

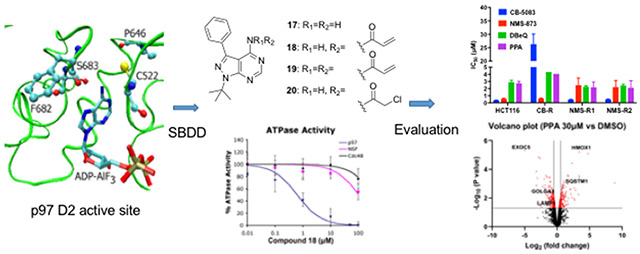
Abstract
Small-molecule inhibitors of p97 are useful tools to study p97 function. Human p97 is an important AAA ATPase due to its diverse cellular functions and implication in mediating the turnover of proteins involved in tumorigenesis and virus infections. Multiple p97 inhibitors identified from previous high-throughput screening studies are thiol-reactive compounds targeting Cys522 in the D2 ATP-binding domain. Thus, these findings suggest a potential strategy to develop covalent p97 inhibitors. We first used purified p97 to assay several known covalent kinase inhibitors to determine if they can inhibit ATPase activity. We evaluated their selectivity using our dual reporter cells that can distinguish p97 dependent and independent degradation. We selected a β-nitrostyrene scaffold to further study the structure-activity relationship. In addition, we used p97 structures to design and synthesize analogues of pyrazolo[3,4-d]pyrimidine (PP). We incorporated electrophiles into a PP-like compound 17 (4-amino-1-tert-butyl-3-phenyl pyrazolo[3,4-d]pyrimidine) to generate eight compounds. A selective compound 18 (N-(1-(tert-butyl)-3-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl)acrylamide, PPA) exhibited excellent selectivity in an in vitro ATPase activity assay: IC50 of 0.6 μM, 300 μM, and 100 μM for wild type p97, yeast Cdc48, and N-ethylmaleimide sensitive factor (NSF), respectively. To further examine the importance of Cys522 on the active site pocket during PPA inhibition, C522A and C522T mutants of p97 were purified and shown to increase IC50 values by 100-fold, whereas replacement of Thr532 of yeast Cdc48 with Cysteine decreased the IC50 by 10-fold. The molecular modeling suggested the hydrogen bonds and hydrophobic interactions in addition to the covalent bonding at Cys522 between WT-p97 and PPA. Furthermore, tandem mass spectrometry confirmed formation of a covalent bond between Cys522 and PPA. An anti-proliferation assay indicated that the proliferation of HCT116, HeLa, and RPMI8226 was inhibited by PPA with IC50 of 2.7 μM, 6.1 μM, and 3.4 μM, respectively. In addition, PPA is able to inhibit proliferation of two HCT116 cell lines that are resistant to CB-5083 and NMS-873, respectively. Proteomic analysis of PPA-treated HCT116 revealed Gene Ontology enrichment of known p97 functional pathways such as the protein ubiquitination and the ER to Golgi transport vesicle membrane. In conclusion, we have identified and characterized PPA as a selective covalent p97 inhibitor, which will allow future exploration to improve the potency of p97 inhibitors with different mechanisms of action.
Keywords: P97, Covalent inhibitor, ATPase Activity, Organic Synthesis, Docking, Anti-proliferative activity, Proteomics
Graphical Abstract
1. Introduction
ATPases associated with diverse cellular activities (AAA+ proteins) are enzymes sharing a common conserved module of approximately 230 amino acid residues [1] and are functionally diverse protein families belonging to the AAA+ protein superfamily of ring-shaped P-loop NTPases. AAA+ proteins catalyze the hydrolysis of a phosphate bond in adenosine triphosphate (ATP), releasing adenosine diphosphate (ADP), inorganic phosphate, and energy to facilitate a variety of cellular processes that are essential for life, including protein folding [2], intracellular transport [3], protein degradation [4], initiation of DNA replication [5], DNA repair [6], DNA remodeling [7], and ion transport [8].
p97, also known as valosin-containing protein (VCP) and Cdc48 (cell division cycle protein 48) in Saccharomyces cerevisiae, Ter94 (transitional endoplasmic reticulum ATPase) in Drosophila melanogaster and VAT (VCP-like ATPase) in Thermoplasma acidophilum, is a hexameric type II AAA+ ATPase. p97 is involved in a wide range of cellular functions, including endoplasmic reticulum-associated degradation (ERAD), membrane fusion, nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) activation, and chromatin-associated processes, which are regulated by ubiquitination. [9] p97 consists of two AAA ATPase domains in tandem, D1, and D2, respectively. A short polypeptide linker (D1-D2 linker) connects the two ATPase domains while the N-D1 linker joins the D1 domain to a large amino-terminal domain (N-domain). The carboxyl-terminus of the D2 domain features a short tail containing ~40 residues. Interaction of p97/Cdc48 with its partners is mostly mediated by the N-domain, although a few proteins bind p97/Cdc48p via the C terminus [10, 11]. Both D1 and D2 domains are homologous in sequence and in structure [12]. The D1 domain has lower intrinsic ATPase activity than does the D2 domain [13], while the D2 domain was thought to underlie p97 function as a mechanochemical transducer, which contributed most of the ATPase activity [14]. Protein substrates released by p97 can be degraded by the 26S proteasome [15, 16]. In addition, p97 is involved in another form of protein degradation and recycling, namely autophagy [17]. The expression levels of p97 are upregulated in many cancers, such as colorectal cancer, pancreatic cancer, thyroid cancer, squamous cell carcinoma, breast cancer, osteosarcoma, gastric carcinoma, and lung cancer [18]. Therefore, p97 is considered a potential anticancer target.
Currently, there are several p97 inhibitors reported (Fig. 1), classified as either ATP-competitive or allosteric inhibitors. The ATP-competitive inhibitor DBeQ (N2, N4-Dibenzylquinazoline-2,4-diamine) mainly targets the D2 domain. DBeQ blocks many biological processes, including degradation of ubiquitin-fusion degradation and ERAD pathway substrates, as well as autophagosome maturation [19-21]. Two DBeQ derivatives with IC50 values of ~100 nM, ML240 and ML241, have been developed. ML240 stimulates the accumulation of LC3-II and rapidly mobilizes the executioner caspases 3 and 7. ML240 also has broad antiproliferative activity toward the NCI-60 panel of cancer cell lines and synergizes with the proteasome inhibitor MG132 to kill multiple colon cancer cell lines. Both ML240 and ML241 have low off-target activity toward a panel of protein kinases and central nervous system targets. [22] CB-5083, derived by structure-activity relationship of the ML240 scaffold, is a selective, ATP-competitive, and orally bioavailable p97 inhibitor, which activates the apoptotic arm of the unfolded protein response and exhibits antitumor activity in several hematological and solid tumor models. CB-5083 entered phase I clinical trials for multiple myeloma in 2015 [23, 24]. However, the trial was terminated due to toxicity caused by an off-target effect. Its successor CB-5339, as the second-generation clinical-grade inhibitor of p97, is in phase I clinical trials for Acute Myeloid Leukemia or Myelodysplastic Syndrome [25]. One of the major challenges of targeted cancer therapy is the emergence of drug-resistant mutations in tumor cells leading to loss of treatment effectiveness. Mutations in p97 that cause resistance to CB-5083 have been identified [23]. Availability of different p97 inhibitors with various binding modes may help overcome this issue. Recently, LC-1028 was reported as a covalent p97 inhibitor with a Michael acceptor consisting of a carbonyl group conjugated with a methyl-capped acetylene group [26]. LC-1028 showed antiproliferative activity in MIA PaCa-2 cells with an EC50 value of 0.436 μM. LC-MS/MS suggested LC-1028 conjugates to C522, indicating that LC-1028 binds to the D2 domain by a covalent bond; the docking model was consistent with this result. However, the selectivity of LC-1028 against WT-97 over other AAA+ ATPases remains unclear. Although there is a much research on the inhibition of p97 and clinical trials are ongoing to evaluate CB-5339, no drug has been approved by the FDA to target p97.
Figure. 1.
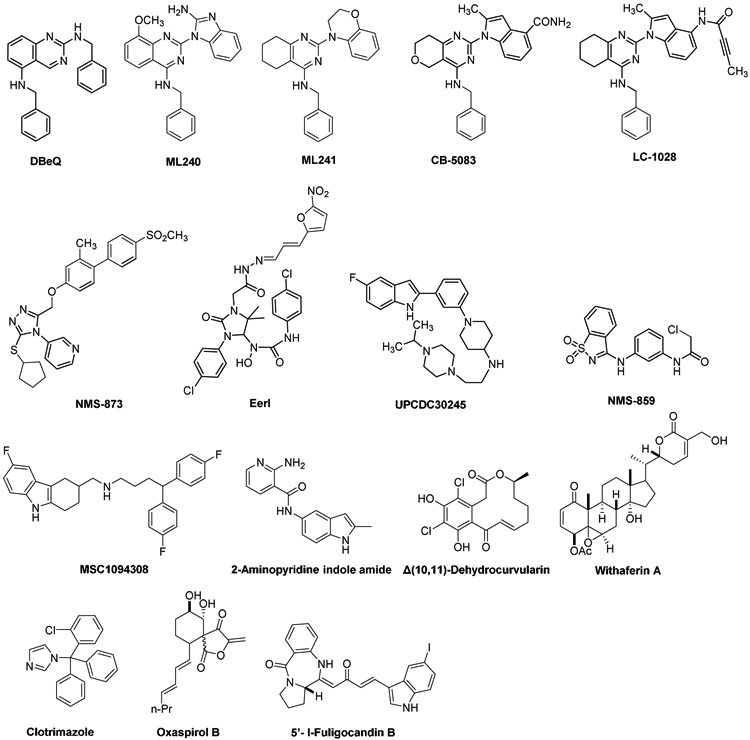
Chemical Structure of Known p97 Inhibitors
Nerviano Medical Sciences and Genentech discovered the allosteric inhibitor NMS-873, an alkylsulfanyl-1,2,4-triazole analog that binds in a tunnel between the D1 and D2 domains [27, 28]. NMS-873 inhibited the ERAD pathway and led to the accumulation of misfolded proteins in the ER. [29] UPCDC30245 is another allosteric p97 inhibitor with an IC50 of 27 nM, which reversibly binds at the interface of the D1 and D2 domains as determined by cryo-electron microscopy [30]. Eeyarestatin I (EerI) serves as a prodrug to convert the 5-nitrofuran-2-yl (NFC) group into a reactive metabolite involved in inhibition of p97-dependent protein degradation in cells even though the exact mechanism of the active metabolite still remains unknown [31, 32]. NMS-859 is a covalent p97 inhibitor, featuring an electrophilic α-chloroacetamide, which selectively modifies Cys522 in the D2 active site with an IC50 of 0.37 μM [28].
MSC1094308 [33] and 2-aminopyridine indole amide [34] are reported as two allosteric p97 inhibitors with IC50 values of 7.2 μM and 0.5 μM, respectively. MSC10944308, with low biochemical potency, showed cellular efficacy at 10 μM. The 2-aminopyridine indole amide exhibited excellent physicochemical properties, including solubility (330 μM in PBS), bidirectional permeability (710 and 460 nm/s), and microsomal stability (human T1/2 > 60 min). Δ(10,11)-Dehydrocurvularin [35] and Withaferin A 27-Acetate [36] covalently modify p97. Δ(10,11)-Dehydrocurvularin inhibited p97 with an IC50 value of 15.3 ± 9.9 μM by covalent bond with Cys522 of p97. Withaferin A 27-Acetate exhibited inhibitory activity against wild-type p97 with an IC50 value of 21 μM, but it didn’t show inhibition against the C522A mutant. However, Withaferin A 27-Acetate exhibited anti-proliferative effects in cancer cell lines at 10-fold lower concentration, suggesting additional modes of action. Clotrimazole [37], Oxaspirol B [38], and 5’-I-Fuligocandin B [39] exhibited p97 inhibition activity with IC50 values of 12 μM, 31.2 ± 3.0 μM and 7.0 μM, respectively. However, their mechanisms of action are unclear. Herein, we report the design, optimization, and biological evaluation of a covalent p97 inhibitor with the potential to be a specific and useful tool to uncover p97’s function and to be further developed to treat cancers.
2. Results and Discussion
2.1. Search for potential irreversible kinase inhibitors to inhibit p97 in vitro and in cells
High throughput (HTS) screens identified multiple hits containing electrophilic groups with submicromolar IC50 values for inhibiting ATPase activity of purified p97 in vitro and turnover of a p97-dependent cellular reporter [21]. C522A p97 is less sensitive to these electrophilic compounds [21, 40]. One of the hits, an aryl alkynyl ketone (Compound 1, JFD02342), inhibited ATPase activity with IC50 of 0.2 μM. However, it also inhibited the turnover of both p97-dependent (UbG76V-GFP) and independent reporters (ODD-Luc) (Table 1). To search for irreversible p97 inhibitors, we evaluated several characterized kinase inhibitors with our in vitro and cell-based p97 HTS assays [41]. Compound 2 is a spleen tyrosine kinase (Syk) inhibitor [42, 43], compounds 4 (cmk), and 5 (fmk) are halomethylketone pyrrolopyrimidine-based p90 ribosomal protein S6 kinase (RSK) inhibitors [44], and compound 6 is a 6-acrylamido-4-anilinoquinazoline-based inhibitor against epidermal growth factor receptor (EGFR) [45]. Compounds 1-3 demonstrated submicromolar IC50, compound 5 was 10-fold less active than the corresponding chloro-substituted compound 4, and compound 6 exhibited IC50 values about 10-18 μM (Table 1). Compound 2 (3,4-methylenedioxy-β-nitrostyrene) [41] was chosen for more detail analysis due to its lower IC50 values in both in vitro ATPase and in cell UbG76V-GFP degradation assays.
Table 1.
Apparent IC50 values of known irreversible inhibitors against p97 and its reporters
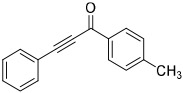
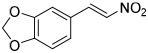
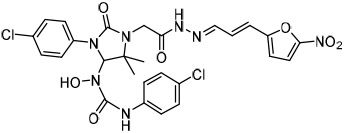
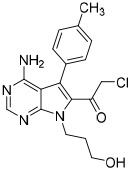
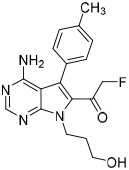
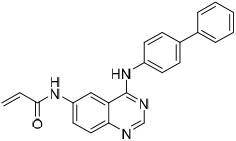
To examine structure-activity relationships of the β-nitrostyrene scaffold, we obtained five and prepared five analogues (compounds 7-16, Table 2). Replacing the nitro group with carboxylic acid (compound 7) led to complete loss of activity, therefore suggesting the nitro group is critical for the observed inhibition. The only other modification that affected both assays by more than 3-fold was the placement of a hydroxyl group in para-substitution to the nitro styrene moiety (compound 9). The requirement for the nitro group and the relative insensitivity of the remainder of the scaffold to modification suggested that the primary mechanism of action was a covalent reaction with Cys522 in the D2 domain active site. The poor activity of Compound 2 towards the C522A mutant and its sensitivity to DTT indicated that the electrophilic attack of Cys522 was indeed critical for the potency of Compound 2. [41] Compound 2 was first reported to be a selective Syk inhibitor [43] with submicromolar IC50; however, our results indicated that it is a potent p97 inhibitor in vitro and in cells, but we were unable to obtain a selective analogue for inhibiting p97 in cells, which is likely due to the high reactivity of the electrophilic moiety. We then sought to design an irreversible scaffold by using x-ray crystal structures of p97 (PDB code1e32).
Table 2.
Apparent IC50 values of compound 2 analogs against p97 and its reporters
| Compounds | Structures | Apparent IC50 (μM) |
||
|---|---|---|---|---|
| UbG76V-GFP | ODD-Luc | p97 | ||
| 7 | 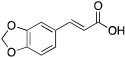 |
> 30 | > 30 | >30 |
| 8 | 1.9 ± 0.9 | 1.8 ± 0.9 | 3.1 ± 1.8 | |
| 9 | 6.8 ± 2.5 | 5 ± 0.8 | 13 ± 5 | |
| 10 | 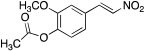 |
5.7 ± 1.5 | 2.3 ± 0.9 | 3.6 ± 2.8 |
| 11 | 4.3 ± 1.6 | 1.6 ± 0.8 | 3.4 ± 1.8 | |
| 12 | 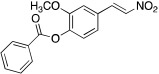 |
5.7 ± 2.8 | 10 ± 3 | 2.0 ± 0.4 |
| 13 | 2.9 ± 0.7 | 3.0 ± 1.0 | 0.7 ± 0.3 | |
| 14 | 1.7 ± 0.7 | 4.0 ± 1.4 | 1.2 ± 0.3 | |
| 15 | 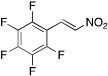 |
2.7 ± 1.0 | 2.1 ± 0.3 | 2.4 ± 0.6 |
| 16 | 1.8 ± 0.8 | 2.0 ± 0.9 | 1.7 ± 0.5 | |
2.2. Structure based drug design to develop a covalent inhibitor of p97
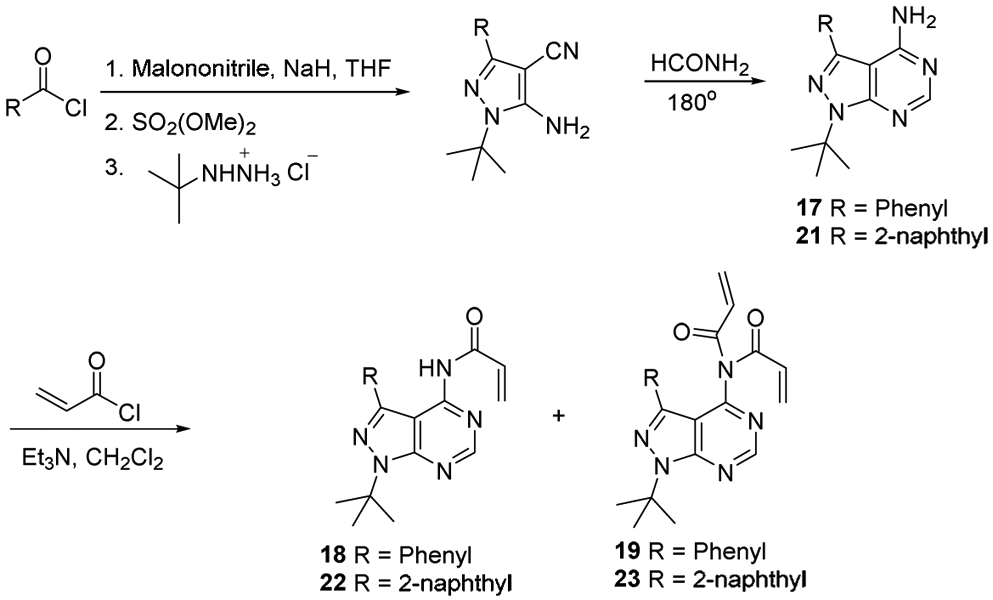
To develop covalent inhibitors of p97, we focused our attention on the D2 ATPase domain. This domain has a cysteine residue at position 522, the side chain of which projects into the active site nearby the amino group (NH2) attached at the C6 position of the purine of a bound ADP-aluminum fluoride complex (Fig. S1a) [46]. We reasoned that an ATP analog carrying an electrophilic substitution at C6 might react with this cysteine residue and inactivate p97. It is noteworthy that a cysteine at position 522 is not essential for p97 ATPase activity, and in fact the budding yeast ortholog of p97, Cdc48, does not have cysteine in this position. Indeed, the presence of cysteine at this position in members of the AAA ATPase family is variable: the D1 domain of NSF has a cysteine in the equivalent position, whereas five of the six AAA ATPase subunits of the 26S proteasome do not (Fig. S1b). To develop an inhibitor capable of reacting with this cysteine, we selected PP (compound 17) as the starting point and optimized this scaffold in a variety of ways in terms of the binding mode of the Src inhibitor pyrazolopyrimidines (PPs) (Fig. 2) [47, 48]. Specifically, we attached various electrophilic groups to the C4 position, including acrylamides (compounds 18, 19, 22, 23, 24, 27, and 28), and a chloroacetamide (compound 20). As negative controls, we prepared compounds that retained an amino group attached at C4 (compounds 17, 21, 25, and 26). All compounds demonstrated inhibitory activity in both ATPase and UbG76V-GFP turnover assays except for the control compounds (Table 3). The most potent inhibitor was compound 18 (PPA), which contains benzene at C3 and a single acrylamide at C4. PPA was 20-fold less potent at inhibiting the p97-independent reporter. Therefore, we selected PPA for a more detailed study.
Figure. 2.
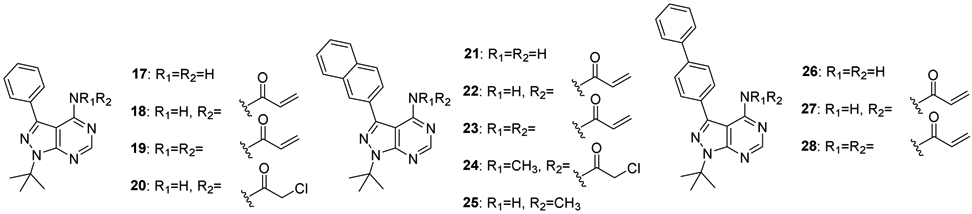
Structures of the pyrazolo[3,4-d]pyrimidine analogs designed for p97 inhibition.
Table 3.
Apparent IC50 values of the pyrazolo[3,4-d]pyrimidine analogs against p97, T532C-Cdc48, and the cellular reporters.
| Compounds | Apparent IC50 (μM)* |
|||
|---|---|---|---|---|
| UbG76V-GFP | ODD-Luc | p97 | T532C-Cdc48 | |
| 17 | > 30 | > 30 | > 100 | > 100 |
| 18 (PPA) | 1.6 ± 0.5 | 35 ± 10 | 0.6 ± 0.2 | 14 ± 5 |
| 19 | 5.4 ± 1.6 | > 30 | 1.1 ± 0.7 | 26 ± 6 |
| 20 | 31 ± 3 | > 30 | 4.5 ± 3.5 | 35 ± 8 |
| 21 | > 30 | > 30 | > 100 | > 100 |
| 22 | 2.0 ± 0.9 | > 30 | 3 ± 1 | 24 ± 7 |
| 23 | 2.5 ± 0.3 | > 30 | 1.3 ± 0.7 | 22 ± 11 |
| 24 | 3.6 ± 0.5 | > 30 | 10 ± 6 | ND |
| 25 | > 30 | > 30 | > 50 | ND |
| 26 | > 30 | > 30 | > 50 | ND |
| 27 | 2.1 ± 0.6 | > 30 | 2.8 ± 1.2 | ND |
| 28 | 6.1 ± 0.7 | > 30 | 0.9 ± 0.2 | ND |
Measurements were carried out in triplicate, and variance is expressed as the standard deviation. ND: not determined.
2.3. Biochemical characterization of PPA (18)
To determine if inhibition by PPA requires a reactive thiol, we determined inhibition of ATPase activity in the presence of dithiothreitol (DTT) and cysteine during the preincubation. The data in Figure S1c demonstrated that the inhibitory effect of PPA could be counteracted by the inclusion of either cysteine or DTT. To evaluate whether PPA modifies cysteine 522, inhibited p97 was digested with trypsin, and peptides were fractionated by HPLC and analyzed by tandem mass spectrometry. The PPA-modified peptide (N14) from the digested p97 sample was eluted using PPA-modified N15-labeled peptide (H2N-GVLFYGP*P*GCGK-OH, where * indicates N15 labeled residues) as an internal control (Fig. S2). Tandem mass spectrometry identified Cys522 as the site of modification, thus confirming the formation of a covalent bond between PPA and p97. To further address whether Cys522 is necessary for inhibition of p97 by PPA, we constructed two mutants in which Cys522 was converted to either alanine or threonine. Furthermore, we constructed a mutant of yeast Cdc48 in which the residue analogous to Cys522, Thr532, was converted to cysteine. Mutation of Cys522 in p97 to either alanine or threonine decreased sensitivity to PPA by more than 100-fold (Table 4). On the other hand, yeast Cdc48, which normally is quite resistant to PPA, became nearly 30-fold more sensitive to the compound upon the introduction of cysteine in place of Thr532. Together, these data indicate that Cys522 is both necessary and sufficient to confer sensitivity to PPA.
Table 4.
Cys522-mediated p97 inhibition by PPA (18) in vitro
| Enzyme | Apparent IC50 (μM) |
|---|---|
| WT-p97 | 0.62 ± 0.25 |
| C522A-p97 | 110 ± 33 |
| C522T-p97 | 82 ± 45 |
| Yeast Cdc48 | 376 ± 95 |
| T532C-yeast Cdc48 | 14 ± 5 |
| Hamster NSF | 105 ± 31 |
| Human 19S ATPase* | 75 ± 19 |
Human Rpt3 contains a cysteine in Walker A motif.
2.4. Docking study of PPA (18)
Although we have confirmed the covalent binding of PPA to Cys522 in Domain 2, it is still unclear whether other interactions between PPA and WT-p97 occur. In order to understand the binding mode of PPA in the active site, we performed the covalent docking study by AutoDock 4.2.6. [49] In addition to the covalent bonding to Cys522 between the terminal carbon atom from the acryl amido group and the thiol group, there were two hydrogen bond interactions between Gly480 and 7-NH, and Gly521 and the amido NH, respectively (Fig. 3A and 3B). In addition, some residues in the active site, such as Ile479, Leu526, Gly684, Ala685, and Thr688 showed hydrophobic interactions with PPA (Fig. 3A).
Figure. 3.

The docking study of PPA into WT-p97. (A) Two-dimensional presentation of the potential interactions between PPA and WT-p97. (B) Three-dimensional presentation of PPA in the active site.
2.5. PPA does not inhibit NSF and 26S proteasome and blocks degradation of p97 substrate irreversibly
To further address the specificity of PPA, we tested its activity against two other AAA ATPases, NSF, and the 19S regulatory particles. Although NSF is known to be sensitive to thiol-reactive agents, it was more than 100-fold less sensitive to PPA than p97 (Fig. 4A, Table 4). Likewise, inhibition of ATP hydrolysis by the 19S regulatory particles was observed only at high concentrations of PPA (IC50 = 75 μM). The results with NSF and the 19S regulatory particles are noteworthy because both the active D1 domain of NSF and the Rpt3 subunit of the 19S regulatory particles have cysteine in the position analogous to cysteine 522 of p97 (Fig. S1b). Moreover, PPA did not inhibit the hydrolysis of a fluorogenic proteasome substrate, LLVY-AMC, with the affinity-purified 26S proteasome from Hek293 cells [50] (Fig. 4B). We used previously established washout assay using UbG76V-GFP reporter to demonstrate PPA irreversibly block degradation of UbG76V-GFP in cells [41]. UbG76V-GFP was degraded after washing out MG132 but it remains stable with PPA treated cells (Fig. 4C). Similarly, we showed that PPA block degradation of an ERAD substrate, TCRα-GFP (α-chain of the T-cell receptor fused to GFP) [51]. We included MG132 (a reversible proteasome inhibitor), YU101 (a covalent proteasome inhibitor) [52], PYR41(a ubiquitin E1 inhibitor) [53], and DBeQ (a reversible p97 inhibitor) [21] as positive controls.
Figure 4.

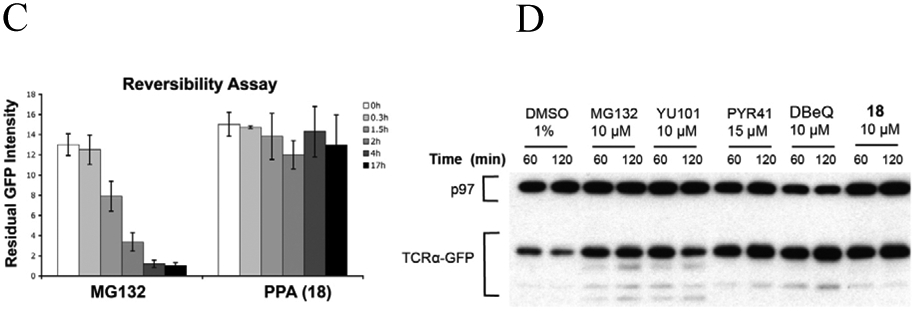
PPA inhibits p97 ATPase activity selectively and irreversibly. (A) Titration curves of the in vitro ATPase assays for PPA inhibition of p97, NSF, and Cdc48. (B) PPA did not inhibit human 26S proteasome activity. (C) Reversibility of PPA inhibition was determined using UbG76V-GFP by washing out test compound and monitoring decay of GFP signal in the presence of cycloheximide for additional 17 h. (D) PPA block degradation of an ERAD substrate, TCRα-GFP stably expressed in HEK293 cells. Cells were incubated with MG132, washed and treated with cycloheximide plus compound for 60 min or 120 min. Samples were immunoblotted with anti-p97 as loading control and anti-GFP antibody to detect TCRα-GFP.
2.6. Anti-proliferative effect of PPA in cancer cells and in CB-5083, NMS-873 resistant colorectal cancer
To explore anti-proliferative effects of these compounds, we used three cancer cell lines and evaluated PPA together with three known p97 inhibitors, CB-5083, NMS-873, and DBeQ. As shown in Fig. 5A, all compounds inhibited proliferation of all three cancer lines. CB-5083 showed the most potent anti-proliferative activity. The IC50 of PPA to HCT116, HeLa, and RPMI8226 were 2.7 μM, 6.1 μM, and 3.4 μM respectively. PPA has comparable potency as DBeQ and less potent compared to CB-5083 and NMS-873. PPA’s cellular activity was consistent with its enzymatic activity against p97. Furthermore, we evaluated its potency toward CB-5083 resistant (CB-R) and NMS-873 resistant (NMS-R) HCT116 cell lines [24], as shown in Fig. 5B, PPA and DBeQ have similar anti-proliferative activities for all three HCT116 lines. CB-5083 is 72-fold less effective in CB-R, whereas NMS-873 is 4.5-fold less effective in NMS-R. The two NMS-873 resistant cell lines we isolated are A530T heterozygous, so the fold resistance is not as high as the homozygous A530T made using CRISPR as described previously [54].
Figure. 5.

Anti-proliferation activity of p97 inhibitors against (A) HCT116, HeLa, and RPMI8226 cell lines. (B) HCT116, CB-5083 resistant HCT116 cell line harboring D649A, T688A p97 mutant (CB-R) and NMS-873 resistant HCT116 cell line harboring A530T p97 mutant (NMS-R). Data are shown as Mean ± SEM taken from four replicate experiments.
2.7. Quantitative Proteomic Analysis of PPA treated HCT116 human colon cancer cells
Label-free quantification using the Orbitrap Eclipse LC-MS/MS was employed to evaluate the effect of PPA (15 μM or 30 μM for 6 hours) on the protein level changes in HCT116 cells. Two independent biological replicates were performed for each group. The proteome analysis using PD 2.4 software identified a total of 5924 proteins from all control and PPA-treated groups. The normalized abundance was used for the following analysis (Fig. S3). We identified 291 proteins change in 15 μM PPA-treated samples (Fig. 6A), and 553 proteins change in 30 μM PPA-treated samples (Fig. 6B and 7A) by limma analysis (p < 0.05). The Venn diagram shows that the level of 58 proteins significantly increased (Fig. 6C, Table S3A) and 46 significantly decreased (Fig. 6D, Table S3B) in both treatment groups. By overlapping with known p97 interaction data from BioGRID database, we found 33 of them significantly increased (Fig. 6C) and 42 of them significantly decreased (Fig. 6D) in either 15 or 30 μM treatment groups. Notably, there are 15 known p97 interacting proteins also showed differentially change in both treatment groups and the log2 fold change values are shown in Table 5. For example, SQSTM1 (also known as p62) [55], a ubiquitin binding protein involved in autophagy, antioxidant response, apoptosis, and regulation of endosomal trafficking, is increased in a concentration-dependent manner (logFC 0.42 vs 0.97, 15 μM vs 30 μM). Another protein GOLGA3 [56], participating in transport of protein, apoptosis, and positioning of the Golgi, is dose-dependently decreased (logFC −0.36 vs −0.70, 15 μM vs 30 μM). These results provide valuable insights for further investigation into the mechanisms by which PPA affects p97 function.
Figure. 6.

Proteomic analysis of PPA treated HCT116 cell line. A-B) Volcano plots illustrate significantly differentially expressed protein in 15 μM (A) and 30 μM (B) PPA treated HCT116 cells (fold change: PPA/DMSO). C-D) Venn diagrams of up-regulated protein (C) and down-regulated (D) proteins (p < 0.05) between 15 μM, 30 μM PPA-treated groups and p97 interactors. The log2FC range of 15 μM is −5.56 to −0.26 and 0.26 to 3.24 and the log2FC range of 30 μM are −5.03 to −0.26 and 0.27 to 8.85.
Figure. 7.

A) Heatmap presentation of differentially regulated proteins (p < 0.05) of DMSO vs. 30 μM PPA-treated groups. B) The biological process analysis of 30 μM PPA-treated group differentially expressed proteins [p < 0.05, and log2 (FC) > 1 or < −1].
Table 5.
Overlapped protein list of differentially expressed proteins by 15 μM and 30 μM PPA-treatment known to be regulated by p97. (p < 0.05)
| Accession | Gene Symbol |
Description | log2 (Fold Change) |
|
|---|---|---|---|---|
| 15 μM | 30 μM | |||
| Q15149 | PLEC | Isoform 3 of Plectin | 0.33 | 0.58 |
| P58107 | EPPK1 | Epiplakin | 0.38 | 0.64 |
| Q13501 | SQSTM1 | Sequestosome-1 | 0.42 | 0.97 |
| Q8TCF1 | ZFAND1 | AN1-type zinc finger protein 1 | 0.49 | 0.42 |
| Q8TDN6 | BRIX1 | Ribosome biogenesis protein BRX1 homolog | 0.52 | 0.70 |
| O43422 | PRKRIR | 52 kDa repressor of the inhibitor of the protein kinase | 0.69 | 0.68 |
| P46736 | BRCC3 | Lys-63-specific deubiquitinase BRCC36 | 0.94 | 0.83 |
| Q13586 | STIM1 | Stromal interaction molecule 1 | 1.49 | 1.65 |
| P11279 | LAMP1 | Lysosome-associated membrane glycoprotein 1 | −0.59 | −0.66 |
| O14519 | CDK2AP1 | Cyclin-dependent kinase 2-associated protein 1 | −0.48 | −0.47 |
| Q14145 | KEAP1 | Kelch-like ECH-associated protein 1 | −0.46 | −1.03 |
| P50748 | KNTC1 | Kinetochore-associated protein 1 | −0.43 | −0.87 |
| Q9Y679 | AUP1 | Ancient ubiquitous protein 1 | −0.39 | −0.43 |
| Q08378 | GOLGA3 | Golgin subfamily A member 3 | −0.36 | −0.70 |
| A0A087WY71 | AP2M1 | AP-2 complex subunit mu | −0.33 | −0.38 |
The 30 μM PPA-treated group had more significantly changed proteins (~2 fold more than 15μM PPA-treated group), which suggests that this group might be more informative as to PPA induced proteomic changes. Therefore, subsequent analyses compared DMSO and 30 μM PPA-treated groups. A set of 110 proteins identified in this analysis was classified as the final differentially expressed proteins (DEPs) between the control and 30 μM PPA-treated groups (listed in Table S2, with p-value<0.05 and fold change >2 (up-regulated) or <0.5 (down-regulated). Subsequently, the 110 DEPs were uploaded to the DAVID Database, and the annotation enrichment analysis was carried out with the complete human gene set as the background. We investigated the biological process, molecular function, cellular component, and functional pathways in the significantly enriched GO terms. The top fourteen relevant changed biological processes are presented in Fig. 7B. The analysis showed these DEPs are primarily related to protein binding, DNA binding and damage response, and mitochondrion. Some p97-related processes, such as protein ubiquitination and ER to Golgi transport vesicle membrane, were also identified, with other processes linked to PPA’s anti-proliferation activity, such as p53 regulation, cell proliferation, and negative regulation of apoptosis. Furthermore, protein-protein interaction (PPI) analysis was performed using the STRING database to understand the interactions between these DEPs in response to treatment with PPA in HCT116 cell lines (Fig. S4). For all PPI networks, the minimum required interaction score was set at medium confidence (0.400). The PPI networks showed that the DEPs have several interactions among themselves, suggesting these proteins are biologically connected. For example, the protein HMOX significantly up-regulated in both 15 and 30 μM PPA-treated groups, was shown to interact with many proteins, such as KEAP1, MSRA, MKLN1, CDKN1A, and FTH1.
3. Conclusion
To develop irreversible p97 inhibitors, we screened six well-characterized kinase inhibitors by our in vitro and cell-based p97 HTS assays. Compounds 1 and 2 were chosen to further optimize their structures to provide 10 compounds (compounds 7-16). Intrigued by the p97 inhibition result of these compounds, we designed and synthesized a series of irreversible p97 inhibitors by introducing a Michael Acceptor on the N-6 position of ADP after analysis of the crystal structure of the p97 complex with ADP-AlF3 (PDB code 1e32). PPA showed potent inhibition against p97 with IC50 of 0.6 ± 0.2 μM. Its irreversible mode of action was confirmed by tandem mass spectrometry and a washout experiment in a p97 dependent degradation assay. An in-silico docking study was performed to validate the feasibility of covalent binding to Cys522. In a panel of Cys522-mediated p97 inhibition assays in vitro, PPA showed up to 100-fold selectivity on wild-type vs. Cys522-mutated p97 with IC50 of 0.62 ± 0.25 μM. In addition, PPA did not show inhibition against NSF and 26S proteasome. In addition, PPA showed potent anti-proliferative effects on HCT116, HeLa, and RPMI8226 cells with IC50 values of 2.7 μM, 6.1 μM, and 3.4 μM, respectively. Moreover, PPA can inhibit growth of CB-5083 and NMS-873 resistant HCT116 cells. Furthermore, proteomic analysis revealed PPA regulated bona fide p97 interacting proteins involved in known p97 functional pathways such as protein ubiquitination and ER to Golgi transport vesicle membranes. Taken together, these observations suggest PPA could serve as a promising starting point to develop an irreversible p97 inhibitor to treat cancer and can be a valuable tool to overcome resistance to other p97 inhibitors with different mode of actions such as ATP competitive and allosteric mechanisms.
4. Materials and Methods
4.1. Chemical Synthesis
4.1.1. Material
Reactions were performed in flame-dried glassware under an argon or nitrogen atmosphere. Solvents were dried by passage through an activated alumina column under argon. Triethylamine (NEt3) was distilled from sodium hydride immediately prior to use. Other commercial reagents were used as received. Thin-layer chromatography (TLC) was performed using E. Merck silica gel 60 F254 precoated plates (0.25 mm) and visualized by UV fluorescence quenching. Silica Flash P60 Academic Silica gel (particle size 0.040-0.063 mm) was used for flash chromatography. 1H and 13C NMR spectra were recorded on either a Varian Mercury 500 spectrometer at 500 MHz and 125 MHz, respectively, or a Varian Mercury 300 spectrometer at 300 or 75 MHz, respectively. 13C spectra are referenced to residual CDCl3 (77.2). 19F NMR spectra were recorded on a Varian 300 (at 282 MHz) and are reported relative to CFCl3 (δ 0.0). Data for 1H and 19F NMR spectra are reported as follows: chemical shift (δ ppm) (multiplicity, coupling constant (Hz), integration). MS were acquired using an Agilent 6200 Series TOF with an Agilent G1978A Multimode source in electrospray ionization (ESI), atmospheric pressure chemical ionization (APCI) or mixed (MM) ionization mode. Some compounds were synthesized in this work and others were obtained from the in-house compound library.
4.1.2. Synthesis of β-Nitro styrenes
β-Nitro styrenes were prepared according to a literature procedure [57] with minor modifications. See below for a representative procedure.
E-1,2-dimethoxy-4-(2-nitrovinyl)benzene (8). A round-bottom flask fitted with a magnetic stir bar and a reflux condenser was flame-dried under vacuum. After cooling to ambient temperature under dry nitrogen, 3,4-dimethoxybenzaldehyde (0.419 g, 2.5 mmol, 1.0 equiv), ammonium acetate (0.170 g, 2.2 mmol, 0.9 equiv), and nitromethane (12.5 mL) were added, and the resulting mixture was heated to reflux and maintained with stirring for 14 hours. The reaction mixture was concentrated under reduced pressure, and the residue was recrystallized from ethanol to yield (8) (0.362 g, 69% yield) as yellow-green flakes: 1H NMR (300 MHz, CDCl3) δ 7.97 (d, J = 13.5 Hz, 1 H), 7.53 (d, J = 13.5 Hz, 1 H), 7.18 (dd, J = 8.7, 2.1 Hz, 1 H), 7.01 (d, J = 2.1 Hz, 1 H), 6.92 (d, J = 8.7 Hz, 1 H), 3.94 (s, 3 H) 3.93 (s, 3 H); MS (EI+) m/z 210.0 (M + H)+.
E-3-(2-nitrovinyl)pyridine (13). Prepared by the above procedure (3-pyridylcarboxaldehyde, 0.25 mL, mmol). Purified by filtration through a short plug (SiO2, 50% EtOAc in hexanes) followed by recrystallization from ethanol to yield (13) (0.112 g, 28% yield) as orange needles: 1H NMR (300 MHz, CDCl3) δ 8.80 (d, J = 2.1 Hz, 1 H), 8.72 (d, J = 4.8, 1.5 Hz, 1 H), 8.01 (d, J = 13.8 Hz, 1 H), 7.87 (ddd, J = 7.8, 2.1, 1.5 Hz, 1 H), 7.62 (d, J = 13.5 Hz, 1 H), 7.41 (dd, J = 7.8, 4.8 Hz, 1 H); HRMS (MM: ESI-APCI) calc’d for C7H7N2O2 [M + H]+: 151.0502, found 151.0503.
E-1-nitro-3-(2-nitrovinyl)benzene (14). Prepared by the above procedure (3-nitrobenzaldehyde, 0.389 g). Purified by flash chromatography (SiO2, 17% EtOAc in hexane), followed by recrystallization from ethanol to yield (14) (0.228 g, 46% yield) as pale yellow powder: 1H NMR (300 MHz, CDCl3) δ 8.42 (t, J = 1.8 Hz, 1 H), 8.35 (ddd, J = 8.1, 1.8, 1.2 Hz, 1 H), 8.05 (d, J = 13.5 Hz, 1 H), 7.87 (dt, J = 7.5, 1.2 Hz, 1 H), 7.68 (t, J = 7.8 Hz, 1 H) 7.66 (d, J = 13.8 Hz, 1 H).
E-pentafluoro(2-nitrovinyl)benzene (15). Prepared by the above procedure (pentafluoro benzaldehyde, 0.538 g, 2.7 mmol). Purified by filtration through a short plug (SiO2, 50% EtOAc in hexanes) followed by flash chromatography (SiO2, 10% EtOAc in hexanes) to yield (15) (0.142 g, 22% yield) as yellow oil: 1H NMR (300 MHz, CDCl3) δ 8.04 (d, J = 13.8 Hz, 1 H), 7.82 (d, J = 13.8 Hz, 1 H); 19F NMR (282 MHz, CDCl3) δ 135.8 (m, 2 F), 146.6 (tt, J = 20.7, 4.6 Hz, 1 F), 159.7 (m, 2 F).
E-2-bromo-1-fluoro-4-(2-nitrovinyl)benzene (16). Prepared by the above procedure (3-bromo-4-fluorobenzaldehyde, 0.500 g, 2.5 mmol). Purified by filtration through a short plug (SiO2, 50% EtOAc in hexanes) followed by recrystallization from ethanol to yield (16) (0.311 g, 51% yield) as yellow microcrystals: 1H NMR (300 MHz, CDCl3) δ 7.91 (d, J = 13.5 Hz, 1 H), 7.77 (dd, J = 6.6, 2.4 Hz, 1 H), 7.51 (d, J = 13.8 Hz, 1 H), 7.49 (m, 1 H), 7.21 (t, J = 8.4 Hz, 1 H); 13C NMR (125 MHz, CDCl3) δ 161.1 (d, J = 255 Hz), 137.7 (d, J = 2.9 Hz), 136.4, 134.2, 129.9 (d, J = 10.6 Hz), 127.8 (d, J = 3.4 Hz), 117.6 (d, J = 22.9 Hz), 110.6 (d, J = 21.9 Hz); 19F NMR (282 MHz, CDCl3) δ 100.3 (dd, J = 11.8, 7.1 Hz, 1 F).
4.1.3. Synthesis of Pyrazolopyrimidine analogues
Pyrazolopyrimidines 17, 21, and 26 were synthesized from the corresponding acid chloride [47, 58].
4.1.3.1. Synthesis of Compounds 18 and 19
Representative Procedure for Acylation of Pyrazolopyrimidines:
To a solution of 4-amino-1-t-butyl-3-phenylpyrazolo[3,4-d] pyrimidine (17) (0.103 g, 0.387 mmol) in CH2Cl2 (4 mL, 0.1 M) was added Et3N (0.043 mL, 0.31 mmol). The solution was cooled in an ice water bath, and acryloyl chloride (0.027 mL, 0.314 mmol) was added dropwise. After stirring for 30 minutes while warming to room temperature, the solution was partially concentrated and purified directly by silica gel column chromatography (10% ethyl acetate/hexanes). In addition to recovered starting material (0.0306 g, 0.1145 mmol, 29.6 % yield), three products were isolated:
N-(1-(tert-butyl)-3-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl)acrylamide (18) as white solid (0.015 g, 0.046 mmol, 14.5 % yield). 1H NMR (300 MHz, d6-DMSO): δ 11.04 (s, 1 H), 8.81 (s, 1 H), 7.61-7.56 (m, 2 H), 7.41-7.29 (m, 3 H), 6.37 (dd, J = 17.2, 10.2 Hz, 1 H), 5.86 (dd, J = 17.2, 1.6 Hz, 1 H), 5.65 (dd, J = 10.2, 1.6 Hz, 1 H), 1.82 (s,9 H). HRMS (EI+) m/z calc’d for C18H19N5O [M]+: 321.1590, found 321.1598. The 13C NMR data with good quality could not be obtained due to poor signal-to-noise as a result of a peak broadening presumed to be a result of the dynamic process that was not investigated further due to the high reactivity of 18.
N-acryloyl-N-(1-(tert-butyl)-3-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl)acrylamide (19) as white solid (0.019 g, 0.051 mmol, 16.2% yield). 1H NMR (300 MHz, CDCl3): δ 8.94 (s, 1 H), 7.50-7.45 (m, 2 H), 7.40-7.35 (m, 3 H), 6.31 (dd, J = 16.7, 2.3 Hz, 2 H), 6.22 (dd, J = 16.7, 9.3 Hz, 2 H), 5.63 (dd, J = 9.3, Hz, 2 H), 1.9 (s, 9 H). 13C NMR (75 MHz, CDCl3): δ 167.2, 155.4, 154.0, 153.5, 142.7, 132.1, 131.0, 129.8, 129.1, 129.0, 128.9, 110.2, 61.4, 29.3. HRMS (EI+) m/z calc’d for C21H21N5O2 [M]+: 375.1695, found 375.1695.
4.1.3.2. Synthesis of compound 20
To a THF (10 mL, 0.1 M) solution of phenyl pyrazolopyrimidine (17) (0.25 g, 0.94 mmol) was Et3N (0.17 mL, 1.2 mmol). Chloroacetic chloride (0.1 mL, 1.2 mmol) was added dropwise. After 12 hr at ambient temperature, the mixture was quenched with H2O (10 mL) and extracted with CHCl3 (3x10 mL). The combined organic layers were dried with MgSO4, filtered, and concentrated. The crude oil was purified by silica gel column chromatography (1:1 hexanes/ethyl acetate) to provide 20 as white solid (0.18 g, 56 % yield). 1H NMR (300 MHz, CDCl3): δ 8.65 (br s, 2 H), 7.69 (br s, 2 H), 7.56-7.42 (m, 3 H), 4.37 (s, 2 H), 1.84 (s, 9 H). Many of the peaks in the 13C NMR spectrum were broadened by a dynamic process that was not investigated further. Poor signal-to-noise prevented acquisition of good 13C NMR spectral data. HRMS (FAB+) m/z calc’d for C17H19ON5Cl [M + H]: 344.1273, found 344.1270.
4.1.3.3. Synthesis of compounds 22 and 23
2-Naphthyl pyrazolopyrimidine (21) (0.1134 g, 0.357 mmol) was acylated as described in the above representative procedure using CH2Cl2 (3 mL), Et3N (0.1 mL, 0.72 mmol) and acryloyl chloride (0.05 mL, 0.547 mmol). Water (10 mL) and NaHCO3 (10 mL, saturated, aq.) were added and extracted with CH2Cl2 (3 x 10 mL). The combined organic layers were dried with Na2SO4, filtered, and concentrated under reduced pressure. The crude yellow oil was purified by silica column chromatography (1:9->1:3->1:0 ethyl acetate/hexanes) to provide three products in addition to recovered starting material (0.0184 g, 0.0466 mmol, 13% yield).
N-(1-(tert-butyl)-3-(naphthalen-2-yl)-1H-pyrazolo[3,4-d]pyrimidin-4-yl)acrylamide (22) as white solid (0.026 g, 0.069 mmol, 19 % Yield). 1H NMR (300 MHz, DMSO) δ 11.23 (br s, 1 H), 8.82 (s, 1 H), 8.20 (s, 1 H), 7.91 (m, 3 H), 7.82 (d, J = 8 Hz, 1 H), 7.50 (m, 2 H), 6.32 (dd, J = 10, 17 Hz, 1 H), 5.73(d, J = 17 Hz, 1 H), 5.51 (d, J = 10 Hz, 1 H), 1.86 (s, 9 H). Many of the peaks in the 13C NMR spectrum were broadened by a dynamic process that was not investigated further. Poor signal-to-noise prevented acquisition of good 13C NMR spectral data. HRMS (FAB+) m/z calc’d for C22H22N5O [M + H]: 372.1819; found 372.1811.
N-acryloyl-N-(1-(tert-butyl)-3-(naphthalen-2-yl)-1H-pyrazolo[3,4-d]pyrimidin-4-yl)acrylamide (23) as white solid (0.066 g, 0.16 mmol, 43 % Yield). 1H NMR (500 MHz, CDCl3) δ 8.94 (s, 1 H), 7.92 (s, 1 H), 7.84 (d, J = 8.4 Hz, 1 H), 7.81 (m, 1 H), 7.76 (m, 1 H), 7.61 (dd, J = 8.4, 1.2 Hz, 1 H), 7.48 (m, 2 H), 6.18 (ABX quartet, 4 H), 5.46 (dd, J = 9.1, 2.8 Hz, 2 H), 1.9 (s, 9 H). 13C NMR (125 MHz, CDCl3) δ 167.3, 155.6, 154.1, 153.6, 142.7, 133.5, 133.4, 131.1, 129.7, 129.5, 128.8, 128.7, 128.4, 127.9, 126.9, 126.8, 126.4, 110.3, 61.8, 29.5. HRMS (FAB+) m/z calc’d for C25H24N5O2 [M + H]: 426.1925; found 426.1930.
4.1.3.4. Synthesis of compounds 24 and 25 [59]
To solution of NaH (0.0055 g, 0.1375 mmol, 1.4 equiv.) and pyrazolopyrimidine 21 (0.0277 g, 0.1 mmol) in THF (3 mL, 0.03 M) was added CH3I (0.006 mL, 0.096 mmol). The solution was heated to 60 °C for δ hr. The solution was purified without workup by silica gel column chromatography (5% ethyl acetate / hexanes) to yield the monomethylated pyrazolopyrimidine (25) as white solid (0.0277 g, 0.0836 mmol, 87% yield). 1H NMR (500 MHz, CDCl3): δ 8.47 (s, 1 H), 8.11 (s, 1 H), 8.01 (d, J = 8.4 Hz, 1 H), 7.93 (m, 2 H), 7.78 (dd, J = 8.4, 1.6 Hz, 1 H), 7.56 (m, 2 H), 5.36 (br s, 1 H), 3.05 (d, J = 4.9 Hz, 3 H), 1.86 (s, 9 H). 13C NMR (125 MHz, CDCl3): δ 158.0, 154.8, 153.9, 141.7, 133.7, 133.4, 131.7, 129.4, 128.4, 128.1, 128.0, 127.0, 126.9, 126.4, 100.4, 60.6, 29.4, 28.0. HRMS (FAB+) m/z calc’d for C20H22N5 [M + H]: 332.1870; found 332.1885.
To a solution of methylated pyrazolopyrimidine 25 (0.0277 g, 0.0836 mmol) in CH2Cl2 (5 mL, 0.02 M) was added Et3N (0.022 mL, 0.16 mmol). The solution was cooled in an ice water bath and to it added acryloyl chloride (0.012 mL, 0.14 mmol). The solution was purified without workup by silica gel column chromatography (5% ethyl acetate/ benzene) to yield 24 as colorless oil (0.0015 g, 0.0039 mmol, 4.7% yield) along with recovered starting material (0.017 g, 62.4%). 1H NMR (500 MHz, CDCl3): δ 8.88 (s, 1 H), 8.05 (d, J = 1.6 Hz, 1 H), 7.91 (d, J = 8.5 Hz, 1 H), 7.86 (m, 2 H), 7.74 (dd, J = 8.5, 1.8 Hz, 1 H), 7.52 (m, 2 H), 6.20 (dd, J = 10.1, 1.9 Hz, 1 H), 6.10 (dd, J = 16.7, 1.9 Hz, 1 H), 5.46 (dd, J = 16.7, 10.1 Hz, 1 H), 3.20 (s, 3 H), 1.93 (s, 9 H). 13C NMR (500 MHz, CDCl3): δ 166.4, 157.7, 155.9, 153.8, 142.6, 133.5, 133.5, 130.0, 128.8, 128.8, 128.7, 128.7, 128.3, 128.0, 127.0, 126.9, 125.9, 108.4, 61.7, 36.3, 29.4. HRMS (FAB+) m/z calc’d for C23H24N5O [M + H]: 386.1975; found 386.1969.
4.1.3.5. Synthesis of compound 27 and 28
Biphenyl pyrazolopyrimidine (26) (0.2 g, 0.58 mmol) was acylated as described in the above representative procedure using CH2Cl2 (10 mL, 0.06 M), Et3N (0.104 mL, 0.75 mmol), and acryloyl chloride (0.04 mL, 0.44 mmol). Purification by silica gel column chromatography (1:3 ethyl acetate/hexanes) yielded two products in addition to recovered starting material (0.042 g, 0.12 mmol, 21% yield). Data for starting pyrazolopyrimidine (26): 1H NMR (300 MHz, CDCl3): δ 8.37 (s, 1 H), 7.77 (m, 4 H), 7.65 (m, 2 H), 7.48 (m, 2 H), 7.39 (m, 1 H), 5.68 (br s, 2 H), 1.85 (s, 9 H). 13C NMR (125 MHz, CDCl3): δ 158.0, 154.7, 154.5, 141.9, 141.8, 140.6, 132.8, 129.2, 129.1, 128.2, 127.9, 127.3, 100.0, 60.7, 29.4. HRMS (FAB+) m/z calc’d for C21H22N5 [M + H]: 344.1870; found 344.1866.
N-(3-([1,1'-biphenyl]-4-yl)-1-(tert-butyl)-1H-pyrazolo[3,4-d]pyrimidin-4-yl)acrylamide (27) as white solid (0.022 g, 0.055 mmol, 9.5% yield). 1H NMR (300 MHz, d6-DMSO): δ 11.10 (br s, 1 H), 8.80 (s, 1 H), 7.68 (br m, 6 H), 7.48 (m, 2 H), 7.37 (m, 1 H), 6.4 (dd, J = 17.1, 10.3 Hz, 1 H), 5.89 (d, J = 17.1 Hz, 1 h), 5.66 (dd, J = 10.3, 1.6 Hz, 1 H), 1.83 (s, 9 H). Many of the peaks in the 13C NMR spectrum were broadened by a dynamic process that was not investigated further. Poor signal-to-noise prevented acquisition of good 13C NMR spectral data. HRMS (FAB+) m/z calc’d. for C24H24N5O [M + H]: 398.1975; found 398.1962.
N-(3-([1,1'-biphenyl]-4-yl)-1-(tert-butyl)-1H-pyrazolo[3,4-d]pyrimidin-4-yl)-N-acryloylacrylamide (28) as white solid (0.030 g, 0.066 mmol, 11.5% yield). 1H NMR (300 MHz, CDCl3) δ 8.95 (s, 1 H), 7.54-7.64 (m, 6 H), 7.47 (m, 2 H), 7.38 (m, 1 H), 6.3 (ABX quartet, JAB = 16.6 Hz, JAX = 3.2 Hz, JBX = 8.5 Hz, 4 H), 5.63 (dd, J = 8.5, 3.2 Hz, 2 H), 1.91 (s, 9 H). 13C NMR (125 MHz, CDCl3) δ 167.23, 155.4, 154.0, 153.5, 142.4, 141.9, 140.7, 131.0, 131.0, 129.8, 129.5, 129.1, 127.8, 127.6, 127.3, 110.1, 61.8, 29.5.
4.2. Protein purification
Plasmids and primers used in this study are listed in Table S1. Proteins were purified as previously described [21, 41]. E. coli BL21 (DE3) containing the plasmid was grown in LB medium containing 50 μg/L ampicillin, which was shaken at 37 °C to an OD600 of 0.5. The cell culture was cooled down to 22 °C and induced with 1 mM isopropyl-beta-D-thiogalactopyranoside (IPTG) and harvested 8–10 h later by centrifugation. The cell pellet (≈6 g from 2 L) was suspended in the 30 mL lysis buffer [100 mM Tris (pH 7.4), 500 mM KCl, 5 mM MgCl2, 20 mM imidazole, 5% glycerol, 2 mM β-mercaptoethanol, and protease inhibitor tablet (Roche)]. The cells (held in an ice bath) were lysed by six 30-s pulses of sonication, separated by 2-min intervals. The lysate was centrifuged at 20,000 × g for 45 min at 4 °C, and the resulting supernatant was loaded onto a Ni-NTA column [5-mL suspension, preequilibrated with wash buffer (50 mL, 50 mM HEPES [pH 7.4], 150 mM KCl, δ mM MgCl2, and 20 mM imidazole)] and incubated at 4 °C with rotation for 30 min. The column was then flushed with wash buffer (100 mL), and His6-tagged p97 was eluted by stepwise application of 10 mL of imidazole elution buffer (50 mM, 100 mM, 150 mM, 200 mM, or 250 mM imidazole in wash buffer). Fractions from the 200-mM and 250-mM imidazole steps were combined and concentrated with an Amicon Ultra-15 centrifugal filter unit (nominal molecular weight limit = 100 kDa). The mixture (0.5 mL of 20 mg/mL) was then fractionated with a gel filtration column (Tricorn Superdex 200; GE Healthcare), eluted with GF buffer [20 mM HEPES (pH 7.4), 250 mM KCl, and 1 mM MgCl2] at 0.5 mL/min flow rate, and fractions corresponding to an apparent molecular weight of 500– 600 kDa were collected and analyzed by 4–12% SDS/PAGE to evaluate purity (Invitrogen). Fractions that contained p97 of ≥95% purity were concentrated to δ mg/mL, exchanged into storage buffer [20 mM HEPES (pH 7.4), 250 mM KCl, 1 mM MgCl2, 5% glycerol, and 1 mM DTT], aliquoted, frozen in liquid nitrogen, and stored at −80 °C.
4.3. ATPase activity
The purified protein was assayed in 50 μL ATPase assay buffer (50 mM Tris pH 7.4, 20 mM MgCl2, 1mM EDTA, 0.5 mM TCEP, and 0.01% Triton X-100) containing 200 μM AtP. After a 60 min-incubation at room temperature, 50 μL Biomol Green reagent (Enzo Life Sciences) was added to stop the reaction, and the absorbance at 635 nm was measured using a BioTek Synergy Neo 2 plate reader. The eight-dose titrations were performed of the compound to the reaction to determine the IC50 values of the compounds. The results were calculated from six replicates using GraphPad Prism 7.0.
4.4. UbG76V-GFP and ODD-Luc degradation
This assay was described previously [41]. UbG76V-GFP/ODD-Luc HeLa cells were seeded as 5000 cells per well per 30 μL in 384 well plate (Greiner 781091). Clear DMEM containing 2.5% FBS, 1% L-Glutamine, and 1% Penicillin-Streptomycin (Thermo Fisher) was used as an assay medium. The 4 μM of MG132 (50 μL of 4 mM MG132 into 50 mL pre-warmed clear DMEM) was prepared, added into each well (25 μL), and incubated at 37°C for 60 min. Each well was washed with 100 μL PBS twice using a Biotek MultiFlo Microplate dispenser and 30 μL CHX/clear DMEM solution (50 μL of 50 mM cycloheximide into 50 mL pre-warmed clear DMEM) was added. 10 μL of compound was added and incubated for 120 min at 37°C. Then each well was washed with 100 μL PBS, and 20 μL of assay medium was dispensed per well. ImageXpress High Content Confocal Screening System was used to read the GFP signal. Next, 10 μL D-luciferin (2.5 mg/mL in PBS) was added, shaken for δ sec, and incubated at r.t. for 10 min. Synergy NEO multi-mode microplate reader was used to read the Luciferase signal. Data was calculated from quadruplicate to obtain the IC50 using GraphPad Prism 7.0.
4.5. Human 26S Proteasome Activity Assay
Human 26S proteasome complex was affinity purified from HEK293 cells that stably express tagged human Rpn 11, as described [50]. Purified human 26S proteasome (19 nM) was incubated with 5, 25, or 50 μM of MG132 or PPA in assay buffer [50 mM Tris (pH 7.4), 20 mM MgCl2, 1 mM EDTA, 0.5 mM TCEP, and 100 μM ATP] for 30 min at room temperature and the fluorogenic proteasome substrate (succinyl-Leu-Leu-Val-Tyr-AMC, 60 μM; Boston Biochem) was added to initiate the reaction. Fluorescence intensity was monitored every 3 min over 30 min. IC50 values of the compounds were calculated from sextuplicate using GraphPad Prism 7.0.
4.6. Western blot
TCRα-GFP (α-chain of the T-cell receptor fused to GFP) 293 cells were treated with 4 μM MG132 for 1 h, washed, and then incubated in the presence of 50 μM cycloheximide plus 10 μM test compound or MG132 (as a positive control) for 60 min or 120 min prior to harvest. Samples were immunoblotted to detect p97, and TCRα-GFP. The level of p97 served as a loading control. Primary antibodies used were anti-p97 (MA3-004, Thermo Scientific), and anti-GFP (Y1030, 2BScientific), ECL reagent (WBKLS0500, MilliporeSigma) and ChemiDoc MP Imaging System (Bio-Rad) was used to image the blots.
4.7. Docking study
The structure of PPA was prepared using Chem3D by CambridgeSoft. The geometry and energy of the structure were optimized using MM2 force field. AutoDock 4.2 was used to identify the binding mode of PPA responsible for the activity. The receptor WT-p97 in PDB format (PDB code: 5FTK) was obtained from the RCSB protein data bank (https://www.rcsb.org/), which was treated by the removal of water and the addition of hydrogen. The docking results were visualized using Ligplot+ [60] and PyMol (version 0.99; Delano Scientific, San Carlos, CA, USA).
4.8. Anti-proliferative assay
Anti-proliferative activity was measured using the CellTiter Glo® Luminescent Cell Viability Assay (Promega G7572) according to the manufacturer’s procedure. RMPI1640 or DMEM containing 5% FBS and 1% Penicillin-Streptomycin was used as the cell viability assay medium. To find the linear relationship between the relative luminescence unit and the number of viable cells, a standard curve for each cell line was generated. Generally, 30 μL of cell suspension was plated in 384-well plates (Greiner 781080) with serial 2-fold dilutions (from 30000 to 284 cells per well). Twenty-four hours after seeding, 8 μL of assay media containing 5% DMSO was added into each well, and the plates were incubated for an additional 48 hr at 37 °C in a 5% CO2 incubator. To test the anti-proliferative activity of p97 inhibitors, cells were seeded at 750 or 3000 cells per well according to the linear range determined from the standard curve of each cell line. Twenty-four hours after seeding, cells were treated with the compounds (three-fold dilution, eight concentrations). After 48 hours of treatment, cell viability was measured by CellTiter Glo, and IC50 values were calculated using the percentage of growth of treated cells versus the DMSO control. The results were analyzed using GraphPad Prism 7.0.
4.9. Proteomics
HCT116 cells were treated with DMSO, 15 μM or 30 μM PPA for 6 h, and pellets were harvested. The LC-MS samples were prepared by following the instructions of Thermo EasyPep Mini MS Sample Prep Kit (REF A4006) and peptide concentration was tested through Pierce Quantitative Fluorometric Peptide Assay (cat# 23290).
The LC-MS/MS experiments were performed using an EASY-nLC 1000 (ThermoFisher Scientific, San Jose, CA) connected to an Orbitrap Eclipse Tribrid mass spectrometer (Thermo Fisher Scientific, San Jose, CA). The sample (1 μg) in 0.1% FA solution was loaded onto an Aurora UHPLC Column (25 cm x 75 μm, 1.6 μm C18, AUR2-25075C18A, Ion Opticks) and separated over 136 min at a flow rate of 0.35 μL/min with the following gradient: 2-6% Solvent B (7.5 min), 6-25% B (82.5 min), 25-40% B (30 min), 40-98% B (1 min), and 98% B (15 min). Solvent A consisted of 97.9% H2O, 2% ACN, and 0.1% formic acid, and solvent B consisted of 19.9% H2O, 80% ACN, and 0.1% formic acid. An MS1 scan was acquired in the Orbitrap at 120,000 resolution with a scan range of 350-1500 m/z. The AGC target was 4x105, and the maximum injection time was 50 mins. Dynamic exclusion was set to exclude features after 1 time for 60 s with a 10-ppm mass tolerance. Higher-energy collisional dissociation (HCD) fragmentation was performed with 35% collision energy after quadruple isolation of features using a 1.6 m/z isolation window, 5x104 AGC target, and 35 ms maximum injection time. MS2 scans were then also acquired by the Orbitrap with 50,000 resolution. Ion source settings were as follows: ion source type, NSI; spray voltage, 2400 V; ion transfer tube temperature, 275 °C. System control and data collection were performed by Xcalibur software.
The proteomic analysis was performed through Proteome Discoverer 2.4 (Thermo Scientific) using the Uniprot human database and the Byonic search algorithm (Protein Metrics). Percolator FDRs were set at 0.001 (strict) and 0.01 (relaxed). Peptide FDRs were set at 0.001 (strict) and 0.01 (relaxed), with medium confidence and a minimum peptide length of 6. Normalization was performed on the total peptide amount. The limma analysis was performed using R studio following the user guide [61]. The volcano figures were plotted by GraphPad Prism 7.0. The Venn diagram was performed using FunRich 3.1. The heatmap figure was plotted by heatmapper (http://www.heatmapper.ca/). The gene ontology analysis was performed by DAVID 6.8 (https://david-d.ncifcrf.gov/). [62] The enrichment dot bubble figure was plotted by http://www.bioinformatics.com.cn. And the protein-protein interaction analysis was performed using the STRING database (http://www.stringdb.org/). [63]
Supplementary Material
Scheme 1.
Synthesis of compound 8
Scheme 2.
Synthetic Route of Pyrazolopyrimidine analogues
Scheme 3.
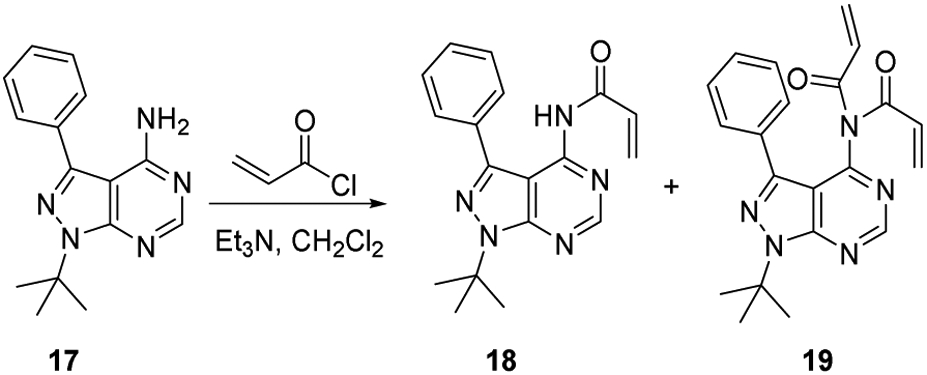
Synthesis of compounds 18 and 19
Scheme 4.
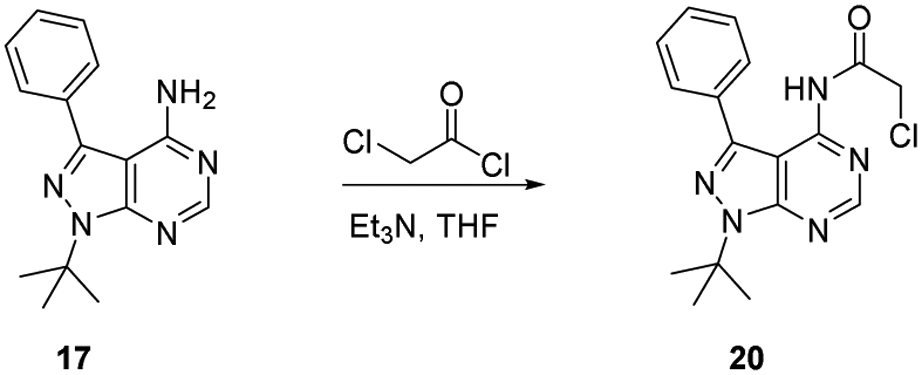
Synthesis of compound 20
Scheme 5.
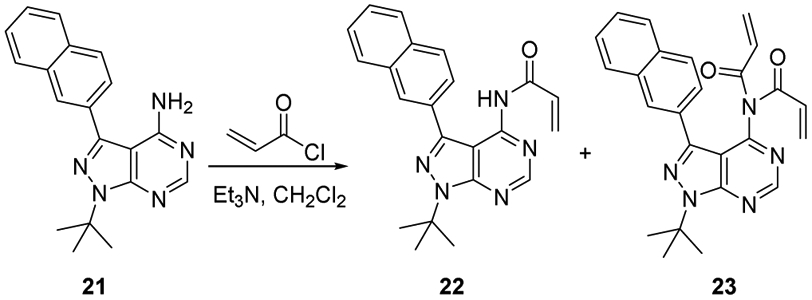
Synthesis of compounds 22 and 23
Scheme 6.
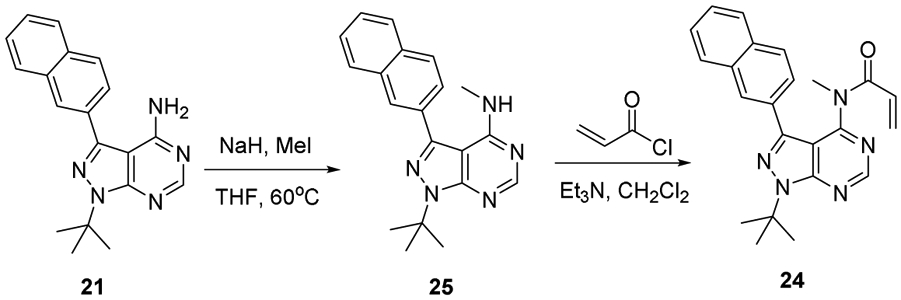
Synthesis of compounds 24 and 25
Scheme 7.
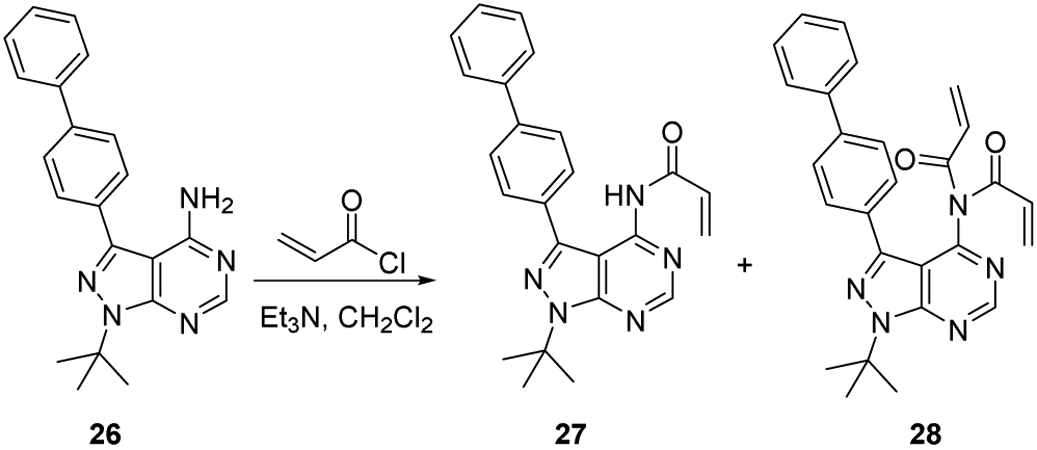
Synthesis of compound 27 and 28
Highlights.
Designed covalent p97 ATPase inhibitors to target the D2 active site.
Identified PPA as a selective p97 covalent inhibitor.
PPA can block growth of CB-5083 and NMS-873 resistant cancer cells.
Proteomic analysis revealed PPA-induced protein level changes.
Acknowledgments
We thank M. S. Cohen, J. Taunton, and K. Shokat for providing compounds 4,5,6, M. Smythe and C. Crews for YU101, A. M. Weissman for PYR-41, C. C. Wu for β-nitrostyrene analogues (compounds 7, 9,10,11,12), Y. Ye at NIDDK/NIH for providing yeast Cdc48 and hamster NSF plasmids. A.C.J. was supported by NIH Grant F32GM082000; A.F.G.G thanks the Natural Sciences and Engineering Research Council (NSERC) of Canada for a PGS D scholarship; R.J.D. was an HHMI Investigator, and this work was funded in part by HHMI; This work was funded in part by the NIH-NINDS (R01NS100815) to T.F.C. and NIH-NIGMS (R01GM080269) to B.M.S. We thank the anonymous reviewers for constructive criticism.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interest
The authors have no conflicts of interest to declare.
Conflict of Interest
We wish to confirm that there are no known conflicts of interest associated with this publication and there has been no significant financial support for this work that could have influenced its outcome.
References
- [1].Confalonieri F, Duguet M, A 200-amino acid ATPase module in search of a basic function, Bioessays, 17 (1995) 639–650. [DOI] [PubMed] [Google Scholar]
- [2].Ranson NA, White HE, Saibil HR, Chaperonins, Biochem. J, 333 (Pt 2) (1998) 233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hirokawa N, Noda Y, Okada Y, Kinesin and dynein superfamily proteins in organelle transport and cell division, Curr. Opin. Cell Biol, 10 (1998) 60–73. [DOI] [PubMed] [Google Scholar]
- [4].Langer T, AAA proteases: cellular machines for degrading membrane proteins, Trends Biochem. Sci, 25 (2000) 247–251. [DOI] [PubMed] [Google Scholar]
- [5].Lee DG, Bell SP, ATPase switches controlling DNA replication initiation, Curr. Opin. Cell Biol, 12 (2000) 280–285. [DOI] [PubMed] [Google Scholar]
- [6].Yang W, Structure and function of mismatch repair proteins, Mutat. Res, 460 (2000) 245–256. [DOI] [PubMed] [Google Scholar]
- [7].Caruthers JM, McKay DB, Helicase structure and mechanism, Curr. Opin. Struct. Biol, 12 (2002) 123–133. [DOI] [PubMed] [Google Scholar]
- [8].Nishi T, Forgac M, The vacuolar (H+)-ATPases--nature's most versatile proton pumps, Nat. Rev. Mol. Cell Biol, 3 (2002) 94–103. [DOI] [PubMed] [Google Scholar]
- [9].Zhang X, Shaw A, Bates PA, Newman RH, Gowen B, Orlova E, Gorman MA, Kondo H, Dokurno P, Lally J, Leonard G, Meyer H, van Heel M, Freemont PS, Structure of the AAA ATPase p97, Mol. Cell, 6 (2000) 1473–1484. [DOI] [PubMed] [Google Scholar]
- [10].Buchberger A, Schindelin H, Hanzelmann P, Control of p97 function by cofactor binding, FEBS Lett., 589 (2015) 2578–2589. [DOI] [PubMed] [Google Scholar]
- [11].Ogura T, Wilkinson AJ, AAA+ superfamily ATPases: common structure--diverse function, Genes Cells, 6 (2001) 575–597. [DOI] [PubMed] [Google Scholar]
- [12].Wang Q, Song C, Yang X, Li CC, D1 ring is stable and nucleotide-independent, whereas D2 ring undergoes major conformational changes during the ATPase cycle of p97-VCP, J. Biol. Chem, 278 (2003) 32784–32793. [DOI] [PubMed] [Google Scholar]
- [13].Song C, Wang Q, Li CC, ATPase activity of p97-valosin-containing protein (VCP). D2 mediates the major enzyme activity, and D1 contributes to the heat-induced activity, J. Biol. Chem, 278 (2003) 3648–3655. [DOI] [PubMed] [Google Scholar]
- [14].Ye Y, Meyer HH, Rapoport TA, Function of the p97-Ufd1-Npl4 complex in retrotranslocation from the ER to the cytosol: dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains, J. Cell Biol, 162 (2003) 71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Platta HW, Debelyy MO, Magraoui FE, Erdmann R, The AAA peroxins Pex1p and Pex6p function as dislocases for the ubiquitinated peroxisomal import receptor Pex5p, Biochem. Soc. Trans, 36 (2008) 99–104. [DOI] [PubMed] [Google Scholar]
- [16].Platta HW, Grunau S, Rosenkranz K, Girzalsky W, Erdmann R, Functional role of the AAA peroxins in dislocation of the cycling PTS1 receptor back to the cytosol, Nat. Cell Biol, 7 (2005) 817–822. [DOI] [PubMed] [Google Scholar]
- [17].Bug M, Meyer H, Expanding into new markets--VCP/p97 in endocytosis and autophagy, J. Struct. Biol, 179 (2012) 78–82. [DOI] [PubMed] [Google Scholar]
- [18].Fessart D, Marza E, Taouji S, Delom F, Chevet E, P97/CDC-48: proteostasis control in tumor cell biology, Cancer Lett., 337 (2013) 26–34. [DOI] [PubMed] [Google Scholar]
- [19].Nishimura N, Radwan MO, Amano M, Endo S, Fujii E, Hayashi H, Ueno S, Ueno N, Tatetsu H, Hata H, Okamoto Y, Otsuka M, Mitsuya H, Matsuoka M, Okuno Y, Novel p97/VCP inhibitor induces endoplasmic reticulum stress and apoptosis in both bortezomib-sensitive and -resistant multiple myeloma cells, Cancer Sci., 110 (2019) 3275–3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Fang CJ, Gui L, Zhang X, Moen DR, Li K, Frankowski KJ, Lin HJ, Schoenen FJ, Chou TF, Evaluating p97 inhibitor analogues for their domain selectivity and potency against the p97-p47 complex, ChemMedChem, 10 (2015) 52–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Chou TF, Brown SJ, Minond D, Nordin BE, Li K, Jones AC, Chase P, Porubsky PR, Stoltz BM, Schoenen FJ, Patricelli MP, Hodder P, Rosen H, Deshaies RJ, Reversible inhibitor of p97, DBeQ, impairs both ubiquitin-dependent and autophagic protein clearance pathways, Proc. Natl. Acad. Sci. U.S.A, 108 (2011) 4834–4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chou TF, Li K, Frankowski KJ, Schoenen FJ, Deshaies RJ, Structure-activity relationship study reveals ML240 and ML241 as potent and selective inhibitors of p97 ATPase, ChemMedChem, 8 (2013) 297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Anderson DJ, Moigne RL, Djakovic S, Kumar B, Rice J, Wong S, Wang J, Yao B, Valle E, Kiss von Soly S, Madriaga A, Soriano F, Menon MK, Wu ZY, Kampmann M, Chen Y, Weissman JS, Aftab BT, Yakes FM, Shawver L, Zhou HJ, Wustrow D, Rolfe M, Targeting the AAA ATPase p97 as an Approach to Treat Cancer through Disruption of Protein Homeostasis, Cancer Cell, 28 (2015) 653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wang F, Li S, Gan T, Stott GM, Flint A, Chou TF, Allosteric p97 Inhibitors Can Overcome Resistance to ATP-Competitive p97 Inhibitors for Potential Anticancer Therapy, ChemMedChem, 15 (2020) 685–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].ClinicalTrails, Study of CB-5339 in Acute Myeloid Leukemia or Myelodysplastic Syndrome, https://clinicaltrials.gov/ct2/show/NCT04402541, July 14 2020.
- [26].Ding R, Zhang T, Wilson DJ, Xie J, Williams J, Xu Y, Ye Y, Chen L, Discovery of Irreversible p97 Inhibitors, J. Med. Chem, 62 (2019) 2814–2829. [DOI] [PubMed] [Google Scholar]
- [27].Polucci P, Magnaghi P, Angiolini M, Asa D, Avanzi N, Badari A, Bertrand J, Casale E, Cauteruccio S, Cirla A, Cozzi L, Galvani A, Jackson PK, Liu Y, Magnuson S, Malgesini B, Nuvoloni S, Orrenius C, Sirtori FR, Riceputi L, Rizzi S, Trucchi B, O'Brien T, Isacchi A, Donati D, D'Alessio R, Alkylsulfanyl-1,2,4-triazoles, a new class of allosteric valosine containing protein inhibitors. Synthesis and structure-activity relationships, J. Med. Chem, 56 (2013) 437–450. [DOI] [PubMed] [Google Scholar]
- [28].Magnaghi P, D'Alessio R, Valsasina B, Avanzi N, Rizzi S, Asa D, Gasparri F, Cozzi L, Cucchi U, Orrenius C, Polucci P, Ballinari D, Perrera C, Leone A, Cervi G, Casale E, Xiao Y, Wong C, Anderson DJ, Galvani A, Donati D, O'Brien T, Jackson PK, Isacchi A, Covalent and allosteric inhibitors of the ATPase VCP/p97 induce cancer cell death, Nat. Chem. Biol, 9 (2013) 548–556. [DOI] [PubMed] [Google Scholar]
- [29].Luo H, Song H, Mao R, Gao Q, Feng Z, Wang N, Song S, Jiao R, Ni P, Ge H, Targeting valosin-containing protein enhances the efficacy of radiation therapy in esophageal squamous cell carcinoma, Cancer Sci., 110 (2019) 3464–3475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Banerjee S, Bartesaghi A, Merk A, Rao P, Bulfer SL, Yan Y, Green N, Mroczkowski B, Neitz RJ, Wipf P, Falconieri V, Deshaies RJ, Milne JL, Huryn D, Arkin M, Subramaniam S, 2.3 Å resolution cryo-EM structure of human p97 and mechanism of allosteric inhibition, Science, 351 (2016) 871–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wang Q, Li L, Ye Y, Inhibition of p97-dependent protein degradation by Eeyarestatin I, J. Biol. Chem, 283 (2008) 7445–7454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wang Q, Shinkre BA, Lee JG, Weniger MA, Liu Y, Chen W, Wiestner A, Trenkle WC, Ye Y, The ERAD inhibitor Eeyarestatin I is a bifunctional compound with a membrane-binding domain and a p97/VCP inhibitory group, PLoS One, 5 (2010) e15479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Pohler R, Krahn JH, van den Boom J, Dobrynin G, Kaschani F, Eggenweiler HM, Zenke FT, Kaiser M, Meyer H, A Non-Competitive Inhibitor of VCP/p97 and VPS4 Reveals Conserved Allosteric Circuits in Type I and II AAA ATPases, Angew. Chem. Int. Ed, 57 (2018) 1576–1580. [DOI] [PubMed] [Google Scholar]
- [34].Alverez C, Bulfer SL, Chakrasali R, Chimenti MS, Deshaies RJ, Green N, Kelly M, LaPorte MG, Lewis TS, Liang M, Moore WJ, Neitz RJ, Peshkov VA, Walters MA, Zhang F, Arkin MR, Wipf P, Huryn DM, Allosteric Indole Amide Inhibitors of p97: Identification of a Novel Probe of the Ubiquitin Pathway, ACS Med. Chem. Lett, 7 (2016) 182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Tillotson J, Bashyal BP, Kang M, Shi T, De La Cruz F, Gunatilaka AA, Chapman E, Selective inhibition of p97 by chlorinated analogues of dehydrocurvularin, Org. Biomol. Chem, 14 (2016) 5918–5921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Tao S, Tillotson J, Wijeratne EMK, Xu YM, Kang M, Wu T, Lau EC, Mesa C, Mason DJ, Brown RV, Clair JJ, Gunatilaka AAL, Zhang DD, Chapman E, Withaferin A Analogs That Target the AAA+ Chaperone p97, ACS Chem. Biol, 10 (2015) 1916–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Segura-Cabrera A, Tripathi R, Zhang X, Gui L, Chou TF, Komurov K, A structure- and chemical genomics-based approach for repositioning of drugs against VCP/p97 ATPase, Sci. Rep, 7 (2017) 44912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Wijeratne EM, Gunaherath GM, Chapla VM, Tillotson J, de la Cruz F, Kang M, U'Ren JM, Araujo AR, Arnold AE, Chapman E, Gunatilaka AA, Oxaspirol B with p97 Inhibitory Activity and Other Oxaspirols from Lecythophora sp. FL1375 and FL1031, Endolichenic Fungi Inhabiting Parmotrema tinctorum and Cladonia evansii, J. Nat. Prod, 79 (2016) 340–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Arai MA, Taguchi S, Komatsuzaki K, Uchiyama K, Masuda A, Sampei M, Satoh M, Kado S, Ishibashi M, Valosin-containing Protein is a Target of 5'-l Fuligocandin B and Enhances TRAIL Resistance in Cancer Cells, ChemistryOpen, 5 (2016) 574–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Noguchi M, Takata T, Kimura Y, Manno A, Murakami K, Koike M, Ohizumi H, Hori S, Kakizuka A, ATPase activity of p97/valosin-containing protein is regulated by oxidative modification of the evolutionally conserved cysteine 522 residue in Walker A motif, J. Biol. Chem, 280 (2005) 41332–41341. [DOI] [PubMed] [Google Scholar]
- [41].Chou TF, Deshaies RJ, Quantitative cell-based protein degradation assays to identify and classify drugs that target the ubiquitin-proteasome system, J. Biol. Chem, 286 (2011) 16546–16554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wang WY, Hsieh PW, Wu YC, Wu CC, Synthesis and pharmacological evaluation of novel beta-nitrostyrene derivatives as tyrosine kinase inhibitors with potent antiplatelet activity, Biochem. Pharmacol, 74 (2007) 601–611. [DOI] [PubMed] [Google Scholar]
- [43].Wang WY, Wu YC, Wu CC, Prevention of platelet glycoprotein IIb/IIIa activation by 3,4-methylenedioxy-beta-nitrostyrene, a novel tyrosine kinase inhibitor, Mol. Pharmacol, 70 (2006) 1380–1389. [DOI] [PubMed] [Google Scholar]
- [44].Cohen MS, Zhang C, Shokat KM, Taunton J, Structural bioinformatics-based design of selective, irreversible kinase inhibitors, Science, 308 (2005) 1318–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Blair JA, Rauh D, Kung C, Yun CH, Fan QW, Rode H, Zhang C, Eck MJ, Weiss WA, Shokat KM, Structure-guided development of affinity probes for tyrosine kinases using chemical genetics, Nat. Chem. Biol, 3 (2007) 229–238. [DOI] [PubMed] [Google Scholar]
- [46].DeLaBarre B, Brunger AT, Complete structure of p97/valosin-containing protein reveals communication between nucleotide domains, Nat. Struct. Biol, 10 (2003) 856–863. [DOI] [PubMed] [Google Scholar]
- [47].Bishop AC, Kung C.-y., Shah K, Witucki L, Shokat KM, Liu Y, Generation of Monospecific Nanomolar Tyrosine Kinase Inhibitors via a Chemical Genetic Approach, J. Am. Chem. Soc, 121 (1999) 627–631. [Google Scholar]
- [48].Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, Pollok BA, Connelly PA, Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation, J. Biol. Chem, 271 (1996) 695–701. [DOI] [PubMed] [Google Scholar]
- [49].Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ, AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility, J. Comput. Chem, 30 (2009) 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Wang X, Chen C-F, Baker PR, Chen P.-l., Kaiser P, Huang L, Mass Spectrometric Characterization of the Affinity-Purified Human 26S Proteasome Complex†, Biochemistry, 46 (2007) 3553–3565. [DOI] [PubMed] [Google Scholar]
- [51].DeLaBarre B, Christianson JC, Kopito RR, Brunger AT, Central pore residues mediate the p97/VCP activity required for ERAD, Mol. Cell, 22 (2006) 451–462. [DOI] [PubMed] [Google Scholar]
- [52].Elofsson M, Splittgerber U, Myung J, Mohan R, Crews CM, Towards subunit-specific proteasome inhibitors: synthesis and evaluation of peptide α', β'-epoxyketones, Chem. Biol, 6 (1999) 811–822. [DOI] [PubMed] [Google Scholar]
- [53].Yang Y, Kitagaki J, Dai RM, Tsai YC, Lorick KL, Ludwig RL, Pierre SA, Jensen JP, Davydov IV, Oberoi P, Li CC, Kenten JH, Beutler JA, Vousden KH, Weissman AM, Inhibitors of ubiquitin-activating enzyme (E1), a new class of potential cancer therapeutics, Cancer Res., 67 (2007) 9472–9481. [DOI] [PubMed] [Google Scholar]
- [54].Her NG, Toth JI, Ma CT, Wei Y, Motamedchaboki K, Sergienko E, Petroski MD, p97 Composition Changes Caused by Allosteric Inhibition Are Suppressed by an On-Target Mechanism that Increases the Enzyme’s ATPase Activity, Cell Chem. Biol, 23 (2016) 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Sanchez-Martin P, Komatsu M, p62/SQSTM1 - steering the cell through health and disease, J. Cell Sci, 131 (2018). [DOI] [PubMed] [Google Scholar]
- [56].Bentson LF, Agbor VA, Agbor LN, Lopez AC, Nfonsam LE, Bornstein SS, Handel MA, Linder CC, New point mutation in Golga3 causes multiple defects in spermatogenesis, Andrology, 1 (2013) 440–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Dowd CS, Herrick-Davis K, Egan C, DuPre A, Smith C, Teitler M, Glennon RA, 1-[4-(3-Phenylalkyl)phenyl]-2-aminopropanes as 5-HT2APartial Agonists, J. Med. Chem, 43 (2000) 3074–3084. [DOI] [PubMed] [Google Scholar]
- [58].Hanefeld U, Rees CW, White AJP, Williams DJ, One-pot synthesis of tetrasubstituted pyrazoles—proof of regiochemistry, J. Chem. Soc., Perkin Trans 1, (1996) 1545–1552. [Google Scholar]
- [59].Guillaumet G, Patriciu O-I, Pillard C, Fînaru A-L, Sãndulescu I, Synthesis of Nitro N,N′-Dipyridinylamines via Oxidative Nucleophilic Substitution of Hydrogen, Synthesis, 2007 (2007) 3868–3876. [Google Scholar]
- [60].Laskowski RA, Swindells MB, LigPlot+: multiple ligand-protein interaction diagrams for drug discovery, J. Chem. Inf. Model, 51 (2011) 2778–2786. [DOI] [PubMed] [Google Scholar]
- [61].Smyth GK, Thorne NP, Wettenhall J, Limma: Linear Models for Microarray Data User's Guide. Software; manual available from http://www.bioconductor.org., 2003. [Google Scholar]
- [62].Huang da W, Sherman BT, Zheng X, Yang J, Imamichi T, Stephens R, Lempicki RA, Extracting biological meaning from large gene lists with DAVID, Curr. Protoc. Bioinformatics, Chapter 13 (2009) Unit 13 11. [DOI] [PubMed] [Google Scholar]
- [63].Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, Jensen LJ, Mering CV, STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets, Nucleic Acids Res., 47 (2019) D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.