

Abstract
Objective
B cells have emerged as a therapeutic target for MS. Anti-CD20 antibodies, which deplete B cells, are effective therapies for MS. However, atacicept (TACI-Fc), which blocks BAFF and APRIL and reduces B cells, unexpectedly exacerbates MS. We tested the hypothesis that B cell maturation antigen (BCMA), a receptor for BAFF and APRIL, plays a role in the paradoxical effects of anti-CD20 antibody and TACI-Fc using experimental autoimmune encephalomyelitis (EAE).
Methods
EAE was induced in wild-type (BCMA+/+) and BCMA-deficient (BCMA−/−) mice with an immunization of rodent myelin oligodendrocyte glycoprotein (MOG)35–55 peptide. Treatment with anti-CD20 antibody, TACI-Fc, and isotype controls was administered by intraperitoneal injections. CNS infiltration was evaluated by histology; immune cell phenotypes were evaluated by flow cytometry; MOG-specific antibodies were determined by ELISA. Mixed bone marrow chimeras and cell culture assays were used to identify the specific subsets of immune cells affected by BCMA deficiency.
Results
First, we found that BCMA−/− mice had more severe EAE compared with BCMA+/+ mice and the increased disease was associated with elevated anti-MOG B-cell responses. Second, we found that anti-CD20 therapy attenuated EAE in BCMA−/− mice but not in BCMA+/+ mice. Third, TACI-Fc attenuated EAE in BCMA+/+ mice but not in BCMA−/− mice. Mixed bone marrow chimeric and cell culture experiments demonstrated that BCMA deficiency elevates inflammatory B-cell responses but inhibits inflammatory responses in macrophages.
Conclusions
BCMA has multifaceted roles during inflammation that affects therapeutic efficacies of anti-CD20 and TACI-Fc in EAE. Our results from BCMA-deficient mice provide insights into the failure of atacicept in MS.
MS is a chronic disease of the CNS that is characterized by inflammation, demyelination, and neuronal damage.1 T cells, B cells, and myeloid cells mediate inflammation in MS. The importance of B cells in driving MS pathology was demonstrated with the successful clinical trials of B cell depletion with the anti-CD20 therapies, rituximab and ocrelizumab.2,3 However, atacicept (TACI-Fc), a recombinant soluble receptor that reduces B cell numbers by blocking both B cell activating factor (BAFF) and a proliferation-inducing ligand (APRIL), was reported to exacerbate disease activity in patients with MS.4 These opposing effects of anti-CD20 and TACI-Fc therapies suggest that B cells have both inflammatory and anti-inflammatory effects in MS; however, the mechanism that drives these disparate results is currently unclear.
BAFF and APRIL are 2 cytokines that play fundamental roles in the development, differentiation, and function of B cells.5–8 BAFF and APRIL signal through the receptors BAFF receptor (BAFFR), transmembrane activator and calcium-modulating cyclophilin ligand interactor (TACI), and B cell maturation antigen (BCMA).9–14 In experimental autoimmune encephalomyelitis (EAE) induced with myelin oligodendrocyte glycoprotein (MOG)35–55 peptide in mice, deficiencies in BAFFR, which cause a developmental blockade early in B cell development, elevate disease severity.12,15,16 This supports the theory that newly developed/immature B cells have regulatory properties in this model of EAE. However, BAFF and APRIL also have effects on B cells at later stages of maturation and on non–B cells, which may also affect EAE.17–19
Unlike BAFFR deficiency, BCMA-deficient mice have no overt defects in the development and homeostasis of B cell populations, although it has been reported that there are effects on antigen presentation and the maintenance of long-lived plasma cells.20,21 Previous studies have identified that BCMA has regulatory properties on inflammation in mouse models of lupus, where BCMA deficiency exacerbates lupus-like disease activity in mice.22 Currently, the function of BCMA in EAE is unclear. Furthermore, it is unknown whether BCMA influences the efficacy of therapies that target B cells. In this present study, we used BCMA−/− mice to explore the function of BCMA in EAE and test whether BCMA deficiency alters the efficacy of the anti-CD20 antibody and TACI-Fc in this disease model.
Methods
Mice
Dr. Loren D. Erickson (University of Virginia) provided BCMA−/− mice. These BCMA−/− were developed on C57BL/6J background by backcrossing for 12 generations, and then, the mice were genotyped using a panel of polymorphic microsatellite markers distributed across the entire genome to confirm B6 genetic background of BCMA−/− mice.20,22 C57BL/6J mice were used as wild-type (BCMA+/+) controls in all experiments. All mice were cohoused in a specific pathogen-free animal facility at Oklahoma Medical Research Foundation. All animal procedures were conducted in strict compliance with the guidelines and approved by the Institutional Animal Care and Use Committee.
Induction and Assessment of EAE
EAE was induced in 8–12-week-old female BCMA−/− mice along with age- and sex-matched BCMA+/+ control mice. Mice were immunized with 150 μg MOG35–55 (Genemed Synthesis Inc., San Antonio, TX) emulsified in complete Freund adjuvant (1.5 mg/mL heat-killed Mycobacterium tuberculosis), followed by an IP injection of 250 ng Bordetella pertussis toxin (List Biological Labs, Inc., Campbell, CA) in phosphate-buffered saline (PBS) at the time of and 2 days after immunization. Clinical signs of EAE were assessed daily with 0–5 scoring range: (1) loss of tail tone, (2) incomplete hind limb paralysis, (3) complete hind limb paralysis, (4) forelimb paralysis, and (5) moribund/dead.
Histology
Mice were perfused with 4% paraformaldehyde (PFA). Spinal cords were dissected from EAE mice, and the tissue was fixed overnight in 4% PFA and then in 20% sucrose and thereafter embedded in a single paraffin block. Five-micrometer-thick tissue sections were stained with hematoxylin and eosin and Luxol fast blue and imaged using a Nikon Eclipse E800M microscope.
Flow Cytometry and Intracellular Staining
Infiltrating cells were isolated from the brain and spinal cord of PBS-perfused mice. For this, CNS homogenates were incubated with collagenase and DNAse for 45 minutes at 37°C, purified by a Percoll gradient, and washed with PBS. Single-cell suspensions were also made from spleen and draining lymph nodes after lysing red blood cells by ACK buffer. Cells were stained with the following reagents in various combinations: CD19 FITC, CD11b-PerCP-Cy5.5, immunoglobulin (Ig) D-PE, Ly-6G-Alexa Fluor 647, AA4.1- PerCP-Cy5.5, CD5-PECy7, CD38-FITC, CD19-PerCP-Cy5.5, CD138-APC, CD138-PE, Ly-6C-FITC, Ly-6G-BV711, CD45.1-BV711, CD45.2-BV711, interleukin (IL)-6-APC, GM-CSF-FITC, Gr1-PE, major histocompatibility complex class II (MHCII)-PECy7, CD4-Pacific blue, B220-APC-efluor780, GL7-FITC, CD1d-APC, IgM-Pacific blue, CD21/35-APC-efluor780, B220-PECy7, Streptavidin-FITC, CD267-APC, GL7-Pacific blue, interferon (IFN)-γ-Alexa488, IL-17A-PE, IL-10-PerCP-Cy5.5, CD4-PECy7, CD11b-PE, and Viability dye-efluor450, CD23-BV711 and CD95-BV711 and Peanut agglutinin-biotin (BioLegend, San Diego, CA; eBioscience, San Diego, CA; BD Biosciences, San Jose, CA; Vector Laboratories Inc., Burlingame, CA). For intracellular staining, cells were stimulated for 5 hours with phorbol 12-myristate 13-acetate (50 ng/mL; Sigma, St. Louis, MO), ionomycin (500 ng/mL; Sigma), and golgi stop (BD Biosciences) containing monensin in complete RPMI medium at 37°C under a 5% CO2 atmosphere, stained, fixed, and permeabilized using buffers (BD Biosciences) and analyzed in flow cytometer. All cells were passed through LSRII flow cytometer, and data were analyzed using FlowJo software (Tree Star Inc., Ashland, OR).
In Vitro MOG35–55 Recall Response
For MOG35–55 recall response, spleens collected on day 10 from BCMA+/+ and BCMA−/− EAE mice were processed into single-cell suspensions and cultured at 2.5 × 106 cells/mL in complete RPMI 1640 with increasing MOG35–55 peptide concentration of 0, 0.1, 1, and 10 μg/mL for 72 hours. IL-6, IL-10, IL-17, IFN-γ, IL-1β, and GM-CSF cytokines from the culture supernatants were assessed by respective ELISA kits (eBiosciences). IL-6 in B cells, T helper cells, and myeloid cells were assessed by intracellular flow cytometry.
Serum and Anti-MOG Ab Detection
Mouse IgG total was measured in plasma with the mouse IgG total ELISA kit (eBioscience). Levels of anti-MOG Abs in mouse plasma were performed using indirect ELISAs. In brief, ELISA plates were coated with 10 μg/mL MOG35–55 peptide. Plates were probed with 1/400 dilutions of serum from individual mice, and reactive Abs were detected using peroxidase-conjugated goat and anti-mouse specific for IgG (Southern Biotech, Birmingham, AL) and developed with tetramethylbenzidine.
Anti-CD20 and TACI-Fc Treatment
The antibody against CD20 along with its isotype control was purchased from BioLegend. TACI-Fc was purchased from BioLegend and Human Fc-G1 from BioXcell. EAE was induced in BCMA+/+ and BCMA−/− mice and treated with either anti-CD20, TACI-Fc, or controls with 2 doses of 250 μg per mice on day 5 and day 10. An isotype control antibody or human Fc-G1 were used as treatment controls for anti-CD20 and TACI-Fc, respectively. Mice were monitored daily for clinical scores, and analysis was performed at the peak of disease (days 16–18).
Flow Cytometry Sorting and Quantitative Real-Time PCR
T cells, B cells, macrophages, and neutrophils were sorted from total splenic cells of EAE wild-type mice using BD FACS Aria cell sorting system (BD Biosciences). For quantitative PCR analysis, RNA was isolated using the RNeasy Micro Kit (Qiagen, Hilden, Germany), and cDNA was generated using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA). To detect BCMA transcript, following primers were used: forward 5′ TGATCCAGTCCCTCATGG 3′ and reverse 5′ GAACTGGTCACGCTTGG 3′. As a housekeeping gene transcript, glyceraldehyde 3-phosphate dehydrogenase was used with the following primer sequences: forward 5′ CTCCCACTCTTCCACCTTCG 3′ and reverse 5′ CCACCACCCTGTTGCTGTAG 3′. Gene expression was assessed using MicroAmp Optical 96-well reaction plates and a 7900HT Fast Real-Time PCR System (Applied Biosystems). Data were analyzed using SDSv2.3 software (Applied Biosystems).
Mixed Bone Marrow Chimeras
Lethally irradiated CD45.1+ BCMA+/+ mice were rescued with mixed 5 × 106 bone marrow cells (50% CD45.1 BCMA+/+ and 50% CD45.2 BCMA−/−). After 8 weeks of reconstitution, EAE was induced, and mice were killed at the peak of disease (day 16 or 17) for analysis.
In Vitro Cell Stimulation With BAFF and APRIL
Whole spleens from healthy mice were homogenized into single-cell suspensions, and CD11b+ cells and B cells were isolated by positive and negative selection, respectively (Miltenyi Biotec Inc., San Diego, CA). B cells were further sorted into discrete subsets using a BD FACS Aria cell sorting system (BD Biosciences).
Purified B cell subsets were stimulated with anti-CD40 antibody (1 μg/mL) + CpG (0.02 μM/mL) at a cell concentration of 2.5 × 106 cells/mL along with BAFF (25 ng/mL) or APRIL (50 ng/mL) in complete RPMI medium at 37°C under a 5% CO2 for 72 hours. CD11b+ myeloid cells were stimulated with lipopolysaccharides (100 ng/mL) at a cell concentration of 2.5 × 106 cells/mL along with BAFF (25 ng/mL) or APRIL (50 ng/mL) in complete RPMI medium at 37°C under a 5% CO2 for 72 hours. ELISAs detected levels of IL-6, IL-10, GM-CSF, and IL-12p40 in the cell culture supernatants (eBiosciences).
Statistical Analysis
Statistics were determined using Prism software v6.0 (GraphPad Software, La Jolla, CA). Data are presented as mean ± SEM, and statistical significance was determined using a 2-tailed Mann-Whitney test, Student t test, or analysis of variance when more than 2 groups were analyzed. For all data sets, differences were considered statistically significant for p < or = 0.05.
Data Availability
Engineered mouse strains described in this article can be made available through a material transfer agreement. TACI-Fc and anti-CD20 antibodies are available for purchase from BioLegend. All data associated with this study are present in the article or in the supplementary materials.
Results
BCMA-Deficient Mice Have Exacerbated EAE
To evaluate the role of BCMA in EAE, we induced disease with MOG35–55 in BCMA-deficient (BCMA−/−) mice and BCMA-sufficient (BCMA+/+) control mice. BCMA−/− mice showed increased EAE severity in comparison to BCMA+/+ mice (figure 1A). At day 15 postinduction of EAE, histologic analysis of spinal cords revealed increased cellular infiltration and increased demyelination in the BCMA−/− mice compared with BCMA+/+ mice (figure 1B and figure e-1, links.lww.com/NXI/A434). We specifically found increased numbers of GM-CSF+ T helper cells (figure 1C), IL-17+ T helper cells (figure e-2A, links.lww.com/NXI/A435), inflammatory macrophages (figure 1D), germinal center (GC) B cells (figure 1E), and plasma cell (figure 1F) infiltrating the spinal cords of the BCMA−/− mice compared with BCMA+/+ mice. Brains from these mice had similar results. The BCMA−/− mice had increased infiltration of IL-17+ T helper cells, GM-CSF+ T helper cells, inflammatory macrophages, and plasma cells compared with the BCMA+/+ mice (figure e-2, B–I). We next compared the MOG-specific cytokine production from BCMA−/− and BCMA+/+ mice from the spleen cells 10 days after induction of EAE. We found that spleen cells from BCMA−/− mice secreted significantly higher levels of the inflammatory cytokines, GM-CSF (figure e-3A, links.lww.com/NXI/A436), IL-6 (figure e-3B), IFN-γ (figure e-3C), and IL-1β (figure e-3D) in response to increasing concentration of MOG35–55 compared with spleens from BCMA+/+ mice. Surprisingly, there were no differences in secretion of the inflammatory IL-17A (figure e-3E). We also found no differences in the secretion of the anti-inflammatory cytokine IL-10 (figure e-3F). Using intracellular flow cytometry, we identified that B cells and T cells but not macrophages were contributing to the elevated IL-6 in the BCMA−/− cultures (figure e-3, G–I).
Figure 1. BCMA Deficiency Exacerbated EAE Disease Severity.

(A) EAE was induced in BCMA−/− mice and BCMA+/+ mice and scored daily for disease severity. Data are pooled from 3 experiments (n = 15–18/group). Nonparametric Mann-Whitney tests were used to determine statistical significance (*p < 0.05 and **p < 0.01). (B) Representative spinal cord sections from BCMA+/+ and BCMA−/− mice harvested at EAE day 15. Sections were stained with hematoxylin and eosin (H&E) and Luxol fast blue, and arrows indicate lesions. Dark purple color indicates infiltrating cells, and blue color indicates the myelin. The presence of (C) GM-CSF+ CD4 T cells, (D) MHCII+GR1+ inflammatory macrophages, and (E) GL7+IgD− germinal center (GC) B cells and (F) CD138+CD38+ plasma cells in the spinal cords of BCMA+/+ and BCMA−/− mice (EAE day 15) was measured by flow cytometry. Data are pooled from 2 experiments (n = 5–10/group). Error bars represent SEM, and Student t tests were used to determine statistical significance (*p < 0.05 and **p < 0.01). BCMA = B cell maturation antigen; EAE = experimental autoimmune encephalomyelitis; Ig = immunoglobulin; MHCII = major histocompatibility complex class II.
BCMA-Deficient Mice Have Increased B Cell Responses During EAE
Previous studies demonstrated that BCMA deficiency alters B cell responses in experimental lupus models.22 Therefore, we assessed whether humoral response to MOG is altered in the BCMA−/− mice compared with BCMA+/+ mice during EAE. We found that by day 15 postinduction of EAE, BCMA−/− mice had significantly higher amounts of anti-MOG IgG in sera compared with BCMA+/+ mice (figure 2A). Total-IgG in the sera was not significantly different in BCMA+/+ and BCMA−/− mice at any time throughout disease (figure 2B).
Figure 2. BCMA Deficiency Increases B Cell Responses Against MOG During EAE.

Serum was collected from BCMA+/+ and BCMA−/− mice on EAE days 0, 10, and 15, and (A) MOG IgG and (B) total-IgG were measured by ELISA (n = 5 per group). At EAE day 15, absolute numbers of (C) transitional (Trans) B cells, (D) regulatory B cells (Bregs), (E) class-switched memory (CSM) B cells, (F) germinal center (GC) B cells, and (G) plasma cells/plasmablasts (PC/PB) in the lymph node and spleen cells from BCMA+/+ and BCMA−/− mice were measured by flow cytometry. (H) The ratio of trans/CSM B cells from lymph nodes and spleens from BCMA+/+ and BCMA−/− mice was determined. Data are pooled from 2 experiments (n = 10/group). +/+ represents BCMA+/+, and −/− represents BCMA−/−. Error bars represent SEM, and Student t tests were used to determine statistical significance. p < 0.05 is statistically significant. BCMA = B cell maturation antigen; EAE = experimental autoimmune encephalomyelitis; Ig = immunoglobulin; MOG = myelin oligodendrocyte glycoprotein.
We next compared BCMA+/+ mice and BCMA−/− mice for alterations in the distribution of B cell populations that would be indicative of a strong B cell response during EAE. Specifically, we assessed the absolute numbers of transitional (Trans) B cells, CD5+ regulatory B cells (Bregs), class-switched memory (CSM) B cells, GC B cells subsets, and plasmablasts/plasma cells (figure e-4, links.lww.com/NXI/A437) in both lymph nodes and spleens in healthy mice and in mice with EAE (days 16–18). In healthy mice, we observed no significant differences in any subset of B cells in the spleens or lymph nodes from BCMA+/+ compared with BCMA−/− mice (figure e-5, links.lww.com/NXI/A438). However, in mice with EAE, there were striking differences in B cell subsets. We found that Trans B cells were significantly reduced in both lymph nodes and spleens of BCMA−/− compared with BCMA+/+ mice (figure 2C). CD5+ Breg numbers were also decreased in lymph nodes compared with BCMA+/+ (figure 2D); however, this difference was not observed in spleens. We observed a significant increase of CSM B cells in lymph nodes of BCMA−/− (figure 2E) but not in spleens. We saw no difference in numbers of GC B cells in lymph nodes or spleens (figure 2F). Finally, we found no alterations of plasmablasts or plasma cells in BCMA−/− compared with BCMA+/+ mice (figure 2G). These data strongly suggest that during EAE, BCMA deficiency acts as brake on the maturation and differentiation of B cells during an immune response. This was revealed when we calculated the ratio of Trans B cells/CSM B cells in both BCMA−/− and BCMA+/+, where there is a significant decrease in the ratio of Trans B cells/CSM B cells in BCMA−/− mice compared with the BCMA+/+ mice in both spleens and lymph nodes during EAE (figure 2H).
These data demonstrate that there are altered B cell populations in BCMA−/− mice during EAE. The most striking result is the reduction of Trans B cells with a maintenance or increase in CSM B cells, GC B cells, and plasmablasts/plasma cells. Our data suggest that BCMA acts to maintain the regulatory transitional B cell population and attenuate downstream B cell responses, which leads to reduced autoimmune inflammation.
Anti-CD20 Treatment Is Effective in BCMA-Deficient Mice
Our data show an association with increased B cell activity with increased disease severity in the BCMA−/− mice. To directly determine whether B cells from the BCMA−/− mice have inflammatory function in EAE, we compared the effects anti-CD20 treatment had on EAE in BCMA−/− mice and in BCMA+/+ mice. We treated mice with anti-CD20 or an isotype control on day 5 and day 10 of EAE, a treatment regimen that has been shown to have no clinical effects in wild-type mice.23 As previously reported, anti-CD20 treatment had no significant effect on EAE severity in the BCMA+/+ mice (figure 3A). The anti-CD20 treatment decreased B cells numbers in the spleen and spinal cord of BCMA+/+ mice but had no effect on the infiltration of macrophages or T helper cells into the spinal cord (figure 3B and figure e-6, links.lww.com/NXI/A439). In contrast to BCMA+/+ mice, this treatment strategy significantly reduced disease severity in BCMA−/− mice (figure 3C). The anti-CD20 treatment decreased B cells numbers in the spleen and spinal cord of BCMA−/− mice and decreased the infiltration of macrophages and T helper cells into the spinal cord (figure 3D and figure e-6).
Figure 3. Anti-CD20 Treatment Ameliorates EAE in BCMA−/− Mice but Not in BCMA+/+ Mice.

(A) BCMA+/+ mice were treated with either isotype control or anti-CD20 on day 5 and day 10 postinduction of EAE and scored daily. Arrows indicate the treatment days. Data are pooled from 2 experiments (n = 10/group). (B) At the peak of the disease, numbers of splenic B cells, spinal cord–infiltrating macrophages, and T helper cells from isotype control and anti-CD20 treated BCMA+/+ mice were assessed by flow cytometry (n = 4/group). (C) BCMA−/− mice were treated with either isotype control or anti-CD20 on day 5 and day 10 postinduction of EAE and scored daily. Arrows indicate the treatment days. Data are pooled from 2 experiments (n = 10/group). (D) At the peak of the disease, numbers of B cells in the spleen and numbers of macrophages and T helper cells in the spinal cord from isotype control and anti-CD20–treated BCMA−/− mice were assessed by flow cytometry (n = 4/group). Error bars represent SEM. Nonparametric Mann-Whitney tests were used for EAE scores (*p < 0.05). Student t tests were used for the flow cytometric data. TACI-Fc treatment ameliorates EAE in BCMA+/+ mice but not in BCMA−/− mice. (E) BCMA+/+ mice were treated with Ig control or TACI-Fc on day 5 and day 10 postinduction of EAE and scored daily. Arrows indicate the treatment days. Data are pooled from 2 experiments (n = 10/group). (F) At the peak of the disease, numbers of splenic B cells, spinal cord–infiltrating macrophages, and T helper cells from control and TACI-Fc treated BCMA+/+ mice were assessed by flow cytometry (n = 5/group). (G) BCMA−/− mice were treated with Ig control or TACI-Fc on day 5 and day 10 postinduction of EAE and scored daily. Arrows indicate the treatment days. Data are pooled from 2 experiments. N = 10/group. (H) At the peak of the disease, numbers of B cells in spleen and numbers of macrophages and T helper cells in the spinal cord of control and TACI-Fc–treated BCMA−/− mice were assessed by flow cytometry (n = 5/group). Error bars represent SEM. Nonparametric Mann-Whitney tests were used for EAE scores (*p < 0.05). Student t tests were used for the flow cytometric data. BCMA = B cell maturation antigen; EAE = experimental autoimmune encephalomyelitis.
These data demonstrate that B cells have an inflammatory role during EAE in BCMA−/− mice and have the potential to drive disease by modulating the functions of inflammatory macrophages and T helper cells and facilitating their infiltration into the CNS.
TACI-Fc Treatment Requires BCMA to Ameliorate EAE
BAFF and APRIL are essential for the development and function of B cells and blocking BAFF and APRIL signaling reduces B cell numbers in both human and mice.24–26 However, a large clinical trial demonstrated that pharmacologic blockade of BAFF and APRIL with TACI-Fc increased inflammatory disease activity in patients with MS (IMP28063, ClinicalTrials.gov identifier: NCT00642902). As BCMA is a receptor of BAFF and APRIL, we assessed the effect of TACI-Fc treatment on the severity of EAE in both BCMA+/+ and BCMA−/− mice. Surprisingly, we found that TACI-Fc treatment, dosed at day 5 and day 10, significantly ameliorates EAE in BCMA+/+ mice (figure 3E). This treatment reduced CNS-infiltrating macrophages but not splenic B cell numbers or CNS-infiltrating CD4+ T cells in the BCMA+/+ mice (figure 3F). In contrast, TACI-Fc treatment had no clinical effects in BCMA−/− mice (figure 3G). This treatment had no effects on splenic B cell numbers or CNS-infiltrating CD4+ T cells, but did elevate the CNS-infiltrating macrophages in the BCMA−/− mice (figure 3H).
The reduction in disease in BCMA+/+ mice by TACI-Fc treatment was quite surprising considering that anti-CD20 treatment had no effect on disease in these mice. This led us to speculate that BAFF and APRIL signaling may have inflammatory effects on non–B cells.
BCMA Directly Affects B Cells and Macrophages in EAE
The disparate efficacies of anti-CD20 and TACI-Fc in the BCMA+/+ and BCMA−/− mice suggest that BCMA is expressed by other immune cells apart from B cells during EAE. Therefore, we assessed the expression of BCMA on FACS-sorted B cells, T cells, macrophages, and neutrophils from C57BL/6 mice with EAE. We found that B cells and macrophages showed elevated expression of BCMA compared with CD4+ T cells and neutrophils (figure 4A).
Figure 4. BCMA Directly Affects B Cells and Myeloid Cells During EAE.

(A) B cells, CD4 T cells, macrophages, and neutrophils were FACS sorted from BCMA+/+ mice with EAE, and quantitative real-time PCR was performed for BCMA expression. Data represent 1/ΔCt of BCMA expression on B cells, CD4 T cells, macrophages, and neutrophils (n = 4/group). Analysis of variance was used for statistics (*p < 0.05). (B) Bone marrow chimeric strategy. Lethally irradiated CD45.1 BCMA+/+ mice were transplanted with equal number of bone marrow cells from CD45.1 BCMA+/+ and CD45.2 BCMA−/− (n = 5). Eight weeks after transplantation, EAE was induced. The percentage of B cells, myeloid cells, and T helper cells derived from donor BCMA+/+ and BCMA−/− cells in were assessed in (C) the blood before inducing EAE and in the (D) blood and (E) spleens 20 days after EAE induction. (F) The percentage of class-switched memory (CSM) B cells, germinal center (GC) B cells, plasmablasts (PB), transitional (Trans) B cells, regulatory B cells (Bregs) derived from donor BCMA+/+, and BCMA−/− cells from spleens during EAE. (G) The percentage of B cells, myeloid cells, and T helper cells derived from donor BCMA+/+ and BCMA−/− cells that have infiltrated the spinal cord during EAE. (H) Percentage of inflammatory macrophages and neutrophils derived from donor BCMA+/+ and BCMA−/− cells that have infiltrated the spinal cord of EAE mice. Error bars represent SEM, and Student t tests were used to determine statistical significance. BCMA = B cell maturation antigen; EAE = experimental autoimmune encephalomyelitis.
These data demonstrate that B cells and macrophages express BCMA. Therefore, we sought to determine the cellular responses directly affected by BCMA deficiency during EAE. To do this, we generated mixed bone marrow chimeric mice by rescuing lethally irradiated CD45.1 BCMA+/+ recipient mice by transplanting equal number of bone marrow cells from CD45.1 BCMA+/+ and CD45.2 BCMA−/− (figure 4B). Eight weeks after bone marrow transplantation, we induced EAE in these mice.
Before EAE induction, we collected blood to assess the ratio of BCMA+/+ (CD45.1+) and BCMA−/− (CD45.2+) in B cell, myeloid cell, and T helper cell populations. We found that BCMA−/− comprised a significantly greater proportion of B cells but not CD11b+ myeloid cells or CD4+ T helper cells (figure 4C). In contrast, at the peak of EAE, we found that both B cells and CD11b+ myeloid populations were skewed toward BCMA−/− but not CD4+ T cells in the blood (figure 4D). Similarly, in the spleens, B cells and myeloid cells, but not T cells, were predominantly BCMA−/− (figure 4E).
We next determined the specific splenic B cell subsets affected by BCMA deficiency. We found that CSM B cells, GC B cells, and plasmablasts all had significantly greater proportion of BCMA−/− compared with BCMA+/+ in spleens (figure 4F). Of interest, this competitive advantage was not extended to the Trans B cells and CD5+ Bregs from spleens as their percentage of BCMA−/− and BCMA+/+ was not significantly different (figure 4F).
Similar to peripheral tissues, we also found that B cells and myeloid cells, but not T cells, had an increased ratio of BCMA−/− to BCMA+/+ in the spinal cords of these chimeric mice (figure 4G). In the myeloid cell compartment, we observed that inflammatory macrophages (GR1+MHCII+), but not neutrophils (GR1+MHCII−), were significantly skewed toward BCMA−/− (figure 4H). Taken together, these data demonstrate that BCMA deficiency has intrinsic effects on the B cell and macrophage responses during EAE but has no effect on T helper cell function.
BCMA Regulates Cytokine Production in Discrete B Cell Subsets
We next assessed the expression of BCMA on different B cell subsets, including plasma cells. We sorted 4 different B cell subsets from the spleens of C57BL/6 mice with EAE by FACS cell sorting. Because of the rarity of certain transitional and regulatory population, we used the following broad gating scheme to determine BCMA transcript expression in B cell subsets. We sorted cells in the CD138+CD19+/− gate that contains plasma cells/plasmablasts, the CD19+IgDhi gate that contains mature B cells, the CD19+IgMhiIgDlo gate that contains Trans B cells/Bregs, and the CD19+IgMloIgDlo gate that contains CSM B cells (figure e-7, links.lww.com/NXI/A440). We found that plasma cells/plasmablasts and CSM B cells expressed significantly higher levels of BCMA compared with CD4+ T cells (figure 5A). Trans B cells and mature B cells did not express BCMA transcript at significantly higher levels to CD4+ T cells (figure 5A).
Figure 5. BAFF and APRIL Signals Through BCMA to Have Anti-inflammatory Functions in B Cells.
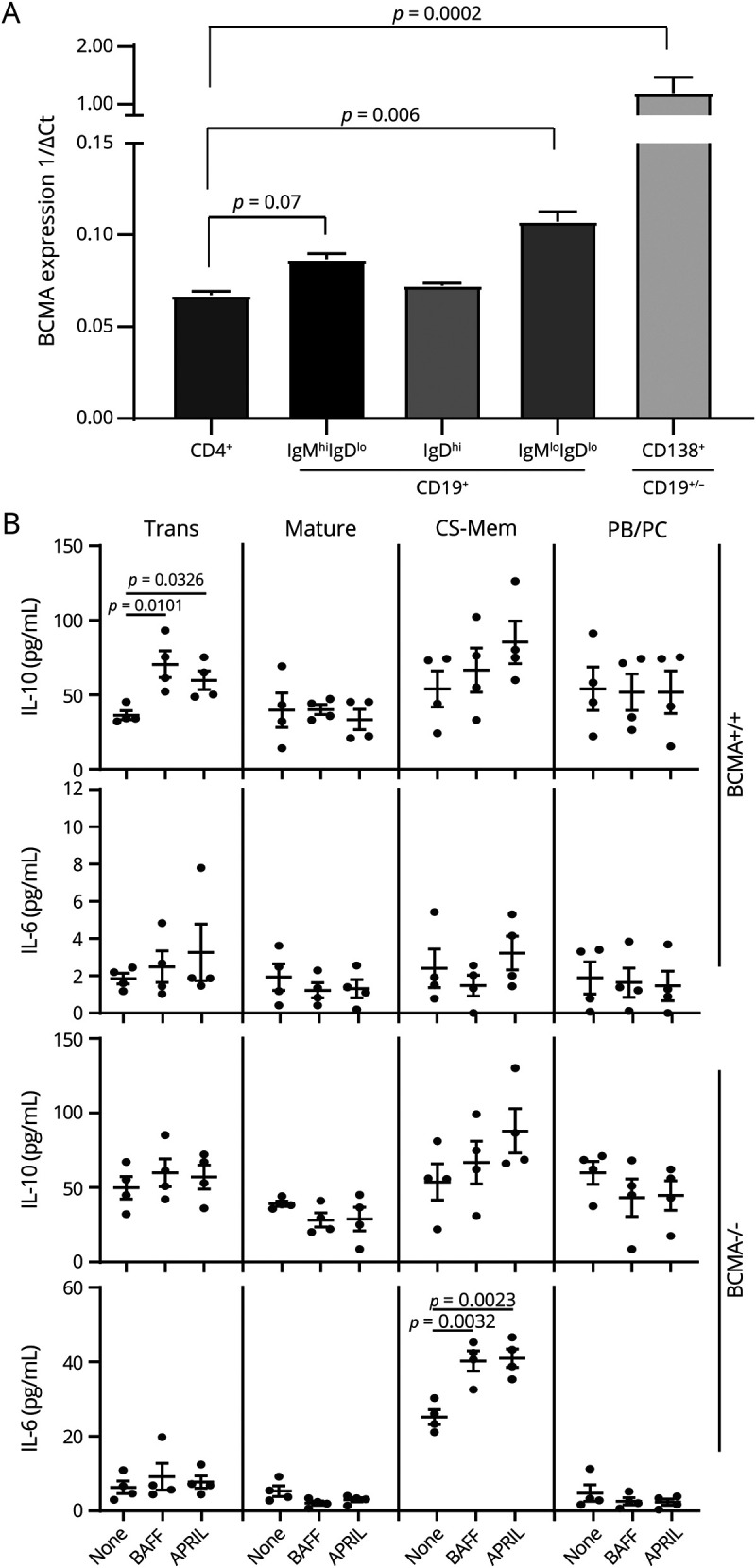
(A) Four different B cell subsets, CD19+IgMhiIgDlo (transitional/regulatory B cells), CD19+IgDhi (mature B cells), CD19+IgMloIgDlo (class-switched memory B cells), CD19+/-CD138+ (plasmablasts/plasma cells), and CD4+ T cells were FACS sorted from BCMA+/+ mice with EAE, and quantitative real-time PCR was performed for BCMA expression. The data represent 1/ΔCt of BCMA expression (n = 4/group). Error bars represent SEM, and analysis of variance was used to determine statistical differences. (B) Trans (transitional/regulatory) B cells, mature B cells, CS-Mem (class-switched memory B cells), and PB/PC (plasmablasts/plasma cells) were FACS sorted from BCMA+/+ and BCMA−/− mice and stimulated with anti-CD40 in the presence or absence of BAFF and APRIL. To get enough numbers of smaller B cell subsets, 5 spleens were pooled together. After 72 hours, cell culture supernatants were collected, and IL-6 and IL-10 secretion was measured by ELISA. Error bars represent SEM, and Student t tests were used to determine statistical significance. p < 0.05 was considered to be statistically significant. APRIL = a proliferation-inducing ligand; BAFF = B cell–activating factor; BCMA = B cell maturation antigen; EAE = experimental autoimmune encephalomyelitis; IL = interleukin.
We next evaluated the effects of in vitro BAFF and APRIL stimulation on these sorted B cell subsets. In cells isolated from BCMA+/+ mice, we found that BAFF or APRIL stimulation elevates IL-10 production by Trans B cells while having no effect on mature B cells, CSM B cells, and plasmablasts/plasma cells (figure 5B). BAFF or APRIL stimulation had no effect on IL-6 production by B cell subsets from BCMA+/+ mice (figure 5B). In BCMA−/− mice, BAFF or APRIL stimulation had no effect on IL-10 production by any B cell subsets but did elevate the production of IL-6 by CSM B cells (figure 5B). We also assessed the secretion of GM-CSF and IL-12p40 on stimulation with BAFF and APRIL and found no differences in BCMA+/+ and BCMA−/− B cells (data not shown).
BCMA Regulates Cytokine Function in Myeloid Cells
The disparate results from the anti-CD20 and TACI-Fc treatment experiment described above provide compelling in vivo evidence that BAFF and/or APRIL stimulation through BCMA is modulating the inflammatory effects of myeloid cells. To understand the cellular mechanisms behind these findings, we designed in vitro assays to compare the effects BAFF and APRIL stimulations have on myeloid cells from BCMA+/+ and BCMA−/− mice. Specifically, we stimulated purified CD11b+ myeloid cells with either BAFF or APRIL and assessed the secretion of the proinflammatory IL-6 and anti-inflammatory IL-10.
We found that CD11b+ cells from BCMA+/+ mice had elevated production of both IL-6 and IL-10 when stimulated with BAFF or APRIL (figure 6A). In BCMA−/− CD11b+ cells, BAFF and APRIL had no significant effect on IL-6 production, but BAFF continued to drive IL-10 production (figure 6B). We also assessed the secretion of IL-12p40 on stimulation with BAFF and APRIL and found no differences in BCMA+/+ and BCMA−/− CD11b+ cells (data not shown).
Figure 6. BAFF and APRIL Signals Through BCMA to Have Proinflammatory Functions in Myeloid Cells.

CD11b+ myeloid cells were MACS sorted from healthy (A) BCMA+/+ and (B) BCMA−/− mice and stimulated with lipopolysaccharides in the presence or absence of BAFF or APRIL (n = 3/group). After 72 hours, supernatants were collected, and IL-6 and IL-10 secretion was measured by ELISA. Error bars represent SEM, and Student t tests were used to determine statistical significance. p < 0.05 was considered to be statistically significant. APRIL = a proliferation-inducing ligand; BAFF = B cell activating factor; BCMA = B cell maturation antigen; EAE = experimental autoimmune encephalomyelitis; IL = interleukin.
Discussion
Until now, no studies have described the function of BCMA deficiency in neuroinflammation. Initially, BCMA was found to be highly expressed by plasma cells and functioned to maintain the survival of antibody producing plasma cells in bone marrow.27 Other studies have shown that GC B cells and memory B cells express BCMA, and this receptor regulates the generation of B cell responses.17,28 In lupus prone mice, BCMA deficiency leads to elevated autoantibody responses and increased morbidity and mortality demonstrating that BCMA regulates autoreactive B cell responses.22 Recent studies suggested that BAFF and APRIL have functional effects on non–B-lineage cell types including myeloid cells, astrocytes, and neurons; yet, the function of BCMA in these non–B cell types is unknown.18
In this study, we have provided evidence for 2 novel mechanisms of BCMA in neuroinflammation. The first mechanism we have identified is that BAFF and APRIL signaling through BCMA has a B cell–intrinsic, anti-inflammatory effect on EAE disease. We find that BCMA−/− mice have exacerbated disease that is associated with increased B cell responses to myelin antigens, which in turn elevates the pathogenicity of the T helper cells within the CNS. In bone marrow chimeras, BCMA deficiency provides a competitive advantage for the development of inflammatory B cell populations (including GC, memory, and plasmablast B cells). During in vitro stimulations, BAFF and APRIL drive the secretion of the anti-inflammatory IL-10 in BCMA+/+ B cells and conversely drive the inflammatory IL-6 in BCMA−/− B cells. Most convincingly, we found that anti-CD20 therapy attenuates EAE in BCMA−/− mice but not BCMA+/+ mice. These data are in accordance with EAE studies from BAFFR-deficient mice. This study shows that deficiency in BAFF signaling exacerbated EAE, which is similar to our results with BCMA-deficient mice.16 Of interest, BAFF−/− and BAFFR−/− mice both have deficiencies in mature B cell development, which is not a defect in BCMA−/− mice.15,20 Therefore, the data from BCMA−/− mice suggest that BAFF and/or APRIL also act downstream of the early developmental checkpoints of B cells and act as a brake on the development and function of inflammatory B cell populations.
The second mechanism is that BCMA has inflammatory effects on myeloid cells during EAE. We detected BCMA gene expression in macrophages. We find that BAFF and APRIL stimulation elevates the secretion of the inflammatory cytokine IL-6 in BCMA+/+ macrophages but not in BCMA−/− macrophages. Finally, we find that TACI-Fc effectively reduces EAE in BCMA+/+ mice but not in BCMA−/− mice and that the reduction of disease in BCMA+/+ mice is correlated with a reduction in infiltrating inflammatory macrophages but not T cells or B cells. These data are in accordance with a recent publication showing that BAFF-deficient mice crossed to B cell–deficient mice have lowered EAE severity.19 Our study provides compelling evidence that the proinflammatory effect of BAFF is specifically through BCMA on macrophages.
The observations from the clinical trials of anti-CD20 and TACI-Fc revealed that there is still a major knowledge gap in the function of BAFF and APRIL in MS and other immune-mediated diseases. Anti-CD20 therapy, which reduces B cell numbers, is an effective therapy for MS.2,29 Blocking BAFF and APRIL also reduces B cell numbers and reduces flares in lupus.30,31 Therefore, it was rational to hypothesize that blocking BAFF and APRIL in MS would be an effective treatment strategy. Paradoxically, TACI-Fc worsened disease activity in patients with MS.4 The similarity between patients with MS and BCMA−/− mice in their therapeutic response to anti-CD20 and TACI-Fc warrants the use of these mice for better assessment of new therapies targeting B cells. Given the multiligand receptor interactions that occur with BAFF and APRIL signaling, it has been difficult to identify mechanisms behind the therapeutic disparities in anti-CD20 and TACI-Fc. Our study provides evidence that BCMA is critical in the therapeutic efficacies of anti-CD20 and TACI-Fc in neuro-autoimmune disorders like MS.
Acknowledgment
The authors thank Dr. Loren Erickson (University of Virginia) for providing the BCMA−/− mice.
Glossary
- APRIL
a proliferation-inducing ligand
- BAFF
B cell activating factor
- BAFFR
BAFF receptor
- BCMA
B cell maturation antigen
- Breg
regulatory B cell
- CSM
class-switched memory
- EAE
experimental autoimmune encephalomyelitis
- GC
germinal center
- IFN
interferon
- Ig
immunoglobulin
- IL
interleukin
- MHCII
major histocompatibility complex class II
- MOG
myelin oligodendrocyte glycoprotein
- PBS
phosphate-buffered saline
- PFA
paraformaldehyde
- TACI
transmembrane activator and calcium-modulating cyclophilin ligand interactor
Appendix. Authors
Study Funding
This study was supported by grants from the NMSS (RG-1602-07722), the NIH (R01AI137047 and R01EY027346), and Merck-KGaA awarded to R.C. Axtell and grants from the NIH (1R01 AI131624-01A1 and R21 AI142186-01), Weill Institute of Neurosciences and the Maisin Foundation awarded to S.S. Zamvil.
Disclosure
R.C. Axtell has consulted Roche, EMD Serono, and Biogen Idec and is on the advisory board for Progentec Diagnostics Inc. S.S. Zamvil is a Deputy Editor of Neurology: Neuroimmunology and Neuroinflammation. The other authors have nothing to disclose. Go to Neurology.org/NN for full disclosures.
References
- 1.Frohman EM, Racke MK, Raine CS. Multiple sclerosis: the plaque and its pathogenesis. N Engl J Med 2006;354:942–955. [DOI] [PubMed] [Google Scholar]
- 2.Hauser SL, Waubant E, Arnold DL, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med 2008;358:676–688. [DOI] [PubMed] [Google Scholar]
- 3.Kappos L, Li D, Calabresi PA, et al. Ocrelizumab in relapsing-remitting multiple sclerosis: a phase 2, randomised, placebo-controlled, multicentre trial. Lancet 2011;378:1779–1787. [DOI] [PubMed] [Google Scholar]
- 4.Kappos L, Hartung HP, Freedman MS, et al. Atacicept in multiple sclerosis (ATAMS): a randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Neurol 2014;13:353–363. [DOI] [PubMed] [Google Scholar]
- 5.Moore PA, Belvedere O, Orr A, et al. BLyS: member of the tumor necrosis factor family and B lymphocyte stimulator. Science 1999;285:260–263. [DOI] [PubMed] [Google Scholar]
- 6.Schneider P, MacKay F, Steiner V, et al. BAFF, a novel ligand of the tumor necrosis factor family, stimulates B cell growth. J Exp Med 1999;189:1747–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stein JV, Lopez-Fraga M, Elustondo FA, et al. APRIL modulates B and T cell immunity. J Clin Invest 2002;109:1587–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mackay F, Schneider P, Rennert P, Browning J. BAFF and APRIL: a tutorial on B cell survival. Annu Rev Immunol 2003;21:231–264. [DOI] [PubMed] [Google Scholar]
- 9.Gross JA, Johnston J, Mudri S, et al. TACI and BCMA are receptors for a TNF homologue implicated in B-cell autoimmune disease. Nature 2000;404:995–999. [DOI] [PubMed] [Google Scholar]
- 10.Marsters SA, Yan M, Pitti RM, Haas PE, Dixit VM, Ashkenazi A. Interaction of the TNF homologues BLyS and APRIL with the TNF receptor homologues BCMA and TACI. Curr Biol 2000;10:785–788. [DOI] [PubMed] [Google Scholar]
- 11.Shu HB, Johnson H. B cell maturation protein is a receptor for the tumor necrosis factor family member TALL-1. Proc Natl Acad Sci USA 2000;97:9156–9161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thompson JS, Bixler SA, Qian F, et al. BAFF-R, a newly identified TNF receptor that specifically interacts with BAFF. Science 2001;293:2108–2111. [DOI] [PubMed] [Google Scholar]
- 13.Thompson JS, Schneider P, Kalled SL, et al. BAFF binds to the tumor necrosis factor receptor-like molecule B cell maturation antigen and is important for maintaining the peripheral B cell population. J Exp Med 2000;192:129–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rennert P, Schneider P, Cachero TG, et al. A soluble form of B cell maturation antigen, a receptor for the tumor necrosis factor family member APRIL, inhibits tumor cell growth. J Exp Med 2000;192:1677–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yan M, Brady JR, Chan B, et al. Identification of a novel receptor for B lymphocyte stimulator that is mutated in a mouse strain with severe B cell deficiency. Curr Biol 2001;11:1547–1552. [DOI] [PubMed] [Google Scholar]
- 16.Kim SS, Richman DP, Zamvil SS, Agius MA. Accelerated central nervous system autoimmunity in BAFF-receptor-deficient mice. J Neurol Sci 2011;306:9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carrillo-Ballesteros FJ, Oregon-Romero E, Franco-Topete RA, et al. B-cell activating factor receptor expression is associated with germinal center B-cell maintenance. Exp Ther Med 2019;17:2053–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krumbholz M, Theil D, Derfuss T, et al. BAFF is produced by astrocytes and up-regulated in multiple sclerosis lesions and primary central nervous system lymphoma. J Exp Med 2005;201:195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stohl W, Banfalvi A. B cell-independent contribution of BAFF to murine autoimmune disease. Clin Immunol 2016;172:111–116. [DOI] [PubMed] [Google Scholar]
- 20.Xu S, Lam KP. B-cell maturation protein, which binds the tumor necrosis factor family members BAFF and APRIL, is dispensable for humoral immune responses. Mol Cell Biol 2001;21:4067–4074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang M, Hase H, Legarda-Addison D, Varughese L, Seed B, Ting AT. B cell maturation antigen, the receptor for a proliferation-inducing ligand and B cell-activating factor of the TNF family, induces antigen presentation in B cells. J Immunol 2005;175:2814–2824. [DOI] [PubMed] [Google Scholar]
- 22.Jiang C, Loo WM, Greenley EJ, Tung KS, Erickson LD. B cell maturation antigen deficiency exacerbates lymphoproliferation and autoimmunity in murine lupus. J Immunol 2011;186:6136–6147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matsushita T, Yanaba K, Bouaziz JD, Fujimoto M, Tedder TF. Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J Clin Invest 2008;118:3420–3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huntington ND, Tomioka R, Clavarino C, et al. A BAFF antagonist suppresses experimental autoimmune encephalomyelitis by targeting cell-mediated and humoral immune responses. Int Immunol 2006;18:1473–1485. [DOI] [PubMed] [Google Scholar]
- 25.Gross JA, Dillon SR, Mudri S, et al. TACI-Ig neutralizes molecules critical for B cell development and autoimmune disease. impaired B cell maturation in mice lacking BLyS. Immunity 2001;15:289–302. [DOI] [PubMed] [Google Scholar]
- 26.Zhang X, Park CS, Yoon SO, et al. BAFF supports human B cell differentiation in the lymphoid follicles through distinct receptors. Int Immunol 2005;17:779–788. [DOI] [PubMed] [Google Scholar]
- 27.O'Connor BP, Raman VS, Erickson LD, et al. BCMA is essential for the survival of long-lived bone marrow plasma cells. J Exp Med 2004;199:91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ng LG, Sutherland AP, Newton R, et al. B cell-activating factor belonging to the TNF family (BAFF)-R is the principal BAFF receptor facilitating BAFF costimulation of circulating T and B cells. J Immunol 2004;173:807–817. [DOI] [PubMed] [Google Scholar]
- 29.Hawker K, O'Connor P, Freedman MS, et al. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double-blind placebo-controlled multicenter trial. Ann Neurol 2009;66:460–471. [DOI] [PubMed] [Google Scholar]
- 30.Dall'Era M, Chakravarty E, Wallace D, et al. Reduced B lymphocyte and immunoglobulin levels after atacicept treatment in patients with systemic lupus erythematosus: results of a multicenter, phase Ib, double-blind, placebo-controlled, dose-escalating trial. Arthritis Rheum 2007;56:4142–4150. [DOI] [PubMed] [Google Scholar]
- 31.Isenberg D, Gordon C, Licu D, Copt S, Rossi CP, Wofsy D. Efficacy and safety of atacicept for prevention of flares in patients with moderate-to-severe systemic lupus erythematosus (SLE): 52-week data (APRIL-SLE randomised trial). Ann Rheum Dis 2015;74:2006–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Engineered mouse strains described in this article can be made available through a material transfer agreement. TACI-Fc and anti-CD20 antibodies are available for purchase from BioLegend. All data associated with this study are present in the article or in the supplementary materials.