

The combination use of TGF-β, M-CSF, and IL-6 represents a promising therapy to repair traumatic brain injury.
Abstract
Traumatic brain injury (TBI) leads to high mortality rate. We aimed to identify the key cytokines favoring TBI repair and found that patients with TBI with a better outcome robustly increased concentrations of macrophage colony-stimulating factor, interleukin-6, and transforming growth factor–β (termed M6T) in cerebrospinal fluid or plasma. Using TBI mice, we identified that M2-like macrophage, microglia, and endothelial cell were major sources to produce M6T. Together with the in vivo tracking of mCherry+ macrophages in zebrafish models, we confirmed that M6T treatment accelerated blood-borne macrophage infiltration and polarization toward a subset of tissue repair macrophages that expressed similar genes as microglia for neuroprotection, angiogenesis and cell migration. M6T therapy in TBI mice and zebrafish improved neurological function while blocking M6T-exacerbated brain injury. Considering low concentrations of M6T in some patients with poor prognostic, M6T treatment might repair TBI via generating a previously unidentified subset of tissue repair macrophages.
INTRODUCTION
Traumatic brain injury (TBI) is a major cause of morbidity and mortality in young adults around the world (1). To date, clinical trials related to TBI treatment have proved discouraging outcome; it may be a result of a failure to well elucidate the complex multifaceted pathogenesis of TBI (2). Mechanical injury to the central nervous system (CNS) leads to a local inflammatory response and recruits immune cells following the insult (3). Macrophages exhibit functional plasticity and heterogeneity under the influence of an inflammatory microenvironment. According to their distinct molecular and biological characteristics, macrophages are classified into proinflammatory and anti-inflammatory macrophages in response to local different cytokines. Inflammatory macrophages are usually induced by interferon-γ and Toll-like receptor or other pathogen-associated molecular pattern stimulation, producing proinflammatory factors such as tumor necrosis factor–α (TNF-α), interleukin-6 (IL-6) and inducible nitric oxide synthase. In contrast, IL-4– and IL-13–induced macrophages exhibit anti-inflammatory activities by up-regulated expression of arginase 1 (Arg1), found in inflammatory zone 1 (Fizz1), chitinase 3-like-3 (Ym1/CHI3l3). Tissue repair macrophages produce vascular endothelial growth factor α (VEGF-α) to facilitate angiogenesis for repair (4, 5), while resolving macrophages produce IL-10 and transforming growth factor–β1 (TGF-β1) to limit inflammation. Both tissue repair macrophages and resolving macrophages have been implicated to favor CNS regeneration and brain repair after injury (5–8). However, the underlying key questions are not fully elucidated, in particular, which key cytokines and growth factors are induced during brain injury, and how could they affect macrophage function for the CNS repair? Answering these questions might help us to find novel biomarkers for potential TBI diagnosis or therapy.
Various cytokines or growth factors are released after brain injury, which can either accelerate or inhibit the repair process. IL-4 and IL-13 are prototypical cytokines favoring tissue repair macrophages in various injury models, including myocardial injury and skin injury. However, it is not well determined whether IL-4 and IL-13 are abundant in the injured brain or any new cytokines play dominant role for the CNS repair. Second, cytokine combination treatment has proven to be more effective with lower levels of toxicity, according to an accumulating amount of practice in cancer immunotherapy. Therefore, administration of combined cytokines/growth factors might offer new therapeutic strategies to treat patients with TBI. Although the functions of various individual cytokines were investigated in brain repair (for instance, TGF-β as a neuroprotective cytokine), treatment of combined cytokines in TBI is not well clarified. Third, previous studies mainly use mouse models to explore cytokine profiling, which might not be recapitulated in patients with TBI. To find the key clue for solving the above concerns, this study examined cytokine/growth factor profiles in plasma and cerebrospinal fluid (CSF) and traced changes in immune cell populations during the whole healing process of patients with TBI.
We have identified that blood circulating monocytes/macrophages were specifically and quickly increased in patients with TBI, together with substantially enhanced concentrations of TGF-β, macrophage colony-stimulating factor (M-CSF), and IL-6 (termed M6T) in both plasma and CSF, in contrast to the barely changed concentrations of IL-4/IL-10, granulocyte macrophage colony-stimulating factor (GM-CSF), and TNF-α. Using CD45.1 or tdTomato+ bone marrow (BM) adoptive transferring model and the in vivo tracking of mCherry+ zebrafish macrophages, we observed blood-borne macrophage migration to sites of injury, some of which displayed microglia-like morphology. M2-like macrophages and microglia were the main cellular source of M6T, which also substantially enhanced production of Arg1, YM-1, Fizz1, and CCL2 in sites of injury. M6T therapy improved neurological function in TBI mice, which educated infiltrating macrophages to display a similar gene expression pattern as local microglia by RNA sequencing (RNA-seq), including a subset of genes functionally related to angiogenesis, neuroprotection, and migration. Consistently, administration of specific antibodies targeting M6T in vivo exacerbates brain injury in TBI mice. Together, we propose a feedback between M6T-enriched microenviroment and macrophage fate changes in sites of injury, which might help us to design a new strategy for TBI therapy.
RESULTS
The enhanced blood monocyte and macrophage populations in patients with TBI
Blood-circulating monocytes, macrophages, and lymphocytes are capable of infiltrating into sites of the injured CNS (3, 9). We asked which immune cell populations might be functionally associated with recovery of patients with TBI. Peripheral blood mononuclear cells (PBMCs) from patients with TBI were isolated by density gradient centrifugation using Ficoll-Paque and monitored by flow cytometric analysis at different time points following injury. We noticed that total numbers (Fig. 1A) and percentages (fig. S1) of lymphocytes from patients with TBI were decreased compared to those of the healthy volunteers (the demographic information of patients with TBI and their controls were summarized in tables S1 to S4; the baseline clinical characteristics of patients with TBI were summarized in tables S5 and S6). Although the percentages of CD8+ T cells to total lymphocytes were slightly reduced at days 0 to 5 after TBI (Fig. 1B), the percentages of CD4+ T cells and CD19+ B cells to total lymphocytes were not substantially affected (Fig. 1, C and D). In contrast, the numbers of CD11b+CD14+ monocytes/macrophages were quickly and substantially increased, even at the day of brain injury (i.e., day 0) [Fig. 1E, the right panels show representative fluorescence-activated cell sorting (FACS) profiles].
Fig. 1. The enhanced blood monocyte and macrophage populations in patients with TBI.
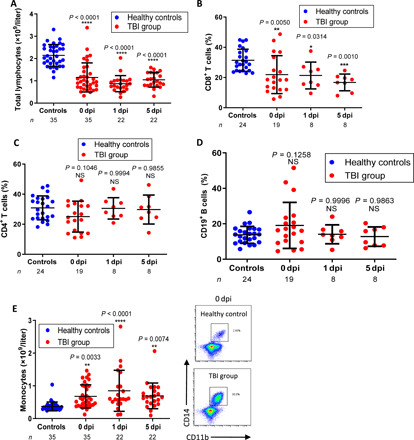
The numbers of lymphocytes (A) or percentages of CD8+ T (B), CD4+ T cells (C), CD19+ B cells (D), and CD11b+CD14+ monocytes (E) in blood collected from healthy volunteers or patients with TBI at the indicated time points were examined by fluorescence-activated cell sorting (FACS). n represents the number of patients. Each symbol represents an individual patient. Statistical significance was determined using one-way analysis of variance (ANOVA) with Dunnett’s multiple comparisons test (A to E) (means ± SD). NS, not significant (P > 0.05); *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
These data promoted us to further ask whether the elevate numbers of circulating monocytes/macrophages migrated into sites of injury, and skewed toward tissue repair macrophages for the recovery of patients with TBI.
Combination of elevated TGF-β1, IL-6, and M-CSF concentrations in CSF and plasma is correlated with outcome of patients with TBI
Various kinds of cytokines and growth factors are generated in response to brain injury and have diverse functions in the repair process. To search which cytokines could account for the enhanced circulating monocytes and educate macrophage for brain repair, we examined concentrations of proinflammatory and anti-inflammatory cytokines, growth factors in plasma, and CSF from patients with TBI. Enzyme-linked immunosorbent assay (ELISA) analyses revealed that TGF-β1 concentrations in plasma were markedly, immediately, and constantly increased from days 0 to 5 in patients with TBI compared to the healthy controls (over 2500 pg/ml; Fig. 2A, left). Unexpectedly, concentrations of plasma IL-10 (Fig. 2A, middle) and IL-4 (less than 3 pg/ml; Fig. 2A, right) were not significantly increased in patients with TBI at various time points. In contrast, IL-6 levels were markedly enhanced in plasma from patients with TBI (Fig. 2B, left). Because TNF-α concentrations were not changed upon TBI (Fig. 2B, right), it suggests that the enhanced concentrations of IL-6 or TGF-β1 were not caused by possible infections in patients with TBI. Both GM-CSF and M-CSF are essential growth factors for monocyte and macrophage differentiation or proliferation. We noticed that M-CSF, but not GM-CSF, was slightly enhanced in plasma of patients with TBI (Fig. 2C).
Fig. 2. Combination of elevated TGF-β1, IL-6, and M-CSF concentrations in CSF and plasma is correlated with outcome of patients with TBI.
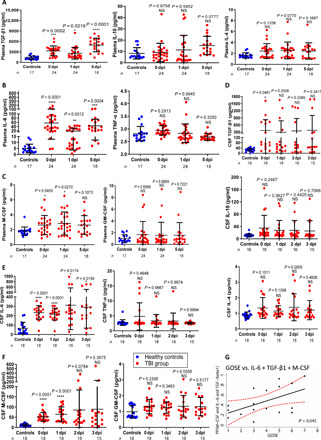
Concentrations of TGF-β1, IL-10, IL-4 (A and D), IL-6, TNF-α (B and E), M-CSF, and GM-CSF (C and F) in the plasma or CSF collected from healthy volunteers or patients with TBI at the indicated time points were measured by ELISA. NS, not significant (P > 0.05); *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. Statistical significance was determined using one-way ANOVA with Dunnett’s multiple comparisons test (A to F) (means ± SD). Each symbol represents an individual patient (A to F). Combination of IL-6, TGF-β1, and M-CSF is positively associated with GOSE (Spearman’s rho = 0.492, P = 0.045) (G). The data were analyzed by the Spearman’s rank correlation test. Predicted probability (PP), which was calculated by binary logistic regression, was used to combine respectively the concentration of M-CSF, IL-6, and TGF-β1 in CSF into one index that used in Spearman’s rank correlation test to assess the association of the index with the percentage of monocytes in patients with TBI at day 0. The black line indicates the fitted line calculated by linear regression, and the red dashed curve indicates the 95% confidence interval (CI) of the fitted line.
CSF is in physical contacted with the brain and provides a connection between the brain and peripheral blood. Considering the difficulties to obtain human brain tissue from patients with TBI, we next tested the production of these cytokines in CSF. Similar to our findings in plasma, we observed a robustly elevated expression of TGF-β1 (the average concentrations 300 pg/ml in patients with TBI versus 20 pg/ml in the controls; Fig. 2D, top) and IL-6 (the average concentrations over 220 pg/ml in patients with TBI versus 50 pg/ml in the controls; Fig. 2E, left) in CSF. In contrast, concentrations of IL-10 or TNF-α were not significantly altered (Fig. 2D, middle, and Fig. 2E, right). Again, IL-4, the classical cytokine for the induction of tissue repair macrophage, was produced at low levels in CSF from patients with TBI (Fig. 2D, bottom), indicating that other cytokines in patients with TBI should be responsible for macrophage polarization. As we noticed, compared to low M-CSF concentrations in plasma, patients with TBI had markedly elevated M-CSF levels in CSF (the average concentrations over 50 pg/ml in patients with TBI versus 10 pg/ml in the controls; Fig. 2F, left). We propose that the injured brain might be the main site to produce cytokines, which then circulated with lower concentrations into the blood vessels. Similar to GM-CSF in plasma, GM-CSF in CSF was low and not substantially changed in patients with TBI (Fig. 2F, right). Together, we uncovered that TGF-β1, IL-6, and M-CSF (but not IL-4 and IL-10) were immediately and significantly elevated in the CSF and plasma of patients with TBI.
Despite this, certain individual patients with TBI did not display high CSF concentrations of TGF-β1, IL-6, or M-CSF. We therefore asked whether the CSF concentrations of the triple cytokines were associated with the outcome of patients with TBI. The 3-month outcome of patients with TBI were assessed by a Glasgow Outcome Scale Extended (GOSE) score ranging from 1 (dead) to 8 (good functional recovery) according to a previous study (10), and patients with TBI were divided into two groups including GOSE 5 to 8 (favorable outcome) and GOSE 1 to 4 (unfavorable outcome). We noticed that CSF concentrations of IL-6 alone, TGF-β1 alone, and M-CSF alone were not associated with GOSE scores of patients with TBI (fig. S2, A to C). In general, IL-6 is regarded as a proinflammatory cytokine that up-regulates the expression of chemokines and adhesion molecules, and a previous study suggests that plasma IL-6 levels at day 1 with a cutoff of 100 ng/ml are positively correlated with severity and infectious complications in brain-injured patients (11). Contrary to these lines of IL-6 function, we identified that IL-6 concentrations in combination with TGF-β1 and M-CSF were positively associated with GOSE scores (Fig. 2G). This provides an interesting hint for further investigation whether the elevated triple cytokines could be used to predict outcome of patients with TBI. Because some patients with TBI did not have high levels of these triple cytokines, we further asked whether administration of the triple cytokines could protect the host from brain injury and elucidate the underline mechanism.
Infiltration of blood-borne macrophages with microglia-like morphology and the enhanced M6T concentrations in the injured brain
To explore how the elevated M6T concentrations contributed to the recovery of patients with TBI, we followed previous studies to generate a TBI mouse model with a closed head injury by a weight-drop device (12–14). TBI mice showed significant brain damage (fig. S3A), edema, and the increased numbers of apoptotic cells [hematoxylin and eosin (H&E) and terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling (TUNEL) staining; fig. S3B] at sites of injury on day 3. The neurological severity score (NSS) was scored from 0 to 10 by 10 individual clinical parameters, including tasks on motor function, alertness, and physiological behavior (13, 14). Higher NSS scores represented worse neurological function. The sham-operated mice underwent identical surgical procedures except for the actual weight drop. Mice showed high NSS scores during 1 to 4 hours after injury and low NSS scores at days 5 to 7 after injury, suggesting that TBI mice gradually recovered neurological function (fig. S3C). A parameter of forelimb grip strength/body weight (FS/BW) showed that the TBI group exhibited decreased forelimb grip strength compared to that of the sham group (fig. S3D).
We next used mass cytometry (also named CyTOF) to achieve a comprehensive examination of the local immune responses at single-cell level. Only a small proportion of CD4+ T cells and CD8+ T were displayed in TBI mice, and CD19+ B cells were rarely detected in both TBI and sham mice (fig. S3E). By contrast, CD11b+ cells were the major immune cells presented in both TBI and sham mice, and a portion of CD11b+CD45+ cells robustly appeared in TBI mice (Fig. 3A). This newly emerged CD11b+CD45+ population also expressed high levels of CD44 and major histocompatibility complex II (MHC-II; Fig. 3B). CD44 has been previously identified as a phagocytic receptor that is able to trigger the ingestion of large particles or apoptotic cells by macrophages (15–17). MHC-II facilitates macrophage phagocytosis and antigen presentation (18). In accordance with these findings, we indeed found that the in vitro M6T-treated peritoneal exudate macrophages (PEMs) enhanced phagocytosis ability (fig. S4C). Immunohistochemical staining of brain sections with antibody against CD68, a monocyte-derived macrophage/microglia marker (19), confirmed the enhanced number of macrophages/microglia in sites of injury at day 3 from TBI mice (Fig. 3C).
Fig. 3. Infiltration of blood-borne macrophages with microglia-like morphology and the enhanced M6T concentrations in the injured brain.
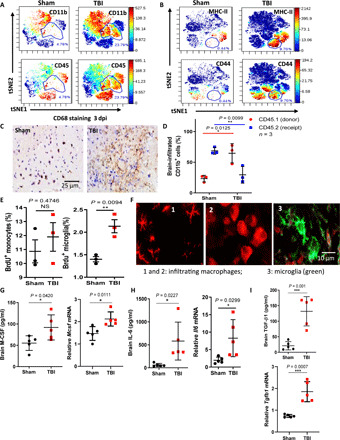
(A and B) Percentages of CD11b+, CD45+, MHC-II+, and CD44+ immune cells in sham or TBI mice brain at 3 day-post-injury (dpi) by CyTOF. (C) Representative images of immunohistochemical staining for CD68+ in sham or TBI mice at 3 dpi. (D) [CD45.1 > CD45.2] chimeric mice were reconstituted by adoptively transfer of RFP+ CD45.1 BM cells into lethally irradiated recipient CD45.2 mice. Following TBI, the percentages of CD45.2+CD11b+ or CD45.1+CD11b+ cells in the injured brain were checked by FACS. (E) 5-bromo-2′-deoxyuridine (BrdU) was injected into TBI mice at 3 dpi, and 24 hours later, cells were harvested for FACS (n = 3). (F) tdTomato+ bone marrow-derived macrophages (BMMs) were transferred into lethally irradiated recipient mice, followed by TBI. Representative images showed morphology changes of infiltrating macrophages in the injured brain sections. (G to I) Concentrations and mRNA levels of M-CSF (G), IL-6 (H), and TGF-β1 (I) in sham or TBI mice brain by ELISA or real-time polymerase chain reaction (RT-PCR) (n = 5). NS, not significant (P > 0.05); *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. Statistical significance was determined using an unpaired Student’s t test (E and G to I) or two-way ANOVA with Sidak’s multiple comparisons test (D). tSNE, t-Distributed Stochastic Neighbor Embedding.
We next investigated whether the enhanced CD11b+CD45+ cells were from infiltration or proliferation of blood-derived monocytes/macrophages and local microglia. Ly6C, a blood-borne monocyte marker, was used to label CD11b+ cells. Ly6C was not expressed in local CD11b+ microglia from the sham mouse brain, while a portion of CD11b+Ly6C+ blood-borne monocytes constantly appeared in brain lesion sites (fig. S3F), suggesting that blood-borne monocytes contributed to elevated numbers of CD11b+CD45+ cells in the injured brain. To compare the proportion of infiltrating cells versus local microglia, we generated BM chimeric mice [CD45.1 > CD45.2] by reconstitution of lethally irradiated CD45.2 mice with BM cells from CD45.1 mice. Circulating monocytes but not microglia in CD45.2 mice were killed by irradiation, and 2 months after transfer, we confirmed BM-derived/blood-borne CD45.1+CD11b+ monocytes in the chimeric mice. Following TBI induction, FACS analysis identified 75% of donor CD45.1+CD11b+ BM-derived/blood-borne monocytes and 25% of local CD45.2+ microglia in sites of injury (Fig. 3D). To further examine whether local microglia and infiltrating monocytes/macrophages proliferated in sites of injury, 5-bromo-2′-deoxyuridine was injected into these mice 1 day before harvesting cells for FACS analysis. We found that local CD45.2+CD11b+ microglia underwent significant proliferation, in contrast to nonproliferative status of infiltrating CD45.1+CD11b+ cells in TBI mice (Fig. 3E).
To trace as well as observe cell morphology of infiltrating macrophages, wild-type recipient mice were lethally irradiated, transferred with tdTomato+ BM cells, followed by the induction of TBI (Fig. 3F). The injured brain sections were prepared for immunohistochemistry staining with Alexa Fluor 488–conjugated anti-F4/80 antibodies. We observed two major populations in sites of brain injury, including a large proportion of F4/80+ tdTomato+ double-positive cells (i.e., infiltrating macrophages) (Fig. 3F and fig. S3G, numbered 1 and 2) versus a small proportion of F4/80+ single-positive cells (i.e., local microglia, numbered 3). Excitingly, F4/80+ tdTomato+ infiltrating macrophages displayed two different morphologies (Fig. 3F and fig. S3G; 1 represents macrophages with a ramified microglia-like morphology; 2 represents macrophages with a round shape). Furthermore, in agreement with our findings in plasma and CSF of patients with TBI, the injured brain of TBI mice substantially produced higher levels of M6T at both mRNA and protein levels (Fig. 3, G to I). Together, we propose that the injured brain promoted local microglia proliferation and attracted BM-derived macrophages, some of which displayed microglia-like morphology.
M2-like macrophage and microglia were major cellular source for both M6T and Arg1 in sites of injury
Next, we investigated the cellular source of M6T and how M6T affected macrophage function. We have followed the methods published in literature to sort endothelial cell (CD45−CD31+) (20), astrocyte (CD45lowCD11b−Ly6C−) (21), inflammatory monocyte (CD45hiCD11b+Ly6C+) (22), and CD206+ M2-like macrophage (CD45hiCD11b+Ly6C−) (23), Iba+ microglia (CD45lowCD11b+Ly6C−) (21), as well as other cells (CD45−CD31−) from sites of injury at day 3 by FACS (fig. S4A). The mRNA expression levels of the triple cytokines and the M2 macrophage makers including Arg1, Ym1, and Fizz1 were examined.
In TBI mice, M-CSF and TGF-β1 were produced mainly by M2-like macrophages, while IL-6 was produced mainly by endothelial cells, inflammatory monocytes, and M2-like macrophages. Compared to the control mice, microglia from TBI mice could also produce low levels of M6T (Fig. 4A). Previous studies have suggested that microglia and neurons can produce M-CSF after brain injury (24), and glial cells produce TGF-β1 in mice with autoimmune encephalomyelitis (25). Together, these studies suggest that upon brain injury, multiple types of cells might produce M6T, including M2-like macrophages and microglia, which generate the M6T-enriched microenviroment.
Fig. 4. M2-like macrophage and microglia were major cellular source for both M6T and Arg1 in sites of injury.
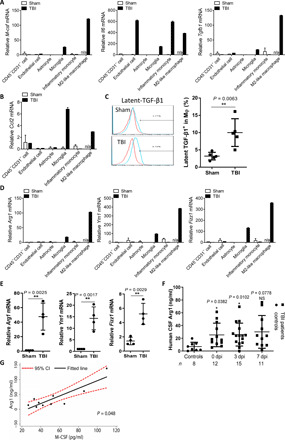
(A, B, and D) Endothelial cells, microglia, astrocytes, inflammatory monocytes, and CD206+ M2-like macrophages from brain at 3 dpi were sorted to check the mRNA expression levels of M-CSF, IL-6, TGF-β1, Arg1, Fizz1, Ym1, and CCL2. n/a indicates no enough cells available. (C) Latent-TGF-β1 expression on macrophages in brain from TBI or sham mice by FACS (n = 5). (E) mRNA levels of Arg-1, Fizz-1, and Ym-1 in TBI or sham mice brain (n = 4). (F) Arg1 concentrations in CSF from patients with TBI by ELISA. (G) Correlation between CSF concentrations of M-CSF and Arg1 from patients with TBI at 0 dpi (Spearman’s rho = 0.636, P = 0.048, Spearman’s rank correlation test). The black line indicates the fitted line calculated by linear regression, and the red dashed curve indicates the 95% CI of the fitted line. NS, not significant (P > 0.05); *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. Data are presented as means ± SD (A to G). Statistical significance was determined using an unpaired Student’s t test (D and E) or one-way ANOVA with Dunnett’s multiple comparisons test (F).
Moreover, we found that microglia and M2-like macrophage were major cellular sources for CCL2 (Fig. 4B). Because the chemokine CCL2 attracts macrophages, this finding was in agreement with the enhanced numbers of infiltrating macrophages in sites of injury (Fig. 3 and fig. S3). Previous studies (26–29) and our data (fig. S4B) found that macrophages could produce TGF-β1 when they engulf apoptotic cells. We observed that infiltrating macrophages in sites of injury significantly enhanced latent TGF-β1 levels on cell surface (Fig. 4C), consistent with these macrophages expressing higher levels of CD44/MHC-II in sites of injury (Fig. 3B) and their increased phagocytosis of apoptotic cells upon M6T treatment (fig. S4C). This could facilitate clearance of apoptotic cells (fig. S3B).
We found that compared to other cell types, CD206+ M2-like macrophages in sites of injury expressed much higher levels of Arg1, Fizz1, and Ym1 (Fig. 4D). In addition, microglia from TBI mice, but not from the control mice, also expressed these M2 markers (Fig. 4D). In agreement with this, the enhanced mRNA levels of Arg1, Fizz1, and Ym1 were confirmed in sites of injury at day 7 (Fig. 4E). This indicates that brain injury could educate the local microglia and infiltrating macrophages for tissue repair.
Arg1 has been confirmed to play critical role for repair in various damaged tissues (30). The relevant data from human patients with TBI showed that, importantly, Arg1 concentrations in CSF from patients with TBI were immediately enhanced after injury and sustained at high levels (Fig. 4F). Arg1 concentrations showed a positive correlation with M-CSF concentrations in CSF from patients with TBI (Fig. 4G). Together, we have identified M2-like macrophage, microglia, or endothelial cell in sites of injury as the major cellular source to generate M6T-enriched microenvironment, which might stimulate infiltrating macrophages and the local microglia to express the M2-macrophage markers Arg1, YM-1, and Fizz1, as well as enhance phagocytosis ability to promote tissue repair.
TGF-β1, M-CSF, and IL-6 favor generation of tissue repair macrophage from murine BM or human PBMC
We next questioned how the elevated triple cytokines could educate BM cells or circulating human PBMC to develop into macrophages and further polarize into tissue repair macrophages. M-CSF is classically used to induce macrophages from the BM after 5 or 7 days (fig. S5A, red dots termed M-CSF). Combination usage of IL-6 alone or together with TGF-β1 could speed up M-CSF–induced macrophage differentiation approximately twofold at day 3 (fig. S5A, green dots termed M6 and blue dots termed M6T). We noticed that M6T or M-CSF plus TGF-β1 could change the round-shaped BM-derived macrophages into a narrow morphology (fig. S5B). This recapitulated the morphology changes of some infiltrating macrophages in sites of injury from TBI mice (Fig. 3F). Compared to M-CSF treatment alone (M), M6 (promoting macrophage development but no morphology change), MT (change morphology, but no strong M2 marker expression), the combination usage of M6T (promoting macrophage development, morphology change, and strong M2 marker expression) significantly enhanced production of Arg1, Ym1, Fizz1, IL-4, and IL-13, which are a group of markers to delineate M2-like macrophages (Fig. 5A). We next expanded these findings using PEMs in a dose-effect analysis (fig. S5C). IL-6 or TGF-β alone induced Arg1 expression in a dose-dependent manner (10 to 40 ng/ml), which could be further enhanced by addition of M-CSF. As noted, the effect to induce Arg1 expression was much better by combination usage of M6 or MT (IL-6 or TGF-β at low concentrations, 10 ng/ml), when compared to the effect by a single cytokine at much higher concentrations (40 ng/ml). These data suggest one of the advantages of M6T combination, i.e., lower concentrations of each single cytokine.
Fig. 5. TGF-β1, M-CSF, and IL-6 favor generation of tissue repair macrophage from murine BM and human PBMCs.
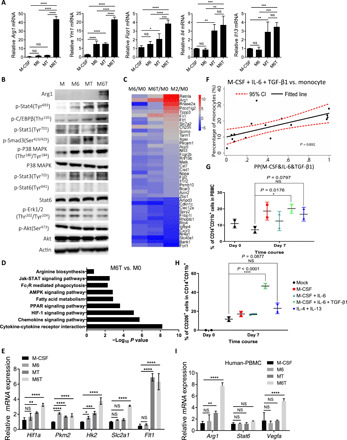
(A and B) BM cells were in vitro cultured with M-CSF, M6, MT, or M6T for 7 days to check the mRNA levels (n = 3) or the phosphorylated and total protein levels of the M2 macrophage–related genes. (C and D) Heatmap showed the differentially expressed genes (DEGs) (C) by RNA-seq on different cell populations described as (A). DEGs were subjected to pathway enrichment analysis with z score. (E) Expression of the interest genes was confirmed by RT-PCR (n = 3). (F) Spearman correlation between M6T concentrations and numbers of blood monocytes in patients with TBI at day 0 (Spearman’s rho = 0.780, P = 0.0002). (G to I) Human PBMCs were stimulated with or without M-CSF, M6, or M6T for 7 days, followed by immunostaining to check the percentages of CD14+CD11b+ (G) or CD206+ cells (H) or the interest genes (I). NS, not significant (P > 0.05); *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. Each point represents data pooled from n = 5 healthy volunteers’ PBMC (G and H). Representative of three or two independent experiments [means ± SD in (A) and (E) to (I)]. Statistical significance was determined using one-way ANOVA with Tukey’s multiple comparisons test (A, E, and I) or with Dunnett’s multiple comparisons test (G and H).
Next, we asked the underline mechanism of how M6T treatment educates macrophages. Although the transcription factors peroxisome proliferator–activated receptor γ (PPARγ) and suppressor of cytokine signaling 1 (SOCS1) have been reported to promote M2 macrophage polarization (31–33), we found that the mRNA expression levels of PPARγ and SOCS1 were not significantly changed in M6T-stimulated macrophages (fig. S5D). STAT (signal transducer and activator of transcription), mitogen-activated protein kinase, and CCAAT/enhancer binding protein β (C/EBPβ) have been reported to regulate M2 macrophages (34–39). We therefore checked the levels of phosphorylated Stat1/3/4/6, ERK1/2 (extracellular signal-regulated kinase 1/2), p38, C/EBPβ, and AKT in M6T-treated macrophages (Fig. 5B). M6T treatment enhanced phosphorylation levels of C/EBPβ and STAT1/STAT4, but not p38, ERK, STAT6, or AKT, when compared to the M, M6, or MT treatment. We next used small interfering RNA to knock down C/EBPβ or STAT4, followed by the treatment with M-CSF or M6T to check Arg1 expression. Reducing C/EBPβ or STAT4 expression could partially reduce the mRNA levels of Arg1 upon M6T treatment. Knocking down both C/EBPβ and STAT1 further decreased Arg1 levels, but still higher than the M-CSF–treated sample (fig. S5E). This might be due to partial knockdown efficiency, or considering the in vitro RNA-seq data in Fig. 5 (C and D) and the in vivo data in Fig. 6H and fig. S6E, these data together support that multiple pathways might be involved.
Fig. 6. M6T induce the in vivo infiltration and neuroprotection function of macrophages with the increased expression of microglia-specific genes.
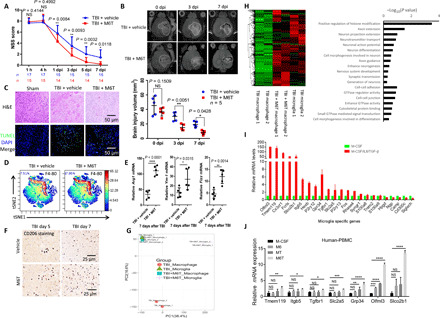
TBI mice were treated with vehicle or M6T to assess NSS [(A) Mann-Whitney U test) or the brain injury volume by MR [(B) two-way ANOVA with Sidak’s multiple comparisons test). At the sites of injury, brain damage and cell apoptosis by H&E staining and TUNEL staining at 3 dpi (C), the percentage of F4/80+ macrophages by CyTOF (D), the mRNA levels of Arg1, Ym1, and Fizz in the isolated CD11b+ cells at day 7 (n = 5) (E), and the numbers of CD206+ cells by immunohistochemistry staining (F) were examined. DAPI, 4′,6-diamidino-2-phenylindole. (G and H) The irradiated recipient mice were transferred with tdTomato+ BM cells, followed by TBI with or without M6T treatment. F4/80+ tdTomato+ infiltrating macrophages versus F4/80+ local microglia were sorted for RNA-seq. The differentially regulated genes in a heatmap and a GO pathway enrichment analysis were shown. (I) The relative expression of the microglia-specific genes was analyzed from the RNA-seq data in Fig. 5C. (J) Human PBMCs were stimulated with M-CSF, M6, MT, or M6T for 7 days to measure expression of the microglia-specific genes (one-way ANOVA with Tukey’s multiple comparisons test, n = 4). tSNE, t-Distributed Stochastic Neighbor Embedding; PC, principal component.
In addition to M2-related markers, we next carried out high-throughput RNA-seq to explore the global transcriptional profile in macrophages after in vitro treatment with M, M6, M6T, or IL-4/IL-13 (the classical M2 macrophages). The heatmap of differentially expressed genes confirmed that the markers of tissue repair macrophages including Arg1, Fn1 (fibronectin), or Retnla (5) and the anti-inflammatory effector Acp5 (tartrate-resistant acid phosphatase) (40) were ranked in the top list in both M6T/M samples and M2/M samples when compared to M6/M samples (Fig. 5C; see arrows). Gene Ontology (GO) pathway enrichment analysis showed that M6T-treated bone marrow-derived macrophages (BMMs) displayed distinct characteristic featured in chemokine signaling, PPAR signaling, cytokine-cytokine receptor interaction, or HIF-1α (hypoxia-inducible factor–1α) signaling, compared to those in M-CSF–treated macrophage (Fig. 5D) or IL-4– and IL-13–treated macrophages (fig. S5F). Consistent with previous studies that HIF-1α and VEGF receptor–1 (VEGFR-1) promote angiogenesis and favor the process of neuron repair (41), we confirmed that Flt-1 (also named VEGFR-1) and HIF-1α signaling proteins including Pkm2 (pyruvate kinase M2), Hk2 (hexokinase 2), Slc2a1 (solute carrier family 2 member 1), and Hif1a were all significantly enhanced in M6T-treated macrophages (Fig. 5E) compared to M-CSF treatment. In particular, we noticed that the expression of Hk2 and Hif1a were indeed further increased by synergistically combination usage of M6T when compared to MT treatment (Fig. 5E). In addition, M6 or M6T also enhanced Pkm2 expression, while Slc2a1 expression was only increased in response to M6T treatment (Fig. 5E). These data emphasize the advantages of M6T combination to induce expression of multiple target genes, which together result in a better recovery in the TBI model. In contrast, M6 or MT might differentially induce only some of these genes. This is consistent with the in vitro RNA-seq data in Fig. 5 (C and D), suggesting that synergistically treated with M6T could induce multiple genes and pathways to support the repair process.
In agreement with that M6T favor macrophage development from murine BMs, we found that M6T concentrations in CSF from patients with TBI were significantly correlated with the enhanced numbers of monocytes (Fig. 5F). The following key question was whether M6T also promoted human circulating blood PBMCs polarization toward tissue repair macrophages. Compared to untreated PBMCs, M6T treatment significantly increased the percentages of CD14+CD11b+ macrophages after 7-day culture (Fig. 5G). Furthermore, populations positive for CD206 (also known as the mannose receptor, a marker for tissue repair macrophages) were greatly elevated in CD14+CD11b+ macrophages after M6T treatment (Fig. 5H). In contrast, IL-4 and IL-13 treatment did not substantially affect the percentages of CD14+CD11b+ or CD206+ macrophages (Fig. 5, G and H). As the controls, M6T treatment did not enhance the percentages of CD3+ T cells or CD9+ B cells in PBMCs (fig. S5G). We next performed real-time polymerase chain reaction (RT-PCR) and confirmed that the tissue repair–associated markers Arg1 and Vegf-a, but not Stat6, were significantly enhanced only when M6T were combined together to treat human PBMCs (Fig. 5I). Together, we propose a novel concept that an elevated production of M-CSF, IL-6, and TGF-β1 in patients with TBI or TBI mice not only promotes macrophage development from BM or PBMCs but also favors generation of tissue repair macrophage.
TGF-β1, M-CSF, and IL-6 induce infiltration and neuroprotection function of macrophages with the increased expression of microglia-specific genes
Although M-CSF, IL-6, and TGF-β1 concentrations were positively associated with the outcome of patients with TBI (Fig. 2G), some individual patients displayed low levels of M6T (Fig. 2, D to F). Could the in vivo combination usage of M-CSF, IL-6, and TGF-β1 (M6T) improve the neurological function and recovery of TBI mice? To address this question, we intraperitoneally injected M6T to TBI mice at 12 and 24 hours after brain injury, followed by one injection each day until day 7. According to the NSS evaluation, M6T-treated TBI mice displayed better neurological function compared to the vehicle-treated TBI mice from day 1 to day 7 after injury (Fig. 6A). Next, the total volume of the injured brain, including the volume of mechanical injury and edema, was determined in a quantitative manner by magnetic resonance imaging (MRI) (Fig. 6B, top images). M6T treatment indeed significantly reduced the volume of the injured area in TBI mice at day 3 and day 7 (Fig. 6B). In agreement with this, TUNEL or H&E staining data confirmed that M6T treatment decreased the number of apoptotic cells with less edema (Fig. 6C).
CyTOF analysis showed that M6T treatment could significantly enhance the percentages of F4/80+CD11b+ in sites of injury compared to the vehicle control (17.95% versus 7.51%; Fig. 6D). However, IL-4 levels were low in sites of injury (slightly increased to 0.65% versus 0.38%; fig. S6, A and B) that was consistent with low IL-4 concentrations in plasma and CSF from patients with TBI (Fig. 2, A and D). In addition, the mRNA expression levels of Arg1, Ym1, and Fizz1 in sites of injury were further enhanced from the in vivo M6T-treated TBI mice compared to the vehicle control (Fig. 6E). In agreement with the enhanced percentages of CD206+ in CD14+CD11b+ macrophages from M6T-treated human PBMCs (Fig. 5I), the number of CD206+ tissue repair macrophage was increased in the injured brain of M6T-treated TBI mice (Fig. 6F).
To further explore the underlying mechanism of the in vivo M6T treatment for neurorepair, we carried out RNA-seq to compare difference of the global transcriptional profile among infiltrating macrophages and local microglia from TBI mice with or without the in vivo M6T treatment. As described in Fig. 4C, we adoptively transferred tdTomato+ BM cells into the lethally irradiated WT recipient mice, followed by the induction of TBI with or without the in vivo M6T treatment. F4/80+ tdTomato+ double-positive cells (i.e., infiltrating macrophages) versus F4/80+ single-positive cells (i.e., local microglia) were sorted for RNA-seq. Macrophages from the in vivo M6T-treated TBI mice displayed a distinguished gene expression pattern to macrophages from the vehicle-treated TBI group, which in contrast showed high similarity as local microglia from the vehicle-treated TBI group (Fig. 6G). Pearson’s correlation analysis showed the significant P values (P values are 1 or 0.99; fig. S6C) between macrophages from M6T-treated TBI mice and microglia from the vehicle-treated TBI mice, which again confirmed their similar gene expression patterns.
Compared to those from the vehicle-treated macrophages, a group of genes were significantly enhanced in the in vivo M6T-treated macrophages, which were similar to those from microglia in a heatmap (twofold changes; Fig. 6H, left). GO or Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis indicated that the in vivo M6T-treated macrophages increased expression of the key genes functionally related to neurorepair or neuroprotection, including the extension and guidance of axons; generation, differentiation, and projection extension of neurons; or neurotransmitter transport (Fig. 6H, right). In addition, M6T-treated macrophages also enhanced the expression of genes related to the cell-cell adhesion/junction, guanosine triphosphatase activity/signal transduction or cytoskeletal protein binding (Fig. 6H, right). This may explain the enhanced infiltration of macrophages into sites of injury, together with the enhanced CCL2 production (Fig. 4B).
Several recent reports have suggested a list of microglia-specific genes that are expressed at much higher levels than in macrophages (42, 43). We analyzed the relative expression of these microglia-specific genes in the in vitro M6T-treated (red bars) versus M-CFS–treated (green bar) macrophages. M6T treatment substantially enhanced expression of most of the microglia-specific genes (Fig. 6I, red bars), including Olfml3 (olfactomedin-like 3, facilitates cell adhesion and intercellular interactions); Slco2b1 (among its related pathways includes blood-brain barrier and immune cell transmigration); TGFbR1 (receptor for TGF-β); CX3CR1 (receptor for the chemokine CX3CL1, mediates cell adhesion and migration); ITGB5 (integrin β5, participates in cell adhesion); SLC2A5 (solute carrier family 2 member 5, a fructose transporter); P2RY12, P2RY13, and GPR34 (paralog of P2RY14) (the family of G-protein–coupled receptors); TMEM119 (transmembrane protein 119; among its related pathways are microglia activation during neuroinflammation: steady-state microglia); and Hexb (mutations in this genes lead to neurodegenerative disorders termed the GM2 gangliosidoses). Also, the in vitro M6T-treated human PBMC significantly increased expression levels of Olfml3, Slco2b1, and Gpr34 compared to that of M, M6, or MT treatment, although expression levels of TMEM119, ITGB5, TGFbR1, and SLC2A5 were not substantially enhanced (values of M-CFS–treated human PBMC were set at 1; Fig. 6J).
In addition, we analyzed significantly enhanced genes in M6T-treated microglia, compared to those of the vehicle control microglia, or macrophages with or without M6T treatment in a heatmap (fig. S6D). GO or KEGG pathway enrichment analysis indicated that M6T-promoted microglia expressed key genes to facilitate phagocytosis/autophagy or clearance of apoptotic cells (fig. S6E), which might contribute to the reduced apoptotic cell numbers in the M6T-treated TBI mice (Fig. 6C). The in vivo M6T treatment also enhanced genes for vasculature development/VEGF production/angiogenesis, as well as neuron regeneration and neuron or axon extension in microglia (fig. S6E), which could benefit the repair process observed in M6T-treated TBI mice (Fig. 6B). These results suggest that not only M2-like macrophages and microglia produce M6T (Fig. 4A), the in vivo M6T-enriched microenvironment could feedback to educate infiltrating macrophages and the local microglia to express a set of genes for improving the neurological outcome.
M6T treatment protects TBI zebrafish for recovery
Zebrafish transparency offers an ideal model to visualize macrophage migration in vivo. We cross-bred the mpeg1:mCherry transgenic zebrafish Tg (mCherry-labeled macrophages) together with the Fli1:GFP transgenic zebrafish Tg [green fluorescent protein (GFP)–labeled endothelium] to real-time trace mcherry+ macrophage migration along the blood vessel into sites of injury. Three-day-old larval fish was punctured with a 25-μm-thin glass tube in brain to construct a TBI model and euthanized 24 hours later. In consistent with our previous data, TBI significantly up-regulated production of M-CSF, IL-6, TGF-β, as well as Arg1 and VEGF-A comparing to the control (Fig. 7A). Furthermore, TGF-β, IL-6, and M-CSF were intra-arterially microinjected into larval fish 1 day before TBI to evaluate M6T-induced effect (Fig. 7, B and C). TGF-β alone was reported previously as a neuroprotector (44, 45). Comparing to TGF-β–treated TBI zebrafish, the combined M6T therapy markedly increased the mRNA levels of Arg1, VEGF-A, and TGF-β in the injured brain (Fig. 7B), suggesting that M6T provides a better protection. The in vivo tracing movies under a fluorescence microscope showed that mcherry+ macrophages migrated along GFP+ blood vessels to the injury brain. Notably, M6T treatment further promoted mCherry+ macrophage accumulation (Fig. 7C, fig. S7A, and movie S1).
Fig. 7. M6T treatment protects TBI zebrafish for recovery, while anti-M6T antibody blockage exacerbates brain injury in TBI mice.
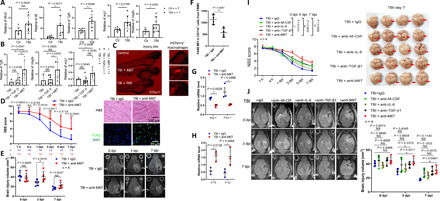
(A and B) Twenty-four hours after TBI, larval fish was euthanized to obtain brain tissue for RT-PCR to evaluate gene expression. (C) The in vivo real-time tracing of mCherry+ macrophages along the GFP+ blood vessels under a fluorescence microscope. (D to J) TBI mice were treated with the specific antibodies blocking M-CSF, IL-6, TGF-β1, or M6T or the isotype control immunoglobulin G (IgG). NSS (D and I) or MR images showing the brain injury volume (E and J) were assessed at the indicated time points. (D) Right: Brain damage and cell apoptosis were examined by H&E and TUNEL staining at 3 dpi. (F) The MFI of F4/80+ staining in CD11b+ cells of brain injury sites were examined by FACS (n = 4). (G and H) The Arg1 mRNA levels were checked by RT-PCR in CD11b+ cells isolated from brain injury sites of TBI mice treated with isotype control or anti-M6T antibodies (n = 3). NS, not significant (P > 0.05); *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. Statistical significance was determined using unpaired Student’s t test (A and H), one-way ANOVA with Tukey’s multiple comparisons test (B), Mann-Whitney U test (D), two-way ANOVA with Sidak’s multiple comparisons test (E) or with Dunnett’s multiple comparisons test (J), or Kruskal-Wallis test with Dunnett’s multiple comparisons test (I). MFI, median fluorescence intensity. Photo credit: Jun Xiao, State Key Laboratory of Cell Biology, SIBCB, Center for Excellence in Molecular Cell Science, Chinese Academy of Sciences; University of Chinese Academy of Sciences.
Antibody blockade targeting M6T exacerbates brain injury in TBI mice
To further evaluate the critical function of M6T for brain recovery, we administered specific antibodies to block the triple cytokines in TBI mice in vivo. Specific antibodies against M-CSF, IL-6, and TGF-β1 were intraperitoneally administered 12 hours before injury and at 12 and 24 hours after injury, followed by daily treatment until the mice were euthanized. According to the NSS scores, blocking antibody–treated TBI mice manifested worse neurological outcomes and more apoptotic cells in the injury sites by TUNEL staining, when compared with the isotype control group (Fig. 7D). Consistent with this, MRI analysis demonstrated a larger brain injury volume (Fig. 7E, right panels are representative images).
In line with the results that M6T treatment could enhance the percentages of F4/80+ cells in the CD11b+ population (Fig. 6D), blocking antibody treatment reduced F4/80 expression in CD11b+ cells in sites of injury (Fig. 7F). We next purified CD11b+ cells from the injured brain and demonstrated that M6T blocking antibody treatment significantly reduced mRNA levels of Arg1 and Fizz1 (Fig. 7G) but enhanced the expression of the proinflammatory markers, including Il12 and Il1β (Fig. 7H) in CD11b+ cells.
Because some reports suggest that TGF-β1 alone has therapeutic effect, we asked whether there was any advantage of using a combination of M6T in vivo. We administered specific antibodies to block each cytokine or the triple cytokines in TBI mice in vivo. When compared with the isotype control or each antibody-treated group, TBI mice treated with the anti-M6T antibodies showed the worst neurological outcomes according to the NSS scores (Fig. 7I) and displayed the largest volume of brain injury according to MRI analysis (Fig. 7J) at each measured time point. While compared to the isotype control group, the anti–TGF-β1–treated group showed worse neurological outcomes (day 5) or larger volume of brain injury (day 3) only at some time point (Fig. 7, I and J). Together, we have used two in vivo vertebrate models, as well as the samples of patients with TBI to propose a feedback regulation between the triple cytokines (i.e., M-CSF, IL-6, and TGF-β1) and M2-like tissue repair macrophage in sites of injury for brain repair (fig. S7B, the model).
DISCUSSION
TBI is becoming an increasingly critical reason for morbidity in young adults but still lacks effective clinical therapeutic strategies. Previous studies have demonstrated that patients with TBI often have accompanying immunodepression, and tissue repair macrophages exist in TBI to facilitate brain recovery (46–49). Our study has unexpectedly showed that the anti-inflammatory cytokine IL-10 and the classical cytokine IL-4 for tissue repair macrophage polarization are produced at relative low concentrations in patients with TBI (Fig. 2, A and D). In contrast, we have identified that the three cytokines M-CSF, IL-6, and TGF-β1 are immediately and constantly elevated in the serum, CSF, and injured brain from TBI patients or mice (Figs. 2 to 4). The in vivo treatment with M6T or their blocking antibodies suggests a protective role of M-CSF, IL-6, and TGF-β1 for brain repair using the TBI mouse model or zebrafish model (Figs. 6 and 7). Notably, multiple TBI patients’ samples have also showed that the combination of elevated M6T concentrations is correlated with better outcomes in patients with TBI (Fig. 2G). Together, our findings open an exciting angle to consider the combination usage of M6T as a new therapeutic strategy as well as for prediction of disease outcome of patients with TBI.
The current understanding of the key cytokines that influence macrophage development and polarization focuses on the balance of GM-CSF and M-CSF. GM-CSF–treated human monocytes can display M1-like macrophages secreting proinflammatory cytokines, and GM-CSF was reported to function as a neuroprotective factor in stroke and neurodegenerative diseases. Nevertheless, we observed that neither the plasma nor the CSF of patients with TBI contains higher levels of GM-CSF compared to those of healthy volunteers (Fig. 2, C and F). By contrast, M-CSF is substantially increased in the CNS and blood of patients with TBI or TBI mice (Fig. 2, C and F). Recently, M-CSF was reported to be an M2-like macrophage stimulus, which promotes the expression of several tissue repair macrophage markers (50). Our study has demonstrated that expression of Arg-1 and Ym-1 can be further increased when M-CSF is combined with IL-6 and, particularly, TGF-β1 (Fig. 5A). In addition, TGF-β alone could increase the mRNA levels Arg1 in PEMs (fig. S5C). In addition, we suggest that the in vivo M6T treatment could also push microglia polarization toward tissue repair microglia in TBI mice via increasing gene expression for the clearance of apoptotic cells, promoting new blood vessel formation, and response to IL-6/IL-4/TGF-β1 (fig. S6E).
M-CSF is also identified as a classical growth and survival factor for macrophage development from BM cells (51, 52), and it usually takes 5 to 7 days for M-CSF to induce BM-derived macrophages. In accordance with previous studies (22, 53), we found that the numbers of monocytes or macrophages are immediately enhanced in the blood or brain (i.e., at day 0 or day 1) upon brain injury (Figs. 1E and 3 and fig. S3). In addition to M-CSF, we propose that IL-6 should be critical to promote this quick response in TBI patients or mice. Brain injury elevates IL-6 production in both serum and CSF to high concentrations (Fig. 2, B and E). IL-6 has been previously regarded as a proinflammatory cytokine and promotes the expression of chemokines and adhesion molecules, which might worsen tissue injury. One supportive study suggests that early serum IL-6 levels might be related with a poor prognosis in brain-injured patients (11). However, our study has provided new evidence that IL-6 treatment accelerates the M-CSF– and TGF-β–induced generation of CD11b+F4/80+ macrophages from murine BM (fig. S5A). This finding is consistent with the previous report that IL-6 promotes monocytes differentiation to macrophages by increasing M-CSFR expression to consume autocrine M-CSF (54). Our GO pathway analysis suggests that upon in vivo M6T treatment, microglia enhance the expression of the key-gene response to IL-4, which is in agreement with a previous work showing that IL-6 induces the expression of the IL-4 receptor and enhances macrophage response to IL-4 in a cell-autonomous manner. A recent elegant study has demonstrated that repopulating microglia is dependent on IL-6 trans-signaling to support neurogenesis (55), which is in agreement with our study showing the neuroprotective effects of the elevated IL-6 in TBI mice (Figs. 2 and 4 to 7). We propose that brain injury immediately induces a high production of M-CSF and IL-6, which cooperate together to substantially promote a quick conversion of BM cells or blood PBMCs into monocytes and macrophages. This is an initial and critical step for repairing the injured CNS because it provides a quick and robust source of monocytes and macrophages. With further education together with TGF-β1, these cells skew toward tissue repair macrophage or microglia-like macrophages, which express key genes related to neuroprotection and the M2 markers upon the in vivo M6T treatment (Fig. 6). These findings together fit with our observation that high concentrations of IL-6 in combination with M-CSF and TGF-β1 are correlated with a better TBI patient outcome (Fig. 2G), and we propose to re-evaluate the in vivo IL-6 function in combination with M-CSF/TGF-β1 as a neuroprotective factor.
Third, we suggest that TGF-β1 is indispensable and should be used together with M-CSF and IL-6 for the generation of tissue repair macrophage (Figs. 4 and 5). Although resting macrophages do not produce TGF-β1, when macrophages engulf apoptotic cells, they indeed produce TGF-β1 (26–28). Our data have showed that the in vitro and in vivo M6T-treated macrophage and human PBMCs as well as the in vivo M6T-treated microglia enhance the cellular response to TGF-β (Figs. 5 and 6), which is responsible for enhanced phagocytosis or clearance of apoptotic cells (fig. S3). This finding suggests that, in an autocrine manner, TGF-β1 is used for the induction of tissue repair macrophage, which then produces more TGF-β1 (Fig. 4). Moreover, we found that the in vivo M6T-treated macrophages expressed a similar gene pattern as the local microglia (Fig. 6 and fig. S6). This finding is in agreement with the TGF-β1 function as a major microglia differentiation factor, and the number of microglia is decreased in the CNS of Tgfb1−/− mice (43). These hints together suggest that the benefit of the combined M-CSF, IL-6, and TGF-β1 might also involve their direct or indirect regulation of microglia function.
Together, this study proposes a model in which (i) in contrast to low concentrations of IL-4/IL-10, brain injury induces high levels of M-CSF/IL-6/TGF-β1 by M2-like macrophage, microglia, or endothelial cell, and the combined M6T concentrations are correlated to TBI outcome. (ii) M6T treatment not only promotes macrophage development from BM or PBMC but also induces polarization of M2-like macrophages that express a subset of microglia-specific genes related to neuroprotection, migration, and angiogenesis. Therefore, a positive feedback between M6T and M2-like macrophage forms to promote brain repair. (iii) M6T therapy in TBI mice and zebrafish improved neurological function, while administration of specific antibodies targeting M6T in vivo exacerbated brain injury in TBI mice. Considering low concentrations of M6T in patients with poor prognostic, our findings implicate a promising application of M6T to repair brain injury (fig. S7B, the model).
MATERIALS AND METHODS
Mice
Wide-type C57BL/6 and 129 mice were purchased from Nanjing Biomedical Research, Institute of Nanjing University. CD45.1 mice are on C57BL/6 background and Rosa26-mTmG mice are on 129 background, which were gifted respectively by Q. Leng [Institute Pasteur of Shanghai, Chinese Academy of Sciences (CAS)] and H. Ping [Shanghai Institute of Biochemistry and Cell Biology (SIBCB), CAS, Shanghai, China]. All animals used in this study were males with an average weight of 28 to 32 g and aged 8 to 16 weeks. Mice were bred under specific pathogen–free conditions at the Animal Care Facility of SIBCB, CAS. The animal experiments were conducted in compliance with the guidance for the care and use of laboratory animals and were approved by the institutional biomedical research ethics committee of SIBCB, CAS.
Sample collection and evaluation of patients with TBI
Samples of PBMC, plasma, and CSF were collected from the patients with TBI who admitted to the level I Neurotrauma Center of Huashan Hospital, Fudan University, from May 2013 to August 2013 and were screened according to the standards below. Patients with TBI with Glasgow Coma Scale (GCS) scores of ≤12 were enrolled. The GCS provides a score in the range of 3 to 15 (a lower number indicates severer brain injury) (56). Exclusion criteria included age <18 years, penetrating head injuries, presence of brain tumors, death within 24 hours, without a surgically placed intraventricular pressure monitoring catheter within 24 hours after injury (only valid for CSF sample collection), polytrauma, extracranial body regions with Abbreviated Injury Scale >2, having infectious disease at admission, blood transfusion within 24 hours after injury, currently taking immunosuppressive drugs, receiving steroid treatment, history of severe organ system insufficiency, an immunocompromised status, or pregnancy. Patients or their legally acceptable representatives who refused to participate in the study were also excluded. All blood and CSF samples from patients with TBI, blood samples from healthy donors, and the CSF control samples from patients with normal pressure hydrocephalus were obtained from Huashan Hospital, Shanghai. The demographic information of patients with TBI and their controls was summarized in tables S1 to S4; the baseline clinical characteristics of patients with TBI were summarized in tables S5 and S6. The study was approved by the Ethics Committee of Huashan Hospital. Written informed consents were obtained from healthy donors, patients, or legally acceptable representatives of patients. Results of clinical routine blood test from 35 patients with TBI and their counterpart control in calculating the absolute number of lymphocytes and monocytes in peripheral blood.
Peripheral blood samples were collected from patients with TBI at different time points after admission. CSF samples from the patients with TBI were collected via intraventricular catheters that had been surgically placed for continuous intracranial pressure monitoring and external ventricular drainage. CSF samples from the control hydrocephalus patients were collected through a ventriculoperitoneal shunt system. PBMCs were isolated by Ficoll density gradient centrifugation. CSF and blood samples were centrifuged at 2000g for 10 min at 4°C and frozen at −70°C until analyzed.
Generation of the TBI mouse model and in vivo treatment
Mice were subjected to experimental closed head injury using a standardized weight-drop device as previously described (12–14). Briefly, after the induction of diethyl ether anesthesia, the skull was exposed by a longitudinal midline scalp incision. The head was fixed, and a 333-g weight was dropped on the skull from a height of 3 cm, inducing a focal blunt injury to the left hemisphere. After the trauma, the surgical incision was closed, and all mice received supporting oxygenation with 100% O2 until fully awake. Analgesia was provided by repeated injections of fentanyl (0.05 mg/kg, ip) every 12 hours. Sham animals underwent the same procedure as TBI mice except for the head trauma.
M-CSF, IL-6, and TGF-β1 [0.02 mg/kg, dissolved in 5% (w/v) dextrose solution] or an equal volume of 5% (w/v) dextrose solution (vehicle) was intraperitoneally administered at 12 and 24 hours after brain injury, followed by daily injection until the mouse was euthanized. Alternatively, specific blocking antibodies against M-CSF, IL-6, and TGF-β1 or the isotype control immunoglobulin G1 [0.04 mg/kg, dissolved in 5% (w/v) dextrose solution] were administered 12 hours before and 12 and 24 hours after brain injury, followed by daily injection until the mouse was euthanized.
Neurological severity score
A standardized 10-point NSS was used for quantification of posttraumatic neurological impairment as described (13, 14). The score consists of 10 individual clinical parameters, including tasks on motor function, alertness, and physiological behavior. NSS was scored from 0 to 10. Sham-operated mice have no neurobehavioral deficits, reflected by a score of 0. A maximum NSS of 10 points indicates severe neurological dysfunction, with failure of all tasks. Task performance was evaluated in a blinded fashion with regard to the animal groups.
MRI analysis
MRI was performed in a 7-tesla horizontal Bruker spectrometer (Karlshure, Germany) run by ParaVision 6.0 software as previously described (57–59). Animals were imaged 6-hour after injury at day 0 and at day 3 or day 7. Anesthetized mice (1.5% isoflurane in a gas mixture of 30% oxygen and 70% nitrous oxide) were placed on a MR-compatible stereotaxic device with built-in small animal monitoring and gating system (SA Instruments, Stony Brook, NY, USA), which is engineered to fit a Bruker four-channel phased-array mouse brain coil and a corresponding transmit volume coil. Whole-brain MRI was performed with acquisition of T2-weighted rapid acquisition with relaxation enhancement (RARE) sequences (imaging parameters: repetition time/echo time = 2500/33 ms; flip angle = 90°; RARE factor = 4, number of averages = 2) with a field of view of 20 mm by 20 mm, matrix of 256 mm by 256 mm and 16 transverse slices (0.5 mm thick). The images were stored offline for identifying key neuropathologies such as hematomas, edema, or gross changes in brain structure. The volumes of the brain injury were calculated using a standard Digital Imaging and Communications in Medicine (DICOM) viewing software (RadiAnt DICOM Viewer Version 3.2.3).
The in vitro generation and treatment of BMMs
BM cells were seeded to 12-well plates at a density of 2 × 106 cells per well and cultured in Dulbecco’s modified Eagle’s medium (DMEM) [supplemented with 10% fetal bovine serum, penicillin (100 U/ml), and streptomycin (100 μg/ml)] with M-CSF (20 ng/ml) alone, or M-CSF + IL-6 (20 ng/ml), or M-CSF + IL-6 + TGF-β1 (20 ng/ml) for 7 days to generate BMMs. On day 7, BMMs were harvested to measure mRNA levels of the tissue repair marker genes by quantitative RT-PCR (qRT-PCR). Alternatively, the microglia cell line BV2 or BMMs generated with M-CSF were respectively stimulated with IL-4 (20 ng/ml) and IL-13 (20 ng/ml) for 24 hours, followed by examining mRNA levels of the tissue repair marker genes by qRT- PCR.
Apoptotic Jurkat T cells (apoJ) were prepared after exposed to ultraviolet irradiation at 254 nm for 10 min and then placed in RPMI 1640 with 10% fetal calf serum (Biosera Products) for 3 hours at 37°C in 5% CO2 (29). BMMs were washed by phosphate-buffered saline (PBS) three times and then cultured in serum-free DMEM for 18 hours with apoJ at a ratio of three per macrophage. TGF-β1 concentrations in the supernatants were measured by ELISA.
Generation of BM chimeras mice
[CD45.1 > CD45.2] and [Rosa26-mTmG > wt] BM chimeras were generated by reconstitution of lethal-irradiated gender-matched receipt mice (8 to10 weeks old) (950 rad) with BM cells from CD45.1 or Rosa26-mTmG mice. Eight weeks after BM transplantation, the chimeric mice were subjected to the induction of TBI.
Isolation of microglia/monocytes in vivo from sites of injury
Brain tissues were collected from PBS-perfused mice and dissociated with Adult Brain Dissociation Kit (Miltenyi Biotec, Germany). Mononuclear cells (including monocytes, lymphocytes, but not including red blood cells and neutrophils) were prepared by 37%/70% Percoll centrifugation (GE Healthcare, Princeton, NJ, USA). Microglia and monocytes were positively isolated by coculture with CD11b+ MicroBeads (Miltenyi Biotec, Germany) according to the manufacturer’s guidelines.
Generation of TBI zebrafish models and the in vivo tracing system
The zebrafish study was approved by the Animal Research Advisory Committee of SIBCB, CAS. Zebrafish were maintained at 28.5°C on a 14-hour light/10-hour dark cycle, according to the guidelines of the Institutional Animal Care and Use Committee. For tracing macrophages in vivo, the transgenic zebrafish Tg(mpeg1:mcherry) was cross-bred with the transgenic zebrafish Tg(Fli1:GFP). Fluorescence microscopy was used to screen and obtain progeny that harbored red and GFP fluorescence in our experiment. To avoid pigment formation in larvae, 0.003% (w/v) phenylthiocarbamide (Sigma-Aldrich, USA) was added to the fish water 24 hours after fertilization. Human cytokines like TGF-β, M-CSF, or IL-6 (0.1 ng) were intra-arterially microinjected into 2-day-old larval fish, and 1 day later, TBI was induced via puncture with a 25-μm-thin glass tube. Zebrafish were fixed in a 60% low-melting agarose 3 hours after TBI, and the in vivo imaging was performed under a fluorescence microscopy to trace macrophage migration from the peripheral to the injury site. In addition, zebrafish were euthanized at 48 hours after TBI to obtain brain tissue for RNA extraction.
Immunohistochemical and immunofluorescence staining of brain tissue
Brain tissues were harvested from PBS-perfused mice and fixed in 4% formaldehyde for 24 hours, and serial coronal 10-μm-thick paraffin-embedded sections were prepared. Brain sections were stained with H&E or TUNEL according to the manufacturer’s protocol for ApoAlert DNA Fragmentation Assay Kit (Clontech). For cryosection, paraformaldehyde-fixed samples were immersed in a 20% sucrose solution at 4°C for 48 hours, followed by embedded in optimal cutting temperature compound for frozen at −20°C. Ten-micrometer-thick cryosections were cut and stained with Alexa Fluor 488–anti-murine F4/80 (Abcam). Images were captured with an Olympus BX51 microscope.
Antibodies, ELISA, and flow cytometry analysis
TGF-β1, M-CSF, and IL-6 cytokines were purchased from PeproTech. Antibodies for flow cytometry were purchased from BD (anti-human CD16), from BioLegend (anti-human LAP/latency-associated peptide TGF-β1), and from eBioscience (anti-human CD4, CD8, CD19, CD11b, and CD14, and anti-mouse CD11b, Ly6C, F4/80, CD45, CD206, CD31, and F4/80). Antibodies for the blocking experiment were purchased from BD (purified rat anti-mouse, human TGF-β1, purified NA/LE rat anti-mouse M-CSF), and from eBioscience (anti-mouse IL-6 functional grade purified). For staining surface markers, cells were suspended in PBS solution supplemented with 1% heat-inactivated fetal calf serum. For intracellular staining, cells were fixed and permeabilized with the Intracellular Staining Kit (eBioscience). Data were acquired on a C6 flow cytometer (BD) and analyzed with FlowJo software. Concentrations of M-CSF, TGF-β1, IL-6, TNF-α, G-CSF, and GM-CSF from TBI patients or mice were detected by ELISA kits from eBioscience or XITANG.
RNA-seq and data processing
RNA was prepared from BMMs after the in vitro treatment with the indicated cytokines. Alternatively, macrophages or microglia were isolated from the in vivo sites of injury of the control TBI mice or the M6T-treated mice. According to the Geo-seq methods (60), RNA-seq library was constructed and sequenced on an Illumina HiSeq 2500 instrument using a 125-base pair paired-end reads setting. Gene expression levels were calculated by Tophat and Cufflinks (61). The differentially regulated genes were identified and selected to generate a heatmap (z score). GO and KEGG pathway enrichment analysis was performed using DAVID (62).
Quantitative real-time PCR
The relative mRNA expression levels of TGF-β1, M-CSF, G-CSF, GM-CSF, IL-6, TNF-α, IL-10, and IL-4 were measured by qRT-PCR. Total RNA was extracted from cells or tissues with TRIzol reagent, and complementary DNA was generated using a Reverse Transcriptase M-MLV kit (Takara). Relative qRT-PCR was performed on a CFX-96 machine (Bio-Rad) with SYBR Green Master Mix (DBI Bioscience).
Mass cytometry/CyTOF assay (antibodies were summarized in table S7)
Immune cells were isolated from the sites of injury in the TBI mice and prepared for cell-staining protocols by Fluidigm. Briefly, cells were stained with 0.5 μM cell-ID cisplatin (Fluidigm, catalog no. 201064) for 2 min, followed by adding 2 ml of MaxPar Cell Staining Buffer (Fluidigm, catalog no. 201068) to stop the reaction. After centrifugation and disposal of the supernatant, the cells were resuspended in MaxPar Cell Staining Buffer to a volume of 50 μl. Fifty microliters of a metal-conjugated surface-marker antibody cocktail was added, and the samples were incubated for 30 min at room temperature. For intracellular staining, cells were fixed in 1 ml of 1× MaxPar Fix I Buffer (Fluidigm, catalog no. 201065) at room temperature for 20 min. After washing twice with 2 ml of MaxPar Perm-S Buffer (Fluidigm, catalog no. 201066), the cells were incubated with 50 μl of an intracellular antibody cocktail for 30 min. After washing twice with 2 ml of MaxPar Cell Staining Buffer, the cells were resuspended in 1 ml of the intercalation solution and incubated overnight at 4°C. After washing twice with 2 ml of MaxPar Cell Staining Buffer, the cell concentration was adjusted to 2.5 to 5 × 105/ml with MaxPar Water (Fluidigm, catalog no. 201069) for the CyTOF assay.
Statistics
Results are expressed as means ± SD. All statistical analyses were assessed with Prism6 software (GraphPad software) or STATA software (version 12.0; STATA Corporation, College Station, TX). Two-tailed Student’s t test, nonparametric Mann-Whitney U test, or one-way/two-way analysis of variance (ANOVA) with Tukey’s or Dunnett’s or Sidak’s multiple comparisons test was used for comparison between different groups. Predicted probability, which was calculated by binary logistic regression, was used to combine respectively the concentration of M-CSF and IL-6, M-CSF and TGF-β1, IL-6 and TGF-β1, or M-CSF, IL-6, and TGF-β1 in CSF into one index that used in Spearman’s rank correlation test to assess the association of the index with the outcome of patients with TBI or the percentage of monocytes in patients with TBI at day 0. P value of less than 0.05 was considered statistically significant (*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001).
Study approval
All procedures of animal experiments were conducted in accordance with the institutional guidelines and were approved by the Institutional Animal Care and Use Committee of SIBCB (protocol no. IBCB0057). Studying human blood and CSF samples was approved by the Ethical Committee of Huashan Hospital, Fudan University.
Acknowledgments
We would like to thank the Animal Core Facility, Core Facility of Molecular Biology (CFMB), and Core Facility of Cell Biology in SIBCB for technical supports and help. We also thank X. Wu (Huashan Hospital) and Y. Chen (Fudan University) for research suggestions. Funding: This work was supported by grants from the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB19000000), National Key R&D Program of China (2018YFA0107900), National Natural Science Foundation of China (81930038, 81825011, and 81630043), the Ministry of Science and Technology of China (2018YFA0800702), and the State Key Laboratory of Cell Biology, SIBCB, CAS (SKL CBKF2013003). H.W. is supported by National Science Foundation for Distinguished Young Scholars. Author contributions: The study was designed by H.W., Z.L., X.X., J.X., N.J., B.W., J.H., B.Z., and J.Z. and performed by Z.L., X.X., J.X., W.L., L.Q., J.C., G.C., S.W., Y.Z., Y.Q., S.L., X.Z., Y.L., and J.L. Results were analyzed by Z.L., X.X., J.X., and W.L. B.W. and J.H. coordinated the project. Z.L., X.X., L.Q., and H.W. wrote the paper with input from the first authors. All authors approved the final version of manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/11/eabb6260/DC1
REFERENCES AND NOTES
- 1.Maas A. I. R., Menon D. K., Adelson P. D., Andelic N., Bell M. J., Belli A., Bragge P., Brazinova A., Büki A., Chesnut R. M., Citerio G., Coburn M., Cooper D. J., Crowder A. T., Czeiter E., Czosnyka M., Diaz-Arrastia R., Dreier J. P., Duhaime A. C., Ercole A., van Essen T. A., Feigin V. L., Gao G., Giacino J., Gonzalez-Lara L. E., Gruen R. L., Gupta D., Hartings J. A., Hill S., Jiang J. Y., Ketharanathan N., Kompanje E. J. O., Lanyon L., Laureys S., Lecky F., Levin H., Lingsma H. F., Maegele M., Majdan M., Manley G., Marsteller J., Mascia L., McFadyen C., Mondello S., Newcombe V., Palotie A., Parizel P. M., Peul W., Piercy J., Polinder S., Puybasset L., Rasmussen T. E., Rossaint R., Smielewski P., Soderberg J., Stanworth S. J., Stein M. B., von Steinbüchel N., Stewart W., Steyerberg E. W., Stocchetti N., Synnot A., Te Ao B., Tenovuo O., Theadom A., Tibboel D., Videtta W., Wang K. K. W., Williams W. H., Wilson L., Yaffe K.; InTBIR Participants and Investigators , Traumatic brain injury: Integrated approaches to improve prevention, clinical care, and research. Lancet Neurol. 16, 987–1048 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Jassam Y. N., Izzy S., Whalen M., McGavern D. B., El Khoury J., Neuroimmunology of traumatic brain injury: Time for a paradigm shift. Neuron 95, 1246–1265 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shechter R., London A., Varol C., Raposo C., Cusimano M., Yovel G., Rolls A., Mack M., Pluchino S., Martino G., Jung S., Schwartz M., Infiltrating blood-derived macrophages are vital cells playing an anti-inflammatory role in recovery from spinal cord injury in mice. PLOS Med. 6, e1000113 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Loke P., Nair M. G., Parkinson J., Guiliano D., Blaxter M., Allen J. E., IL-4 dependent alternatively-activated macrophages have a distinctive in vivo gene expression phenotype. BMC Immunol. 3, 7 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gordon S., Martinez F. O., Alternative activation of macrophages: Mechanism and functions. Immunity 32, 593–604 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Glod J., Kobiler D., Noel M., Koneru R., Lehrer S., Medina D., Maric D., Fine H. A., Monocytes form a vascular barrier and participate in vessel repair after brain injury. Blood 107, 940–946 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shechter R., Miller O., Yovel G., Rosenzweig N., London A., Ruckh J., Kim K.-W., Klein E., Kalchenko V., Bendel P., Lira S. A., Jung S., Schwartz M., Recruitment of beneficial M2 macrophages to injured spinal cord is orchestrated by remote brain choroid plexus. Immunity 38, 555–569 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fleming J. C., Norenberg M. D., Ramsay D. A., Dekaban G. A., Marcillo A. E., Saenz A. D., Pasquale-Styles M., Dietrich W. D., Weaver L. C., The cellular inflammatory response in human spinal cords after injury. Brain 129, 3249–3269 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Mildner A., Schmidt H., Nitsche M., Merkler D., Hanisch U.-K., Mack M., Heikenwalder M., Brück W., Priller J., Prinz M., Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat. Neurosci. 10, 1544–1553 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Lu J., Marmarou A., Lapane K., Turf E., Wilson L., A method for reducing misclassification in the extended Glasgow Outcome Score. J. Neurotrauma 27, 843–852 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Woiciechowsky C., Schöning B., Cobanov J., Lanksch W. R., Volk H.-D., Docke W.-D., Early IL-6 plasma concentrations correlate with severity of brain injury and pneumonia in brain-injured patients. J. Trauma 52, 339–345 (2002). [DOI] [PubMed] [Google Scholar]
- 12.Xiong Y., Mahmood A., Chopp M., Animal models of traumatic brain injury. Nat. Rev. Neurosci. 14, 128–142 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stahel P. F., Shohami E., Younis F. M., Kariya K., Otto V. I., Lenzlinger P. M., Grosjean M. B., Eugster H.-P., Trentz O., Kossmann T., Morganti-Kossmann M. C., Experimental closed head injury: Analysis of neurological outcome, blood-brain barrier dysfunction, intracranial neutrophil infiltration, and neuronal cell death in mice deficient in genes for pro-inflammatory cytokines. J. Cereb. Blood Flow Metab. 20, 369–380 (2000). [DOI] [PubMed] [Google Scholar]
- 14.Flierl M. A., Stahel P. F., Beauchamp K. M., Morgan S. J., Smith W. R., Shohami E., Mouse closed head injury model induced by a weight-drop device. Nat. Protoc. 4, 1328–1337 (2009). [DOI] [PubMed] [Google Scholar]
- 15.Vachon E., Martin R., Plumb J., Kwok V., Vandivier R. W., Glogauer M., Kapus A., Wang X., Chow C.-W., Grinstein S., Downey G. P., CD44 is a phagocytic receptor. Blood 107, 4149–4158 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Hart S. P., Rossi A. G., Haslett C., Dransfield I., Characterization of the effects of cross-linking of macrophage CD44 associated with increased phagocytosis of apoptotic PMN. PLOS ONE 7, e33142 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Korin B., Ben-Shaanan T. L., Schiller M., Dubovik T., Azulay-Debby H., Boshnak N. T., Koren T., Rolls A., High-dimensional, single-cell characterization of the brain’s immune compartment. Nat. Neurosci. 20, 1300–1309 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Harding C. V., Geuze H. J., Class II MHC molecules are present in macrophage lysosomes and phagolysosomes that function in the phagocytic processing of Listeria monocytogenes for presentation to T cells. J. Cell Biol. 119, 531–542 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cha S.-J., Park K., Srinivasan P., Schindler C. W., van Rooijen N., Stins M., Jacobs-Lorena M., CD68 acts as a major gateway for malaria sporozoite liver infection. J. Exp. Med. 212, 1391–1403 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crouch E. E., Doetsch F., FACS isolation of endothelial cells and pericytes from mouse brain microregions. Nat. Protoc. 13, 738–751 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Rothhammer V., Borucki D. M., Tjon E. C., Takenaka M. C., Chao C.-C., Ardura-Fabregat A., de Lima K. A., Gutiérrez-Vázquez C., Hewson P., Staszewski O., Blain M., Healy L., Neziraj T., Borio M., Wheeler M., Dragin L. L., Laplaud D. A., Antel J., Alvarez J. I., Prinz M., Quintana F. J., Microglial control of astrocytes in response to microbial metabolites. Nature 557, 724–728 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hsieh C. L., Kim C. C., Ryba B. E., Niemi E. C., Bando J. K., Locksley R. M., Liu J., Nakamura M. C., Seaman W. E., Traumatic brain injury induces macrophage subsets in the brain. Eur. J. Immunol. 43, 2010–2022 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jin X., Ishii H., Bai Z., Itokazu T., Yamashita T., Temporal changes in cell marker expression and cellular infiltration in a controlled cortical impact model in adult male C57BL/6 mice. PLOS ONE 7, e41892 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takeuchi A., Miyaishi O., Kiuchi K., Isobe K., Macrophage colony-stimulating factor is expressed in neuron and microglia after focal brain injury. J. Neurosci. Res. 65, 38–44 (2001). [DOI] [PubMed] [Google Scholar]
- 25.Luo J., Ho P. P., Buckwalter M. S., Hsu T., Lee L. Y., Zhang H., Kim D.-K., Kim S.-J., Gambhir S. S., Steinman L., Wyss-Coray T., Glia-dependent TGF-β signaling, acting independently of the TH17 pathway, is critical for initiation of murine autoimmune encephalomyelitis. J. Clin. Invest. 117, 3306–3315 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fadok V. A., Bratton D. L., Konowal A., Freed P. W., Westcott J. Y., Henson P. M., Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J. Clin. Invest. 101, 890–898 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Freire-de-Lima C. G., Xiao Y. Q., Gardai S. J., Bratton D. L., Schiemann W. P., Henson P. M., Apoptotic cells, through transforming growth factor-β, coordinately induce anti-inflammatory and suppress pro-inflammatory eicosanoid and NO synthesis in murine macrophages. J. Biol. Chem. 281, 38376–38384 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Perruche S., Zhang P., Liu Y., Saas P., Bluestone J. A., Chen W., CD3-specific antibody-induced immune tolerance involves transforming growth factor-β from phagocytes digesting apoptotic T cells. Nat. Med. 14, 528–535 (2008). [DOI] [PubMed] [Google Scholar]
- 29.Huynh M.-L., Fadok V. A., Henson P. M., Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-β1 secretion and the resolution of inflammation. J. Clin. Invest. 109, 41–50 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lasch M., Caballero-Martinez A., Troidl K., Schloegl I., Lautz T., Deindl E., Arginase inhibition attenuates arteriogenesis and interferes with M2 macrophage accumulation. Lab. Invest. 96, 830–838 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Yao Q., Liu J., Zhang Z., Li F., Zhang C., Lai B., Xiao L., Wang N., Peroxisome proliferator-activated receptor γ (PPARγ) induces the gene expression of integrin αVβ5 to promote macrophage M2 polarization. J. Biol. Chem. 293, 16572–16582 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Charo I. F., Macrophage polarization and insulin resistance: PPARγ in control. Cell Metab. 6, 96–98 (2007). [DOI] [PubMed] [Google Scholar]
- 33.McCormick S. M., Gowda N., Fang J. X., Heller N. M., Suppressor of cytokine signaling (socs)1 regulates interleukin-4 (IL-4)-activated insulin receptor substrate (IRS)-2 tyrosine phosphorylation in monocytes and macrophages via the proteasome. J. Biol. Chem. 291, 20574–20587 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fu C., Jiang L., Xu X., Zhu F., Zhang S., Wu X., Liu Z., Yang X., Li S., STAT4 knockout protects LPS-induced lung injury by increasing of MDSC and promoting of macrophage differentiation. Respir. Physiol. Neurobiol. 223, 16–22 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Ruffell D., Mourkioti F., Gambardella A., Kirstetter P., Lopez R. G., Rosenthal N., Nerlov C., A CREB-C/EBPβ cascade induces M2 macrophage-specific gene expression and promotes muscle injury repair. Proc. Natl. Acad. Sci. U.S.A. 106, 17475–17480 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang O., Zhang J., Atorvastatin promotes human monocyte differentiation toward alternative M2 macrophages through p38 mitogen-activated protein kinase-dependent peroxisome proliferator-activated receptor γ activation. Int. Immunopharmacol. 26, 58–64 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Jiang X., Zhou T., Xiao Y., Yu J., Dou S., Chen G., Wang R., Xiao H., Hou C., Wang W., Shi Q., Feng J., Ma Y., Shen B., Li Y., Han G., Tim-3 promotes tumor-promoting M2 macrophage polarization by binding to STAT1 and suppressing the STAT1-miR-155 signaling axis. Oncoimmunology 5, e1211219 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li B., Tan T.-B., Wang L., Zhao X.-Y., Tan G.-J., p38MAPK/SGK1 signaling regulates macrophage polarization in experimental autoimmune encephalomyelitis. Aging (Albany NY) 11, 898–907 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiménez-Garcia L., Herránz S., Luque A., Hortelano S., Critical role of p38 MAPK in IL-4-induced alternative activation of peritoneal macrophages. Eur. J. Immunol. 45, 273–286 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Bune A. J., Hayman A. R., Evans M. J., Cox T. M., Mice lacking tartrate-resistant acid phosphatase (Acp 5) have disordered macrophage inflammatory responses and reduced clearance of the pathogen, Staphylococcus aureus. Immunology 102, 103–113 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anderson J., Sandhir R., Hamilton E. S., Berman N. E. J., Impaired expression of neuroprotective molecules in the HIF-1α pathway following traumatic brain injury in aged mice. J. Neurotrauma 26, 1557–1566 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hickman S. E., Kingery N. D., Ohsumi T. K., Borowsky M. L., Wang L.-c., Means T. K., El Khoury J., The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 16, 1896–1905 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Butovsky O., Jedrychowski M. P., Moore C. S., Cialic R., Lanser A. J., Gabriely G., Koeglsperger T., Dake B., Wu P. M., Doykan C. E., Fanek Z., Liu L., Chen Z., Rothstein J. D., Ransohoff R. M., Gygi S. P., Antel J. P., Weiner H. L., Identification of a unique TGF-β–dependent molecular and functional signature in microglia. Nat. Neurosci. 17, 131–143 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boche D., Cunningham C., Gauldie J., Perry V. H., Transforming growth factor-β1-mediated neuroprotection against excitotoxic injury in vivo. J. Cereb. Blood Flow Metab. 23, 1174–1182 (2003). [DOI] [PubMed] [Google Scholar]
- 45.Ma M., Ma Y., Yi X., Guo R., Zhu W., Fan X., Xu G., Frey W. H. II, Liu X., Intranasal delivery of transforming growth factor-beta1 in mice after stroke reduces infarct volume and increases neurogenesis in the subventricular zone. BMC Neurosci. 9, 117 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bronchard R., Albaladejo P., Brezac G., Geffroy A., Seince P.-F., Morris W., Branger C., Marty J., Early onset pneumonia: Risk factors and consequences in head trauma patients. Anesthesiology 100, 234–239 (2004). [DOI] [PubMed] [Google Scholar]
- 47.Ewig S., Torres A., El-Ebiary M., Fàbregas N., Hernández C., González J., Nicolás J. M., Soto L., Bacterial colonization patterns in mechanically ventilated patients with traumatic and medical head injury. Incidence, risk factors, and association with ventilator-associated pneumonia. Am. J. Respir. Crit. Care Med. 159, 188–198 (1999). [DOI] [PubMed] [Google Scholar]
- 48.Fàbregas N., Torres A., Pulmonary infection in the brain injured patient. Minerva Anestesiol. 68, 285–290 (2002). [PubMed] [Google Scholar]
- 49.Ishikawa K., Tanaka H., Shiozaki T., Takaoka M., Ogura H., Kishi M., Shimazu T., Sugimoto H., Characteristics of infection and leukocyte count in severely head-injured patients treated with mild hypothermia. J. Trauma 49, 912–922 (2000). [DOI] [PubMed] [Google Scholar]
- 50.Martinez F. O., Gordon S., The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 6, 13 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hamilton J. A., Colony-stimulating factors in inflammation and autoimmunity. Nat. Rev. Immunol. 8, 533–544 (2008). [DOI] [PubMed] [Google Scholar]
- 52.Wang Y., Szretter K. J., Vermi W., Gilfillan S., Rossini C., Cella M., Barrow A. D., Diamond M. S., Colonna M., IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat. Immunol. 13, 753–760 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kumar A., Alvarez-Croda D.-M., Stoica B. A., Faden A. I., Loane D. J., Microglial/macrophage polarization dynamics following traumatic brain injury. J. Neurotrauma 33, 1732–1750 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chomarat P., Banchereau J., Davoust J., Palucka A. K., IL-6 switches the differentiation of monocytes from dendritic cells to macrophages. Nat. Immunol. 1, 510–514 (2000). [DOI] [PubMed] [Google Scholar]
- 55.Willis E. F., MacDonald K. P. A., Nguyen Q. H., Garrido A. L., Gillespie E. R., Harley S. B. R., Bartlett P. F., Schroder W. A., Yates A. G., Anthony D. C., Rose-John S., Ruitenberg M. J., Vukovic J., Repopulating microglia promote brain repair in an IL-6-dependent manner. Cell 180, 833–846.e16 (2020). [DOI] [PubMed] [Google Scholar]
- 56.King J. T. Jr., Carlier P. M., Marion D. W., Early Glasgow Outcome Scale scores predict long-term functional outcome in patients with severe traumatic brain injury. J. Neurotrauma 22, 947–954 (2005). [DOI] [PubMed] [Google Scholar]
- 57.Rodriguez O., Schaefer M. L., Wester B., Lee Y.-C., Boggs N., Conner H. A., Merkle A. C., Fricke S. T., Albanese C., Koliatsos V. E., Manganese-enhanced magnetic resonance imaging as a diagnostic and dispositional tool after mild-moderate blast traumatic brain injury. J. Neurotrauma 33, 662–671 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fricke S. T., Rodriguez O., Vanmeter J., Dettin L. E., Casimiro M., Chien C. D., Newell T., Johnson K., Ileva L., Ojeifo J., Johnson M. D., Albanese C., In vivo magnetic resonance volumetric and spectroscopic analysis of mouse prostate cancer models. Prostate 66, 708–717 (2006). [DOI] [PubMed] [Google Scholar]
- 59.Tian Y., Wang S. S., Zhang Z., Rodriguez O. C., Petricoin E. III, Shih I.-M., Chan D., Avantaggiati M., Yu G., Ye S., Clarke R., Wang C., Zhang B., Wang Y., Albanese C., Integration of network biology and imaging to study cancer phenotypes and responses. IEEE/ACM Trans. Comput. Biol. Bioinform. 11, 1009–1019 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen J., Suo S., Tam P. P. L., Han J.-D. J., Peng G., Jing N., Spatial transcriptomic analysis of cryosectioned tissue samples with Geo-seq. Nat. Protoc. 12, 566–580 (2017). [DOI] [PubMed] [Google Scholar]
- 61.Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D. R., Pimentel H., Salzberg S. L., Rinn J. L., Pachter L., Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huang D. W., Sherman B. T., Lempicki R. A., Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/11/eabb6260/DC1