 0.1 M KOH in 99% ethanol. Tgs were analyzed using Free Glycerol Reagent and Glycerol Standards (Sigma-Aldrich) to construct the standard curve. The glycerol concentration (triolein equivalents) was measured by spectrophotometry (SAFAS-MONACO) at λ = 540 nm and determined by extrapolation from the standard curve. Total Tg content was expressed as milligrams per gram of liver tissue.
Lipase activity measurements
LPL and HL were determined in tissue samples of BAT and liver, respectively. Tissue samples were quickly dissected and immediately frozen in liquid nitrogen. Thereafter, frozen tissues were homogenized in nine volumes of a buffer containing 0.025 M ammonia, 1% Triton X-100, 0.1% SDS, and protease inhibitor cocktail tablets (cOmplete Mini, Roche Diagnosis). Homogenates were centrifuged at 10,621g for 15 min at 4°C. The supernatant was incubated, as previously described (47), using a 3H-triolein–labeled intralipid as a substrate. Samples were gently mixed with scintillation liquid (Optiphase, PerkinElmer Inc.) and analyzed using a scintillation counter (PerkinElmer, Waltham, MA, Wallac, Win Spectral). Enzyme activity was normalized by tissue weight and expressed as milli–international units per milligram of tissue.
BDH-1 activity measurements
BDH-1 activity was determined in liver tissue. Tissue samples were quickly dissected and immediately frozen in liquid nitrogen. Thereafter, 10 to 20 mg of frozen tissue were homogenized in 500 μl of lysis buffer according to the manufacturer’s instructions (Biomedical Research Service Center University at Buffalo, State University of New York, USA). Tissue lysates were centrifuged at 20,817g for 5 min, and supernatant was used for protein determination. All samples were adjusted to a concentration of 2 mg/ml and loaded in quadruplicate in plane (uncoated) 96-well plate placed on ice. Thereafter, one set of duplicate samples was treated with control solution (containing one part of double-distilled water and nine parts of BDH assay solution), and the other set of duplicates was treated with the enzyme substrate (containing one part of 10× BDH substrate and nine parts of BDH assay solution). Plates were covered and incubated at 37°C in a temperature-controlled environment without CO2, as specified by the manufacturer’s protocol. After 60-min incubation, absorbance was measured by spectrophotometry (Envision 2103 Multilabel Reader, PerkinElmer) at λ = 492 nm. Thereafter, subtraction of the reading of the control wells from the reading of the reaction wells for each sample was performed, and enzyme activity was determined, as indicated in the manufacturer’s protocol, using the following equation: BDH-1 activity in μmol of reduced form of nicotinamide adenine dinucleotide (NADH)/(L × min) = Δoptical density × 12.96. Obtained data were corrected by the microgram of protein in each well, and activity units were expressed as μmol of NADH/(L × min)/μg of proteins.
Size exclusion chromatography analysis
Lipid profiles were obtained by size exclusion chromatography in plasma from mice under nonfasting conditions as previously described (48). Plasma lipoproteins were separated by size class using a LaChrom Elite high-performance liquid chromatography system (Hitachi) and Superose 6 PC 3.2/300 gel column (GE Healthcare). Plasma Tg concentrations were determined using a coupled enzymatic reaction system (glycerol-3-phosphate oxidase–phenol + aminophenazone method, Roche). The Tgs associated to the different lipoprotein classes were measured as area under the curve using EZChrom Elite software (Agilent Technologies).
Plasma biochemistry
For biochemical determinations in plasma, blood samples from nonfasted mice were obtained at the end of the experiments, collected in Microvette CB 300 K2 EDTA tubes (SARSTEDT AG & Co. KG), and kept on ice. Thereafter, blood samples were centrifuged at 2500g for 15 min at 4°C; plasma was collected and preserved at −80°C until use. Plasma insulin, leptin, and adiponectin were measured using mouse-specific ELISA kits (Phoenix Pharmaceuticals for leptin and Crystal Chem for insulin and adiponectin). For glucagon determination, blood samples were collected in a separate set of Microvette CB 300 K2 EDTA tubes (SARSTEDT AG & Co. KG), supplemented with aprotinin (500 kIU/ml) kept on ice. Immediately after, blood samples were centrifuged as specified above and obtained plasma was preserved at −80°C until use. Plasma glucagon levels were determined using a mouse-specific glucagon ELISA kit (Crystal Chem). Plasma transaminases, AST and ALT, were measured with mouse-specific commercial ELISA kits (MyBiosource). Non-esterified fatty acids were measured by colorimetry using a coupled enzymatic reaction system (acyl-CoA synthase–acyl-CoA oxidase method, Cell Biolabs Inc.) and ACAC with a nonenzymatic colorimetric assay (Abcam). BHB measurements were performed using an enzymatic colorimetric assay based on BHB dehydrogenase (Abcam). LDL and HDL (high-density lipoprotein) cholesterol were determined by the Trinder reaction using colorimetric assays based on a modified polyvinyl sulfonic acid (PVS) and polyethylene glycol methyl ether (PEGME) coupled classic precipitation method (Crystal Chem).
RNA isolation and quantitative real-time polymerase chain reaction
Tissues were quickly dissected out and put in RNA stabilization solution (RNAlater, Invitrogen by Thermo Fisher Scientific). After RNA stabilization, samples were preserved at −80°C until use. Total RNA was isolated using the RNeasy Lipid Tissue Mini Kit according to the manufacturer’s protocol (Qiagen). On-column digestion of DNA was performed during RNA purification using RNase-Free DNase I set (Qiagen). Total RNA concentrations were determined using NanoPhotometer P330 (IMPLEN). One microgram of total RNA was used for complementary DNA (cDNA) preparation using the High Capacity cDNA Reverse Transcription Kit (Life Technologies). Quantitative real-time polymerase chain reaction (qRT-PCR) was performed in a QuantStudio 5 PCR system thermal cycler (Applied Biosystems) with Power-Up SYBR Green PCR Master Mix (Applied Biosystems). As control genes for liver target transcripts, we used β-actin, TATA-binding protein (TBP), and hypoxanthine phosphoribosyltransferase 1 (HPRT), and for BAT target transcripts, we used TBP and hydroxymethylbilane synthase (HMBS). Analysis of gene expression was performed with the ΔΔCt method. The relative transcript levels of each target gene were normalized to each control gene and to the geometric mean of the cycle threshold (Ct) of all control genes used for qRT-PCR analyses in liver and BAT. The results are expressed as mRNA levels relative to Scr-treated mice. Primer sequences are available in table S1.
Western blotting
For determination of circulating apoCIII, plasma was albumin-depleted using AlbuSorb according to the manufacturer’s protocol (Biotech Support Group LLC) and resuspended in 0.1% (v/v) trifluoroacetic acid. For hepatic apoCIII, liver samples were homogenized in ice-cold buffer containing 0.42 mM NaCl, 20 mM Hepes (pH 7.9), 1 mM Na4P2O7, 1 mM EDTA, 1 mM EGTA, 1 mM dithiothreitol, 20% (v/v) glycerol, 20 mM sodium fluoride, 1 mM Na3O4V, and Halt protease and phosphatase inhibitor cocktail (1:100). Tubes containing homogenates were exposed to thermal shock at −80°C in liquid nitrogen and thawed to 37°C in a water bath three consecutive times. Thereafter, homogenates were centrifuged at 10,000g for 20 min at 4°C, and supernatants were collected. Protein concentrations were determined by Bradford assay (Bio-Rad). Equal amounts of protein (12.5 μg for plasma and 30 μg for liver) were loaded onto a 4 to 12% bis-tris gel (Invitrogen). For immunoblotting, membranes were blocked with 5% (w/v) bovine serum albumin in 0.1% (v/v) Tween 20 (Sigma-Aldrich) in PBS. After blocking, membranes were probed overnight with either a rabbit polyclonal anti-apoCIII primary antibody (1:200; LSBio) or a mouse monoclonal anti–BDH-1 antibody (1:1000; Thermo Fisher Scientific). After incubation with either anti-rabbit immunoglobulin G (IgG)–peroxidase complexes for apoCIII protein determination (1:4000 for plasma and 1:2000 for liver; GE Healthcare) or anti-mouse IgG–peroxidase complexes (1:1000; GE Healthcare), membranes were exposed to commercial enhanced chemiluminescence reagents (Clarity Western ECL Substrate, Bio-Rad), and blots were developed with a luminescent image analyzer (ChemiDoc Touch Imaging System, Bio-Rad). Transferrin was detected as a control protein in the membranes loaded with plasma samples by using a rabbit polyclonal anti-transferrin primary antibody (1:1000; LSBio). Anti-rabbit IgG–peroxidase complexes (GE Healthcare) for transferrin detection were used at 1:4000 dilution. β-Actin was detected as a control protein in those membranes loaded with liver samples used for determination of apoCIII protein levels by using a mouse monoclonal anti–β-actin primary antibody (1:8000; Sigma-Aldrich). Anti-mouse IgG–peroxidase complexes (GE Healthcare) for β-actin detection were used at 1:10,000 dilution. γ-Tubulin was detected as a control protein in those membranes loaded with liver samples and tested for BDH-1 protein levels. After detection of protein levels, membranes were stripped by applying 5% (v/v) acetic acid in double-distilled water and incubated for 5 min. Subsequently, membranes were washed, blocked overnight, and thereafter probed with a mouse monoclonal anti–γ-tubulin primary antibody (1:4000; Thermo Fisher Scientific). Anti-mouse IgG–peroxidase complexes (Cell Signaling Technology) for γ-tubulin detection were used at 1:1000 dilution. Obtained images were quantified using ImageJ software (National Institutes of Health). Details on primary and secondary antibodies used are available in table S2.
Statistical analysis
All statistical analyses were performed using GraphPad Prism 5.0. Nonparametric two-tailed Mann-Whitney U t test was used when two groups were compared, and one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test was used when comparing multiple groups. The metabolic parameters VO2, VCO2, energy expenditure, and in vivo FAO were analyzed by generalized linear modeling as previously described (49), which is equivalent to analysis of covariance (ANCOVA). The nonparametric Friedman test with repeated measures followed by Dunn’s post hoc test was used for statistical analyses of RER. Statistical significance was defined as P < 0.05.
Acknowledgments
We thank R. Crooke and M. Graham (now retired) from IONIS for support. Funding: This work was supported by the Swedish Diabetes Association, Funds of Karolinska Institutet, Swedish Research Council, Novo Nordisk Foundation, Family Erling-Persson Foundation, Strategic Research Program in Diabetes at Karolinska Institutet, Family Knut and Alice Wallenberg Foundation, Stichting af Jochnick Foundation, Skandia Insurance Company Ltd., Diabetes and Wellness Foundation, Bert von Kantzow Foundation, Svenska Diabetesstiftelsen, AstraZeneca, Swedish Association for Diabetology, and ERC-EYLETS 834860. Author contributions: Conceptualization: I.V.-A., K.Å., P.-O.B., and L.J.-B.; methodology: I.V.-A., K.Å., N.M., and P.R.-L.; investigation: I.V.-A., K.Å., N.M., P.R.-L., G.B., M.B., E.F.A., M.E., and F.L.; visualization: A.K. and S.K.N.; supervision: I.V.-A., P.-O.B., and L.J.-B.; project administration: P.-O.B. and L.J.-B.; funding acquisition: P.-O.B. and L.J.-B.; writing (original draft): I.V.-A., P.-O.B., and L.J.-B.; writing (review and editing): I.V.-A., K.Å., N.M., P.R.-L., G.B., A.K., E.F.A., M.E., F.L., S.K.N., P.-O.B., and L.J.-B. Competing interests: P.-O.B. is cofounder and chief executive officer of Biocrine, a biotech company that is focusing on apolipoprotein CIII as a potential drug target in diabetes. L.J.-B. is consultant for the same company and has participated in advisory boards for Novo Nordisk, AstraZeneca, and Sanofi. The authors declare no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/11/eabc2931/DC1
REFERENCES AND NOTES
- 1.Åvall K., Ali E., Leibiger I. B., Leibiger B., Moede T., Paschen M., Dicker A., Daré E., Köhler M., Ilegems E., Abdulreda M. H., Graham M., Crooke R. M., Tay V. S., Refai E., Nilsson S. K., Jacob S., Selander L., Berggren P.-O., Juntti-Berggren L., Apolipoprotein CIII links islet insulin resistance to β-cell failure in diabetes. Proc. Natl. Acad. Sci. U.S.A. 112, E2611–E2619 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Khetarpal S. A., Zeng X., Millar J. S., Vitali C., Somasundara A. V. H., Zanoni P., Landro J. A., Barucci N., Zavadoski W. J., Sun Z., de Haard H., Toth I. V., Peloso G. M., Natarajan P., Cuchel M., Lund-Katz S., Phillips M. C., Tall A. R., Kathiresan S., DaSilva-Jardine P., Yates N. A., Rader D., A human APOC3 missense variant and monoclonal antibody accelerate apoC-III clearance and lower triglyceride-rich lipoprotein levels. Nat. Med. 23, 1086–1094 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kinnunen P. K., Ehnolm C., Effect of serum and C-apoproteins from very low density lipoproteins on human postheparin plasma hepatic lipase. FEBS Lett. 65, 354–357 (1976). [DOI] [PubMed] [Google Scholar]
- 4.Lee S. J., Campos H., Moye L. A., Sacks F. M., LDL containing apolipoprotein CIII is an independent risk factor for coronary events in diabetic patients. Arterioscler. Thromb. Vasc. Biol. 23, 853–858 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Gordts P. L., Nock R., Son N.-H., Ramms B., Lew I., Gonzales J. C., Thacker B. E., Basu D., Lee R. G., Mullick A. E., Graham M. J., Goldberg I. J., Crooke R. M., Witztum J. L., Esko J. D., ApoC-III inhibits clearance of triglyceride-rich lipoproteins through LDL family receptors. J. Clin. Invest. 126, 2855–2866 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Larsson M., Allan C. M., Jung R. S., Heizer P. J., Beigneux A. P., Young S. G., Fong L. G., Apolipoprotein C-III inhibits triglyceride hydrolysis by GPIHBP1-bound LPL. J. Lipid Res. 58, 1893–1902 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aalto-Setälä K., Fisher E. A., Chen X., Chajek-Shaul T., Hayek T., Zechner R., Walsh A., Ramakrishnan R., Ginsberg H. N., Breslow J. L., Mechanism of hypertriglyceridemia in human apolipoprotein (apo) CIII transgenic mice. Diminished very low density lipoprotein fractional catabolic rate associated with increased apo CIII and reduced apo E on the particles. J. Clin. Invest. 5, 1889–1900 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kohan A. B., Apolipoprotein C-III: A potent modulator of hypertriglyceridemia and cardiovascular disease. Curr. Opin. Endocrinol. Diabetes Obes. 22, 119–125 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jørgensen A. B., Frikke-Schmidt R., Nordestgaard B. G., Tybjærg-Hansen A., Loss-of-function mutations in APOC3 and risk of ischemic vascular disease. N. Engl. J. Med. 371, 32–41 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Petersen K. F., Dufour S., Hariri A., Nelson-Williams C., Foo J. N., Zhang X. M., Dziura J., Lifton R. P., Shulman G. I., Apolipoprotein C3 gene variants in nonalcoholic fatty liver disease. N. Engl. J. Med. 362, 1082–1089 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perry R. J., Samuel V. T., Petersen K. F., Shulman G. I., The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 510, 84–91 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Altomonte J., Cong L., Harbaran S., Richter A., Xu J., Meseck M., Dong H. H., Foxo1 mediates insulin action on apoC-III and triglyceride metabolism. J. Clin. Invest. 114, 1493–1503 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Béliard S., Nogueira J. P., Maraninchi M., Lairon D., Nicolay A., Giral P., Portugal H., Vialettes B., Valéro R., Parallel increase of plasma apoproteins C-II and C-III in type 2 diabetic patients. Diabet. Med. 26, 736–739 (2009). [DOI] [PubMed] [Google Scholar]
- 14.Caron S., Verrijken A., Mertens I., Samanez C. H., Mautino G., Haas J. T., Duran-Sandoval D., Prawitt J., Francque S., Vallez E., Muhr-Tailleux A., Berard I., Kuipers F., Kuivenhoven J. A., Biddinger S. B., Taskinen M. R., Van Gaal L., Staels B., Transcriptional activation of apolipoprotein CIII expression by glucose may contribute to diabetic dyslipidemia. Arterioscler. Thromb. Vasc. Biol. 31, 513–519 (2011). [DOI] [PubMed] [Google Scholar]
- 15.Duivenvoorden I., Teusink B., Rensen P. C., Romijn J. A., Havekes L. M., Voshol P. J., Apolipoprotein C3 deficiency results in diet-induced obesity and aggravated insulin resistance in mice. Diabetes 54, 664–671 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Graham M. J., Lee R. G., Bell T. A., Fu W., Mullick A. E., Alexander V. J., Singleton W., Viney N., Geary R., Su J., Baker B. F., Burkey J., Crooke S. T., Crooke R. M., Antisense oligonucleotide inhibition of apolipoprotein C-III reduces plasma triglycerides in rodents, nonhuman primates, and humans. Circ. Res. 112, 1479–1490 (2013). [DOI] [PubMed] [Google Scholar]
- 17.Groenendijk M., Cantor R. M., de Bruin T. W., Dallinga-Thie G. M., The apoAI-CIII-AIV gene cluster. Atherosclerosis 157, 1–11 (2001). [DOI] [PubMed] [Google Scholar]
- 18.Tschöp M. H., Speakman J. R., Arch J. R. S., Auwerx J., Brüning J. C., Chan L., Eckel R. H., Farese R. V. Jr., Galgani J. E., Hambly C., Herman M. A., Horvath T. L., Kahn B. B., Kozma S. C., Maratos-Flier E., Müller T. D., Münzberg H., Pfluger P. T., Plum L., Reitman M. L., Rahmouni K., Shulman G. I., Thomas G., Kahn C. R., Ravussin E., A guide to analysis of mouse energy metabolism. Nat. Methods 9, 57–63 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Richard D., Monge-Roffarello B., Chechi K., Labbé S. M., Turcotte E. E., Control and physiological determinants of sympathetically mediated brown adipose tissue thermogenesis. Front. Endocrinol. 3, 36 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Szentirmai É., Kapás L., The role of the brown adipose tissue in β3-adrenergic receptor activation-induced sleep, metabolic and feeding responses. Sci. Rep. 7, 958 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bartelt A., Bruns O. T., Reimer R., Hohenberg H., Ittrich H., Peldschus K., Kaul M. G., Tromsdorf U. I., Weller H., Waurisch C., Eychmüller A., Gordts P. L., Rinninger F., Bruegelann K., Freund B., Nielsen P., Merkel M., Heeren J., Brown adipose tissue activity controls triglyceride clearance. Nat. Med. 17, 200–205 (2011). [DOI] [PubMed] [Google Scholar]
- 22.Garcia-Arcos I., Hiyama Y., Drosatos K., Bharadwaj K. G., Hu Y., Son N. H., O’Byrne S. M., Chang C. L., Deckelbaum R. J., Takahashi M., Westerterp M., Obunike J. C., Jiang H., Yagyu H., Blaner W. S., Goldberg I. J., Adipose-specific lipoprotein lipase deficiency more profoundly affects brown than white fat biology. J. Biol. Chem. 288, 14046–14058 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stern J. H., Rutkowski J. M., Scherer P. E., Adiponectin, leptin, and fatty acids in the maintenance of metabolic homeostasis through adipose tissue crosstalk. Cell Metab. 23, 770–784 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reyes-Soffer G., Horenstein R. B., Holleran S., Matveyenko A., Thomas T., Nandakumar R., Ngai C., Karmally W., Ginsberg H. N., Ramakrishnan R., Pollin T. I., Effects of APOC3 heterozygous deficiency on plasma lipid and lipoprotein metabolism. Arterioscler. Thromb. Vasc. Biol. 39, 63–72 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramms B., Patel S., Nora C., Pessentheiner A. R., Chang M. W., Green C. R., Golden G. J., Secrest P., Krauss R. M., Metallo C. M., Benner C., Alexander V. J., Witztum J. L., Tsimikas S., Esko J. D., Gordts P. L. S. M., ApoC-III ASO promotes tissue LPL activity in the absence of ApoE-Mediated TRL clearance. J. Lipid Res. 60, 1379–1395 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Merkel M., Weinstock P. H., Chajek-Shaul T., Radner H., Yin B., Breslow J. L., Goldberg I. J., Lipoprotein lipase expression exclusively in liver. A mouse model for metabolism in the neonatal period and during cachexia. J. Clin. Invest. 102, 893–901 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Teong Ong K., Mashek M. T., Young Bu S., Greenberg A. S., Mashek D. G., Adipose triglyceride lipase is a major hepatic lipase that regulates triacylglycerol turnover and fatty acid signaling and partitioning. Hepatology 53, 116–126 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Teong Ong K., Mashek M. T., Davidson N. O., Mashek D. G., Hepatic ATGL mediates PPAR-α signaling and fatty acid channeling through an L-FABP independent mechanism. J. Lipid Res. 55, 808–815 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Souza-Mello V., Peroxisome proliferator-activated receptors as targets to treat non-alcoholic fatty liver disease. World J. Hepatol. 7, 1012–1019 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lusk G., Analysis of the oxidation of mixtures of carbohydrate and fat: A correction. J. Biol. Chem. 59, 2 (1924). [Google Scholar]
- 31.Bruss M. D., Khambatta C. F., Ruby M. A., Aggarwal I., Hellerstein M. K., Calorie restriction increases fatty acid synthesis and whole body fat oxidation rates. Am. J. Physiol. Endocrinol. Metab. 298, E108–E116 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Laperrousaz E., Denis R. G., Kassis N., Contreras C., López M., Luquet S., Cruciani-Guglielmacci C., Magnan C., Lipoprotein lipase expression in hypothalamus is involved in the central regulation of thermogenesis and the response to cold exposure. Front. Endocrinol. 9, 103 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanders F. W., Griffin J. L., De novo lipogenesis in the liver in health and disease: More than just a shunting yard for glucose. Biol. Rev. Camb. Philos. Soc. 91, 452–468 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Puchalska P., Crawford P. A., Multi-dimensional roles of ketone bodies in fuel metabolism, signaling, and therapeutics. Cell Metab. 25, 262–284 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sacks F. M., The crucial roles of apolipoproteins E and C-III in apoB lipoprotein metabolism in normolipidemia and hypertriglyceridemia. Curr. Opin. Lipidol. 26, 56–63 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li H., Han Y., Qi R., Wang Y., Zhang X., Yu M., Tang Y., Wang M., Shu Y. N., Huang W., Liu X., Rodrigues B., Han M., Liu G., Aggravated restenosis and atherogenesis in ApoCIII transgenic mice but lack of protection in ApoCIII knockouts: The effect of authentic triglyceride-rich lipoproteins with and without ApoCIII. Cardiovasc. Res. 107, 579–589 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Borén J., Packard C. J., Taskinen M. R., The roles of ApoC-III on the metabolism of triglyceride-rich lipoproteins in humans. Front. Endocrinol. 28, 474 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koskensalo K., Raiko J., Saari T., Saunavaara V., Eskola O., Nuutila P., Saunavaara J., Parkkola R., Virtanen K. A., Human brown adipose tissue temperature and fat fraction are related to its metabolic activity. J. Clin. Endocrinol. Metab. 102, 1200–1207 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Zvintzou E., Lhomme M., Chasapi S., Filou S., Theodoropoulos V., Xapapadaki E., Kontush A., Spyroulias G., Tellis C. C., Tselepis A. D., Constantinou C., Kypreos K. E., Pleiotropic effects of apolipoprotein C3 on HDL functionality and adipose tissue metabolic activity. J. Lipid Res. 58, 1869–1883 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cannon B., Nedergaard J., Brown adipose tissue: Function and physiological significance. Physiol. Rev. 84, 277–359 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Cannon B., Nedergaard J., Nonshivering thermogenesis and its adequate measurement in metabolic studies. J. Exp. Biol. 214, 242–253 (2011). [DOI] [PubMed] [Google Scholar]
- 42.Fernández-Verdejo R., Ravussin E., Speakman J. R., Galgani J. E., Progress and challenges in analyzing rodent energy expenditure. Nat. Methods 16, 797–799 (2019). [DOI] [PubMed] [Google Scholar]
- 43.Than N. N., Newsome P. N., A concise review of non-alcoholic fatty liver disease. Atherosclerosis 239, 192–202 (2015). [DOI] [PubMed] [Google Scholar]
- 44.Tinsley F. C., Taicher G. Z., Heiman M. L., Evaluation of a quantitative magnetic resonance method for mouse whole body composition analysis. Obes. Res. 12, 150–160 (2004). [DOI] [PubMed] [Google Scholar]
- 45.Paschen M., Moede T., Valladolid-Acebes I., Leibiger B., Moruzzi N., Jacob S., García-Prieto C. F., Brismar K., Leibiger I. B., Berggren P.-O., Diet-induced β-cell insulin resistance results in reversible loss of functional β-cell mass. FASEB J. 33, 204–218 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Norris A. W., Chen L., Fisher S. J., Szanto I., Ristow M., Jozsi A. C., Hirshman M. F., Rosen E. D., Goodyear L. J., Gonzalez F. J., Spiegelman B. M., Kahn C. R., Muscle-specific PPARgamma-deficient mice develop increased adiposity and insulin resistance but respond to thiazolidinediones. J. Clin. Invest. 112, 608–618 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bergö M., Olivecrona G., Olivecrona T., Forms of lipoprotein lipase in rat tissues: In adipose tissue the proportion of inactive lipase increases on fasting. Biochem. J. 313, 893–898 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Parini P., Johansson L., Bröijersén A., Angelin B., Rudling M., Lipoprotein profiles in plasma and interstitial fluid analyzed with an automated gel-filtration system. Eur. J. Clin. Invest. 36, 98–104 (2006). [DOI] [PubMed] [Google Scholar]
- 49.J. H. Zar, Biostatistical Analysis (Prentice-Hall, ed. 4, 1999), chap. 18. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/11/eabc2931/DC1