

Abstract
Treatment resistance leads to cancer patient mortality. Therapeutic approaches that employ synthetic lethality to target mutational vulnerabilities in key tumor cell signaling pathways have proven effective in overcoming therapeutic resistance in some cancers. Yet, tumors are organs composed of malignant cells residing within a cellular and noncellular stroma. Tumor evolution and resistance to anticancer treatment are mediated through a dynamic and reciprocal dialogue with the tumor microenvironment (TME). Accordingly, expanding tumor cell synthetic lethality to encompass contextual synthetic lethality has the potential to eradicate tumors by targeting critical TME circuits that promote tumor progression and therapeutic resistance. In this Review, we summarize current knowledge about the TME and discuss its role in treatment. We outline the concept of tumor cell–specific synthetic lethality and describe therapeutic approaches to expand this paradigm to leverage TME synthetic lethality to improve cancer therapy.
Introduction
Solid tumors are organs with a complex organization that fosters tumor cell growth, survival, invasion, and evolution (1, 2). The tumor organ is composed of cancer cells, noncancerous stromal cells (fibroblasts, adipocytes, nerves, and endothelial cells as well as resident and infiltrating immune cells), and an extracellular matrix (ECM) with associated soluble factors that collectively contribute to cancer development, modulate treatment response, and ultimately participate in the evolution of treatment-resistant, metastatic tumors (3, 4). These noncancerous stromal cells and noncellular components are collectively referred to as the tumor microenvironment (TME). The composition and behavior of the TME are dictated by genetic and epigenetic elements of the cancer cells that collaborate through bidirectional communication with the TME to create a functional cancerous tissue. Within the context of this cancerous tissue, therapy-resistant tumors arise through their ability to subvert this dynamism toward their continued survival and regrowth after treatment (5, 6). This tumor organ homeostasis permits the development of drug-resistant, immune-resistant tumors.
Cytotoxic chemotherapy has been used successfully to treat many cancers. However, drug resistance and off-target toxicities remain major challenges that all too often lead to tumor recurrence and patient mortality. These challenges have motivated the search for patient-specific targeted treatments with a lower propensity for drug resistance and fewer off-target toxicities. Tailored therapeutic strategies are matched to a patient’s tumor biopsy phenotype and driver mutations. One exciting class of personalized cancer therapy exploits the concept of synthetic lethality, wherein cancer cells with a mutation in a key survival pathway are uniquely sensitive to therapeutic inhibition of a related survival pathway (7). A synthetic lethal therapeutic exploits the relationship between proliferation/viability and survival pathway redundancy; cancer cells achieve proliferation and viability at the expense of losing survival pathway redundancy. Synthetic lethal therapeutics limit off-target toxicity because healthy cells, which lack the tumorigenic mutation, remain insensitive to the therapeutic. Additionally, the high differential sensitivity of cancer cells and noncancer cells to synthetic lethal therapeutics creates a large therapeutic window that could decrease drug dosing and further limit toxicity (8). Despite encouraging success using synthetic lethal cancer therapies, this approach has been limited primarily because only a minority of cancer-associated mutations are understood well enough to identify and develop such targeted treatments. Complicating this issue and contributing to the emergence of treatment resistance is the high heterogeneity of tumors. These heterogeneous cell types are loosely organized into semifunctional tumor tissues with coordinated behavior that lends itself to contextual TME lethality.
Resistance to antitumor treatment can arise either from the emergence of individual tumor cells that harbor mutations that provide survival and proliferative advantages to individual tumor cells or from TME factors that provide context-dependent resistance cues (5). While much work has already been done to understand clonal selection of individual cancer cells, there is an urgent need to better understand how the TME fosters treatment resistance. Indeed, while some cancer cells in therapy-resistant tumors demonstrate tissue-specific treatment resistance, they can exhibit elevated sensitivity when treated as isolated cells. TME factors that promote treatment resistance include hypoxia, the tumor-associated vasculature and ECM, and a protumor immune infiltrate (5, 9). Thus, analogous to the emergence of cancer cell–specific dependence on specific survival signaling pathways, the tumor organ’s growth and survival are exquisitely reliant on key TME factors. Identifying and targeting these critical treatment-stimulated TME growth and survival factors has high potential to improve patient care.
Contextual synthetic lethality, a concept first proposed by Bristow and colleagues, is a nongenetic form of synthetic lethality, where an abnormal structure or function of the TME sensitizes cancer cells to therapy. For example, Bristow and colleagues showed that hypoxic cancer cells, which are deficient in DNA repair, are more sensitive to therapeutic inhibition of DNA damage pathways (10). This example highlights how hypoxia in the TME sensitizes cancer cells to contextually synthetic lethal therapeutics, despite also driving invasive phenotypes and cytotoxic drug resistance (Figure 1). In this Review we describe how cancer cell dependence on tumor-specific TME factors constitutes unique therapeutic vulnerabilities that, if targeted, could synergize with specific tumor cell–targeted therapies to improve cancer patient treatment response and prevent the emergence of treatment-resistant, lethal tumors.
Figure 1. Sensitizing cancer cells with contextually synthetic lethal therapeutics.
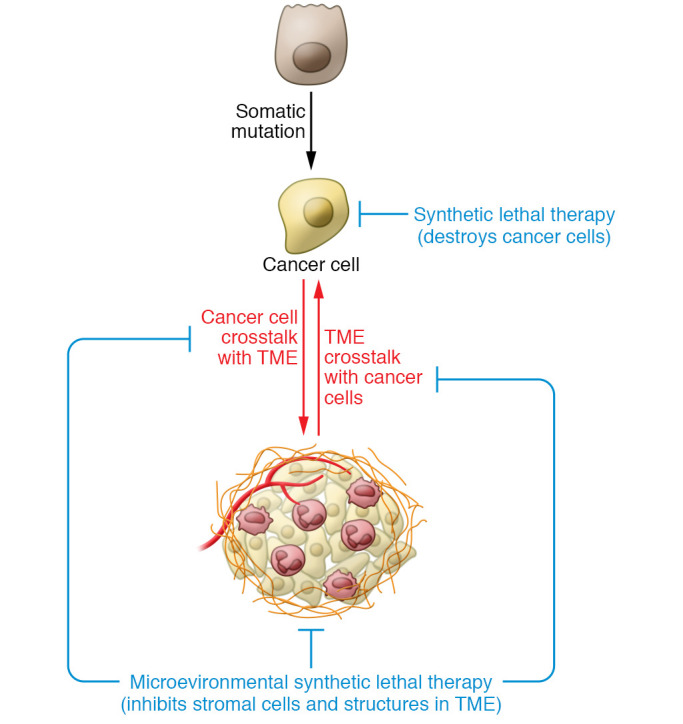
Cancer develops within a TME to form an organ that engages in bidirectional communication to promote tumor evolution. Contextually synthetic lethal therapies interrupt this system by targeting stromal cells or molecules within the TME to arrest tumor progression and improve treatment responses that ultimately eradicate the cancer. As an example, hypoxia in the TME inhibits DNA damage repair pathways, making hypoxic cancer cells more sensitive poly-ADP ribose polymerase (PARP) inhibitors. In contrast, conventional synthetic lethal therapies target cancer cells based only on mutational vulnerabilities.
Overview of the tumor microenvironment and drug resistance
Tumors are composed of a population of cancer cells that are genetically, phenotypically, and spatially heterogeneous, and this heterogeneity correlates with drug resistance (11, 12). There are regions of abnormal and poor vascularization in tumors such that some cancer cells experience low nutrient and hypoxic conditions (13, 14). Further, leaky arterial vessels and compressed drainage vessels create a high interstitial pressure that further limits nutrient availability (9). Differential access to nutrients could explain why cancer cell proliferation is spatially heterogeneous as indicated by phospho–histone H3 staining of human breast tumors (luminal A, luminal B, and HER2+ subtypes; ref. 15). Because most cytotoxic chemotherapeutics target dividing cells, cancers with a higher frequency of proliferating cells are generally thought to be more drug sensitive. Indeed, cancer cells grown in three-dimensional spheroids are more resistant to cytotoxic chemotherapeutics, as this culture format typically shows reduced proliferation rates and decreased glycolysis (16, 17). However, in vivo, fast-growing regions of cancer cells within the breast cancer mouse model MMTV-PyMT, denoted by greater levels of the phospho–histone H3 proliferation marker, feature increased expression of hypoxia-associated genes and greater resistance to cytotoxic chemotherapy (15).
Cancer cell heterogeneity and drug resistance are also promoted by the ECM, which provides location-specific cues to cancer cells. ECM is categorized into two groups — the basement membrane, which separates the epithelial layer from the mesenchyme, and the interstitial ECM, which forms the bulk of the tissue ECM and provides structural support and is where the stromal cells reside. Invasive epithelial tumors disrupt healthy ECM organization by cleaving and remodeling the basement membrane to invade into the interstitial ECM. As tumors evolve they exhibit substantial interstitial heterogeneity that is most evident at the invasive edge where the ECM is stiffest because of increased levels of reorganized, oriented collagen bundles that project perpendicularly from the tumor core (4, 18–20). ECM composition, organization, and cross-linking are mediated by stromal cells and stimulated by infiltrating immune cells. The cells that populate the TME are specified by the genotype of the tumor cells such that different tumor subtypes develop distinct ECM phenotypes (21, 22). Mesenchymal fibroblasts transdifferentiate into cancer-associated fibroblasts (CAFs) and are the primary cell type that deposit, remodel, and cross-link the interstitial ECM (23). This CAF-instructed ECM provides critical biochemical cues that foster tumor cell growth, survival, and invasion through ligation of ECM receptors including integrins, discoidins, and syndecans (18, 24–27). In particular, laminin, which is a component of the basement membrane, induces β4 integrin signaling and promotes resistance to drug-induced apoptosis (17). A dense, stiffened, and cross-linked ECM can also create a hostile tumor environment by impeding the vasculature to induce hypoxia and restrict drug delivery (28). The ECM also sequesters cytokines, growth factors, and morphogens. These factors are released by CAF-secreted metalloproteins and cathepsins to stimulate tumor cell growth, survival, and invasion and recruit inflammatory cells to the tumor. Importantly, the ECM is highly heterogeneous with respect to its biochemical properties, mechanical features, and associated soluble factors. This heterogeneity creates gradients that can markedly modulate tumor cell invasion and metastasis (29). ECM-dense regions within tumors can also present ECM-bound immune-inhibitory cytokines that compromise the efficacy of cytotoxic and immune therapies (30).
A heterogeneous population of immune cell types in tumors contributes to cancer progression and drug resistance. While immune cells have the ability to destroy cancer cells (31), developing tumors can evade the immune system. Spatial heterogeneity within a tumor, which is reflected by tumor subtype (32), contributes to immune suppression by impeding direct interactions between immune cells and cancer cells. Poor tumor vascularity (33) and a dense ECM likely contribute to reduce immune cell infiltration (34). Not surprisingly, tumors with few infiltrating leukocytes or spatially segregated immune infiltrates have poorer outcomes, and these tumors are classified as “immune excluded” and “compartmentalized” (35–37). Indeed, patients whose ER+ breast tumors demonstrated high lymphocyte spatial heterogeneity when treated with endocrine therapy had higher recurrence rates, emphasizing the importance of immune cell distribution to therapy response (38).
Tumors build their organ and foster drug resistance by coordinating multiple cell types through cell-cell communication. In many healthy tissues, structural cells (epithelial cells, endothelial cells, and fibroblasts) produce cytokines to recruit immune cells (39). Cancer cells subvert this function to produce, or induce stromal cells to produce, cytokines that affect angiogenesis, hematopoiesis, and immune cell recruitment to establish an immunosuppressive microenvironment (40–45). These cytokines also participate in new signaling networks established by aberrant gene expression in malignant cells. For example, CCL2 promotes CCR2+ cancer stem cell self-renewal and promotes tumor growth (46, 47).
The tumor microenvironment inhibits therapeutic response
While chemotherapy, radiation, and surgery are effective mainstream treatments that improve patient survival, all too frequently patients present with recurrent disease that leads to their mortality. Immunotherapies have emerged as treatments with impressive results and cancer patient cures. Unfortunately, not all tumor types are amenable to these therapies, and even in tumor types that show effective remission, some patients do not respond. Although tumor-intrinsic mechanisms drive drug resistance, the TME is now recognized as an additional major contributor to drug resistance. In order to improve therapy for patients not well served by current treatments, a better understanding of how the TME promotes drug resistance is needed.
Radiation and surgery induce inflammation and tumor-promoting immunity.
Radiation and surgery both induce tissue damage that stimulates a wound healing response mediated by local and systemic inflammation (48). This treatment-induced acute inflammation in turn increases risk of local tumor recurrence and promotes metastatic dissemination and progression (49, 50). For instance, following surgery, patients have higher levels of immunosuppressive neutrophils and monocytes that are recruited to the wound (51). Experimental studies have demonstrated that inducing a surgical wound at a site distal to a primary mouse breast tumor promotes tumor growth by stimulating a systemic inflammatory response. Wounding mobilizes immunosuppressive neutrophils and monocytes via increased levels of IL-6, G-CSF, and CCL2 (52). Surgical resection of tumors rapidly returns local and systemic immunity to a healthy state (53), indicating that persisting cancer cells likely play a key role in driving the tumor-promoting inflammatory response. Surgical patients receiving perioperative nonsteroidal antiinflammatory drugs experience lower rates of breast cancer recurrence, which demonstrates that controlling tissue inflammation can improve long-term patient outcomes (52, 54). The degree and nature of tumor inflammation depend on patient-specific factors. For example, triple-negative breast cancer patients with a high neutrophil/lymphocyte ratio after radiation therapy have increased risk of recurrence and decreased survival (55). To harness the inflammatory response to surgery or radiation as a contextual synthetic lethality, polarization of immune cells to antitumor phenotypes could increase infiltration of cytotoxic immune cells in the TME.
Immune response to chemotherapy.
Cytotoxic chemotherapy seeks to induce apoptotic cell death in cancer cells. Nevertheless, and unfortunately, heterogeneity in the cancer cell population allows for persistence of drug-resistant cells. A patient’s immune response can enhance chemotherapy to eradicate resistant cancer cells (56). Cytotoxic chemotherapy induces apoptotic cells to release their cellular contents, including cytokines and intracellular proteins, that drive local inflammation of innate effector cells (57) and generate cancer-specific neoantigens (56). The chemokine CCL2 is released by cancer cells treated with doxorubicin and attracts CCR2+ monocytes and other myeloid cells involved in antigen presentation to the tumor (58). These antigen-presenting cells present cancer cell peptides by MHC class II and lead to T cell–directed killing of persisting cancer cells (59). In both breast cancer patients and mouse models, a type I interferon response improves efficacy of doxorubicin treatment, which is produced by dying cancer cells and recruited myeloid cells (60). Interestingly, genetic deletion of Mmp9 also improves doxorubicin treatment, suggesting a link between inflammation, ECM remodeling, and response to chemotherapy (61). Yet, the plasticity of myeloid cells can lead to an immunosuppressive TME that inhibits immune clearance after chemotherapy.
Patients with residual disease after chemotherapy have greater myeloid infiltration (62), suggesting a functional link between inflammation and chemotherapy response (63). In mice, paclitaxel or cisplatin therapy increases recruitment of tumor-promoting macrophages by stimulating the production of CSF-1 by cancer cells. A CSF-1R antagonist improves response to paclitaxel by blocking macrophage recruitment (64). Deletion of Ccr2 decreases monocyte recruitment to tumors and improves doxorubicin and cisplatin treatment in the MMTV-PyMT mouse model. The fact that CCR2+ myeloid cells can both enhance and inhibit immune clearance after cytotoxic chemotherapy suggests that CCR2+ myeloid cells are a heterogeneous population of cells with potential for tumor-promoting and tumor-suppressing activities (61).
Controlling the immune response to chemotherapy may harness immune clearance while limiting immune suppression. New chemotherapy regimens for existing drugs have enhanced efficacy that may in part limit tumor-promoting inflammation. Low-dose metronomic chemotherapy improves therapeutic response by decreasing tumor-promoting inflammation and CAF activation (65–67). Incorporating microenvironmental and inflammatory responses to an adaptive therapy regimen, in which drug dosing is varied as a function of patient response, could further balance the benefits of cytotoxic chemotherapy with the costs of tumor-promoting inflammation (68).
Cancer cells resist immune clearance during immunotherapy.
Developing tumors create an immunosuppressive environment that permits their progression. While driver mutations in cancer cells promote carcinogenesis, these mutations also create neoantigens that can be recognized by the immune system for destruction. Cancer cells remodel their cell surface to evade immune destruction, and also secrete factors that create a tolerogenic microenvironment that inhibits immune clearance (69). Immunotherapies that block immune effector checkpoints, especially antibodies blocking PD-1, PD-L1, and CTLA-4 (reviewed in ref. 70), overcome the immunosuppressive TME. Checkpoint therapy has transformed the treatment of cancers, including melanoma, hepatocellular carcinoma, and lung cancer, but has not found success in multiple tumor types (71). Cancers that are recalcitrant to immunotherapy utilize additional mechanisms to inhibit immune surveillance.
Immune destruction of cancer cells requires recognition of changes to the cell surface, relative to healthy cells. All human cells express MHC class I molecules that present intracellular peptides to allow for immune surveillance of mutant or infected cells. While complete loss of MHC class I marks cancer cells for destruction by NK cells (72), downregulation of MHC class I proteins through lysosomal degradation (73) and allelic loss (74, 75) can dampen immune recognition of cancer cells and decrease response to checkpoint inhibitors (73, 76). Further, cancer cells inhibit immune activation by upregulating immune checkpoint proteins including PD-L1 (77), CD47 (78), and CD24 (79). In addition to surface proteins, cancer cells evade immune destruction via a hypersialylated glycocalyx, the outermost layer of protein and glycan polymers that covers all human cells. Sialoglycans directly interact with Siglec receptors on immune cells including T cells (80, 81), NK cells (82–84), and myeloid cells (85). These Siglec receptors have cytosolic immunoreceptor tyrosine-based inhibition motif (ITIM) domains that lead to immune suppression and block cytotoxic reactions toward the hypersialylated cancer cells (86). Immune suppression by cancer cell sialic acids could decrease immunotherapy efficacy. Remodeling of the cancer cell surface to remove sialic acid improves NK cell–mediated cancer cell killing (87) and controls tumor growth in a Siglec-E–dependent manner in a mouse model (85).
Environmental stress promotes drug resistance.
In addition to cellular forms of drug resistance, a tumor-promoting metabolite milieu fosters drug resistance. Tumors have increased acidity, higher concentrations of reactive oxygen species, and lower nutrient levels relative to healthy tissue. This harsh microenvironment collectively causes cellular stress in cancer and stromal cells (88). More aggressive tumors express a heat-shock factor 1 (HSF1) transcriptional program in cancer cells and CAFs near necrotic regions (89, 90). Increased glycolysis by cancer cells acidifies the TME by increasing the concentration of lactate, which stimulates differentiation of macrophages into an immunosuppressive state (91). Cancer cells also respond to environmental stress by increasing autophagy levels, which improves cellular viability in nutrient-poor conditions (92). Cancer cells under high environmental stress are more drug resistant. For example, HSF1 positively regulates the expression of drug resistance genes (93–95), and higher levels of autophagy are linked with increased drug resistance (92).
Microenvironmental synthetic lethality for cancer therapy
Synthetic lethal cancer therapies take advantage of differential drug sensitivity of cancer cells relative to healthy cells (8). The first and currently only approved synthetic lethal cancer therapy is poly-ADP ribose polymerase (PARP) inhibitors for BRCA1- and BRCA2-mutant ovarian cancers (96). PARP, BRCA1, and BRCA2 contribute to DNA repair, making cancer cells carrying BRCA mutations more sensitive to PARP inhibitors (97, 98). Interestingly, PARP inhibitors have not been as successful in treating breast cancers with BRCA mutations. This lack of success suggests that successful synthetic lethal therapies depend on context outside of the two genes involved in the synthetic lethality (7).
Moving beyond a cancer cell–focused view of synthetic lethal therapy, many new therapies seek to shape the protumor microenvironment to an antitumor phenotype, which in turn leads to cancer cell death. The protumor microenvironment supports tumor viability with abnormal structures and functions that differ from healthy tissue. Analogous to cancer-associated mutations, protumor elements of the TME are unique, and thus tumors are specifically sensitive to targeting of these elements. In this way, therapy targeting the TME uses a principle of nongenetic synthetic lethality. Many key features of the protumor microenvironment are different compared with healthy tissue, opening up microenvironmental synthetic lethal therapy.
Tumor structure and biochemistry create contextual synthetic lethalities.
Tumor organs have unique properties that are required for progression. These properties create contextual synthetic lethalities, as healthy organs do not feature these properties and are thus not sensitive to therapeutic inhibition. For example, therapies that target tumorigenic vasculature and ECM could enable immune control and greatly improve patient survival (99).
Inhibiting angiogenesis to normalize vasculature.
Tumors need to grow new blood vessels in order to feed rapidly dividing cancer cells (100). Cancer cells rely on glycolysis, as opposed to oxidative phosphorylation, requiring greater nutrient supply and glucose flux (69). Yet many tumors are abnormally and poorly vascularized, which causes nutrient-poor and hypoxic regions within tumors (101). Tumor hypoxia promotes aggressive cancer cell phenotypes and fosters drug resistance (102). However, hypoxic cancer cells are deficient in DNA damage repair (103), representing a contextual synthetic lethality to inhibition of cellular detection of DNA damage. Indeed, hypoxia-induced deficiencies in DNA damage repair sensitize these cells to PARP inhibitors (10, 104). Poor tumor vasculature also causes high interstitial fluid pressure that resists convection of cancer therapeutics from the blood into the core of a tumor (9).
To relieve hypoxia and normalize the abnormal vasculature of tumors, vascular endothelial growth factor (VEGF) antagonists are used to block neoangiogenesis. However, high-dose therapy can lead to complete disruption of tumor vasculature that induces hypoxia, creating a niche for cancer stem cells and immunosuppressive immune cells. Lower-dose therapy that normalizes tumor vasculature has shown greater clinical success (Figure 2A and ref. 13). The success of antiangiogenesis therapy may depend on microenvironmental factors. For example, patients with obesity benefit less from anti-VEGF therapy. In obese mice, anti-VEGF therapy does not inhibit tumor growth due to increased IL-6 and FGF-2 expression in adipocyte and myeloid cells in hypoxic adipocyte-rich regions. Inhibition of IL-6 or FGF-2 improved response to anti-VEGF therapy in obese mice (105).
Figure 2. Targeting contextual synthetic lethalities in the TME.

(A) Low-dose antiangiogenesis therapy normalizes tumor vasculature. (B) Leaky vasculature enables affinity targeting of ECM components enriched in tumors. (C) Small-molecule drugs educate myeloid cells to antitumor phenotypes by differentiation or repolarization. Nanoparticle (NP) encapsulation improves delivery to phagocytes. (D) Enzymatic hydrolysis of ECM or inhibition of CAFs debulks tumor ECM, normalizing tumor structure and vascularity.
Tumorigenic ECM presents ligands for localization of therapies.
The combination of leaky vasculature and tumor-specific ECM allows for affinity targeting of the TME. In breast cancer, tumors have increased deposition of fibrillar collagen that leads to denser and stiffer tissue and is associated with poorer prognoses (4, 19). Increased stiffness and abnormal structure coupled with leaky vasculature make tumor-associated collagen a unique epitope. For example, affinity targeting of probes to tumor-associated collagen enables diagnostic detection of tumors and micrometastases (106). Further, drug delivery using collagen affinity has shown promise in multiple immuno- and chemotherapeutic drugs in preclinical mouse models (Figure 2B and refs. 107, 108). Tumor localization of IL-2 or IL-12 by linkage to the collagen-binding protein lumican leads to tumor rejection by increasing tumor infiltration of cytotoxic T cells (109). Likewise, multiple immunotherapies (anti–CTLA-4, anti–PD-L1, IL-2, and IL-12) linked to the collagen-binding von Willebrand factor A3 domain selectively localize these protein therapeutics to the TME and lead to tumor rejection in mouse models (110, 111). Thus, leaky vasculature coupled with tumor-specific ECM represents a contextual synthetic lethality for drug localization.
Like collagen, fibrin and fibronectin are also distinct in the TME, opening possibilities for drug delivery (106, 112). Antibody fragments and peptides identified using phage display are selectively enriched in the TME in multiple tumor types in mice (113–116). The L19 antibody, which binds to the cancer-associated extra domain B splice variant of fibronectin, localizes IL-2, IL-12, and TNF to tumors and improves treatment response in glioblastoma multiforme (GBM) in patients in a phase I/II clinical trial. GBM is poorly infiltrated by immune cells, and these immunocytokines enhance immune infiltration and cancer cell death (117).
Manipulating tumor-promoting stromal cell functions.
High infiltration of tumor-promoting stromal cell phenotypes could enable contextual synthetic lethalities through polarization of these cells to tumor-suppressing phenotypes. In this section we describe therapies that directly stimulate immune cell polarization and recruitment of cytotoxic effector T cells. In addition, other therapies may have previously unknown immunostimulatory side effects. For example, DNA methyltransferase inhibitors permit expression of tumor antigens and endogenous retrovirus transcripts that drive a cytotoxic immune response, demonstrating how therapies that target the cancer cell epigenome also affect the TME (118). We focus this section on therapies that target myeloid cells and CAFs, and refer the reader to excellent reviews on targeting of lymphoid cells (119, 120).
Phenotypically plastic tumor-associated macrophages can acquire antitumor functions.
Tumor-associated macrophages (TAMs) are highly abundant in many cancers, and their high abundance could be therapeutically harnessed as a contextual synthetic lethality if they are polarized to antitumor phenotypes (Figure 2C and ref. 121). Selective modification of monocyte gene expression using the class IIa histone deacetylase (HDAC) inhibitor TMP195 polarizes TAMs to a phagocytic antitumor phenotype that enhances efficacy of carboplatin, paclitaxel, and anti–PD-1 therapies (122). Lysosomal activity may also determine TAM phenotype, as the lysosomal inhibitor chloroquine polarizes macrophages to antitumor phenotypes and enhances T cell control of melanoma tumors in mice (123). Combination therapy of hydroxychloroquine with doxorubicin improves therapeutic efficacy in a non–small cell lung cancer mouse model by reprogramming immunosuppressive TAMs to an antitumor phenotype marked by increased levels of MHC class II and decreased levels of CD206. These reprogramed TAMs increase infiltration of tumor-killing CD8+ T cells (124). Targeting of TAM reprogramming can also be achieved by direct targeting of protumor CD206+ TAMs. The host defense peptide synthetic analog RP-182 binds to the CD206 receptor on tumor-promoting TAMs and activates RAC1/CDC42 signaling. CD206 activation reprograms TAMs to an antitumor phenotype characterized by increased phagocytosis of cancer cells that results in decreased tumor growth in a pancreatic cancer mouse model (125).
TAMs have a propensity to phagocytose particles in the size range of approximately 0.1–1 μm, acting to endocytose nanoparticle therapeutics. Nanoencapsulation of the TLR7/8 agonist R848 improves macrophage-specific drug delivery and drives TAM expression of genes associated with antitumor activity, which improves survival in mice bearing MC38 xenografts (126). The FDA-approved iron deficiency nanoparticle drug ferumoxytol induces reactive oxygen species–mediated cytotoxic activities in TAMs toward cancer cells that decrease tumor growth and metastatic burden in multiple mouse models (127). As a result of the phagocytic activity of TAMs, they can accumulate nanoencapsulated chemotherapeutics to create a high concentration of cytotoxic drugs within the TME. In the 4T1 breast cancer mouse model, liver metastases are decreased by a nanoencapsulated platinum that accumulates in TAMs (128). This effect was enhanced when tumor-bearing mice were first treated with radiation to increase TAM infiltration (129), a known side effect of radiation therapy (48). Thus, tumors with high TAM infiltration have a contextual synthetic lethality to nanoparticle-based therapies.
Inducing antitumor phenotypes in tumor-mobilized immunosuppressive myeloid cells.
Myeloid cells are recruited to the tumor and can support an immunosuppressive microenvironment that is drug resistant. An immature and heterogeneous population of these cells, known as myeloid-derived suppressor cells, serves to inhibit cytotoxic activities of immune effector cells against cancer cells (130). The spectrum of cell types within this population makes depletion difficult. Instead, therapy could be enhanced by targeting repolarization or differentiation of immunosuppressive myeloid cells to antitumor phenotypes (Figure 2C). Control of myeloid polarization can be achieved by manipulation of the differentiation of myeloid progenitors using a bone marrow–homing nanoparticle therapeutic bearing the immunostimulatory muramyl tripeptide. In a mouse model, this nanoparticle therapy increases myelopoiesis, decreases the number of TAMs, and potentiates anti–PD-1 and anti–CTLA-4 therapy (131). Also, β-glucan induces a potent antitumor neutrophil phenotype through epigenetic rewiring during granulopoiesis (132). Outside of the bone marrow, myeloid cells continue to differentiate and polarize. Monocytes are differentiated into antitumor TAMs by a prodrug of 6-diazo-5-oxo-l-norleucine (DON) that blocks glutamine metabolism in myeloid-derived suppressor cells, resulting in decreased tumor growth and metastatic burden in tumor-bearing mice (133). These examples show how the propensity of some tumors to be highly infiltrated by immunosuppressive myeloid cells can thus be turned into a microenvironmental synthetic lethality. Polarizing myeloid cell progenitors to a potent antitumor phenotype, which are then recruited to the tumor, induces an immunostimulatory TME that improves patient outcomes.
Targeting CAFs to remodel ECM.
CAFs exist in a unique cell state that could allow for contextual synthetic lethal therapeutic targeting (23). Fibroblast activation protein (FAP) is a CAF marker that is of considerable clinical interest, but has yet to be realized in an approved therapy (134). For example, depletion of FAP+ CAFs using a FAP vaccine decreases collagen density in tumors and improves response to doxorubicin in mice (135). But targeting CAFs for depletion may counterintuitively promote tumor growth in certain contexts, likely owing to FAP expression on other stromal cell types, including cancer-associated pericytes (136). In pancreatic ductal adenocarcinoma (PDAC), which is CAF rich and highly fibrotic, genetic depletion of α-smooth muscle actin–positive (αSMA+) myofibroblasts in a mouse model leads to increased tumor invasion, decreased survival, and increased CTLA-4 expression in tumors. This example shows that CAFs and tumorigenic ECM have multiple tumor-suppressing activities. Combining αSMA+ myofibroblast depletion with gemcitabine treatment does not improve therapeutic response. However, combination of αSMA+ myofibroblast depletion with anti–CTLA-4 therapy improves therapeutic response, showing that the interplay between CAFs and treatment depends on context (137). As another example, while deletion of Sonic hedgehog (Shh) in cancer cells decreases the number of CAFs, it also decreases the number of infiltrating immune cells, leading to greater tumor growth in a mouse PDAC model (138). It is likely that the heterogeneity of CAFs within a tumor points to a spectrum of tumor-promoting and -suppressing activities that may be lost by depletion therapies.
Instead of targeting CAF depletion, inhibition of tumor-promoting CAF activities may hold greater promise (Figure 2D). Proliferation of CAFs is inhibited by antagonism of Smoothened of the hedgehog pathway, leading to decreased density of collagen I. Inhibition of CAFs in turn improves therapeutic response to gemcitabine by increasing tumor vascularity and intratumoral concentration of gemcitabine in a PDAC mouse model (139). Combination of a Smoothened antagonist with docetaxel showed promising results in phase I clinical trials when given to triple-negative breast cancer patients with high levels of cancer cell–CAF paracrine hedgehog signaling (140). In the MMTV-PyMT model of breast cancer, cancer cells were maintained in a tamoxifen-resistant state by CAFs. Antibody neutralization of cancer cell–derived PDGF-CC blocked PDGFR signaling in CAFs and sensitized these tumors to tamoxifen. These results demonstrate that response to hormone therapy is in part determined by CAF activity (141). Instead of inhibiting CAF function, ECM components can be targeted directly. Debulking hyaluronan with hyaluronidase improves therapeutic response to gemcitabine by enhancing vascularity in a PDAC mouse model (142). In these examples, fibrotic, CAF-rich tumors are vulnerable to chemo- or immune therapy when the tumor ECM is therapeutically normalized. However, this vulnerability must be balanced with potential loss of tumor-suppressive ECM activities.
Conclusion and future directions
The TME plays an important role in tumorigenesis and response to therapy. Clinicians can use therapies that target microenvironmental synthetic lethalities in combination with traditional cytotoxic chemotherapy, surgery, immunotherapy, and radiation to improve survival in patients who currently lack effective treatments. Because these TME-targeted therapies are directed toward protumor microenvironmental features, there are fewer off-target effects and less potential for drug resistance. Treatment of the TME, analogous to targeted therapy, will need to be personalized to treat the specific vulnerabilities of a patient’s tumor. Ideally, TME properties could be discerned with blood measurements of surrogates for tumor inflammation, such as cytokines or circulating immune cell counts. Finally, understanding the evolution of the TME, especially in recurrence, will bring us closer to making cancer a disease that we die with, not from.
Acknowledgments
This work is dedicated to the memory of Zena Werb. This work was supported by an American Cancer Society–2017 Seattle Gala Paddle Raise Postdoctoral Fellowship (PF-18-118-01-CDD to KJM) and NIH National Cancer Institute funding (R35CA242447-01A1 and R01CA227942 to VMW). Figures were created with BioRender (BioRender.com).
Version 1. 03/15/2021
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2021, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2021;131(6):e143765.https://doi.org/10.1172/JCI143765.
Contributor Information
Kevin J. Metcalf, Email: Kevin.Metcalf@ucsf.edu.
Alaa Alazzeh, Email: aalazzeh@dons.usfca.edu.
Zena Werb, Email: zena.werb@ucsf.edu.
References
- 1.Egeblad M, et al. Tumors as organs: complex tissues that interface with the entire organism. Dev Cell. 2010;18(6):884–901. doi: 10.1016/j.devcel.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bissell MJ, Radisky D. Putting tumours in context. Nat Rev Cancer. 2001;1(1):46–54. doi: 10.1038/35094059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balkwill FR, et al. The tumor microenvironment at a glance. J Cell Sci. 2012;125(23):5591–5596. doi: 10.1242/jcs.116392. [DOI] [PubMed] [Google Scholar]
- 4.Winkler J, et al. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat Commun. 2020;11(1):5120. doi: 10.1038/s41467-020-18794-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Trédan O, et al. Drug resistance and the solid tumor microenvironment. JNCI J Natl Cancer Inst. 2007;99(19):1441–1454. doi: 10.1093/jnci/djm135. [DOI] [PubMed] [Google Scholar]
- 6.Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature. 2013;501(7467):346–354. doi: 10.1038/nature12626. [DOI] [PubMed] [Google Scholar]
- 7.Huang A, et al. Synthetic lethality as an engine for cancer drug target discovery. Nat Rev Drug Discov. 2020;19(1):23–38. doi: 10.1038/s41573-019-0046-z. [DOI] [PubMed] [Google Scholar]
- 8.O’Neil NJ, et al. Synthetic lethality and cancer. Nat Rev Genet. 2017;18(10):613–623. doi: 10.1038/nrg.2017.47. [DOI] [PubMed] [Google Scholar]
- 9.Nia HT, et al. Physical traits of cancer. Science. 2020;370(6516):eaaz0868. doi: 10.1126/science.aaz0868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan N, et al. Contextual synthetic lethality of cancer cell kill based on the tumor microenvironment. Cancer Res. 2010;70(20):8045–8054. doi: 10.1158/0008-5472.CAN-10-2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gerlinger M, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zardavas D, et al. Clinical management of breast cancer heterogeneity. Nat Rev Clin Oncol. 2015;12(7):381–394. doi: 10.1038/nrclinonc.2015.73. [DOI] [PubMed] [Google Scholar]
- 13.Jain RK. Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell. 2014;26(5):605–622. doi: 10.1016/j.ccell.2014.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Serganova I, et al. Molecular imaging of temporal dynamics and spatial heterogeneity of hypoxia-inducible factor-1 signal transduction activity in tumors in living mice. Cancer Res. 2004;64(17):6101–6108. doi: 10.1158/0008-5472.CAN-04-0842. [DOI] [PubMed] [Google Scholar]
- 15.Tiede S, et al. Multi-color clonal tracking reveals intra-stage proliferative heterogeneity during mammary tumor progression. Oncogene. 2020;40(1):12–27. doi: 10.1038/s41388-020-01508-4. [DOI] [PubMed] [Google Scholar]
- 16.Zahir N, Weaver VM. Death in the third dimension: apoptosis regulation and tissue architecture. Curr Opin Genet Dev. 2004;14(1):71–80. doi: 10.1016/j.gde.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 17.Weaver VM, et al. β4 Integrin-dependent formation of polarized three-dimensional architecture confers resistance to apoptosis in normal and malignant mammary epithelium. Cancer Cell. 2002;2(3):205–216. doi: 10.1016/S1535-6108(02)00125-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Levental KR, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139(5):891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Acerbi I, et al. Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integr Biol. 2015;7(10):1120–1134. doi: 10.1039/c5ib00040h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lopez JI, et al. In situ force mapping of mammary gland transformation. Integr Biol. 2011;3(9):910–921. doi: 10.1039/c1ib00043h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. doi: 10.1038/s41563-020-00849-5. Maller O, et al. Tumour-associated macrophages drive stromal cell-dependent collagen crosslinking and stiffening to promote breast cancer aggression [published online November 30, 2020]. Nat Mater . [DOI] [PMC free article] [PubMed]
- 22.Laklai H, et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat Med. 2016;22(5):497–505. doi: 10.1038/nm.4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sahai E, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer. 2020;20(3):174–186. doi: 10.1038/s41568-019-0238-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kechagia JZ, et al. Integrins as biomechanical sensors of the microenvironment. Nat Rev Mol Cell Biol. 2019;20(8):457–473. doi: 10.1038/s41580-019-0134-2. [DOI] [PubMed] [Google Scholar]
- 25.et al. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8(3):241–254. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 26.Borza CM, Pozzi A. Discoidin domain receptors in disease. Matrix Biol. 2014;34:185–192. doi: 10.1016/j.matbio.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xian X, et al. Syndecans as receptors and organizers of the extracellular matrix. Cell Tissue Res. 2009;339(1):31–46. doi: 10.1007/s00441-009-0829-3. [DOI] [PubMed] [Google Scholar]
- 28.Hussain S, et al. Cancer drug resistance: a fleet to conquer. J Cell Biochem. 2019;120(9):14213–14225. doi: 10.1002/jcb.28782. [DOI] [PubMed] [Google Scholar]
- 29.Oudin MJ, Weaver VM. Physical and chemical gradients in the tumor microenvironment regulate tumor cell invasion, migration, and metastasis. Cold Spring Harb Symp Quant Biol. 2016;81:189–205. doi: 10.1101/sqb.2016.81.030817. [DOI] [PubMed] [Google Scholar]
- 30.Hynes RO. The extracellular matrix: not just pretty fibrils. Science. 2009;326(5957):1216–1219. doi: 10.1126/science.1176009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Swann JB, Smyth MJ. Immune surveillance of tumors. J Clin Invest. 2007;117(5):1137–1146. doi: 10.1172/JCI31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ali HR, et al. Imaging mass cytometry and multiplatform genomics define the phenogenomic landscape of breast cancer. Nat Cancer. 2020;1(2):163–175. doi: 10.1038/s43018-020-0026-6. [DOI] [PubMed] [Google Scholar]
- 33.Chen Q, et al. Tumor microvasculature as a barrier to antitumor immunity. Cancer Immunol Immunother. 2003;52(11):670–679. doi: 10.1007/s00262-003-0425-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Salmon H, et al. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J Clin Invest. 2012;122(3):899–910. doi: 10.1172/JCI45817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Keren L, et al. A structured tumor-immune microenvironment in triple negative breast cancer revealed by multiplexed ion beam imaging. Cell. 2018;174(6):1373–1387.e19. doi: 10.1016/j.cell.2018.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jackson HW, et al. The single-cell pathology landscape of breast cancer. Nature. 2020;578(7796):615–620. doi: 10.1038/s41586-019-1876-x. [DOI] [PubMed] [Google Scholar]
- 37.Binnewies M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24(5):541–550. doi: 10.1038/s41591-018-0014-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heindl A, et al. Relevance of spatial heterogeneity of immune infiltration for predicting risk of recurrence after endocrine therapy of ER+ breast cancer. JNCI J Natl Cancer Inst. 2018;110(2):166–175. doi: 10.1093/jnci/djx137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krausgruber T, et al. Structural cells are key regulators of organ-specific immune responses. Nature. 2020;583(7815):296–302. doi: 10.1038/s41586-020-2424-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Casbon AJ, et al. Invasive breast cancer reprograms early myeloid differentiation in the bone marrow to generate immunosuppressive neutrophils. Proc Natl Acad Sci U S A. 2015;112(6):E566–575. doi: 10.1073/pnas.1424927112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hagerling C, et al. Immune effector monocyte–neutrophil cooperation induced by the primary tumor prevents metastatic progression of breast cancer. Proc Natl Acad Sci U S A. 2019;116(43):21704–21714. doi: 10.1073/pnas.1907660116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Waight JD, et al. Tumor-derived G-CSF facilitates neoplastic growth through a granulocytic myeloid-derived suppressor cell-dependent mechanism. PLoS One. 2011;6(11):e27690. doi: 10.1371/journal.pone.0027690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morales JK, et al. GM-CSF is one of the main breast tumor-derived soluble factors involved in the differentiation of CD11b-Gr1- bone marrow progenitor cells into myeloid-derived suppressor cells. Breast Cancer Res Treat. 2010;123(1):39–49. doi: 10.1007/s10549-009-0622-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dolcetti L, et al. Hierarchy of immunosuppressive strength among myeloid-derived suppressor cell subsets is determined by GM-CSF. Eur J Immunol. 2010;40(1):22–35. doi: 10.1002/eji.200939903. [DOI] [PubMed] [Google Scholar]
- 45.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9(3):162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsuyada A, et al. CCL2 mediates cross-talk between cancer cells and stromal fibroblasts that regulates breast cancer stem cells. Cancer Res. 2012;72(11):2768–2779. doi: 10.1158/0008-5472.CAN-11-3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fang WB, et al. CCL2/CCR2 chemokine signaling coordinates survival and motility of breast cancer cells through Smad3 protein- and p42/44 mitogen-activated protein kinase (MAPK)-dependent mechanisms. J Biol Chem. 2012;287(43):36593–36608. doi: 10.1074/jbc.M112.365999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huet E, et al. Stroma in normal and cancer wound healing. FEBS J. 2019;286(15):2909–2920. doi: 10.1111/febs.14842. [DOI] [PubMed] [Google Scholar]
- 49.Schaue D, et al. Radiation and inflammation. Semin Radiat Oncol. 2015;25(1):4–10. doi: 10.1016/j.semradonc.2014.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Demicheli R, et al. Tumor dormancy and surgery-driven interruption of dormancy in breast cancer: learning from failures. Nat Clin Pract Oncol. 2007;4(12):699–710. doi: 10.1038/ncponc0999. [DOI] [PubMed] [Google Scholar]
- 51.Bartal I, et al. Immune perturbations in patients along the perioperative period: alterations in cell surface markers and leukocyte subtypes before and after surgery. Brain Behav Immun. 2010;24(3):376–386. doi: 10.1016/j.bbi.2009.02.010. [DOI] [PubMed] [Google Scholar]
- 52.Krall JA, et al. The systemic response to surgery triggers the outgrowth of distant immune-controlled tumors in mouse models of dormancy. Sci Transl Med. 2018;10(436):eaan3464. doi: 10.1126/scitranslmed.aan3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Allen BM, et al. Systemic dysfunction and plasticity of the immune macroenvironment in cancer models. Nat Med. 2020;26(7):1125–1134. doi: 10.1038/s41591-020-0892-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Retsky M, et al. Reduction of breast cancer relapses with perioperative non-steroidal anti-inflammatory drugs: new findings and a review. Curr Med Chem. 2013;20(33):4163–4176. doi: 10.2174/09298673113209990250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sherry AD, et al. Systemic inflammation after radiation predicts locoregional recurrence, progression, and mortality in stage II-III triple-negative breast cancer. Int J Radiat Oncol. 2020;108(1):268–276. doi: 10.1016/j.ijrobp.2019.11.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Galluzzi L, et al. The secret ally: immunostimulation by anticancer drugs. Nat Rev Drug Discov. 2012;11(3):215–233. doi: 10.1038/nrd3626. [DOI] [PubMed] [Google Scholar]
- 57.Guerriero JL, et al. DNA alkylating therapy induces tumor regression through an HMGB1-mediated activation of innate immunity. J Immunol. 2011;186(6):3517–3526. doi: 10.4049/jimmunol.1003267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ma Y, et al. CCL2/CCR2-dependent recruitment of functional antigen-presenting cells into tumors upon chemotherapy. Cancer Res. 2014;74(2):436–445. doi: 10.1158/0008-5472.CAN-13-1265. [DOI] [PubMed] [Google Scholar]
- 59.Ma Y, et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity. 2013;38(4):729–741. doi: 10.1016/j.immuni.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 60.Sistigu A, et al. Cancer cell–autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med. 2014;20(11):1301–1309. doi: 10.1038/nm.3708. [DOI] [PubMed] [Google Scholar]
- 61.Nakasone ES, et al. Imaging tumor-stroma interactions during chemotherapy reveals contributions of the microenvironment to resistance. Cancer Cell. 2012;21(4):488–503. doi: 10.1016/j.ccr.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ruffell B, et al. Leukocyte composition of human breast cancer. Proc Natl Acad Sci U S A. 2012;109(8):2796–2801. doi: 10.1073/pnas.1104303108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.DeNardo DG, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011;1(1):54–67. doi: 10.1158/2159-8274.CD-10-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shree T, et al. Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Genes Dev. 2011;25(23):2465–2479. doi: 10.1101/gad.180331.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chan TS, et al. Metronomic chemotherapy prevents therapy-induced stromal activation and induction of tumor-initiating cells. J Exp Med. 2016;213(13):2967–2988. doi: 10.1084/jem.20151665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Browder T, et al. Antiangiogenic scheduling of chemotherapy improves efficacy against experimental drug-resistant cancer. Cancer Res. 2000;60(7):1878–1886. [PubMed] [Google Scholar]
- 67.Pasquier E, et al. Metronomic chemotherapy: new rationale for new directions. Nat Rev Clin Oncol. 2010;7(8):455–465. doi: 10.1038/nrclinonc.2010.82. [DOI] [PubMed] [Google Scholar]
- 68.Zhang J, et al. Integrating evolutionary dynamics into treatment of metastatic castrate-resistant prostate cancer. Nat Commun. 2017;8(1):1816. doi: 10.1038/s41467-017-01968-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 70.Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359(6382):1350–1355. doi: 10.1126/science.aar4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Haslam A, Prasad V. Estimation of the percentage of US patients with cancer who are eligible for and respond to checkpoint inhibitor immunotherapy drugs. JAMA Netw Open. 2019;2(5):e192535. doi: 10.1001/jamanetworkopen.2019.2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Morvan MG, Lanier LL. NK cells and cancer: you can teach innate cells new tricks. Nat Rev Cancer. 2016;16(1):7–19. doi: 10.1038/nrc.2015.5. [DOI] [PubMed] [Google Scholar]
- 73.Yamamoto K, et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature. 2020;581(7806):100–105. doi: 10.1038/s41586-020-2229-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McGranahan N, et al. Allele-specific HLA loss and immune escape in lung cancer evolution. Cell. 2017;171(6):1259–1271. doi: 10.1016/j.cell.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rooney MS, et al. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160(1–2):48–61. doi: 10.1016/j.cell.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rodig SJ, et al. MHC proteins confer differential sensitivity to CTLA-4 and PD-1 blockade in untreated metastatic melanoma. Sci Transl Med. 2018;10(450):eaar3342. doi: 10.1126/scitranslmed.aar3342. [DOI] [PubMed] [Google Scholar]
- 77.Dong H, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8(8):793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 78.Majeti R, et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell. 2009;138(2):286–299. doi: 10.1016/j.cell.2009.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Barkal AA, et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature. 2019;572(7769):392–396. doi: 10.1038/s41586-019-1456-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Stanczak MA, et al. Self-associated molecular patterns mediate cancer immune evasion by engaging Siglecs on T cells. J Clin Invest. 2018;128(11):4912–4923. doi: 10.1172/JCI120612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Perdicchio M, et al. Tumor sialylation impedes T cell mediated anti-tumor responses while promoting tumor associated-regulatory T cells. Oncotarget. 2016;7(8):8771–8782. doi: 10.18632/oncotarget.6822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hudak JE, et al. Glycocalyx engineering reveals a Siglec-based mechanism for NK cell immunoevasion. Nat Chem Biol. 2014;10(1):69–75. doi: 10.1038/nchembio.1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jandus C, et al. Interactions between Siglec-7/9 receptors and ligands influence NK cell-dependent tumor immunosurveillance. J Clin Invest. 2014;124(4):1810–1820. doi: 10.1172/JCI65899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cohen M, et al. Sialylation of 3-methylcholanthrene–induced fibrosarcoma determines antitumor immune responses during immunoediting. J Immunol. 2010;185(10):5869–5878. doi: 10.4049/jimmunol.1001635. [DOI] [PubMed] [Google Scholar]
- 85.Gray MA, et al. Targeted glycan degradation potentiates the anticancer immune response in vivo. Nat Chem Biol. 2020;16(12):1376–1384. doi: 10.1038/s41589-020-0622-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Macauley MS, et al. Siglec-mediated regulation of immune cell function in disease. Nat Rev Immunol. 2014;14(10):653–666. doi: 10.1038/nri3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Xiao H, et al. Precision glycocalyx editing as a strategy for cancer immunotherapy. Proc Natl Acad Sci U S A. 2016;113(37):10304–10309. doi: 10.1073/pnas.1608069113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cubillos-Ruiz JR, et al. Tumorigenic and immunosuppressive effects of endoplasmic reticulum stress in cancer. Cell. 2017;168(4):692–706. doi: 10.1016/j.cell.2016.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mendillo ML, et al. HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell. 2012;150(3):549–562. doi: 10.1016/j.cell.2012.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Scherz-Shouval R, et al. The reprogramming of tumor stroma by HSF1 is a potent enabler of malignancy. Cell. 2014;158(3):564–578. doi: 10.1016/j.cell.2014.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Colegio OR, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513(7519):559–563. doi: 10.1038/nature13490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sui X, et al. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis. 2013;4(10):e838–e838. doi: 10.1038/cddis.2013.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Vilaboa NE, et al. Regulation of multidrug resistance 1 (MDR1)/P-glycoprotein gene expression and activity by heat-shock transcription factor 1 (HSF1) J Biol Chem. 2000;275(32):24970–24976. doi: 10.1074/jbc.M909136199. [DOI] [PubMed] [Google Scholar]
- 94.Mun G-I, et al. Decreased expression of FBXW7 by ERK1/2 activation in drug-resistant cancer cells confers transcriptional activation of MDR1 by suppression of ubiquitin degradation of HSF1. Cell Death Dis. 2020;11(5):1–14. doi: 10.1038/s41419-020-2600-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Desai S, et al. Heat shock factor 1 (HSF1) controls chemoresistance and autophagy through transcriptional regulation of autophagy-related protein 7 (ATG7) J Biol Chem. 2013;288(13):9165–9176. doi: 10.1074/jbc.M112.422071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lord CJ, Ashworth A. PARP inhibitors: synthetic lethality in the clinic. Science. 2017;355(6330):1152–1158. doi: 10.1126/science.aam7344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bryant HE, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 98.Farmer H, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 99.Jain RK. Normalizing tumor microenvironment to treat cancer: bench to bedside to biomarkers. J Clin Oncol. 2013;31(17):2205–2218. doi: 10.1200/JCO.2012.46.3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Weis SM, Cheresh DA. Tumor angiogenesis: molecular pathways and therapeutic targets. Nat Med. 2011;17(11):1359–1370. doi: 10.1038/nm.2537. [DOI] [PubMed] [Google Scholar]
- 101.Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407(6801):249–257. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 102.Vaupel P, Mayer A. Hypoxia in cancer: significance and impact on clinical outcome. Cancer Metastasis Rev. 2007;26(2):225–239. doi: 10.1007/s10555-007-9055-1. [DOI] [PubMed] [Google Scholar]
- 103.Bindra RS, et al. Regulation of DNA repair in hypoxic cancer cells. Cancer Metastasis Rev. 2007;26(2):249–260. doi: 10.1007/s10555-007-9061-3. [DOI] [PubMed] [Google Scholar]
- 104.Hegan DC, et al. Inhibition of poly(ADP-ribose) polymerase down-regulates BRCA1 and RAD51 in a pathway mediated by E2F4 and p130. Proc Natl Acad Sci U S A. 2010;107(5):2201–2206. doi: 10.1073/pnas.0904783107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Incio J, et al. Obesity promotes resistance to anti-VEGF therapy in breast cancer by up-regulating IL-6 and potentially FGF-2. Sci Transl Med. 2018;10(432):eaag0945. doi: 10.1126/scitranslmed.aag0945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhou Z, Lu Z-R. Molecular imaging of the tumor microenvironment. Adv Drug Deliv Rev. 2017;113:24–48. doi: 10.1016/j.addr.2016.07.012. [DOI] [PubMed] [Google Scholar]
- 107.Ishihara J, et al. Matrix-binding checkpoint immunotherapies enhance antitumor efficacy and reduce adverse events. Sci Transl Med. 2017;9(415):eaan0401. doi: 10.1126/scitranslmed.aan0401. [DOI] [PubMed] [Google Scholar]
- 108.Liang H, et al. A collagen-binding EGFR single-chain Fv antibody fragment for the targeted cancer therapy. J Control Release. 2015;209:101–109. doi: 10.1016/j.jconrel.2015.04.029. [DOI] [PubMed] [Google Scholar]
- 109.Momin N, et al. Anchoring of intratumorally administered cytokines to collagen safely potentiates systemic cancer immunotherapy. Sci Transl Med. 2019;11(498):eaaw2614. doi: 10.1126/scitranslmed.aaw2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ishihara J, et al. Targeted antibody and cytokine cancer immunotherapies through collagen affinity. Sci Transl Med. 2019;11(487):eaau3259. doi: 10.1126/scitranslmed.aau3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mansurov A, et al. Collagen-binding IL-12 enhances tumour inflammation and drives the complete remission of established immunologically cold mouse tumours. Nat Biomed Eng. 2020;4(5):531–543. doi: 10.1038/s41551-020-0549-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Raavé R, et al. Chemotherapeutic drug delivery by tumoral extracellular matrix targeting. J Control Release. 2018;274:1–8. doi: 10.1016/j.jconrel.2018.01.029. [DOI] [PubMed] [Google Scholar]
- 113.Pilch J, et al. Peptides selected for binding to clotted plasma accumulate in tumor stroma and wounds. Proc Natl Acad Sci U S A. 2006;103(8):2800–2804. doi: 10.1073/pnas.0511219103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Simberg D, et al. Biomimetic amplification of nanoparticle homing to tumors. Proc Natl Acad Sci U S A. 2007;104(3):932–936. doi: 10.1073/pnas.0610298104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhou Z, et al. MRI detection of breast cancer micrometastases with a fibronectin-targeting contrast agent. Nat Commun. 2015;6(1):7984. doi: 10.1038/ncomms8984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Neri D, et al. Targeting by affinity-matured recombinant antibody fragments of an angiogenesis associated fibronectin isoform. Nat Biotechnol. 1997;15(12):1271–1275. doi: 10.1038/nbt1197-1271. [DOI] [PubMed] [Google Scholar]
- 117.Weiss T, et al. Immunocytokines are a promising immunotherapeutic approach against glioblastoma. Sci Transl Med. 2020;12(564):eabb2311. doi: 10.1126/scitranslmed.abb2311. [DOI] [PubMed] [Google Scholar]
- 118.Jones PA, et al. Targeting the cancer epigenome for therapy. Nat Rev Genet. 2016;17(10):630–641. doi: 10.1038/nrg.2016.93. [DOI] [PubMed] [Google Scholar]
- 119.Chiossone L, et al. Natural killer cells and other innate lymphoid cells in cancer. Nat Rev Immunol. 2018;18(11):671–688. doi: 10.1038/s41577-018-0061-z. [DOI] [PubMed] [Google Scholar]
- 120.Thommen DS, Schumacher TN. T cell dysfunction in cancer. Cancer Cell. 2018;33(4):547–562. doi: 10.1016/j.ccell.2018.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Solinas G, et al. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J Leukoc Biol. 2009;86(5):1065–1073. doi: 10.1189/jlb.0609385. [DOI] [PubMed] [Google Scholar]
- 122.Guerriero JL, et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature. 2017;543(7645):428–432. doi: 10.1038/nature21409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chen D, et al. Chloroquine modulates antitumor immune response by resetting tumor-associated macrophages toward M1 phenotype. Nat Commun. 2018;9(1):873. doi: 10.1038/s41467-018-03225-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Li Y, et al. Hydroxychloroquine induced lung cancer suppression by enhancing chemo-sensitization and promoting the transition of M2-TAMs to M1-like macrophages. J Exp Clin Cancer Res. 2018;37(1):259. doi: 10.1186/s13046-018-0938-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Jaynes JM, et al. Mannose receptor (CD206) activation in tumor-associated macrophages enhances adaptive and innate antitumor immune responses. Sci Transl Med. 2020;12(530):eaax6337. doi: 10.1126/scitranslmed.aax6337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rodell CB, et al. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat Biomed Eng. 2018;2(8):578–588. doi: 10.1038/s41551-018-0236-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zanganeh S, et al. Iron oxide nanoparticles inhibit tumour growth by inducing pro-inflammatory macrophage polarization in tumour tissues. Nat Nanotechnol. 2016;11(11):986–994. doi: 10.1038/nnano.2016.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Miller MA, et al. Tumour-associated macrophages act as a slow-release reservoir of nano-therapeutic Pt(IV) pro-drug. Nat Commun. 2015;6(1):8692. doi: 10.1038/ncomms9692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Miller MA, et al. Radiation therapy primes tumors for nanotherapeutic delivery via macrophage-mediated vascular bursts. Sci Transl Med. 2017;9(392):eaal0225. doi: 10.1126/scitranslmed.aal0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Engblom C, et al. The role of myeloid cells in cancer therapies. Nat Rev Cancer. 2016;16(7):447–462. doi: 10.1038/nrc.2016.54. [DOI] [PubMed] [Google Scholar]
- 131.Priem B, et al. Trained immunity-promoting nanobiologic therapy suppresses tumor growth and potentiates checkpoint inhibition. Cell. 2020;183(3):786–801. doi: 10.1016/j.cell.2020.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kalafati L, et al. Innate immune training of granulopoiesis promotes anti-tumor activity. Cell. 2020;183(3):771–785. doi: 10.1016/j.cell.2020.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Oh M-H, et al. Targeting glutamine metabolism enhances tumor-specific immunity by modulating suppressive myeloid cells. J Clin Invest. 2020;130(7):3865–3884. doi: 10.1172/JCI131859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gieniec KA, et al. Cancer-associated fibroblasts—heroes or villains? Br J Cancer. 2019;121(4):293–302. doi: 10.1038/s41416-019-0509-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Loeffler M, et al. Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J Clin Invest. 2006;116(7):1955–1962. doi: 10.1172/JCI26532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Cremasco V, et al. FAP delineates heterogeneous and functionally divergent stromal cells in immune-excluded breast tumors. Cancer Immunol Res. 2018;6(12):1472–1485. doi: 10.1158/2326-6066.CIR-18-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Özdemir BC, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25(6):719–734. doi: 10.1016/j.ccr.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Rhim AD, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25(6):735–747. doi: 10.1016/j.ccr.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Olive KP, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324(5933):1457–1461. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Cazet AS, et al. Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nat Commun. 2018;9(1):2897. doi: 10.1038/s41467-018-05220-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Roswall P, et al. Microenvironmental control of breast cancer subtype elicited through paracrine platelet-derived growth factor-CC signaling. Nat Med. 2018;24(4):463–473. doi: 10.1038/nm.4494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Provenzano PP, et al. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21(3):418–429. doi: 10.1016/j.ccr.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]