

Abstract
Inhibitors of cAMP-phosphodiesterase 4 (PDE4) exert a number of promising therapeutic benefits, but adverse effects, in particular emesis and nausea, have curbed their clinical utility. Here, we show that PAN-selective inhibition of PDE4, but not inhibition of PDE3, causes a time- and dose-dependent accumulation of chow in the stomachs of mice fed ad libitum without changing the animal’s food intake or the weight of their intestines, suggesting that PDE4 inhibition impairs gastric emptying. Indeed, PDE4 inhibition induced gastric retention in an acute model of gastric motility that traces the passage of a food bolus through the stomach over a 30 min time period. In humans, abnormal gastric retention of food is known as gastroparesis, a syndrome predominated by nausea (>90% of cases) and vomiting (>80% of cases). We thus explored the abnormal gastric retention induced by PDE4 inhibition in mice under the premise that it may represent a useful correlate of emesis and nausea. Delayed gastric emptying was produced by structurally distinct PAN-PDE4 inhibitors including Rolipram, Piclamilast, Roflumilast and RS25344, suggesting it is a class effect. PDE4 inhibitors induced gastric retention at similar or below doses commonly used to induce therapeutic benefits (e.g. 0.04 mg/kg Rolipram), thus mirroring the narrow therapeutic window of PDE4 inhibitors in humans. YM976, a PAN-PDE4 inhibitor that does not efficiently cross the blood-brain barrier, induced gastroparesis only at significantly higher doses (≥1 mg/kg). This suggests that PDE4 inhibition may act in part through effects on the autonomic nervous system regulation of gastric emptying, and that PDE4 inhibitors that are not brain-penetrant may have an improved safety profile. The PDE4 family comprises four subtypes, PDE4A, B, C and D. Selective ablation of any of these subtypes in mice did not induce gastroparesis per se, nor did it protect from PAN-PDE4 inhibitor-induced gastroparesis, indicating that gastric retention may result from the concurrent inhibition of multiple PDE4s. Thus, potentially, any of the four PDE4 subtypes may be targeted individually for therapeutic benefits without inducing nausea or emesis. Acute gastric retention induced by PDE4 inhibition is alleviated by treatment with the widely used prokinetic Metoclopramide, suggesting a potential of this drug to alleviate the side effects of PDE4 inhibitors. Finally, given that the cause of gastroparesis remains largely idiopathic, our findings open the possibility that a physiologic or pathophysiologic downregulation of PDE4 activity/expression may be causative in a subset of patients.
Keywords: gastric retention, emesis, nausea, PDE4D, xylazine/ketamine anesthesia
Introduction
cAMP is a ubiquitous second messenger that transduces the action of a plethora of extracellular signals, from hormones and neurotransmitters, to odorants and pH, into a diverse set of cellular responses1–4. The cellular concentration of cAMP is determined by the equilibrium between the rate of its synthesis by adenylyl cyclases, and the rate of its hydrolysis and inactivation by cyclic nucleotide phosphodiesterases (PDEs)4. The mammalian PDEs comprise a large group of isoenzymes that are encoded by 21 genes, and are divided into eleven PDE families by their sequence homology, substrate specificity and pharmacological properties5,6.
The PDE4 family is the largest and most complex PDE family, and arguably most widely expressed throughout the body. It comprises four genes, PDE4A, PDE4B, PDE4C and PDE4D7–9. Each PDE4 gene is expressed as several protein variants that are generated via use of alternative promoters and transcription start sites or alternative splicing, so that in total, likely more than 25 PDE4 proteins are expressed in humans and other mammals. While individual PDE4 protein variants often exhibit a unique cell- and tissue distribution, essentially all cells and tissues express a barcode of PDE4 isoforms. As a result of this ubiquitous presence of PDE4 in the body, PAN-selective inhibition of PDE4 produces a large number of effects of potentially high therapeutic value10–13 including potent anti-inflammatory effects14–16, improvement of memory and cognition17–20, as well as cardiovascular21–23, metabolic24 and antineoplastic25–27 effects.
However, despite major research and development efforts, only a handful of PDE4 inhibitors are now approved for clinical use including Roflumilast (chronic obstructive pulmonary disease), Apremilast (psoriasis) and Crisaborole (atopic dermatitis)11,14,16,28. The main impediment to a wider clinical utility and commercial success are adverse gastrointestinal effects including nausea, emesis and diarrhea that limit the therapeutic window. The main approach to mitigate these side effects is to inhibit only a fraction of the total PDE4 in the body. For some indications, the local delivery of a PDE4 inhibitor to the target site, while limiting its systemic exposure, is a suitable approach. The topical application of Crisaborole for dermatitis15 or clinical trials exploring inhaled preparations of PDE4 inhibitors for inflammatory lung disease are examples of that29. If a local delivery is not feasible, PDE4 inhibitor development is geared towards selectively targeting specific PDE4 subtypes or distinct conformational states of PDE413,30,31.
Using genetic and immunologic approaches, it was first established in the 1990s that the Rolipram-sensitive PDE4 activity found in many cells and tissues is comprised of multiple protein variants generated from distinct PDE4 genes32–34. Subsequent studies showed that genetic ablation of individual PDE4 subtypes in mice35,36 or siRNA-mediated knockdown in cells37,38 produces clearly distinct effects, suggesting that individual PDE4 enzymes serve unique and non-overlapping physiological roles and generally cannot functionally replace or compensate for each other. Since then, major progress has been made in deciphering the unique roles of individual PDE4 subtypes and protein variants in many cells and tissues, as well as their unique post-translational regulation and recruitment into macromolecular signaling complexes that underpin the unique roles of individual PDE4 isoforms even if they are expressed in the same cell9,21,39,40. As a result, considerable knowledge as to the specific PDE4 subtypes and variants that represent promising targets for development of novel therapeutics is available11–15,17,19,26,31,18. Conversely, which PDE4 subtypes and variants are involved in mediating the adverse effects of PDE4 inhibitors remains ill understood. As a result, it is unclear which portion of PDE4 in the body must be avoided in the development of PDE4 inhibitors with an improved safety profile. This is largely due to an inherent difficulty in assessing the role of individual PDE4s in emesis and nausea in animal models. Despite development of inhibitors with some subtype selectivity41–46, there are no highly selective inhibitors for individual PDE4 subtypes available to date that would allow conclusive testing of this question in animals or humans. Genetic inactivation of individual PDE4 subtypes in mice and rats47,48 revealed many potentially therapeutic benefits of targeting specific PDE4s. However, as these species are anatomically unable to vomit, these models cannot be used to study emesis directly. Expression of various PDE4 isoforms in the emesis centers of the brain, such as the area postrema, has been explored49–51. However, there is insufficient evidence for the idea that inhibition of PDE4 within these regions is predominantly or exclusively responsible for the side effect profile of PAN-PDE4 inhibitors.
The effect of PDE4 inhibitors in a model of xylazine/ketamine-induced anesthesia in various animal species has received particular attention and has since largely shaped opinion around the emetic potential of these drugs. In ferrets, treatment with PAN-PDE4 inhibitors mimics the effects of α2-adrenoceptor antagonists, in that both induce vomiting as well as shorten the duration of α2-adrenoceptor-dependent, xylazine/ketamine-induced anesthesia52. Moreover, treatment with α2-adrenoceptor agonists alleviated vomiting induced by PDE4 inhibitors in ferrets, leading to the hypothesis that PDE4 inhibitor-induced emesis is due to α2-adrenoceptor antagonism52,53. The xylazine/ketamine anesthesia test has since been used extensively to assess the emetic potential of PDE4 inhibitors in species that are not able to vomit, such as rats and mice54,55. In a key paper, it was reported that genetic ablation of PDE4D shortens xylazine/ketamine-induced anesthesia in mice, thus mimicking the effects of PAN-PDE4 inhibitors, whereas ablation of PDE4B did not55. The authors thus proposed that inhibition of PDE4D is responsible for the emetic effects of PAN-PDE4 inhibitors. This has led to significant efforts to develop PDE4 inhibitors selective for PDE4B over PDE4D in expectation of an improved safety profile45,56–59; at the same time largely forgoing any therapeutic benefits potentially derived from inhibition of PDE4D. However, while the xylazine/ketamine-anesthesia test is a reliable measure for α2-adrenoreceptor antagonism, it does have limitations as a predictor of emetic potential, as shown recently60. Moreover, at least one study has shown that inhibitors with some selectivity for PDE4D exhibited reduced vomiting compared to PAN-PDE4 inhibitors in several species46. Thus, in our opinion, the association of individual PDE4 subtypes with emesis and/or nausea remains inconclusive.
During a cancer xenograft study that involved treatment of mice with PAN-PDE4 inhibitors for an extended period, we noted that PDE4 inhibitor-treated mice had substantially enlarged stomachs due to accumulation of dry food. In humans, abnormal gastric retention of food is a well-established symptomatic disorder termed gastroparesis, which is defined by delayed gastric emptying in the absence of mechanical obstruction61–63. Patients present with a constellation of symptoms of which nausea (>90%), vomiting (>80%), and early satiety and post-prandial fullness (60%) are most frequent62–66. Although the cause of gastroparesis is idiopathic in the majority of cases67, diabetes, post gastric surgery (e.g. vagal nerve injury), various neurologic (e.g. Parkinson’s disease), endocrine and eating disorders as well as medications such as opioids and anticholinergics are established causes61–63,68. Given the strong association of gastroparesis with nausea and emesis in humans, we further explored the abnormal gastric retention of food we observed in mice treated with PDE4 inhibitors under the premise that it may represent a useful correlate of nausea and emesis in humans and hence provide a tool to further delineate the role of individual PDE4s in mediating the established adverse effects of PDE4 inhibitors.
MATERIAL AND METHODS
Drugs
Piclamilast69,70 (RP73401; 3-(Cyclopentyloxy)-N-(3,5-dichloropyridin-4-yl)-4-methoxybenzamide), Rolipram71 (4-(3-cyclopentyloxy-4-methoxyphenyl)pyrrolidin-2-one), Roflumilast28,72 (3-(cyclopropylmethoxy)-N-(3,5-dichloropyridin-4-yl)-4-(difluoromethoxy)benzamide), Cilostamide73 (N-cyclohexyl-N-methyl-4-[(2-oxo-1H-quinolin-6-yl)oxy]butanamide), Metoclopramide (4-amino-5-chloro-N-[2-(diethylamino)ethyl]-2-methoxybenzamide), Clonidine (N-(2,6-dichlorophenyl)-4,5-dihydro-1H-imidazol-2-amine) and Yohimbine (methyl (1S,15R,18S,19R,20S)-18-hydroxy-1,3,11,12,14,15,16,17,18,19,20,21-dodecahydroyohimban-19-carboxylate) were from Cayman Chemical (Ann Arbor, MI), YM97674 (4-(3-chlorophenyl)-1,7-diethylpyrido[2,3-d]pyrimidin-2-one) was from Tocris/Bio-Techne (Minneapolis, MN) and RS2534475 (1-(3-nitrophenyl)-3-(pyridin-4-ylmethyl)pyrido[2,3-d]pyrimidine-2,4-dione) was obtained from Santa Cruz Biotech (Santa Cruz, CA). All PDE inhibitors, Clonidine and Yohimbine were initially dissolved in DMSO, subsequently diluted into phosphate-buffered saline (PBS), pH 7.4, containing final concentrations of 5% DMSO and 5% Cremophor EL (Millipore Sigma, St. Louis, MO) and were applied by intraperitoneal injection (100 μl per 20 g body weight). Metoclopramide was dissolved in 1 % methylcellulose in water and administered via oral gavage.
Animals
Female nude athymic mice (Crl:NU(NCr)-Foxn1nu; 490 (Homozygous)) were obtained from Charles River Laboratories (Wilmington, MA). Beginning at day 6 after placement of a xenograft under the skin at their flanks, mice were injected intraperitoneally (i.p.) twice daily with Piclamilast (5 mg/kg) or solvent for the following 14 days. Upon euthanasia and post-mortem dissection of the animals, the stomachs were then extracted and weighed. Wild type C57BL/6 mice for experimentation were generated in-house using breeders obtained from Charles River Laboratories (Wilmington, MA). Mice deficient in PDE4A76, PDE4B36 and PDE4D35 mice were generated by Drs. S.-L. Catherine Jin and Marco Conti (Stanford University, CA; also see48) and kindly distributed via the Mutant Mouse Resource and Research Centers (MMRRC, http://www.mmrrc.org, PDE4A stock ID# 034793-UCD, PDE4B stock ID# 034682-UCD, PDE4D stock ID# 034588-UCD) of the University of California at Davis. PDE4C knockout mice (Pde4ctm1.1(KOMP)Wtsi/J) were generated by the National Institutes of Health (NIH) Knockout Mouse Program (KOMP; www.komp.org) and kindly distributed via the KOMP repository at the University of California at Davis. PDE4C knock-out mice were generated through targeted deletion of ~4500 nucleotides starting at position 70745158 and ending at position 70749713 of chromosome 8 (Genome Build GRCm38; see http://www.informatics.jax.org/allele/MGI:5585589 for details) resulting in the removal of exons 4 to 16 of mouse PDE4C1 (NP_001297394.1), thereby removing both the upstream conserved regions 1 and 2 (UCR1/2) as well as a major portion of the catalytic domain of PDE4C (please see Supplementary Fig. 1 for a description of the PDE4C knock-out mouse; additional details are available on the website of the Mutant Mouse Regional Resource Centers (MMRRC; http://www.mmrrc.org; Stock number 049025-UCD). All PDE4 knockout mouse colonies were maintained on a C57BL/6 background by Het/Het breeding and homozygous PDE4-KO mice were compared to their respective wildtype littermates. All mice were group housed 4 mice per cage with ad libitum access to food and water and were maintained in a temperature-controlled (22–23°C) vivarium with a 12-h light/dark cycle. Adult mice ≥10 weeks of age and of either sex were used for experimentation by equally dividing cage littermates into experimental groups. They are indicated by filled circles (females) and open squares (males), respectively. Experimenters were blinded to the identity of the injected drugs until data acquisition and analysis were completed. All experiments and procedures were conducted in accordance with the guidelines described in the Guide for the Care and Use of Laboratory Animals (National Institutes of Health, Bethesda, MD, USA) and were approved by the University of South Alabama Institutional Animal Care and Use Committee.
Measurement of stomach weight in mice fed ad libitum
Mice were single housed for one week prior to the experiment. They were then injected intraperitoneally (i.p.) twice daily with test drugs or solvent controls as indicated, while food consumption was measured. Mice were euthanized at the indicated time points using EUTHASOL® Euthanasia Solution (Patterson Veterinary, Greeley, CO) followed by cervical dislocation, and stomachs and intestines were then extracted and weighed.
Measurement of acute gastric retention
Mice were housed on wire racks to prevent ingestion of feces and fasted overnight while having free access to water. To prepare the food bolus, DietGel® Recovery (ClearH2O, Portland, ME) was dissolved to 40% w/v in water by heating, subsequently cooled to 37°C and mixed with 2 mg/ml Fluorescein isothiocyanate (FITC)-Dextran (Millipore Sigma, St. Louis, MO). Unless indicated otherwise, the food bolus was administered via oral gavage at 30 min after i.p. injection of test drugs or solvent controls. Mice were euthanized at the indicated times after food administration, the stomach and small intestines were extracted, minced with scissors and suspended in 5 ml of 2x phosphate-buffered saline by vortexing. Solids were allowed to settle for 10 min and 150 ul of supernatants were transferred to 96-well plates for measurement of FITC fluorescence (excitation at 485/20 nm, emission at 528/20 nm) using a Biotek Synergy 2 (Biotek Instruments, Winooski, VT) plate reader. Gastric retention of food was calculated as the amount of fluorescence (relative fluorescence units) recovered in stomach or intestine extracts as a fraction of the total fluorescence of the administered food bolus.
Statistics
Data are expressed as the mean ± SEM and Student’s t-test or one-way ANOVA followed by Tukey’s post hoc test was used to determine differences between two, or more than two groups, respectively. Differences were considered significant if p<0.05.
RESULTS
Treatment with the PDE4 inhibitor Piclamilast induces retention of food in the stomach of mice
As part of a cancer xenograft study, female nude athymic mice were treated with the PAN-PDE4 inhibitor Piclamilast69,70 (RP73401; 5 mg/kg, i.p., twice daily) for 14 days. During post-mortem dissection of the carcasses, we noted an unexpected, substantial increase in the size and weight of the stomachs of Piclamilast-treated animals, compared to solvent controls (Fig. 1A/B). This ≥3-fold weight increase results from the accumulation of rodent chow in the stomachs of Piclamilast-treated mice. The effect of Piclamilast treatment on chow accumulation in the stomach did not require drug treatment for 14 days. As shown in Fig. 1C/D, treatment with the same dose of Piclamilast, 5 mg/kg, induced retention of food in the stomachs of C57BL/6 mice fed ad libitum in as little as 24 h, and a comparable ≥3-fold increase in stomach weight was reached within 72 h. Finally, Piclamilast-induced chow accumulation in the stomach was observed in both female and male mice (Supplementary Fig. 2A).
Fig. 1. Treatment with the PAN-PDE4 inhibitor Piclamilast triggers a time-dependent accumulation of food in the stomach of mice fed ad libitum.

(A/B) Female athymic nude mice were treated with the PDE4 inhibitor Piclamilast (5 mg/kg, i.p., twice daily) for 14 days as part of a xenograft study. Post-mortem dissections of the animals revealed the accumulation of large amounts of chow in the stomachs of Piclamilast-treated mice compared to solvent controls (Mock). Representative images are shown in (A). The graph in (B) represents the mean ± SEM. *** indicates p<0.001 as determined using Student’s t-test. (C/D) Female C57BL/6 mice were treated with the PDE4 inhibitor Piclamilast (5 mg/kg, i.p., twice daily) for up to 72 h while having free access to food and water. After euthanasia, stomachs were extracted and weighed. The graph in (C) represents the mean ± SEM. *** indicates p<0.001 using one-way ANOVA with Tukey’s post hoc test. Representative images are shown in (D).
Gastric retention of food is a class effect of PAN-PDE4 inhibitors
To determine whether the increased stomach weights are an effect unique to Piclamilast, or a class effect of PAN-PDE4 inhibitors, we next treated mice fed ad libitum with a number of structurally distinct PAN-PDE4 inhibitors including Rolipram71, Roflumilast28,72 and RS2534475, as well as the PDE3-selective inhibitor Cilostamide73. As shown in Fig. 2, treatment with any of the PAN-PDE4 inhibitors (all at 5 mg/kg, i.p., twice daily for 72 h) resulted in significant increases in stomach weights, whereas treatment with the PDE3 inhibitor Cilostamide (10 mg/kg, i.p., twice daily for 72 h) did not, suggesting that abnormal food accumulation in the stomachs of mice is a class effect of PAN-PDE4 family-selective inhibitors.
Fig. 2. Retention of food in the stomach of mice is a class effect of PAN-PDE4 inhibitors.
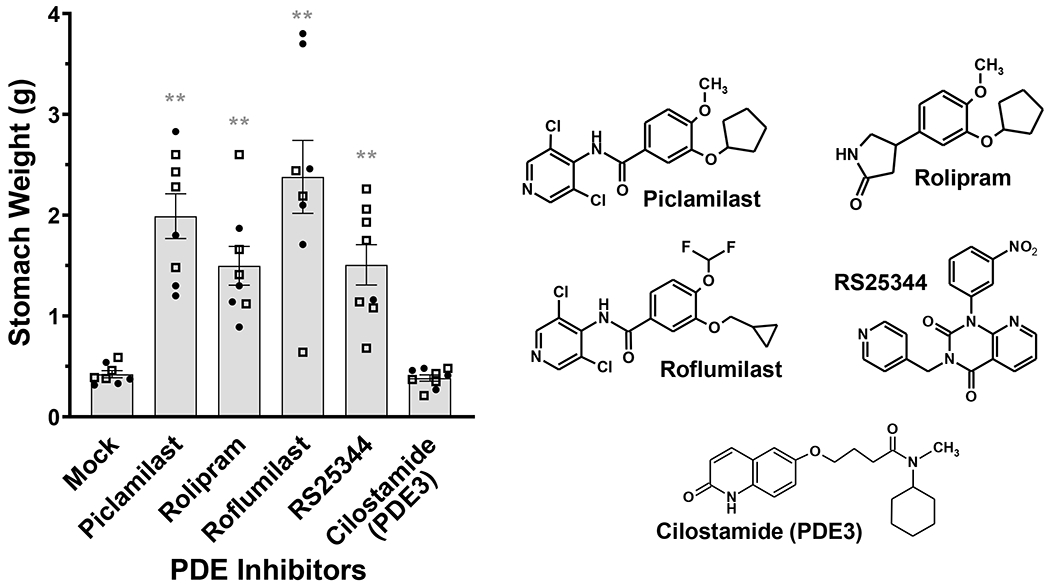
C57BL/6 mice were treated with the structurally distinct PAN-PDE4 inhibitors Piclamilast, Rolipram, Roflumilast and RS25344 (each 5 mg/kg, i.p., twice daily) as well as the PDE3 inhibitor Cilostamide (10 mg/kg, i.p., twice daily) or solvent controls (Mock) for 72 h while animals had free access to food and water. After euthanasia, stomachs were extracted and weighed. Data represent the mean ± SEM. Female mice are represented as filled circles (●), males as open squares (□). Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test and is indicated as ** (p<0.01). The chemical structures of the PDE4 inhibitors tested are shown for comparison.
In mice fed ad libitum, Piclamilast dose-dependently increased the weight of their stomachs (Fig. 3A) but had no effect on either the weight of the intestines (Fig. 3B) or food consumption (Fig. 3C). In addition, we did not observe intestinal obstruction during dissection of any of the animals in this study. This suggests that food accumulation in the stomachs of Piclamilast-treated mice is due to abnormal retention of food in the stomach, but not due to back-up of food retained in the intestines or effects of PDE4 inhibition on appetite.
Fig. 3. PDE4 inhibition reduces gastric emptying, without increasing food accumulation in the intestines or increasing food intake.

Male C57BL/6 mice were treated with the indicated doses of the PDE4 inhibitor Piclamilast (i.p., twice daily) for 72 h while animals had free access to food and water. After euthanasia, stomachs and intestines were extracted and weighed (A/B) and dry food consumed over this time period was measured (C). Data represent the mean ± SEM. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test and is indicated as *** (p<0.001) and * (p<0.05).
PAN-PDE4 inhibition delays stomach emptying in an acute model of gastric retention
To confirm this idea and to limit the impact of pharmacokinetics on further mechanistic studies, we established an acute model of gastric retention. In brief, a food bolus of 200 μl DietGel® traced with FITC-dextran is given to fasted mice via oral gavage. The mice are euthanized at different time intervals after food application, the stomachs and small intestines extracted and homogenized, and the amount of FITC-dextran retained in these tissues is assessed by fluorescence microscopy or spectroscopy. As shown in Fig. 4A/B, the majority of the food bolus (~80%) passes through the stomach within 20 to 30 min of food delivery and is then found in the small intestines or has entered the cecum (Fig. 4A, compare bottom/left and center tissues). Conversely, treatment with the PAN-PDE4 inhibitor Piclamilast (1 mg/kg, i.p., 30 min prior to food bolus) causes a major delay in gastric emptying (Fig. 4A, top/right tissue) with >50% of the food bolus remaining in the stomach even at 60 min after food application (Fig. 4C), and this Piclamilast-induced delay in gastric emptying was observed in both female and male mice (Supplementary Fig. 2B). This confirms the hypothesis that chow accumulation in the stomachs of mice fed ad libitum (Fig. 1–3) is due to impaired gastric emptying.
Fig. 4. Development of an assay to assess acute gastric retention.

Fasted C57BL6 mice of either sex were injected (i.p.) with the PDE4 inhibitor Piclamilast (1 mg/kg, i.p.) or solvent control (Mock). Thirty minutes later, a food bolus of 200 μl Clear H2O® Recovery diet gel diluted in water and mixed with 2 mg/ml FITC-dextran was delivered by oral gavage. Animals were euthanized at the indicated time points, the stomach and small intestine were extracted and the FITC-labeled diet gel present in the tissues assessed by fluorescence imaging (A) or the measurement of fluorescence intensity in homogenized tissues using a plate reader (B/C). (A) Shown are composite FITC-fluorescence/light images of the gastrointestinal tracts of solvent-treated mice (Mock) extracted 2 min after food bolus delivery (left tissue) or 32 min after food administration (middle tissue). The tissue on the right was extracted from a Piclamilast-treated (1 mg/kg, i.p.) mouse at 32 min after food administration. (B) Shown is a time-course of FITC-fluorescence distribution between stomachs and small intestines of solvent-treated mice (n≥6). (C) Comparison of the gastric retention of the food bolus in the stomachs of solvent (Mock)- and Piclamilast (1 mg/kg, i.p.)-treated mice as assessed by FITC-fluorescence (n ≥ 6).
Mirroring findings in the ad libitum model (Fig. 2), treatment with a number of structurally distinct PDE4 inhibitors including Piclamilast, Rolipram, Roflumilast or RS25344 (all 1 mg/kg, i.p., 30 min prior to food bolus), but not the PDE3-selective inhibitor Cilostamide (1 mg/kg, i.p.), increased gastric retention, confirming it as a class effect of PAN-PDE4 inhibitors. Dose-response curves revealed that as little as 0.04 mg/kg Rolipram (Fig. 5B) or 0.2 mg/kg Piclamilast (Fig. 5C) significantly increases gastric retention. Most published studies use the same or higher doses of these PDE4 inhibitors to produce therapeutic effects such as anti-inflammatory, memory- and cognition enhancing, or antineoplastic effects 14,18,77 as well as emesis52,54,77,78 in animal models. Thus, doses that effectively induce abnormal gastric retention parallel the doses required for most therapeutic benefits as well as adverse effects of PDE4 inhibitors in mice and other animals.
Fig. 5. PAN-PDE4 inhibition causes acute gastroparesis.

Mice were injected (i.p.) with the indicated doses of PDE inhibitors or solvent control (Mock). Thirty minutes later, a 200 μl food bolus traced with FITC-dextran was delivered by oral gavage and 30 min after that the animals were euthanized, the stomach extracted, and FITC retained in the stomach measured via fluorescence spectroscopy. Female mice are represented as filled circles (●), males as open squares (□). (A) Shown is the effect of the brain-penetrant PAN-PDE4 inhibitors Piclamilast, Rolipram, Roflumilast and RS25344, as well as the poorly brain-penetrant PAN-PDE4 inhibitor YM976, and the PDE3-selective inhibitor Cilostamide (all 1 mg/kg). (B/C/D) Shown are dose response curves for the PAN-PDE4 inhibitors Rolipram (B), Piclamilast (C), and YM976 (D). The chemical structure of YM976 is shown above the graph in (D). Data represent the mean ± SEM of FITC fluorescence retained in the stomach as % of total fluorescence of the food bolus. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test and is indicated as * (p<0.05), ** (p<0.01) and *** (p<0.001).
Prior studies have shown that the PAN-PDE4 inhibitor YM976 has a similar potency in inhibiting cAMP hydrolysis by PDE4 compared to Piclamilast or Roflumilast, but is distinguished by its poor brain penetrance74. As shown in Figs. 5A/D, YM976 exhibits a reduced potency in inducing gastric retention compared to the brain-penetrant compounds tested, suggesting that PDE4 inhibitor-induced gastric retention may be due, at least in part, to inhibition of PDE4 in the brain.
Concurrent, but not subtype-selective ablation of PDE4s induces gastroparesis
The PDE4 family comprises four subtypes, PDE4A, B, C and D, and prior studies utilizing the genetic ablation of PDE4A76, PDE4B36 or PDE4D35 in mice48, or using siRNA-mediated knockdown of individual PDE4 isoforms in cells37,38 have shown that individual PDE4 subtypes play unique and non-overlapping roles in the body. To determine whether one of the four PDE4 subtypes is predominantly associated with impaired gastric emptying, we compared acute gastric retention in mice deficient in PDE4A (Fig. 6A), PDE4B (Fig. 6B), PDE4C (Fig. 6C) or PDE4D (Fig. 6D) to their respective wildtype littermates. As shown in Figs. 6A–D, genetic ablation of any of the four PDE4 subtypes per se did not increase gastric retention. To exclude the possibility that compensatory changes may have obscured the role of a specific PDE4 subtype, the effect of PDE4 inhibitor treatment on gastric retention was then tested in each knockout mouse line. As shown in Figs. 6A to D, treatment with Piclamilast (1 mg/kg) increased gastric retention in each of the four PDE4 knockout mouse lines to levels similar to those in their wildtype littermates. These findings suggest that inactivation of any individual PDE4 subtype does not cause gastric retention per se, which instead likely results from the concurrent inhibition of multiple PDE4 subtypes. Which PDE4 subtypes must be concurrently inhibited and how these PDE4 subtypes are involved in the regulation of gastric retention remains to be delineated.
Fig. 6. Selective ablation of individual PDE4 subtypes does not produce gastroparesis, nor does it protect from PAN-PDE4 inhibitor-induced gastroparesis.

A 200 μl food bolus traced with FITC-dextran was delivered by oral gavage into mice. The animals were euthanized 30 min afterwards, the stomach extracted, and FITC retained in the stomach measured via fluorescence spectroscopy. Female mice are represented as filled circles (●), males as open squares (□). (A-D) Mice deficient in either PDE4A (A), PDE4B (B), PDE4C (C) or PDE4D (D) do not exhibit gastric retention compared to their respective wildtype littermates (two left columns of each graph; Mock), nor are they protected from the effect of treatment with the PAN-PDE4 inhibitor Piclamilast (1 mg/kg, i.p.) shown in the two right columns of each graph. Data represent the mean ± SEM of FITC fluorescence retained in the stomach as % of total fluorescence of the food bolus. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test. In all four PDE4 knockout mouse lines (A-D), there were no significant differences (p>0.05) in gastric retention between knockout mice and their respective wildtype littermates either under solvent-treated or Piclamilast-treated groups. Conversely, for each individual genotype, gastric retention in Piclamilast-treated mice was significantly greater than in solvent-treated mice (p<0.001).
Role of α2-adrenoceptor signaling in PDE4 inhibitor-induced gastroparesis in mice
Given that in humans, nausea and emesis are predominant symptoms of abnormal gastric retention, and given that they are also the main adverse effects of PAN-PDE4 inhibitor treatment, we further explored the idea that PDE4 inhibitor-induced gastric retention in mice may serve as a correlate of emesis in humans. Up until now, PDE4-inhibitor-induced α2-adrenoceptor antagonism, which shortens the duration of xylazine/ketamine-induced anesthesia, has been widely used to assess the emetic potential of PDE4 inhibitors in animal species that cannot vomit, in particular in mice and rats54,55. Thus, we wished to determine whether α2-adrenoceptor antagonism may also mediate PDE4 inhibitor-induced gastric retention. As shown in Fig. 7A, treatment with Clonidine, an α2-adrenoceptor agonist, potently induced gastric retention, thus paralleling the effect of PDE4 inhibition, whereas treatment with Yohimbine, an α2-adrenoceptor antagonist, did not. These findings are in contradiction to the xylazine/ketamine anesthesia test, in which α2-adrenoceptor antagonists, such as Yohimbine, parallel the effect of PDE4 inhibitors to shorten anesthesia. Thus, these data suggest that distinct signaling cascades must mediate the effects of PDE4 inhibitors on gastric retention and xylazine/ketamine-induced anesthesia.
Fig. 7. Effect of α2-adrenoceptor agonism/antagonism or treatment with the prokinetic Metoclopramide on PDE4 inhibitor-induced gastroparesis.

A 200 μl food bolus traced with FITC-dextran was delivered by oral gavage into mice. The animals were euthanized 30 min afterwards, the stomach extracted, and FITC retained in the stomach measured via fluorescence spectroscopy. Female mice are represented as filled circles (●), males as open squares (□). (A) Shown is the effect of the α2-adrenoceptor agonist Clonidine, the α2-adrenoceptor antagonist Yohimbine, or solvent controls (Mock), all injected i.p. 30 min prior to delivery of the food bolus, on acute gastric retention in female C57BL6 mice. (B) Shown is the effect of treatment with the prokinetic Metoclopramide (10 mg/kg, o.g.) delivered 30 min prior to PDE4 inhibitor injection (Rolipram, 0.2 mg/kg, i.p.) on acute gastric retention. Data represent the mean ± SEM of FITC fluorescence retained in the stomach as % of total fluorescence of the food bolus. Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test and is indicated as * (p<0.05), ** (p<0.01) and *** (p<0.001).
The prokinetic Metoclopramide alleviates PDE4 inhibitor-induced gastroparesis
Should gastroparesis be a critical cause of PDE4-inhibitor induced nausea and emesis in humans, then treatment of delayed gastric emptying should also alleviate these symptoms. To this end, we tested whether Metoclopramide, a prokinetic acting via D2-dopamine receptor antagonism and 5-hydroxytryptamine (5-HT)4-serotonin receptor agonism, that is widely used in the treatment of gastroparesis in humans and animals61,62, may also alleviate PDE4 inhibitor-induced gastroparesis. Indeed, as shown in Fig. 7B, pre-treatment with Metoclopramide (10 mg/kg, o.g., 30 min before PDE4 inhibitor treatment) significantly reduced PDE4 inhibitor-induced gastric retention.
DISCUSSION
Increased gastric retention is a class effect of PAN-PDE4 inhibitors in mice
We show here that PAN-selective inhibition of PDE4, but not inhibition of PDE3, causes a time- (Fig. 1) and dose- (Fig. 3) dependent accumulation of chow in the stomachs of mice fed ad libitum. This phenotype is produced by several structurally distinct PAN-PDE4 inhibitors, suggesting it is a class effect of these drugs (Fig. 2). After 3 days of treatment, the stomachs of PDE4 inhibitor-treated mice increased in weight ≥3-fold over controls, whereas no abnormal accumulation of food in the intestines was observed (Fig. 3B), nor did the mice eat more (Fig. 3C). Together, this suggests that food accumulation in the stomachs of PDE4 inhibitor-treated mice is due to abnormal retention of food in their stomachs, and this idea is broadly confirmed in an acute model of gastric retention that tracks the passage of a food bolus through the stomach over a 30 min time period (Figs. 4 and 5).
In several unrelated studies, in which we have treated mice with high doses of PDE4 inhibitors including Piclamilast, Rolipram and Roflumilast (all ≥5 mg/kg, i.p. twice daily) for up to 14 days, we never observed an animal to die as a result of PDE4 inhibitor treatment, suggesting that their stomachs do not increase to the point of rupture, nor did stomach weights increase much further than the weight reached within 3 days (for example, compare Figs. 1B and 1C). This suggests that within 3 days of PDE4-inhibitor treatment, the maximal capacity of the stomach to hold food (e.g. fundic accommodation) is reached. As mice are not able to vomit, but continue to eat similar amounts of food (Fig. 3C), this suggests that the pressure of the accumulated food in the stomach overcomes the cause of PDE4 inhibitor-induced gastric retention after the 3-day time point, so that any additional food eaten is passively pushed through the stomach. It is somewhat surprising that the mice keep eating despite the substantial accumulation of food in their stomachs. Clearly, in the regulation of their appetite, any discomfort resulting from the gastric accumulation of food is superseded by the metabolic demands of the body.
PDE4-inhibitor induced gastroparesis as a correlate of emesis and nausea
In humans, abnormal gastric retention of food (in the absence of mechanical obstruction) is known as gastroparesis, a syndrome predominated by nausea and vomiting62–66. Given that nausea and emesis are also the most common and significant side effects of PDE4 inhibition in humans and animals, it is possible that these adverse effects may result (in part or as a whole) from delayed gastric emptying induced by PDE4 inhibition. If so, treatment with prokinetics, such as Metoclopramide, which relieve PDE4 inhibitor-induced gastroparesis (Fig. 7B), should also alleviate drug-induced emesis and nausea. Whether or not treatment with PDE4 inhibitors causes gastroparesis in humans has not been tested to our knowledge. However, two lines of reasoning suggest that PDE4 inhibitor induced gastroparesis may represent a useful correlate of emesis and nausea in mice.
First, in humans and animal species that can vomit, systemic administration of PDE4 inhibitors is generally associated with a narrow therapeutic window in that similar doses produce both various therapeutic effects (e.g. anti-inflammatory benefits) as well as emesis and nausea. Similarly, we show here that doses as low as 0.04 mg/kg Rolipram (Fig. 5B) and 0.2 mg/kg Piclamilast (Fig. 5C) induce acute gastric retention in mice, whereas a dose of 1 mg/kg Piclamilast induces gastric retention in the ad libitum model (Fig. 3A), which is at or below the doses generally used to produce anti-inflammatory benefits in these animals79–84.
Second, several observations suggest that central nervous system effects contribute to PDE4 inhibitor-induced emesis30,85–88 and that the ability to cross the blood-brain barrier enhances the emetic potential of PDE4 inhibitors. For example, Robichaud and colleagues have shown that the brain-penetrant tachykinin NK1-receptor blocker CP-99994 protects from PDE4 inhibitor-induced emesis in ferrets, whereas L-743310, an NK1-receptor blocker that does not cross the blood-brain barrier, had no effect89. Moreover, as reported by Aoki et.al.74, the low emetogenicity of the PAN-PDE4 inhibitor YM976 is likely due to its limited brain penetrance. We show here that YM976 also exhibits a reduced potency to induce gastric retention compared to the brain-penetrant PAN-PDE4 inhibitors tested (see Figs. 5A to D), thus providing another correlation between emetic potential and induction of gastroparesis in mice.
Potential correlation of PDE4 levels and gastroparesis
Given their critical roles in the regulation of a myriad of cAMP signals, the expression, subcellular location and activity of the PDE4 enzymes themselves is tightly regulated9,40. Among a variety of mechanisms that regulate PDE4 functions (reviewed here 9,40) are a number of mechanisms that suppress PDE4 activity. Acutely, phosphorylation of PDE4s at a conserved C-terminal site by the extracellular signaling regulated kinase (ERK) has been shown to inhibit the activity of so-called PDE4 long forms 90–92. In addition, distinct PDE4 isoforms are down-regulated in a number of pathologic conditions, such as in heart failure or atrial fibrillation93–96, in neurologic conditions such as depression 97, in cancer98 during ageing93, or in response to stress99 or drug treatment 100. In this context, while in the present study, gastroparesis was produced by decreasing PDE4 activity via pharmacologic means, our findings also open the possibility that a physiologic or pathophysiologic downregulation of PDE4 expression and/or activity may be the cause of gastroparesis in a subset of patients, given that the majority of cases remain idiopathic to date67. Moreover, if a reduction in PDE4 activity produces gastroparesis, one may speculate that activation of PDE4, such as via treatment with small-molecule PDE4 activators101, may alleviate gastroparesis in some patients.
Mechanisms of PDE4 inhibitor-induced gastroparesis
Digestion of food and emptying into the intestines is a complex process requiring the synchronized contraction of a series of muscles that is finely tuned and coordinated by sensory, hormonal, and enteric- and autonomic nervous system regulations61–63. How PDE4 inhibition induces gastroparesis at the tissue-, cell- and molecular level remains to be delineated. At the tissue level, increased accommodation of the fundus, impairment of antral motility or pyloric relaxation, as well as duodenal dysmotility, may all potentially contribute to the observed phenotype. In a similar vein, it remains to be delineated to which extent direct effects (e.g. perhaps via inhibition of PDE4 in smooth muscle or the interstitial cells of Cajal) or indirect effects (e.g. PDE4 inhibition affecting sensory or neuronal regulation) contribute to the observed phenotype; though, as discussed above, the low potency of the not brain-penetrant PDE4 inhibitor YM976 suggests a critical contribution of central autonomic nervous system regulation. The reduced emetogenicity of YM976 (see 74) may also suggest that the emetic potential of PDE4 inhibitors is not predominated by inhibition of PDE4 in the emesis centers of the brain (such as the area postrema) as these regions are not protected by the blood-brain barrier. Hence, development of PDE4 inhibitors with poor brain penetrance may alleviate the side effects of these drugs and enhance their therapeutic window for indications that target the periphery.
Given the observation that ablation of any single PDE4 gene in mice does not induce gastric retention, nor protect from gastric retention induced by PAN-PDE4 inhibition, we propose that a minimum of two distinct PDE4 subfamilies must be inhibited, and that the effects of their inhibition act in an additive or synergistic manner to produce gastroparesis. It is possible that inhibition of two (or more) PDE4s within the same cell may synergize to elevate cAMP levels past a certain threshold to induce gastric retention, or that inhibition of distinct PDE4s in different cells and tissues produces additive or synergistic physiologic effects that, in their aggregate, but not each by themselves, produce gastroparesis. Indeed, it has been shown using the siRNA-dependent knockdown of distinct PDE4s that the concurrent inactivation of multiple PDE4 isoforms may generate a phenotypic output that is distinct from that engendered by inactivation of any single PDE4 species, and vice versa37.
As an alternative explanation to requiring the concurrent inactivation of multiple PDE4s as discussed above, the absence of gastroparesis in any of the four PDE4 knockout mouse lines may theoretically also be explained by one PDE4 subtype compensating for the loss of another. While we cannot exclude this possibility, we consider this less likely given the large of body of data suggesting that PDEs are functionally not interchangeable. While cells and animals do adapt to the loss of a PDE, compensatory mechanism generally alter downstream cAMP-signaling effectors or change cAMP production, such as for example at the level of G protein-coupled receptor (GPCR) signaling35,48,102. Conversely, although all PDE4 isoforms share a highly conserved catalytic domain, and thus exhibit similar kinetics for cAMP hydrolysis, regions N- and C-terminal of the catalytic domain are less conserved among the four PDE4 genes, and the far N-termini of PDE4s are often encoded by variant-specific first exons, and are thus unique to individual PDE4 protein variants. These region often mediate critical protein/protein or protein/lipid interactions that serve to target the respective variant to specific subcellular compartments and/or signaling complexes39,40. As a result, individual PDE4 variants control distinct subcellular pools of cAMP signaling, which is the foundation of their unique and non-overlapping cellular and physiologic functions, and hence cannot functionally replace each other.
Curiously, while we report here that inhibition PDE4 causes a substantial delay in gastric emptying, prior studies have shown that the prokinetic effects of serotonin 5-HT4 receptor agonism are enhanced by PDE4 inhibition in porcine, though not in mouse, gastric circular muscle, suggesting that not all cAMP signals resulting from PDE4 inhibition are inherently wired to delay gastric emptying103,104.
Role of PDE4D in mediating the side effects of PAN-PDE4 inhibitors
There is a general consensus in the field that PDE4 inhibitors with an improved safety profile must target only a portion of the total PDE4 in the body. One approach to achieve this is to develop inhibitors with high selectivity for individual PDE4 subtypes. This begs the question of which PDE4 subtype (or subtypes) mediate(s) the adverse effects of PAN-PDE4 inhibitors, primarily nausea and emesis, and must thus be avoided. Opinion has largely been shaped by a previous study that used the duration of xylazine/ketamine-induced anesthesia in mice as a correlate of emesis resulting from PDE4 inhibition. In this study, Robichaud and colleagues reported that genetic ablation of PDE4D shortens xylazine/ketamine-induced anesthesia in mice, thus mimicking the effects of treatment with PAN-PDE4 inhibitors, whereas ablation of PDE4B did not55, and that treatment with PAN-PDE4 inhibitors had no further effects on the duration of xylazine/ketamine-induced anesthesia in PDE4D-KO mice. The authors thus proposed that inhibition of PDE4D, but not PDE4B, is mediating the emetic effects of PAN-PDE4 inhibitors, which has led to significant efforts to develop PDE4 inhibitors selective for PDE4B over PDE4D in expectation of an improved safety profile45,56–59; while at the same time largely forgoing any therapeutic benefits potentially derived from inhibition of PDE4D.
Our current findings challenge this paradigm and suggest an alternative correlation of PDE4 subtypes with emesis and nausea. We show that ablation of any individual PDE4 subtype per se does not induce gastroparesis (Fig. 6). This does not exclude a role for PDE4D in gastroparesis, as PDE4D may be one of the PDE4 subtypes that must be concurrently inhibited to produce the phenotype. However, it suggests that if PDE4 inhibitor-induced gastroparesis in mice accurately reflects the role of individual PDE4 subtypes in emesis and nausea in humans, then selective inhibition of any individual PDE4 subtype, including the selective inhibition of PDE4D, may be free of these adverse effects and may be targeted for therapeutic benefits. In line with this idea, inhibitors with some selectivity for PDE4D exhibited reduced emetic effects compared to PAN-selective PDE4 inhibitors in a prior study46. Whether gastroparesis or the duration of xylazine/ketamine-induced anesthesia in mice accurately predicts the role of PDE4 subtypes in inducing emesis in humans remains to be established. As suggested by Prickaerts and colleagues60, while the xylazine/ketamine-anesthesia test is a reliable measure for α2-adrenoreceptor antagonism, it may have limitations as a predictor of emetic potential60. For example, there are critical species differences in that α2-adrenoceptor antagonism induces emesis in ferrets, whereas the opposite, namely agonism at α2-adrenoceptors, induces emesis in cats and dogs105–109. In addition, some drugs known to cause emesis in humans (e.g. Imipramine) lengthen, rather than shorten, the duration of xylazine/ketamine-anesthesia in rodents60. While the xylazine/ketamine-induced anesthesia test may have limitations, it also remains to be shown whether PDE4 inhibition induces gastroparesis, and thereby induces emesis and nausea, in humans. It is not surprising, however, that the two models produce a different pattern of PDE4 subtype involvement in mice given that PDE4 inhibitors induce antagonism at α2-adrenoreceptor to shorten xylazine/ketamine-induced anesthesia, whereas agonism, but not antagonism at α2-adrenoreceptor induces gastroparesis (Fig. 7A).
Supplementary Material
Acknowledgements:
We are grateful to the entire staff of the Department of Comparative Medicine at the University of South Alabama for providing excellent care of the animals, their advice on experimental design, and help with experimentation, as well as to Dr. Jonathan Scammell for advice on statistics. This work was supported in part by funds from the Cystic Fibrosis Foundation (RICHTE16GO), the National Institutes of Health (HL076125, HL141473, HL066299) and a Research and Scholarly Development Grant from the University of South Alabama Office of Research and Economic Development.
Abbreviations:
- DMSO
Dimethyl sulfoxide
- ERK
Extracellular signal-regulated kinase
- FITC
Fluorescein isothiocyanate
- 5-HT4
5-Hydroxytryptamine receptor 4
- i.p.
intraperitoneal
- KO
Knockout
- NK1
Neurokinin-1 receptor
- PBS
Phosphate-buffered saline
- PDE
cyclic nucleotide phosphodiesterase
- PDE4
cAMP phosphodiesterase 4
- WT
Wildtype
REFERENCES
- 1.Kato A, Touhara K. Mammalian olfactory receptors: pharmacology, G protein coupling and desensitization. Cell Mol Life Sci. 2009;66(23):3743–3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ludwig MG, Vanek M, Guerini D, et al. Proton-sensing G-protein-coupled receptors. Nature. 2003;425(6953):93–98. [DOI] [PubMed] [Google Scholar]
- 3.Seuwen K, Ludwig MG, Wolf RM. Receptors for protons or lipid messengers or both? J Recept Signal Transduct Res. 2006;26(5–6):599–610. [DOI] [PubMed] [Google Scholar]
- 4.Sassone-Corsi P The cyclic AMP pathway. Cold Spring Harb Perspect Biol. 2012;4(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Conti M, Beavo J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem. 2007;76:481–511. [DOI] [PubMed] [Google Scholar]
- 6.Omori K, Kotera J. Overview of PDEs and their regulation. Circ Res. 2007;100(3):309–327. [DOI] [PubMed] [Google Scholar]
- 7.Conti M, Richter W, Mehats C, Livera G, Park JY, Jin C. Cyclic AMP-specific PDE4 phosphodiesterases as critical components of cyclic AMP signaling. J Biol Chem. 2003;278(8):5493–5496. [DOI] [PubMed] [Google Scholar]
- 8.Bolger GB, Conti M, Houslay MD. Cellular Functions of PDE4 Enzymes. In: Beavo JAFSHaHMD, ed. Cyclic Nucleotide Phosphodiesterases in Health and Disease. Boca Raton: CRC Press; 2007:99–130. [Google Scholar]
- 9.Houslay MD, Adams DR. PDE4 cAMP phosphodiesterases: modular enzymes that orchestrate signalling cross-talk, desensitization and compartmentalization. Biochem J. 2003;370(Pt 1):1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maurice DH, Ke H, Ahmad F, Wang Y, Chung J, Manganiello VC. Advances in targeting cyclic nucleotide phosphodiesterases. Nat Rev Drug Discov. 2014;13(4):290–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baillie GS, Tejeda GS, Kelly MP. Therapeutic targeting of 3’,5’-cyclic nucleotide phosphodiesterases: inhibition and beyond. Nat Rev Drug Discov. 2019;18(10):770–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Houslay MD, Schafer P, Zhang KY. Keynote review: phosphodiesterase-4 as a therapeutic target. Drug Discov Today. 2005;10(22):1503–1519. [DOI] [PubMed] [Google Scholar]
- 13.Zhang KY, Ibrahim PN, Gillette S, Bollag G. Phosphodiesterase-4 as a potential drug target. Expert Opin Ther Targets. 2005;9(6):1283–1305. [DOI] [PubMed] [Google Scholar]
- 14.Jin SL, Ding SL, Lin SC. Phosphodiesterase 4 and its inhibitors in inflammatory diseases. Chang Gung Med J. 2012;35(3):197–210. [DOI] [PubMed] [Google Scholar]
- 15.Li H, Zuo J, Tang W. Phosphodiesterase-4 Inhibitors for the Treatment of Inflammatory Diseases. Front Pharmacol. 2018;9:1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Page CP, Spina D. Phosphodiesterase inhibitors in the treatment of inflammatory diseases. Handb Exp Pharmacol. 2011(204):391–414. [DOI] [PubMed] [Google Scholar]
- 17.Blokland A, Schreiber R, Prickaerts J. Improving memory: a role for phosphodiesterases. Curr Pharm Des. 2006;12(20):2511–2523. [DOI] [PubMed] [Google Scholar]
- 18.Richter W, Menniti FS, Zhang HT, Conti M. PDE4 as a target for cognition enhancement. Expert Opin Ther Targets. 2013;17(9):1011–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghavami A, Hirst WD, Novak TJ. Selective phosphodiesterase (PDE)-4 inhibitors: a novel approach to treating memory deficit? Drugs R D. 2006;7(2):63–71. [DOI] [PubMed] [Google Scholar]
- 20.Reneerkens OA, Rutten K, Steinbusch HW, Blokland A, Prickaerts J. Selective phosphodiesterase inhibitors: a promising target for cognition enhancement. Psychopharmacology (Berl). 2009;202(1–3):419–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Houslay MD, Baillie GS, Maurice DH. cAMP-Specific phosphodiesterase-4 enzymes in the cardiovascular system: a molecular toolbox for generating compartmentalized cAMP signaling. Circ Res. 2007;100(7):950–966. [DOI] [PubMed] [Google Scholar]
- 22.Maurice DH. Subcellular signaling in the endothelium: cyclic nucleotides take their place. Curr Opin Pharmacol. 2011;11(6):656–664. [DOI] [PubMed] [Google Scholar]
- 23.Yasmeen S, Akram BH, Hainsworth AH, Kruuse C. Cyclic nucleotide phosphodiesterases (PDEs) and endothelial function in ischaemic stroke. A review. Cell Signal. 2019;61:108–119. [DOI] [PubMed] [Google Scholar]
- 24.Wu C, Rajagopalan S. Phosphodiesterase-4 inhibition as a therapeutic strategy for metabolic disorders. Obes Rev. 2016;17(5):429–441. [DOI] [PubMed] [Google Scholar]
- 25.Peng T, Gong J, Jin Y, et al. Inhibitors of phosphodiesterase as cancer therapeutics. Eur J Med Chem. 2018;150:742–756. [DOI] [PubMed] [Google Scholar]
- 26.Cooney JD, Aguiar RC. Phosphodiesterase 4 inhibitors have wide-ranging activity in B-cell malignancies. Blood. 2016;128(25):2886–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tiwari S, Dong H, Kim EJ, Weintraub L, Epstein PM, Lerner A. Type 4 cAMP phosphodiesterase (PDE4) inhibitors augment glucocorticoid-mediated apoptosis in B cell chronic lymphocytic leukemia (B-CLL) in the absence of exogenous adenylyl cyclase stimulation. Biochem Pharmacol. 2005;69(3):473–483. [DOI] [PubMed] [Google Scholar]
- 28.Giembycz MA, Field SK. Roflumilast: first phosphodiesterase 4 inhibitor approved for treatment of COPD. Drug Des Devel Ther. 2010;4:147–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moretto N, Caruso P, Bosco R, et al. CHF6001 I: a novel highly potent and selective phosphodiesterase 4 inhibitor with robust anti-inflammatory activity and suitable for topical pulmonary administration. J Pharmacol Exp Ther. 2015;352(3):559–567. [DOI] [PubMed] [Google Scholar]
- 30.Souness JE, Rao S. Proposal for pharmacologically distinct conformers of PDE4 cyclic AMP phosphodiesterases. Cell Signal. 1997;9(3–4):227–236. [DOI] [PubMed] [Google Scholar]
- 31.Srivani P, Usharani D, Jemmis ED, Sastry GN. Subtype selectivity in phosphodiesterase 4 (PDE4): a bottleneck in rational drug design. Curr Pharm Des. 2008;14(36):3854–3872. [DOI] [PubMed] [Google Scholar]
- 32.Iona S, Cuomo M, Bushnik T, et al. Characterization of the rolipram-sensitive, cyclic AMP-specific phosphodiesterases: identification and differential expression of immunologically distinct forms in the rat brain. Mol Pharmacol. 1998;53(1):23–32. [DOI] [PubMed] [Google Scholar]
- 33.Bolger GB, McPhee I, Houslay MD. Alternative splicing of cAMP-specific phosphodiesterase mRNA transcripts. Characterization of a novel tissue-specific isoform, RNPDE4A8. J Biol Chem. 1996;271(2):1065–1071. [DOI] [PubMed] [Google Scholar]
- 34.McPhee I, Pooley L, Lobban M, Bolger G, Houslay MD. Identification, characterization and regional distribution in brain of RPDE-6 (RNPDE4A5), a novel splice variant of the PDE4A cyclic AMP phosphodiesterase family. Biochem J. 1995;310 (Pt 3):965–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jin SL, Richard FJ, Kuo WP, D’Ercole AJ, Conti M. Impaired growth and fertility of cAMP-specific phosphodiesterase PDE4D-deficient mice. Proc Natl Acad Sci U S A. 1999;96(21):11998–12003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jin SL, Conti M. Induction of the cyclic nucleotide phosphodiesterase PDE4B is essential for LPS-activated TNF-alpha responses. Proc Natl Acad Sci U S A. 2002;99(11):7628–7633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huston E, Lynch MJ, Mohamed A, et al. EPAC and PKA allow cAMP dual control over DNA-PK nuclear translocation. Proc Natl Acad Sci U S A. 2008;105(35):12791–12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lynch MJ, Baillie GS, Mohamed A, et al. RNA silencing identifies PDE4D5 as the functionally relevant cAMP phosphodiesterase interacting with beta arrestin to control the protein kinase A/AKAP79-mediated switching of the beta2-adrenergic receptor to activation of ERK in HEK293B2 cells. J Biol Chem. 2005;280(39):33178–33189. [DOI] [PubMed] [Google Scholar]
- 39.Houslay MD. Underpinning compartmentalised cAMP signalling through targeted cAMP breakdown. Trends Biochem Sci. 2010;35(2):91–100. [DOI] [PubMed] [Google Scholar]
- 40.Conti M, Mika D, Richter W. Perspectives on: Cyclic nucleotide microdomains and signaling specificity: Cyclic AMP compartments and signaling specificity: Role of cyclic nucleotide phosphodiesterases. J Gen Physiol. 2014;143(1):29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aspiotis R, Deschênes D, Dubé D, et al. The discovery and synthesis of highly potent subtype selective phosphodiesterase 4D inhibitors. Bioorg Med Chem Lett. 2010;20(18):5502–5505. [DOI] [PubMed] [Google Scholar]
- 42.Bruno O, Fedele E, Prickaerts J, et al. GEBR-7b, a novel PDE4D selective inhibitor that improves memory in rodents at non-emetic doses. Br J Pharmacol. 2011;164(8):2054–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Donnell AF, Dollings PJ, Butera JA, et al. Identification of pyridazino[4,5-b]indolizines as selective PDE4B inhibitors. Bioorg Med Chem Lett. 2010;20(7):2163–2167. [DOI] [PubMed] [Google Scholar]
- 44.Goto T, Shiina A, Murata T, et al. Identification of the 5,5-dioxo-7,8-dihydro-6H-thiopyrano[3,2-d]pyrimidine derivatives as highly selective PDE4B inhibitors. Bioorg Med Chem Lett. 2014;24(3):893–899. [DOI] [PubMed] [Google Scholar]
- 45.Hagen TJ, Mo X, Burgin AB, Fox D, Zhang Z, Gurney ME. Discovery of triazines as selective PDE4B versus PDE4D inhibitors. Bioorg Med Chem Lett. 2014;24(16):4031–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burgin AB, Magnusson OT, Singh J, et al. Design of phosphodiesterase 4D (PDE4D) allosteric modulators for enhancing cognition with improved safety. Nat Biotechnol. 2010;28(1):63–70. [DOI] [PubMed] [Google Scholar]
- 47.Schaefer TL, Braun AA, Amos-Kroohs RM, Williams MT, Ostertag E, Vorhees CV. A new model of Pde4d deficiency: genetic knock-down of PDE4D enzyme in rats produces an antidepressant phenotype without spatial cognitive effects. Genes Brain Behav. 2012;11(5):614–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jin SLC, Richter W, Conti M. Insights into the physiological functions of PDE4 derived from knockout mice. in Cyclic Nucleotide Phosphodiesterases in Health and Disease (Beavo JA, Francis SH, Houslay MD, ed), P 323–346, CRC Press, Boca Raton, FL. 2006. [Google Scholar]
- 49.Lamontagne S, Meadows E, Luk P, et al. Localization of phosphodiesterase-4 isoforms in the medulla and nodose ganglion of the squirrel monkey. Brain Res. 2001;920(1–2):84–96. [DOI] [PubMed] [Google Scholar]
- 50.Miro X, Perez-Torres S, Puigdomenech P, Palacios JM, Mengod G. Differential distribution of PDE4D splice variant mRNAs in rat brain suggests association with specific pathways and presynaptical localization. Synapse. 2002;45(4):259–269. [DOI] [PubMed] [Google Scholar]
- 51.Mori F, Pérez-Torres S, De Caro R, et al. The human area postrema and other nuclei related to the emetic reflex express cAMP phosphodiesterases 4B and 4D. J Chem Neuroanat. 2010;40(1):36–42. [DOI] [PubMed] [Google Scholar]
- 52.Robichaud A, Savoie C, Stamatiou PB, Tattersall FD, Chan CC. PDE4 inhibitors induce emesis in ferrets via a noradrenergic pathway. Neuropharmacology. 2001;40(2):262–269. [DOI] [PubMed] [Google Scholar]
- 53.Huang Z, Ducharme Y, Macdonald D, Robichaud A. The next generation of PDE4 inhibitors. Curr Opin Chem Biol. 2001;5(4):432–438. [DOI] [PubMed] [Google Scholar]
- 54.Robichaud A, Savoie C, Stamatiou PB, et al. Assessing the emetic potential of PDE4 inhibitors in rats. Br J Pharmacol. 2002;135(1):113–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Robichaud A, Stamatiou PB, Jin SL, et al. Deletion of phosphodiesterase 4D in mice shortens alpha(2)-adrenoceptor-mediated anesthesia, a behavioral correlate of emesis. J Clin Invest. 2002;110(7):1045–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Azam MA, Tripuraneni NS. Selective Phosphodiesterase 4B Inhibitors: A Review. Sci Pharm. 2014;82(3):453–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kranz M, Wall M, Evans B, et al. Identification of PDE4B Over 4D subtype-selective inhibitors revealing an unprecedented binding mode. Bioorg Med Chem. 2009;17(14):5336–5341. [DOI] [PubMed] [Google Scholar]
- 58.Naganuma K, Omura A, Maekawara N, et al. Discovery of selective PDE4B inhibitors. Bioorg Med Chem Lett. 2009;19(12):3174–3176. [DOI] [PubMed] [Google Scholar]
- 59.Suzuki O, Mizukami K, Etori M, et al. Evaluation of the therapeutic index of a novel phosphodiesterase 4B-selective inhibitor over phosphodiesterase 4D in mice. J Pharmacol Sci. 2013;123(3):219–226. [DOI] [PubMed] [Google Scholar]
- 60.Nelissen E, van Goethem NP, Bonassoli VT, et al. Validation of the xylazine/ketamine anesthesia test as a predictor of the emetic potential of pharmacological compounds in rats. Neurosci Lett. 2019;699:41–46. [DOI] [PubMed] [Google Scholar]
- 61.Krishnasamy S, Abell TL. Diabetic Gastroparesis: Principles and Current Trends in Management. Diabetes Ther. 2018;9(Suppl 1):1–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Grover M, Farrugia G, Stanghellini V. Gastroparesis: a turning point in understanding and treatment. Gut. 2019;68(12):2238–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pasricha PJ, Parkman HP. Gastroparesis: definitions and diagnosis. Gastroenterol Clin North Am. 2015;44(1):1–7. [DOI] [PubMed] [Google Scholar]
- 64.Soykan I, Sivri B, Sarosiek I, Kiernan B, McCallum RW. Demography, clinical characteristics, psychological and abuse profiles, treatment, and long-term follow-up of patients with gastroparesis. Dig Dis Sci. 1998;43(11):2398–2404. [DOI] [PubMed] [Google Scholar]
- 65.Parkman HP, Hallinan EK, Hasler WL, et al. Nausea and vomiting in gastroparesis: similarities and differences in idiopathic and diabetic gastroparesis. Neurogastroenterol Motil. 2016;28(12):1902–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hasler WL, Wilson LA, Parkman HP, et al. Factors related to abdominal pain in gastroparesis: contrast to patients with predominant nausea and vomiting. Neurogastroenterol Motil. 2013;25(5):427–438, e300–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Parkman HP. Idiopathic gastroparesis. Gastroenterol Clin North Am. 2015;44(1):59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hasler WL, Wilson LA, Nguyen LA, et al. Opioid Use and Potency Are Associated With Clinical Features, Quality of Life, and Use of Resources in Patients With Gastroparesis. Clin Gastroenterol Hepatol. 2019;17(7):1285–1294.e1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Raeburn D, Underwood SL, Lewis SA, et al. Anti-inflammatory and bronchodilator properties of RP 73401, a novel and selective phosphodiesterase type IV inhibitor. Br J Pharmacol. 1994;113(4):1423–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Souness JE, Maslen C, Webber S, et al. Suppression of eosinophil function by RP 73401, a potent and selective inhibitor of cyclic AMP-specific phosphodiesterase: comparison with rolipram. Br J Pharmacol. 1995;115(1):39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Thompson WJ. Cyclic nucleotide phosphodiesterases: pharmacology, biochemistry and function. Pharmacol Ther. 1991;51(1):13–33. [DOI] [PubMed] [Google Scholar]
- 72.Hatzelmann A, Morcillo EJ, Lungarella G, et al. The preclinical pharmacology of roflumilast--a selective, oral phosphodiesterase 4 inhibitor in development for chronic obstructive pulmonary disease. Pulm Pharmacol Ther. 2010;23(4):235–256. [DOI] [PubMed] [Google Scholar]
- 73.Bender AT, Beavo JA. Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev. 2006;58(3):488–520. [DOI] [PubMed] [Google Scholar]
- 74.Aoki M, Fukunaga M, Sugimoto T, et al. Studies on mechanisms of low emetogenicity of YM976, a novel phosphodiesterase type 4 inhibitor. J Pharmacol Exp Ther. 2001;298(3):1142–1149. [PubMed] [Google Scholar]
- 75.Saldou N, Obernolte R, Huber A, et al. Comparison of recombinant human PDE4 isoforms: interaction with substrate and inhibitors. Cell Signal. 1998;10(6):427–440. [DOI] [PubMed] [Google Scholar]
- 76.Hansen RT, Conti M, Zhang HT. Mice deficient in phosphodiesterase-4A display anxiogenic-like behavior. Psychopharmacology (Berl). 2014. [DOI] [PubMed] [Google Scholar]
- 77.Ukita T, Sugahara M, Terakawa Y, et al. Novel, potent, and selective phosphodiesterase-4 inhibitors as antiasthmatic agents: synthesis and biological activities of a series of 1-pyridylnaphthalene derivatives. J Med Chem. 1999;42(6):1088–1099. [DOI] [PubMed] [Google Scholar]
- 78.Heaslip RJ, Evans DY. Emetic, central nervous system, and pulmonary activities of rolipram in the dog. Eur J Pharmacol. 1995;286(3):281–290. [DOI] [PubMed] [Google Scholar]
- 79.Sisson TH, Christensen PJ, Muraki Y, et al. Phosphodiesterase 4 inhibition reduces lung fibrosis following targeted type II alveolar epithelial cell injury. Physiol Rep. 2018;6(12):e13753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nio Y, Tanaka M, Hirozane Y, et al. Phosphodiesterase 4 inhibitor and phosphodiesterase 5 inhibitor combination therapy has antifibrotic and anti-inflammatory effects in mdx mice with Duchenne muscular dystrophy. Faseb j. 2017;31(12):5307–5320. [DOI] [PubMed] [Google Scholar]
- 81.Seimetz M, Parajuli N, Pichl A, et al. Cigarette Smoke-Induced Emphysema and Pulmonary Hypertension Can Be Prevented by Phosphodiesterase 4 and 5 Inhibition in Mice. PLoS One. 2015;10(6):e0129327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sun JG, Deng YM, Wu X, et al. Inhibition of phosphodiesterase activity, airway inflammation and hyperresponsiveness by PDE4 inhibitor and glucocorticoid in a murine model of allergic asthma. Life Sci. 2006;79(22):2077–2085. [DOI] [PubMed] [Google Scholar]
- 83.Schmitz T, Souil E, Hervé R, et al. PDE4 Inhibition Prevents Preterm Delivery Induced by an Intrauterine Inflammation. 2007. [DOI] [PubMed] [Google Scholar]
- 84.Ariga M, Neitzert B, Nakae S, et al. Nonredundant function of phosphodiesterases 4D and 4B in neutrophil recruitment to the site of inflammation. J Immunol. 2004;173(12):7531–7538. [DOI] [PubMed] [Google Scholar]
- 85.Zhang HT, Zhao Y, Huang Y, et al. Antidepressant-like effects of PDE4 inhibitors mediated by the high-affinity rolipram binding state (HARBS) of the phosphodiesterase-4 enzyme (PDE4) in rats. Psychopharmacology (Berl). 2006;186(2):209–217. [DOI] [PubMed] [Google Scholar]
- 86.Zhao Y, Zhang HT, O’Donnell JM. Antidepressant-induced increase in high-affinity rolipram binding sites in rat brain: dependence on noradrenergic and serotonergic function. J Pharmacol Exp Ther. 2003;307(1):246–253. [DOI] [PubMed] [Google Scholar]
- 87.Zhao Y, Zhang HT, O’Donnell JM. Inhibitor binding to type 4 phosphodiesterase (PDE4) assessed using [3H]piclamilast and [3H]rolipram. J Pharmacol Exp Ther. 2003;305(2):565–572. [DOI] [PubMed] [Google Scholar]
- 88.Hirose R, Manabe H, Nonaka H, et al. Correlation between emetic effect of phosphodiesterase 4 inhibitors and their occupation of the high-affinity rolipram binding site in Suncus murinus brain. Eur J Pharmacol. 2007;573(1–3):93–99. [DOI] [PubMed] [Google Scholar]
- 89.Robichaud A, Tattersall FD, Choudhury I, Rodger IW. Emesis induced by inhibitors of type IV cyclic nucleotide phosphodiesterase (PDE IV) in the ferret. Neuropharmacology. 1999;38(2):289–297. [DOI] [PubMed] [Google Scholar]
- 90.Baillie GS, MacKenzie SJ, McPhee I, Houslay MD. Sub-family selective actions in the ability of Erk2 MAP kinase to phosphorylate and regulate the activity of PDE4 cyclic AMP-specific phosphodiesterases. Br J Pharmacol. 2000;131(4):811–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hoffmann R, Baillie GS, MacKenzie SJ, Yarwood SJ, Houslay MD. The MAP kinase ERK2 inhibits the cyclic AMP-specific phosphodiesterase HSPDE4D3 by phosphorylating it at Ser579. EMBO J. 1999;18(4):893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Houslay MD, Baillie GS. The role of ERK2 docking and phosphorylation of PDE4 cAMP phosphodiesterase isoforms in mediating cross-talk between the cAMP and ERK signalling pathways. Biochem Soc Trans. 2003;31(Pt 6):1186–1190. [DOI] [PubMed] [Google Scholar]
- 93.Molina CE, Leroy J, Richter W, et al. Cyclic adenosine monophosphate phosphodiesterase type 4 protects against atrial arrhythmias. J Am Coll Cardiol. 2012;59(24):2182–2190. [DOI] [PubMed] [Google Scholar]
- 94.Abi-Gerges A, Richter W, Lefebvre F, et al. Decreased expression and activity of cAMP phosphodiesterases in cardiac hypertrophy and its impact on beta-adrenergic cAMP signals. Circ Res. 2009;105(8):784–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Richter W, Xie M, Scheitrum C, Krall J, Movsesian MA, Conti M. Conserved expression and functions of PDE4 in rodent and human heart. Basic Res Cardiol. 2011;106(2):249–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lehnart SE, Wehrens XH, Reiken S, et al. Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell. 2005;123(1):25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fujita M, Hines CS, Zoghbi SS, et al. Downregulation of brain phosphodiesterase type IV measured with 11C-(R)-rolipram positron emission tomography in major depressive disorder. Biol Psychiatry. 2012;72(7):548–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Henderson DJ, Byrne A, Dulla K, et al. The cAMP phosphodiesterase-4D7 (PDE4D7) is downregulated in androgen-independent prostate cancer cells and mediates proliferation by compartmentalising cAMP at the plasma membrane of VCaP prostate cancer cells. Br J Cancer. 2014;110(5):1278–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ryan S, Li C, Menigoz A, et al. Repeated shock stress facilitates basolateral amygdala synaptic plasticity through decreased cAMP-specific phosphodiesterase type IV (PDE4) expression. Brain Struct Funct. 2018;223(4):1731–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Polesskaya OO, Smith RF, Fryxell KJ. Chronic nicotine doses down-regulate PDE4 isoforms that are targets of antidepressants in adolescent female rats. Biol Psychiatry. 2007;61(1):56–64. [DOI] [PubMed] [Google Scholar]
- 101.Omar F, Findlay JE, Carfray G, et al. Small-molecule allosteric activators of PDE4 long form cyclic AMP phosphodiesterases. Proc Natl Acad Sci U S A. 2019;116(27):13320–13329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mehats C, Jin SL, Wahlstrom J, Law E, Umetsu DT, Conti M. PDE4D plays a critical role in the control of airway smooth muscle contraction. FASEB J. 2003;17(13):1831–1841. [DOI] [PubMed] [Google Scholar]
- 103.Lefebvre RA, Van Colen I, Pauwelyn V, De Maeyer JH. Synergistic effect between 5-HT4 receptor agonist and phosphodiesterase 4-inhibitor in releasing acetylcholine in pig gastric circular muscle in vitro. Eur J Pharmacol. 2016;781:76–82. [DOI] [PubMed] [Google Scholar]
- 104.Priem E, Van Colen I, De Maeyer JH, Lefebvre RA. The facilitating effect of prucalopride on cholinergic neurotransmission in pig gastric circular muscle is regulated by phosphodiesterase 4. Neuropharmacology. 2012;62(5–6):2126–2135. [DOI] [PubMed] [Google Scholar]
- 105.Hikasa Y, Takase K, Saito K, Ogasawara S. Antagonism of the emetic action of xylazine by alpha-adrenoceptor blocking agents. Eur J Pharmacol. 1986;130(3):229–235. [DOI] [PubMed] [Google Scholar]
- 106.Hikasa Y, Takase K, Ogasawara S. Evidence for the involvement of alpha 2-adrenoceptors in the emetic action of xylazine in cats. Am J Vet Res. 1989;50(8):1348–1351. [PubMed] [Google Scholar]
- 107.Hikasa Y, Ogasawara S, Takase K. Alpha adrenoceptor subtypes involved in the emetic action in dogs. J Pharmacol Exp Ther. 1992;261(2):746–754. [PubMed] [Google Scholar]
- 108.Hikasa Y, Akiba T, Iino Y, Matsukura M, Takase K, Ogasawara S. Central alpha-adrenoceptor subtypes involved in the emetic pathway in cats. Eur J Pharmacol. 1992;229(2–3):241–251. [DOI] [PubMed] [Google Scholar]
- 109.Japundzić-Zigon N, Samardzić R, Beleslin DB. Clonidine-induced emesis: a multitransmitter pathway concept. Pharmacol Res. 1997;35(4):287–297. [DOI] [PubMed] [Google Scholar]
- 111.Blanchard E, Zlock L, Lao A, et al. Anchored PDE4 regulates chloride conductance in wild-type and ΔF508-CFTR human airway epithelia. FASEB J. 2014;28(2):791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.