

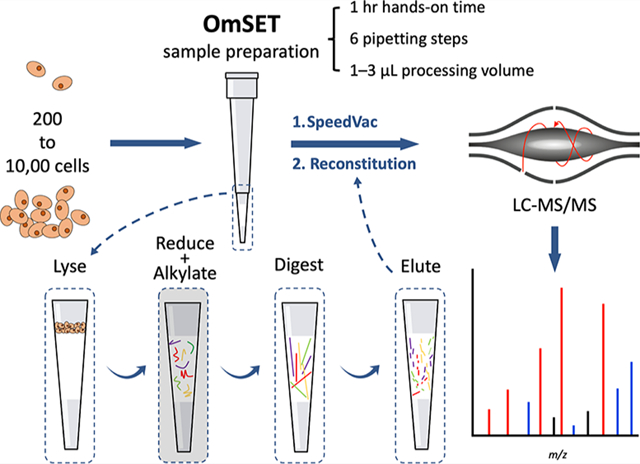
Abstract
In-depth LC–MS-based proteomic profiling of limited biological and clinical samples, such as rare cells or tissue sections from laser capture microdissection or microneedle biopsies, has been problematic due, in large, to the inefficiency of sample preparation and attendant sample losses. To address this issue, we developed on-microsolid-phase extraction tip (OmSET)-based sample preparation for limited biological samples. OmSET is simple, efficient, reproducible, and scalable and is a widely accessible method for processing ~200 to 10,000 cells. The developed method benefits from minimal sample processing volumes (1–3 μL) and conducting all sample processing steps on-membrane within a single microreactor. We first assessed the feasibility of using micro-SPE tips for nanogram-level amounts of tryptic peptides, minimized the number of required sample handling steps, and reduced the hands-on time. We then evaluated the capability of OmSET for quantitative analysis of low numbers of human monocytes. Reliable and reproducible label-free quantitation results were obtained with excellent correlations between protein abundances and the amounts of starting material (R2 = 0.93) and pairwise correlations between sample processing replicates (R2 = 0.95) along with the identification of approximately 300, 1800, and 2000 protein groups from injected ~10, 100, and 500 cell equivalents, resulting from processing approximately 200, 2000, and 10,000 cells, respectively.
Keywords: sample preparation, on-membrane digestion, micro-SPE tip, limited samples, label-free quantitation, bottom-up nanoLC–MS-based proteomics, OmSET (on-microsolid-phase extraction tip)
Graphical Abstract
INTRODUCTION
Many biological and clinical samples, such as rare cells or fine-needle aspiration biopsies, are available in scarce amounts. Circulating tumor cells (CTCs), for example, can act as a therapeutic monitoring tool1,2 and are expected to provide insights into the metastatic processes of cancer3 but exist within the range of approximately 5 to 1300 cells/mL whole blood.4,5 From tissue slices, laser capture microdissection can be used to probe phenotypically distinct cell populations6 whose unique proteomes would otherwise be hidden within ensemble results from a bulk collection of cells and has been reported for a proteomic application with as few as 100 cells.7 Additionally, fine-needle aspiration biopsy is a commonplace clinical sampling method that can collect on the order of 20,000 cells8 for various types of interrogation, including proteomic profiling.9 Proteomic analysis of quantity-limited biological and clinical samples is a critical pursuit.
Mass spectrometry (MS)-based bottom-up proteomics has been well established as an efficient tool to study diverse biological and clinical samples. Deep proteome coverage of minimal amounts of tryptic peptides has been enabled by recent advances in liquid chromatography (LC)–MS methodologies and instrumentation. Specifically, low zeptomole sensitivity can be achieved, using advanced nanoflow LC systems, improved LC–MS interfaces, MS ion optics and mass analyzers, and refined data processing algorithms.10–13 Many proteins within mammalian somatic cells exist at these levels and have even been characterized at the single-cell level.14–19 Although major advancements have been made to these downstream processes, the sensitivity of the overall platform is considerably limited by the efficiency of sample processing upstream.
Sample processing for bottom-up proteomics is a multistep procedure, typically consisting of cell/tissue lysis, protein extraction and denaturation, disulfide bond reduction, cysteine alkylation, and proteolytic digestion steps. The main challenges preventing efficient recovery of peptides available at scarce amounts are nonspecific adsorption to exposed surfaces during these multiple sample processing steps and required sample transfers20,21 and losses during cleanup steps that include the removal of incompatible salts and detergents.21 Several groups have reported on different strategies to improve recovery of limited biological samples, including using chemically passivated or low retention surfaces,14,19,22 minimizing sample volume and contact surface area,7,10,23–27 and integrating contaminant removal devices so that all steps can be performed within a single vessel.10,28–32 For samples >1000 cells, the most widely used protocols are in-StageTip (iST) sample processing,28 filter-aided sample preparation (FASP),29 sample processing within a kinked microreactor tip,23 and single-pot solid-phase-enhanced sample preparation (SP3).30 For low numbers of cells, several techniques were reported, including single-cell proteomics by mass spectrometry (SCoPE-MS and SCoPE2) (single-cell),14,19 nano- and microdroplet processing in one-pot for trace samples (nanoPOTS and μPOTS) (<100 cells),7,24 nanoliter-scale oil–air droplet (OAD) chip-based sample preparation (<100 cells),33 integrated proteome analysis device (iPAD) (<100 cells),17,25 digital microfluidics (DMF) chip-based sample preparation (<500 cells),32 and in-line sample preparation for efficient cellular proteomics (ISPEC) (<1000 cells).16 These ultrasensitive systems, however, are often difficult to perform and/or require expensive or in-house custom-built instrumentation or devices. Accordingly, a simple and efficient sample preparation workflow for the analysis of hundreds to thousands of cells is needed to bridge the gap between ultrasensitive low cell number approaches and more widely used microgram-scale methods.
One such workflow, a miniaturized FASP method called MICRO-FASP,27 suits these needs. In MICRO-FASP, cell lysis, reduction, and alkylation are performed in solution in a low microliter-scale volume. The sample is then transferred to a homemade micropipette tip-based MICRO-FASP reactor containing a molecular weight cutoff (MWCO) ultrafiltration membrane for cleanup and tryptic digestion before elution directly into an LC autosampler vial. Sample losses during MICRO-FASP are minimal because low microliter-scale volumes are used, and the sample cleanup device is semi-integrated.
On-microsolid-phase extraction tip (OmSET) sample preparation, first described by our group with its initial workflow in 2015,10 is conceptually similar to MICRO-FASP; however, there are two important differences. First, OmSET utilizes a C18 membrane, which, in contrast with an MWCO membrane, is suboptimal for sample processing with lysis buffers containing detergents. Thus, MICRO-FASP may be preferred for hard-to-lyse samples. Second, the contact surface area is reduced in OmSET by eliminating the sample transfer step. Sample losses due to nonspecific adsorption are therefore expectedly decreased. OmSET builds specifically on techniques and findings reported by Kulak et al.28 and Ethier et al.,26 iST and proteomic reactor-based sample processing, respectively. Our approach benefits from minimized sample processing volumes (1–3 μL), the performance of all steps conducted on-membrane within a single microreactor, and no additional sample cleanup steps. Further, the simple protocol is highly scalable, affordable, and accessible as it requires only basic tools available in most proteomics laboratories.
In this work, we set out to improve peptide recovery for quantity-limited samples by OmSET sample preparation and, simultaneously, to reduce the hands-on time for the workflow. We first assessed the feasibility of using micro-SPE tips for nanogram-level amounts of tryptic peptides and simplified the established protocol to just 1 h prior to an overnight digestion step. Next, we evaluated the capability of our method for label-free quantitative analysis of samples consisting of 200 to 10,000 mammalian cells. Importantly, we demonstrated that quantitation is reliable and reproducible with excellent correlations between protein abundances and the amounts of starting material (R2 = 0.93 ± 0.09; mean ± SD) and pairwise correlations between sample processing replicates (R2 = 0.95 ± 0.02) and that sample losses are minimal with the identification of approximately 300, 1800, and 2000 protein groups from injections equivalent to approximately 10, 100, and 500 cells, respectively, using a conventional LC–MS setup equipped with a 75 μm inner-diameter in-house packed C18 column coupled to an Orbitrap Fusion Lumos mass spectrometer. Protein group identifications were increased to approximately 450 and 2000 for 10 and 100 cells, respectively, with the integration of spectral library searching into the data processing workflow.
EXPERIMENTAL SECTION
Materials and Reagents
Empore C18 SPE discs were obtained from 3 M (St. Paul, MN). LC–MS grade solvents (methanol, water, and acetonitrile (ACN)) and formic acid (FA) as well as phosphate-buffered saline (PBS), thiourea, tris(2-carboxyethyl)phosphine (TCEP), Gibco penicillin–streptomycin (P/S), and HeLa protein digest standard (P/N 88328) were products of Thermo Fisher Scientific (Waltham, MA). Urea, iodoacetamide (IAA), and trypan blue solution were purchased from Sigma-Aldrich (St. Louis, MO). Lysyl endopeptidase (Lys-C) was obtained from Wako Chemicals (Richmond, VA). Trypsin Gold was purchased from Promega (Madison, WI). Ammonium bicarbonate (ABC) was from Honeywell Fluka (Charlotte, NC). The RPMI 1640 medium with l-glutamine was a product of Corning (Corning, NY). Premium fetal bovine serum (FBS) was obtained from R&D Systems (Minneapolis, MN).
U937 Cell Culture
Human monocyte U937 cells (ATCC, Manassas, VA) were grown in the RPMI 1640 medium with l-glutamine supplemented with 10% FBS and 1% P/S at 37 °C and 5% CO2. Cell density was maintained between 1 × 105 and 2 × 106 viable cells/mL. For experiments, cells were collected by centrifugation at 300g for 5 min, washed three times with ice cold 1× PBS, and finally resuspended in ice cold 1× PBS. Cell count and viability were determined using trypan blue staining and an improved Neubauer hemocytometer (Paul Marienfeld, Germany).
Packing of Micro-SPE Tips
Micro-SPE tips were packed in the format of StageTips as previously described.34 Briefly, two punches of a C18 solid-phase extraction disc (0.5 mm depth) were taken with a 20 gauge blunt tip needle (0.6 mm inner diameter). The cores were then pushed into a 10 μL pipette tip with a plunger, taken from a 10 μL Hamilton syringe, until strong resistance was met, and they sat snugly against the walls of the tip.
Assessment of Recovery Efficiency for Micro-SPE Tips
HeLa protein digest standard was prepared to 1, 5, 10, 50, 100, and 500 ng/μL in 1% FA. As a control, an aliquot of the starting material was saved and diluted 10-fold (to match eluate and flow-through samples) for analysis. Each concentration of protein digest (1 uL) was loaded on to micro-SPE tips in duplicate (pre-equilibrated with methanol followed by water, as described below) before adding 9 μL of 1% FA. Loading was completed by centrifugation at 5000g for 30 s. The flow-through was captured directly into a glass LC vial insert and saved for analysis. Peptides were then eluted from the tips with 20 μL of 65% ACN in 0.1% FA and centrifuged at 5000g for 3 min directly into a glass LC vial insert. The eluate was evaporated using a SpeedVac until ~1 μL remained before reconstitution to a total volume of 10 μL with 1% FA aided by sonication for 30 s. For LC–MS analysis, 1 μL, corresponding to 1/10th of the sample, was injected (e.g., from 1 ng of starting material, 0.1 ng was injected for analysis) for analysis on an Orbitrap Fusion Lumos mass spectrometer; triplicate LC–MS runs were performed for each sample.
OmSET Sample Preparation
All centrifugation steps were performed at 5000g. Spin times are described as used in the conducted experiments; however, specific times in other applications may depend on the amount of material loaded and acceleration/brake parameters of the specific centrifuge used. Micro-SPE tips were first equilibrated with two volumes of 50 μL of methanol followed by two volumes of 50 μL of water flushed through by centrifugation for 1 min each. Aliquots of a U937 cell suspension (estimated ~200 to 500 pg protein/cell)35 corresponding to 0 (5 μL of PBS), approximately 200 (estimated by serial dilution), 2000, and 10,000 cells were then loaded directly into the tips in duplicate. PBS was removed by centrifugation for 1 min and replaced with 3 μL of lysis buffer containing 10 M urea, 2.5 M thiourea, 6 mM TCEP, and 30 mM ABC (pH 8). The lysis buffer was partially driven into the membrane by centrifugation for 8 s and was in contact with the cells for 10 min, shaking at 300 RPM at room temperature. Following protein extraction and denaturation, proteins were exposed to 1 μL of 50 mM TCEP, 10 mM IAA, and 25 mM ABC (pH 8), driven partially into the membrane by centrifugation for 5 s, for simultaneous reduction and alkylation at room temperature in the dark for 30 min. The remaining chaotropic, reducing, and alkylating reagents were removed by washing the micro-SPE tips with 20 μL of 25 mM ABC (pH 8) by spinning for 50 s. Then, Lys-C and trypsin in 25 mM ABC (pH 8) were driven into the membrane by centrifugation for 5 s at an estimated enzyme to substrate ratio of 1:10 (w/w) each or 1:1 for 200 cells followed by 15 μL of 25 mM ABC (pH 8) added on top to retain moisture in the column (Figure S1). The digestion reaction proceeded overnight, shaking gently at 300 RPM, at 45 °C. The next day, remaining ABC was flushed through by centrifugation for 10 s before digested peptides were eluted with 20 μL of 65% ACN in 0.1% FA directly into a glass LC vial insert by centrifugation for 3 min. Samples were evaporated in a SpeedVac, set to 35 °C, until ~1 μL remained, and reconstituted to a total volume of 20 μL with 1% FA followed by sonication for 30 s in the ultrasonication bath to promote solubilization of peptides. Finally, using a gel loading tip, one half of each sample was transferred to a separate glass LC vial insert and stored at −80 °C for LC–MS analysis at a later date. For LC–MS analysis, 1 μL, corresponding to a 1/20th aliquot of each sample, was injected (e.g., for ~200 cells processed, an estimated ~10 cell equivalent was injected) three times for triplicate LC–MS runs on an Orbitrap Fusion Lumos mass spectrometer.
In-Solution Digestion
Aliquots (1 μL) of U937 cell suspension in PBS corresponding to 0, approximately 200, 2000, and 10,000 cells were dispensed into low protein binding tubes (1.5 mL) in duplicate. Proteins were extracted with 8 M urea, 2 M thiourea, 5 mM TCEP, and 25 mM ABC (pH 8) for 10 min at room temperature shaking at 300 RPM in a total volume of 5 μL. Next, proteins were reduced and alkylated with 50 mM TCEP and 20 mM IAA (pH 8) for 30 min at room temperature in the dark in a total volume of 10 μL. The total volume was then brought to 100 μL with 20 mM ABC (pH 8), and Lys-C and trypsin were added at enzyme to substrate ratios of 1:10 (w/w) each. The digestion reaction proceeded overnight, shaking gently at 300 RPM, at 45 °C. The next day, digestion was halted by lowering the pH value with 1 μL of FA. Samples were evaporated until ~1 μL remained before reconstitution in a total volume of 20 μL of 1% FA followed by sonication for 30 s. Finally, samples were transferred to glass LC vial inserts for LC–MS analysis, which was performed as described for samples prepared by OmSET sample preparation.
Comparison of Digestion Sequences
U937 cells (2000) were processed using OmSET (as described) with adjustments made to the digestion steps. In triplicates, digestion was performed with either a single addition of a Lys-C (estimated enzyme to substrate ratio of 1:10) and trypsin (1:10) mixture (“simult”), an addition of a Lys-C (1:10) and trypsin (1:10) mixture followed by another addition of trypsin (1:20) after 1 h (“trypsin readd”), or a pretreatment with Lys-C (1:10) for 30 min before the addition of trypsin (1:10) (“Lys-C preadd”). The “simult” sequence followed the protocol from the OmSET sample preparation section. For Lys-C pretreatment (“Lys-C preadd”) and trypsin readdition (“trypsin readd”) sequences, the membranes were kept wetted with 5 μL of 25 mM ABC (pH 8) during the 30 min and 1 h digestion steps. Following the short incubation, ABC was removed by spinning for 5 s at 2000g before the final enzyme addition and 15 μL of 25 mM ABC (pH 8) layered on top of the membrane. For LC–MS analysis, a 1/10th aliquot was injected twice for duplicate LC–MS runs on a Q Exactive HF-X mass spectrometer.
LC–MS/MS
NanoLC was performed using a Dionex UltiMate 3000 RSLCnano system (Thermo). Separation was achieved with an 18 cm long × 75 μm inner-diameter in-house prepared nanoLC column with a pulled tip and packed with 1.9 μm ReproSil-Pur 120 Å C18-AQ beads (Dr. Maisch, Ammerbuch, Germany). Digested lysates and standards were loaded using an autosampler for 15 min (Fusion Lumos) or 20 min (Q Exactive HF-X) in 1% solvent B (0.1% FA in ACN) at a flow rate of 200 nL/min (Orbitrap Fusion Lumos) or 150 nL/min (Q Exactive HF-X) before a 60 min linear gradient from 1 to 25% solvent B was applied at a flow rate of 150 nL/min (both instruments). For samples processed in solution, a 10 min isocratic step at 1% solvent B was added before the gradient for sample cleanup/desalting. Following the gradient, the mobile-phase composition was changed from 25 to 80% solvent B over 2 min. The column was then washed with 80% solvent B for 2 min before re-equilibrating with 99% solvent A (0.1% FA in water) for another 15 min. At least three technical replicates were used across the study. LC and MS parameters were controlled using Xcalibur software (Thermo).
The analytical column was interfaced to a Q Exactive HF-X or an Orbitrap Fusion Lumos mass spectrometer (both Thermo) via a Nanospray Flex ion source (Thermo). A spray voltage of 2.0 kV was applied to generate a stable nanoelectrospray, while the temperature of the ion transfer tube/heated capillary was kept at 275 °C. Both instruments were configured in data-dependent acquisition (DDA) mode and using positive polarity. All data were acquired in profile and centroid modes for MS1 and MS2, respectively. Other MS parameters were varied between the Q Exactive HF-X and the Orbitrap Fusion Lumos to optimize sensitivity and performance. The Q Exactive HF-X was used for method development experiments where sample amounts were moderately low (i.e., comparison of digestion sequences and micro-SPE tip packing style experiments), while the Fusion Lumos was used to achieve higher sensitivity levels and better proteome coverage for experiments including extremely limited sample amounts, such as down to 1 ng of HeLa digest and down to ~10 cell equivalent injections (i.e., HeLa recovery and analysis of proteome coverage for U937 cell experiments).
For the Q Exactive HF-X, full MS1 scans were acquired in the range of 375 to 1500 m/z at a resolution of 120,000 (at 200 m/z) with the automatic gain control (AGC) target set to 3 × 106, maximum injection time to 50 ms, and funnel RF level at 45. The top 20 most intense precursor ions, with charge states 2 through 6, were selected for higher-energy collisional dissociation (HCD) fragmentation. The normalized collision energy was set to 28%. MS2 scans were collected at a resolution of 45,000, and the isolation window was set to 1.4 m/z. The maximum ion injection time was 86 ms with an AGC target of 1 × 106, and the intensity threshold was kept at 2.3 × 104. A fixed first mass of 100 m/z was used. Dynamic exclusion was set to 15 s. The peptide match was set to preferred, and isotope exclusion was on.
For the Orbitrap Fusion Lumos, full MS1 scans were acquired in the range of 375 to 1500 m/z at a resolution of 120,000 (at 200 m/z). The AGC target was set to 4 × 105 for a maximum ion injection time of 50 ms. The RF lens was set to 30%. Monoisotopic peak determination was set to Peptide. MS2 scans were acquired on the linear ion trap set to rapid scan mode, isolation window to 1.6 m/z, and the maximum ion injection time to 35 ms with an AGC target of 3 × 104. The intensity threshold was set to 5 × 103. The HCD collision energy was set to 28%. Dynamic exclusion was set to 30 s. The highest abundance peaks were analyzed by MS2 for a cycle time of 3 s, and ions were injected using parallelization mode. Only ions with a charge state of 2 through 7 were considered for MS2. A fixed first mass of 110 m/z was used.
Data Analysis
Raw LC–MS/MS files were analyzed using Proteome Discoverer v2.3 software (Thermo). LC–MS raw files were searched against the reviewed UniProt human database (release 2020_01 containing 20,302 sequences) with the SEQUEST-HT search engine (Thermo). Contaminants were filtered out using a combined, nonredundant FASTA database (276 sequences) created from MaxQuant’s built-in contaminant database and the common Repository of Adventitious Proteins (cRAP) database.36 Cysteine carbamidomethylation was set as a fixed modification, while methionine oxidation and N-terminal acetylation were set as variable modifications. The precursor mass tolerance was set to 10 ppm and the fragment mass tolerance to 0.02 Da for Orbitrap-acquired MS2 and 0.6 Da for ion trap-acquired MS2. Only fully tryptic peptides with a minimum length of six amino acid residues and up to two missed cleavages were considered. At least one high-confidence peptide sequence was required for protein identifications, and a false discovery rate (FDR) filter of ≤1% was applied with the Percolator module. Protein semiquantification was achieved by label-free quantification (LFQ) analysis in Proteome Discoverer v2.3 software. An FDR of ≤1% was applied for all peptide-level identifications, unique and razor peptides were used for quantitation, and precursor abundance was determined based on the intensity. No normalization was applied.
For the proof-of-concept spectral matching searches, a spectral library was built using the BiblioSpec v2.0 spectral library construction tool from DDA SEQUEST-HT-searched data acquired from three injections of a bulk U937 cell digest on the Orbitrap Fusion Lumos. The BiblioSpec library was then converted to a SpectraST format (*.sptxt) as Proteome Discoverer only accepts SpectraST format libraries. The library was next imported into Proteome Discoverer where the MSPepSearch node was run in parallel with the SEQUEST-HT search engine. The peptide sequence match score threshold was set to 400 for identifications resulting from spectral library searches. For further description of the spectral library construction and format conversion, please see the Supplementary methods.
Gene ontology (GO) analysis was performed in FunRich (v3.1.3)37 using the Gene Ontology database (downloaded April 2020). Venn diagrams were created using the BioVenn web application.38 Violin and box plots were generated using the BoxPlotR web tool.39
RESULTS AND DISCUSSION
Micro-SPE tips, often termed StageTips, have been used for final sample cleanup in proteomics for almost two decades.40 The device simply consists of a pipette tip with an inserted C18 particle-loaded polytetrafluoroethylene (PTFE) membrane disc. Kulak et al. first adapted a StageTip to be used for the entire sample processing workflow and termed the method iST.28 In iST, all sample preparation steps are performed in solution within the tip vessel above the SPE membrane, which is then used for final sample cleanup following digestion. Additionally, iST is commercially available in a kit format for use with down to 1 μg of sample. Li et al. reported the first use of a micro-SPE tip for on-membrane sample preparation, where all processing steps are instead performed within the SPE membrane.10 They processed just 2000 MCF-7 cells and showed excellent peptide recovery in combination with ultrasensitive porous layer open-tubular nanoLC–MS. We sought to retain the major advantages of the on-membrane protocol, namely, minimized sample volume and decreased exposure to irreversibly binding surfaces, while, simultaneously, reducing the number of sample handling steps and hands-on time. We additionally focused on widening the range of viable sample inputs to include low numbers of cells (i.e., <1000 cells).
Assessment of Feasibility for Limited Samples
To first assess the feasibility of using micro-SPE tips to process low numbers of cells, we investigated the recovery efficiency for low to mid-nanogram amounts of tryptic peptides. The starting material (as a control), flow-through fractions, and eluates were compared for six amounts of HeLa protein digest standard, spanning between 1 and 500 ng, and loaded on the tip (0.6 mm diameter × 1.0 mm length membrane) (Figure 1A). Expectedly, when comparing label-free quantitation (LFQ) abundances from the starting material and eluate fractions, relative sample losses were the highest at the lowest sample loading amount (1 ng loaded on tip 56 ± 18% (mean ± SD) recovery of summed peptide intensity) and the lowest at the largest sample loading amount (500 ng loaded on tip 93 ± 5% recovery of summed peptide intensity) (Figure 1B). Chemical passivation of micro-SPE tip surfaces may help to prevent losses, especially for lower sample amounts,14 although we did not passivate in this study and used tips only one time before discarding. Numbers of protein group and peptide group identifications followed a similar trend (50 ± 12 and 113 ± 2% recovery of peptide group identifications from 1 ng and 500 ng loaded on tip, respectively) (Figure 1C,D, respectively). Conceivably, the increase in peptide group identifications at the largest sample loading amount is due to a cleanup/enrichment effect of the micro-SPE tips when the HeLa protein digest was additionally desalted and depleted of highly hydrophobic and highly hydrophilic peptides. We hypothesize that the depleted short, highly hydrophilic peptides, which were possibly not retained on the SPE membrane and eluted in flow-through fractions, or highly hydrophobic peptides, which were possibly irreversibly retained on the SPE membrane, could have resulted in detrimental ion suppression and ion co-isolation effects during the LC–MS analysis. Peptide group identifications from flow-through fractions were, on average across all loading amounts, just 17 ± 21 peptide groups. Comparison of ion density maps from the starting material, eluate, and flow-through fractions further suggests that sample losses in the flow-through fraction during loading were minimal (Figure S2). Notably, in agreement with our hypothesis, flow-through fractions were composed mainly of short hydrophilic peptides, and peptide matches were mostly identified within the first 10 min of the 60 min long LC elution gradient.
Figure 1.
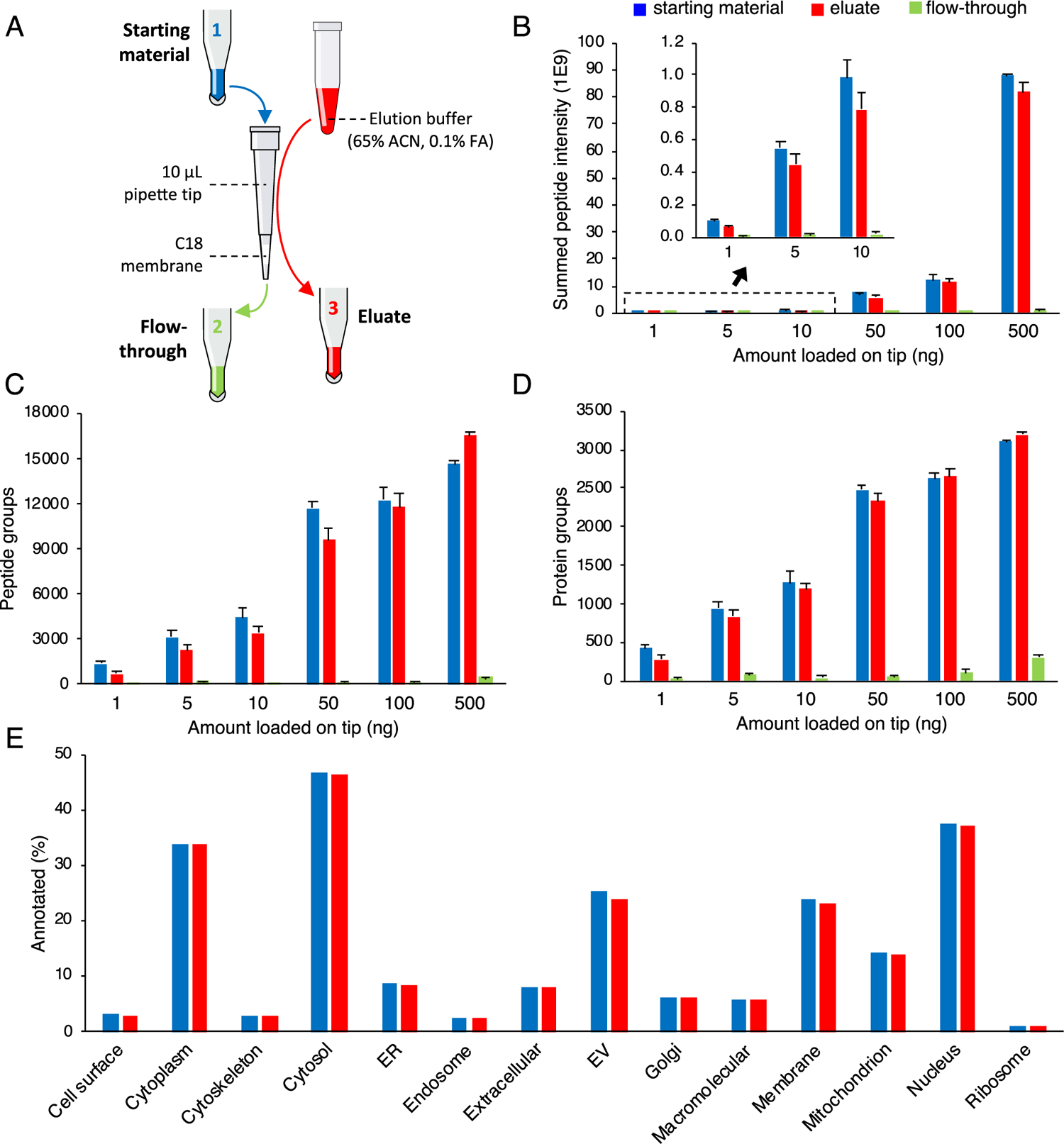
Recovery of nanogram amounts of HeLa digest standard loaded onto and eluted from micro-SPE tips. Schematic representation of the experimental workflow (A). Summed peptide LFQ intensities (B) and numbers of peptide group (C) and protein group identifications (D) for 1/10th aliquots from the starting material (control), eluate, and flow-through fractions of varying nanogram amounts of HeLa digest standard loaded and eluted from micro-SPE tips. Error bars show standard deviation from micro-SPE tip duplicates and LC–MS technical triplicates (N = 6 in total). Results of GO-term enrichment analysis conducted using proteomic profiling data (E). Recovery of proteins pertinent to specific cellular components was compared across all loading amounts for the starting material and eluate.
Eluate protein lists were further analyzed by GO annotation for the cellular component and compared against the starting material across all loading amounts (Figure 1D) and for each loading amount (Figure S3). We found that micro-SPE tips did not lead to preferential losses of any particular protein classes. Additionally, when protein lists were directly contrasted, Venn diagrams comparing protein identifications in the starting material and eluates were highly comparable to Venn diagrams created from LC–MS technical replicates of the same sample (Figures S4 and S5), further strengthening the case for unbiased recovery of nanogram amounts of peptides from micro-SPE tips.
These findings, in combination, prompted our belief that the current micro-SPE tip configuration is suitable for unbiased sample processing of cell amounts close to or above 200 U937 cells (or >50 ng of sample input).35 At 50 ng of input, we observed ~80% recovery in peptide group identifications, which we assumed as an acceptable level of loss for such low sample amounts while using an easily employable sample processing platform. For cell amounts greater than 10,000 cells, we have observed sporadic clogging in preliminary experiments and recommend larger tips with a larger surface area of the membrane and the diameter of the tip opening (e.g., 100 μL tips). This upper cell loading level (2–5 μg), which we evaluated in this study, is well below the binding capacity of 9–18 μg we estimated for the used micro-SPE tips based on a published report.40
Simplification of the Workflow
Reduction and alkylation have traditionally been separated into two steps, but these reactions have been shown to proceed simultaneously when nonthiol reducing agents are used (e.g., TCEP).41,42 So, to simplify the workflow in line with other similar sample processing protocols,23,28 reduction and alkylation were combined into a single step using TCEP and iodoacetamide. Consequently, the workflow was shortened by 35 min, and the number of pipetting steps was reduced to just six.
We next examined proteolytic digestion steps. Lys-C and trypsin are often used in combination to improve proteome coverage.43 Further, common instruction suggests pretreating samples with Lys-C before the addition of trypsin. However, we sought to reduce the number of sample handling steps and amount of hands-on time for the protocol; thus, three sequences of enzyme additions prior to overnight digestion were tested: single addition of a Lys-C (estimated enzyme to substrate ratio of 1:10) and trypsin (1:10) mixture (“simult”), addition of a Lys-C (1:10) and trypsin (1:10) mixture followed by another addition of trypsin (1:20) after 1 h (“trypsin readd”), and pretreatment with Lys-C (1:10) for 30 min before addition of trypsin (1:10) (“Lys-C preadd”).
We found that numbers of protein group and peptide group identifications did not differ significantly between the tested digestion sequences (Figure 2A,B). Similarly, analysis of GO-term cellular component annotations did not reveal any variations in identified protein classes between the tested digestion sequences (Figure S6). When the digestion efficiency was indirectly assessed based on the percentage tryptic miscleavages for peptide sequence matches (Figure 2C), the workflow with the simultaneous addition of enzymes (“simult”) (85.33 ± 1.39%) was found to be moderately improved over the trypsin readdition (“trypsin readd”) (80.09 ± 0.42%; P = 0.006) and the Lys-C pretreatment (“Lys-C preadd”) (76.98 ± 4.03%; P < 0.001) sequences at 0 miscleavage. For this reason, with the goal of simplifying the workflow in mind, we incorporated the simultaneous digestion sequence (“simult”) into the OmSET protocol.
Figure 2.
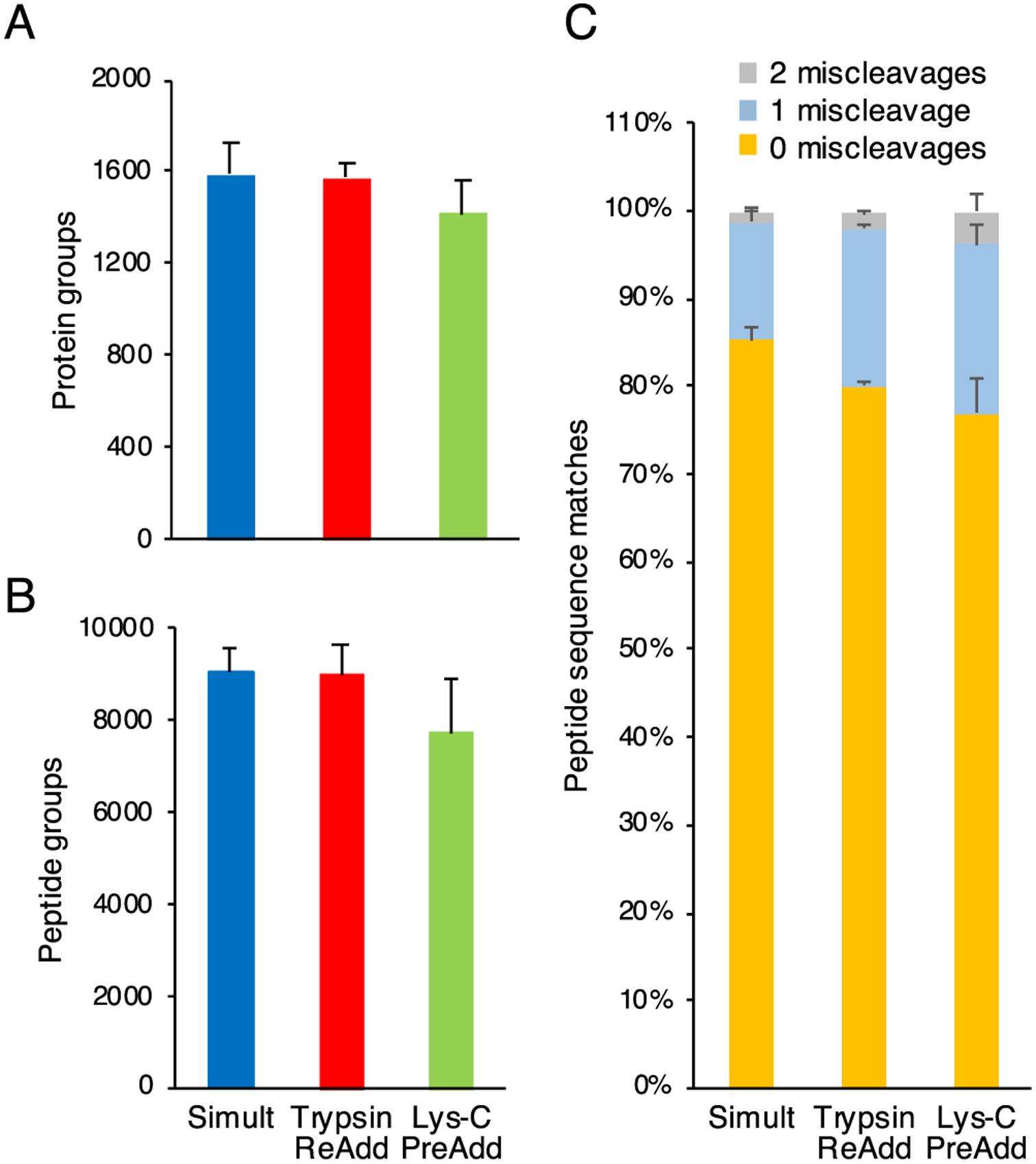
Comparison of Lys-C and trypsin addition sequences by OmSET sample preparation for processing 2000 U937 cells. Numbers of protein groups (A) and peptide groups (B) identified from aliquots equivalent to ~200 injected cells for three tested sequences of enzyme additions: addition of Lys-C (estimated enzyme to substrate ratio of 1:10) and trypsin (1:10) simultaneously (“simult”), addition of Lys-C (1:10) and trypsin (1:10) simultaneously followed by a readdition of trypsin (1:20) after 1 h (“trypsin readd”), and pretreatment with Lys-C (1:10) for 30 min before the addition of trypsin (1:10) (“Lys-C preadd”). Error bars indicate standard deviation from sample processing triplicates with LC–MS technical duplicates each (N = 6 in total). Percentages of 0 (yellow), 1 (light blue), and 2 miscleavages (gray) for peptide sequence matches are shown in panel (C). The max number of shown miscleavages was 2.
By combining reducing and alkylating steps and by streamlining proteolysis steps, the OmSET workflow was slimmed down to just six pipetting steps and only 1 h in length prior to overnight digestion (Figure 3). Critically, this minimization of the number of sample handling steps greatly reduces opportunities for losses, contamination, and pipetting errors to occur.
Figure 3.
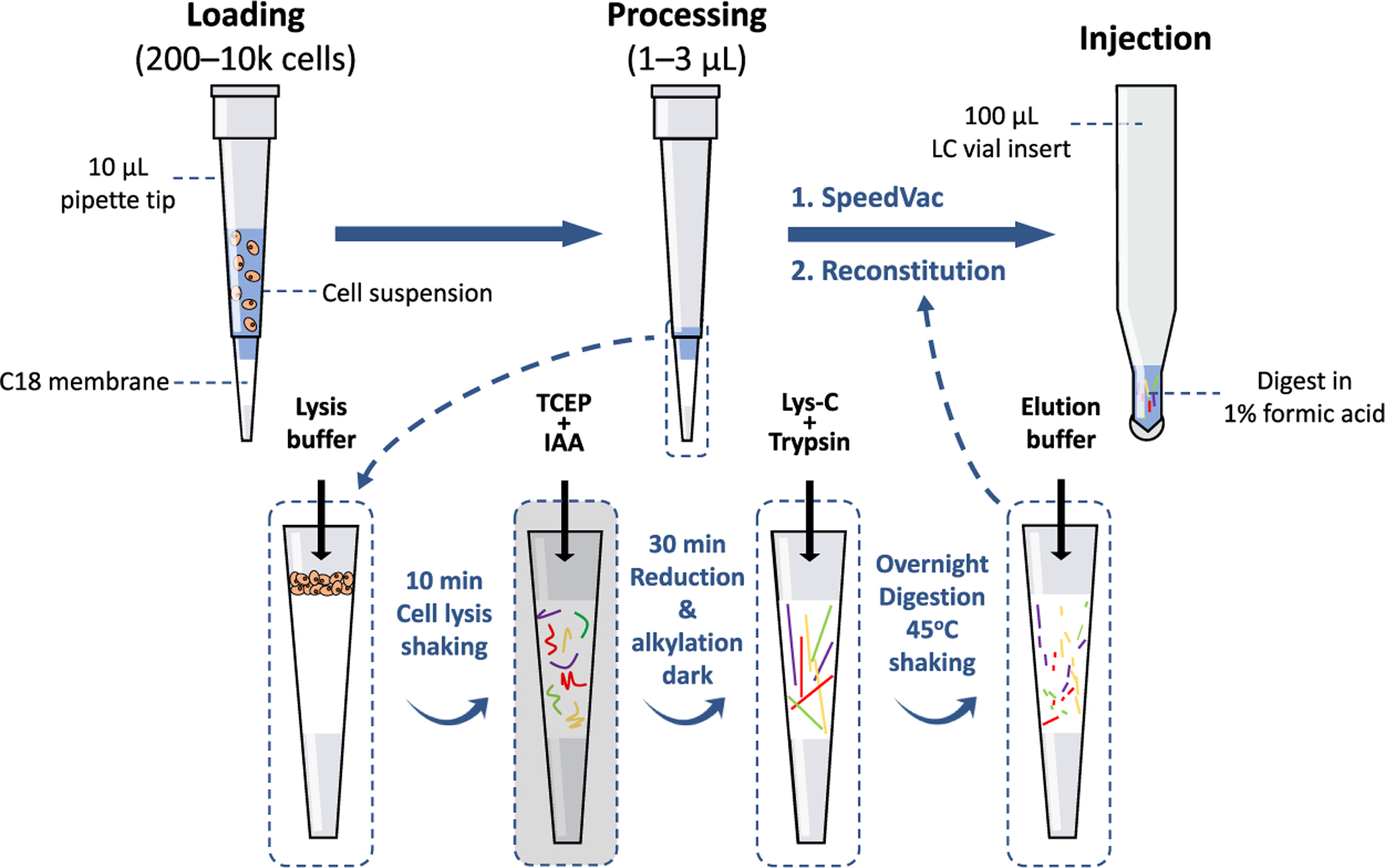
OmSET sample preparation workflow. Sample is loaded directly onto the micro-SPE tip, cells are lysed, and proteins are extracted and denatured during a 10 min lysis step with urea-based lysis buffer. Next, disulfide bonds are reduced and cysteines are alkylated for 30 min in the dark. Protein digestion is then performed gently shaking overnight at 45 °C before elution the following morning. Finally, peptides are dried down and reconstituted in an MS-friendly sample loading buffer for injection and subsequent LC–MS analysis.
Tips and Tricks to Ensure Successful Performance of OmSET
In an effort to determine the optimal method for packing micro-SPE tips, we examined four tip packing styles by varying two elements: how many layers of the membrane were transferred into the tip at a time and the firmness of the packing. Protein and peptide group identifications were comparable between all four styles (Figure S7). Thus, with no significant differences observed between packing styles, we recommend using the method that leads to the greatest personal tip-to-tip reproducibility. In our observation, that method is to core both membrane punches, one after the other using the syringe needle before transferring both excised membrane discs to the tip simultaneously and packing firmly until strong resistance is met (style 1). With some practice, approximately 95% of the micro-SPE tips can be packed with reproducible performance while producing about four micro-SPE tips per minute. Please note that in the current protocol, reproducibility of micro-SPE tip packing is the most critical step for achieving reproducible sample processing. Tip-to-tip packing consistency is necessary to maintain a similar flow rate across tips. Without proper consideration, there is a risk for more loosely packed tips to become dry during centrifugation steps, leading to reduced peptide recovery and sample-to-sample reproducibility (data not shown). Often, irregular tips can be easily caught and discarded during the equilibration step, which warrants that valuable samples are not wasted during sample processing.
Because digestion is performed overnight at 45 °C, a significant concern is tips running dry if proper considerations are not made. To minimize evaporation, we add a 15 μL plug of 25 mM ABC (pH 8) to the micro-SPE tip, which ensures full saturation of the membrane throughout the entire incubation. Additionally, we add a volume of ABC to the bottom tube of the micro-SPE tip assembly such that the outlet of the tip is placed ~5 mm above the liquid (~200 μL for a 1.5 mL tube) (Figure S1). Also, as a final precaution, we incubate micro-SPE tips in an Eppendorf ThermoMixer with the lid snugly fitted and open microcentrifuge tubes containing deionized water in the remaining available wells to increase the humidity of the system. Other humidified thermoregulated mixing chambers can be potentially used.
Analysis of 200, 2000, and 10,000 U937 Cells
With the protocol simplified and optimized, we next evaluated the OmSET sample preparation workflow for processing of limited biological samples. Human monocyte U937 cells were chosen as a model system for their moderate size (~10 μm on average)44 and straightforward growth requirements. We estimate that individual U937 cells contain ~200 to 500 pg of protein based on a published report.35 Cells were counted in suspension, and volumes corresponding to approximately 200, 2000, and 10,000 cells were loaded onto micro-SPE tips. Cells were then lysed, and proteins were extracted and denatured by exposure to a urea-based lysis buffer for 10 min, simultaneously reduced and alkylated for 30 min, and finally digested using Lys-C and trypsin proteases overnight. A 1/20th aliquot of the resulting sample was analyzed by conventional nanoLC–MS with a 60 min long elution gradient. For example, estimated ~10 cell equivalents were injected for ~200 cells processed.
Average values of 727 ± 147, 7710 ± 669, and 9393 ± 1002 peptide groups were identified from approximately 10, 100, and 500 cell equivalent injections, respectively, corresponding to 311 ± 45, 1828 ± 105, and 2008 ± 73 protein group identifications, respectively, using conventional database searching software, without matching LC–MS features between runs, at an FDR of ≤1% and after removing common contaminants (Figure 4A,B). Although the improvement in the number of identifications is modest from 100 to 500 cells, the difference in the signal intensity is apparent from base ion chromatographs (Figure S8) and ion density maps (Figure S9). From blank samples (micro-SPE tips loaded with PBS containing no cells and processed identically to other tips), 74 ± 12 peptide groups and 23 ± 6 protein groups were identified, indicating that the levels of carryover and contamination are low. Overlap of protein group identifications was very substantial between the three cell loading levels (Figure 4E), with just 3.0 and 8.4% of the protein groups from 10 and 100 cells, respectively, unique to their level. Of note, just one protein group was unique to the blank sample. Protein group identifications for the three analyzed cell loading levels increased to 466 ± 58, 1923 ± 88, and 2061 ± 51, respectively, when spectral library searching was integrated into the data processing workflow to assess the feasibility of using spectral library searching in the profiling of limited samples (Figure 4A,B and Table S1). This corresponds to gains of 49.8 ± 9.5, 5.2 ± 0.4, and 2.6 ± 0.1%, respectively. Despite the modesty of gains for the 100 and 500 cell levels, which we attribute to insufficient depth of our pilot spectral library, the addition of library searching greatly increased the number of identifications at the 10 cell level and additionally served to improve confidence in identifications at all levels. We expect that searches against a more informative and structurally rich spectral library will lead to more pronounced gains in the sensitivity of profiling such samples.
Figure 4.
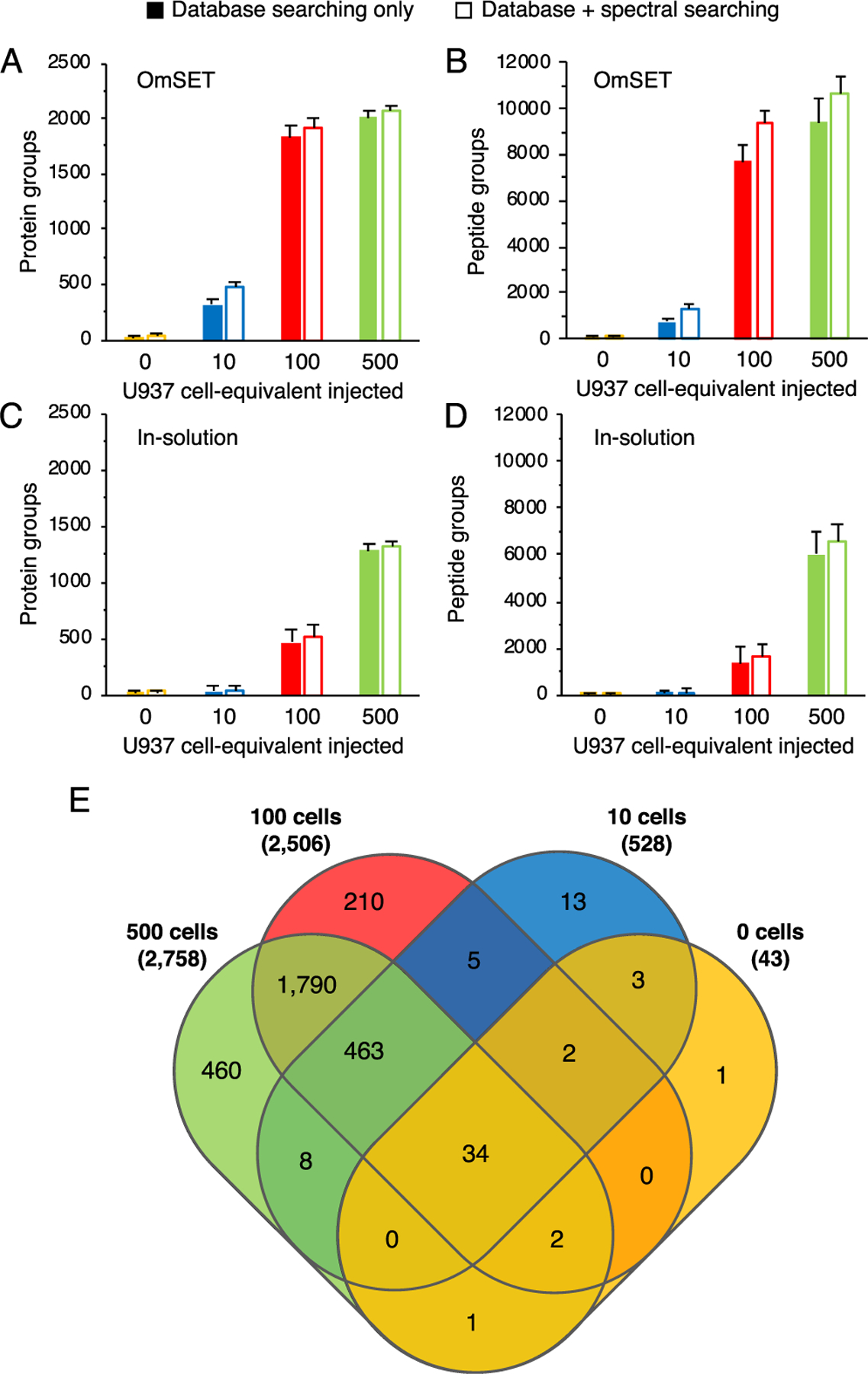
Evaluation of depth of proteomic profiling for 10 to 500 U937 cells by OmSET sample preparation. Numbers of protein groups (A) and peptide groups (B) for 0, 10, 100, and 500 cell equivalents injected from 0, 200, 2000, and 10,000 cells processed, respectively. An aliquot of PBS was processed in parallel and used as a negative control (0 cell). Solid bars represent identifications using database searching only, while outlined bars show identifications using database and spectral library searching in combination. Error bars show standard deviation from triplicate LC–MS runs of sample processing duplicates (N = 6 in total). Numbers of protein groups (C) and peptide groups (D) identified from equivalent samples prepared using a conventional in-solution approach. Venn diagram (E) shows the overlap in protein group identifications between the three cell loading amounts and the blank sample processed using OmSET.
To further assess the value of OmSET sample preparation, we compared proteome coverage achieved using the developed approach against a conventional in-solution sample preparation approach (Figure 4C,D). For the conventional approach, all sample processing steps were conducted in solution in low protein binding Eppendorf tubes (1.5 mL) and with a total processing volume of 100 μL. The nanoLC–MS conditions used were the same as used for samples prepared by OmSET except for an added sample cleanup/desalting step (10 min at 1% solvent B) before the gradient was applied. Using database searching only, average values of 471 ± 71 and 1279 ± 144 protein groups were identified from ~100 and 500 cell equivalent injections from 2000 and 10,000 U937 cells processed, respectively, corresponding to gains of ~4-fold and ~1.5-fold for OmSET over the conventional in-solution approach, respectively (Figure 4C). No meaningful protein identifications were produced from 10 cell equivalent injections of the samples resulted from 200 cells processed in vials as the number of identifications was on par with blank samples containing zero cells (29 ± 7 vs 26 ± 5 protein groups from 10 and 0 cells, respectively). The improvement in performance from OmSET over the conventional in-solution approach, across the range of 200–10,000 cells processed, demonstrates the contribution of the OmSET sample preparation platform to advancing depth of proteomic profiling for scarce samples.
To inspect for biases in types of proteins identified, we compared GO annotations for pertinent cellular components between OmSET and in-solution digestion. Overall, the recovery of proteins of different cellular components was similar for the compared sample processing techniques. In-solution digestion resulted in slightly more proteins annotated for cytosol (+4.2%) and extracellular vesicle (+6.9%) than OmSET (Figure S10). We hypothesized that this bias might be driven by differences in physiochemical properties, so we further compared the size, hydrophobicity (GRAVY),45 and isoelectric point of identified proteins (Figure S11). We found that OmSET resulted in slightly fewer proteins with a pI of 5 but slightly more proteins with pIs of 9 and 10 than in-solution digestion, while the distributions of hydrophobicity and molecular weight values were seemingly unchanged. These results confirmed that the C18 membrane used in OmSET effectively retained both hydrophilic and hydrophobic proteins. When sufficient sample is available, several alternative sample preparation techniques and proteolytic enzymes can be used to achieve maximum and unbiased proteome coverage.46,47
The sensitivity of proteomic profiling based on the developed approach also exceeds the sensitivity levels of most other published techniques, which use alternative sample preparation platforms for processing limited samples (Table S2). To the best of our knowledge, the current state-of-the-art techniques that resulted in deeper profiling sensitivity at similar sample amounts were digital microfluidics (DMF) chip-based sample preparation at the level of 500 cells (2500 protein groups from 500 Jurkat cells),32 nanoPOTS (3056 and 1517 protein groups from 137–141 and 10–14 HeLa cells, respectively),7 microPOTS (2064 and 1252 protein groups from 91–93 and 21–28 HeLa cells, respectively),24 and focused acoustics-assisted (AFA) sample preparation (2512 and 1802 protein groups from 122 and 50 MCF-7 cells, respectively).10 These systems, however, require highly specialized and/or expensive equipment and/or may be difficult to implement in other laboratories. In comparison to the similar MICRO-FASP platform (~1650 and ~500 protein groups from 500 and 100 MCF-7 cells, respectively),27 OmSET resulted in more protein group identifications; however, it is important to note the differences in starting amounts, cell types, LC column, gradient length, mass spectrometer, MS parameters, and data processing software used (Tables S2 and S3).
While it is virtually impossible to benchmark the profiling sensitivity levels achieved by different techniques because of the discrepancies between samples, analytical instrumentation, and utilized methods, our simplistic, scalable, and affordable method demonstrates ultrahigh profiling sensitivity levels approaching the current records in the field. Depth of proteome coverage for OmSET can be further improved using a longer gradient and ultranarrow bore nanoLC columns that enable ultralow flow rates as well as ion mobility spectrometry approaches interfaced with such LC–MS techniques.10,13,48,49 However, we focus on accessibility of the approach presented in this report using a more conventional and robust nanoLC–MS setup with a relatively short gradient (i.e., 60 min).
To evaluate the capacity of OmSET sample preparation for unbiased and reliable qualitative analysis, we used an LFQ method to extract intensity values for identified proteins (i.e., LFQ protein abundance values). First, we inspected the LFQ data for biases of the OmSET approach against low or high abundance proteins at different sample amounts. Protein LFQ abundances were normalized, and kernel density estimations were plotted to visualize distributions of abundances for quantified proteins. In comparison to in-solution digestion, OmSET resulted in quantification of fewer low abundance proteins relative to other abundances at the 500 cell loading level, while at the 100 cell level, quantification of low abundance proteins was similar (Figure S12). Interestingly, as the sample amount was reduced, OmSET seemingly resulted in quantification of greater distributions of low abundance proteins, indicating that OmSET sample preparation may be slightly more biased against lower abundance proteins at larger sample amounts than in-solution digestion. This difference, however, is not critical when the boost in proteome coverage is considered.
Next, to illustrate the capability of OmSET for dependable quantitative analysis, we compared LFQ protein abundances between replicates and across sample amounts. Pairwise correlations performed between LC–MS technical replicates and between sample processing replicates resulted in mean R2 values of 0.99 ± 0.01 and 0.95 ± 0.02, respectively (Figure S13). We then plotted LFQ intensity values as a function of the number of cells injected per analysis to determine trends for each of the 453 commonly identified proteins across the three cell input levels and found an excellent overall correlation (R2 = 0.93 ± 0.09), corroborating our estimates for the numbers of cells loaded (Figure 5). Observed R2 values, in all comparisons, indicated significant correlation and, thus, signified that the platform is well suited for reliable and reproducible quantitative analysis of biological and clinical samples at such scarce amounts.
Figure 5.
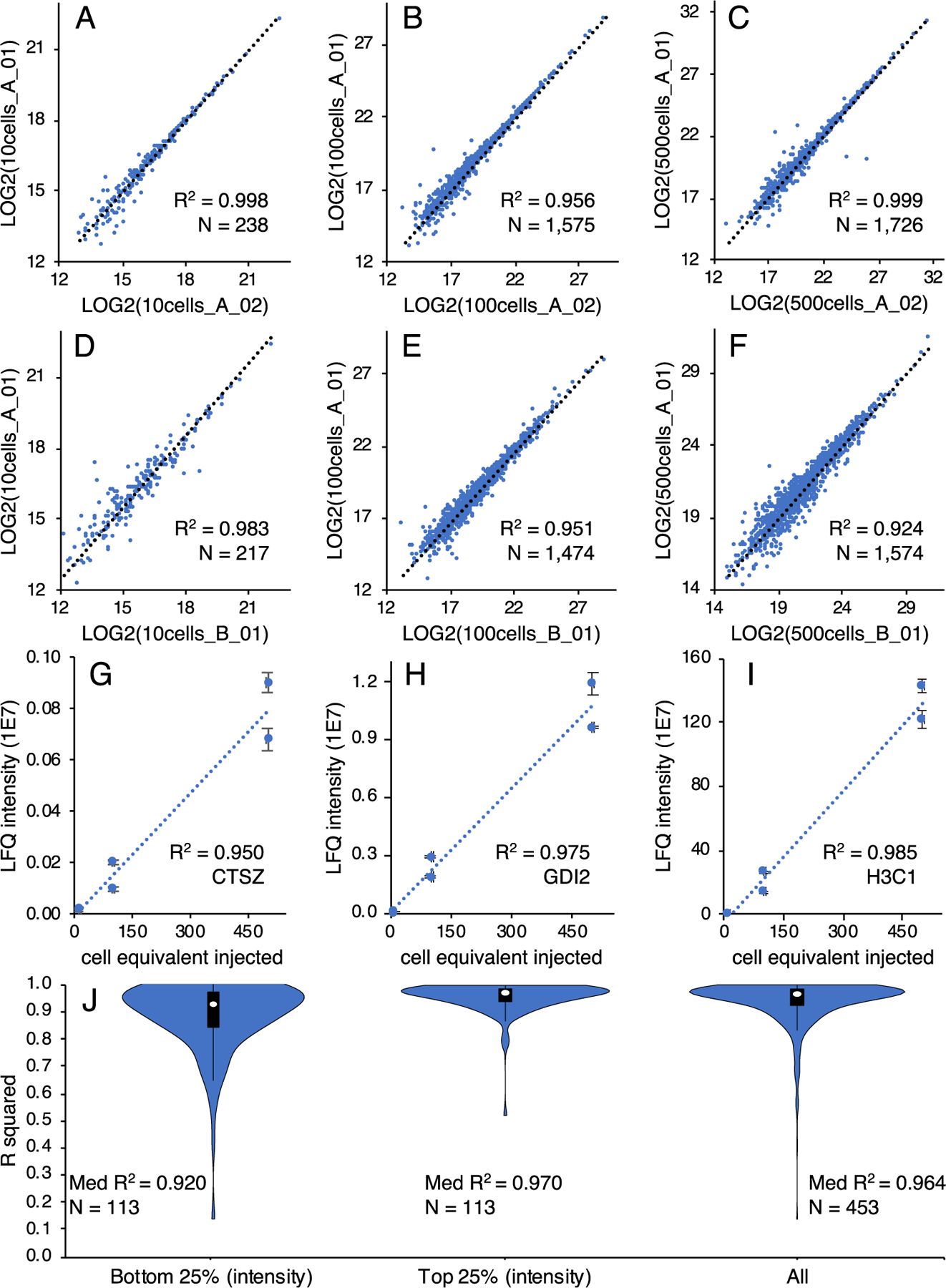
Comparison of label-free quantitation results from ~10, ~100, and ~500 U937 cells. Pairwise correlations of protein abundances for LC–MS runs of the same sample for aliquots equivalent to ~10 cells (A), ~100 cells (B), and ~500 cells (C). Pairwise correlations of protein abundances for LC–MS runs of different sample processing replicates for aliquots equivalent to ~10 cells (D), ~100 cells (E), and ~500 cells (F). Examples of low (G), medium (H), and high (I) abundance proteins from the comparison of protein abundances to the number of cells analyzed. Gene names are given for each protein example. Distribution of R2 values from the bottom 25%, top 25%, and all abundances for protein abundances compared to the number of cells analyzed (J). R2 values are given as mean ± SD.
CONCLUSIONS
In this work, we optimized and characterized the OmSET sample preparation technique for accessible use in the in-depth proteomic profiling of quantity-limited biological and clinical samples. Our processing platform benefits from minimal sample volumes and the performance of all sample processing steps, including sample cleanup, within a single microreactor. Moreover, the only equipment required to perform OmSET—StageTips and a microcentrifuge—is already commercially available and in use in many Proteomic laboratories. We simplified the protocol to require only six pipetting steps and to take just 1 h prior to an overnight digestion step to perform, thus, further reducing opportunities for contamination, pipetting errors, and sample losses. In this work, we aimed to provide descriptions of all experimentally critical tips and tricks to make the developed technique as broadly accessible as possible.
Importantly, we evaluated our system with both commercially available digested cell lysate standards and in-house cultured mammalian cells and found that sample losses were minimal. Implementing a pilot spectral library into the data processing workflow increased the number of identifications significantly for low numbers of cells. Our results represent an advancement in the field of ultrasensitivity proteomic profiling techniques in the area of sample preparation for limited samples 200–10,000 cells while simultaneously maintaining a high level of accessibility. We additionally demonstrated the capability of our platform for reliable and reproducible protein quantitation.
The OmSET workflow shows considerable potential for scaling and automation with straightforward adaptations to the protocol. A 96-tip format device has already been described for use with iST sample preparation.28 We expect that, with use of this device or implementation of an improved design in a multiwell format, up to 384 samples could be processed in parallel with a minor time extension from the current, lower-throughput version of the workflow. Additionally, facile integration of isobaric tag labeling to our protocol with little additional sample losses can be adapted as on-membrane TMT labeling using StageTips has already been reported for limited samples.23
We believe that the particular combination of sensitivity and simplicity could propel OmSET sample preparation to widespread use for a variety of proteomic applications for profiling of quantity-limited biological and clinical samples.
Supplementary Material
ACKNOWLEDGMENTS
The human monocyte U937 cell line was kindly provided by Prof. Nikolai Slavov, Northeastern University. We are thankful to Christopher Kostas for his help with data cleanup and to Alan Zimmerman and Kendall Johnson for their assistance with experiments. This work was supported by the NIH under award numbers R01GM120272 (A.R.I.), R01CA218500 (A.R.I.), and R35GM136421 (A.R.I.) and also by the Dana-Farber Cancer Institute/Northeastern University Joint Program in Cancer Drug Development Award (A.R.I.). We acknowledge Thermo Fisher Scientific for their support through a Technology Alliance Partnership program.
ABBREVIATIONS
- SPE
solid-phase extraction
- OmSET
on-microsolid-phase extraction tip
- CTCs
circulating tumor cells
- iST
in-StageTip
- SCoPE-MS
single-cell proteomics by mass spectrometry
- nanoPOTS
nanodroplet processing in one-pot for trace samples
- μPOTS
microdroplet processing in one-pot for trace samples
- OAD
nanoliter-scale oil–air droplet
- iPAD
integrated proteome analysis device
- DMF
digital microfluidics
- ISPEC
in-line sample preparation for efficient cellular proteomics
- ACN
acetonitrile
- FA
formic acid
- TCEP
tris(2-carboxyethyl)phosphine
- P/S
penicillin–streptomycin
- IAA
iodoacetamide
- Lys-C
lysol endopeptidase
- ABC
ammonium bicarbonate
- FBS
fetal bovine serum
- DDA
data-dependent acquisition
- AGC
automatic gain control
- HCD
higher-energy collisional dissociation
- cRAP
common Repository of Adventitious Proteins
- FDR
false discovery rate
- LFQ
label-free quantitation
- PTFE
polytetrafluoroethylene
- GO
gene ontology
- AFA
focused acoustics-assisted
- GRAVY
grand average of hydropathy
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.0c00890.
Supplementary methods; Figure S1, diagram of micro-SPE tip setup during digestion; Figure S2, ion density maps of the starting material, eluate, and flow-through from HeLa protein digest loaded on micro-SPE tips; Figure S3, GO-term annotation for cellular component of the starting material, eluate, and flow-through from HeLa protein digest loaded on micro-SPE tips; Figure S4, Venn diagrams showing overlap of protein IDs from the starting material and eluates from HeLa protein digest loaded on micro-SPE tips; Figure S5, Venn diagrams showing overlap of proteins identified between representative technical and sample processing duplicates from HeLa protein digest loaded on micro-SPE tips; Figure S6, GO-term annotation comparing cellular components of digestion sequences; Figure S7, numbers of protein group and peptide group IDs for different micro-SPE tip packing styles; Figure S8, representative base ion chromatographs for U937 cells processed using OmSET; Figure S9, representative ion density maps for U937 cells processed using OmSET; Figure S10, GO-term annotation comparing cellular components for OmSET and in-solution digestion; Figure S11, distribution of proteins identified using OmSET and in-solution digestion according to physiochemical properties; Figure S12, distribution of proteins quantified using OmSET and in-solution digestion according to LFQ protein abundance; Figure S13, box plots showing distributions of pairwise correlations for runs of U937 cells processed using OmSET; Table S1, numbers of protein and peptide group identifications from U937 cells processed using OmSET; Table S2, published results from alternative sample processing platforms; and Table S3, comparison of LC–MS/MS conditions between OmSET and MICRO-FASP sample preparation platforms (PDF)
Protein group and peptide group IDs from HeLa protein digest standards and U937 cells processed using OmSET and in-solution digestion with integrated spectral library searching and protein LFQ results from HeLa protein digest standards and U937 cells processing using OmSET and in-solution digestion (ZIP)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jproteome.0c00890
The authors declare no competing financial interest.
The mass spectrometry files and Proteome Discoverer searches have been deposited to the PRIDE Archive (http://www.ebi.ac.uk/pride/archive/) via the PRIDE partner repository with the data set identifier PXD022313.
REFERENCES
- (1).Cristofanilli M; Budd GT; Ellis MJ; Stopeck A; Matera J; Miller MC; Reuben JM; Doyle GV; Allard WJ; Terstappen LWMM; Hayes DF Circulating Tumor Cells, Disease Progression, and Survival in Metastatic Breast Cancer. New Eng. J. Med 2004, 351, 781–791. [DOI] [PubMed] [Google Scholar]
- (2).Riethdorf S; Fritsche H; Müller V; Rau T; Schindlbeck C; Rack B; Janni W; Coith C; Beck K; Jänicke F; Jackson S; Gornet T; Cristofanilli M; Pantel K Detection of circulating tumor cells in peripheral blood of patients with metastatic breast cancer: a validation study of the CellSearch system. Clin. Cancer Res 2007, 13, 920–928. [DOI] [PubMed] [Google Scholar]
- (3).Krebs MG; Hou JM; Ward TH; Blackhall FH; Dive C Circulating tumour cells: their utility in cancer management and predicting outcomes. Ther. Adv. Med. Oncol 2010, 2, 351–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Nagrath S; Sequist LV; Maheswaran S; Bell DW; Irimia D; Ulkus L; Smith MR; Kwak EL; Digumarthy S; Muzikansky A; Ryan P; Balis UJ; Tompkins RG; Haber DA; Toner M Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature 2007, 450, 1235–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Yoon HJ; Kim TH; Zhang Z; Azizi E; Pham TM; Paoletti C; Lin J; Ramnath N; Wicha MS; Hayes DF; Simeone DM; Nagrath S Sensitive capture of circulating tumour cells by functionalized graphene oxide nanosheets. Nat. Nanotechnol 2013, 8, 735–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Roulhac PL; Ward JM; Thompson JW; Soderblom EJ; Silva M; Moseley MA; Jarvis ED Microproteomics: Quantitative Proteomic Profiling of Small Numbers of Laser-Captured Cells. Cold Spring Harbor Protoc 2011, 2011, pdb-prot5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Zhu Y; Piehowski PD; Zhao R; Chen J; Shen Y; Moore RJ; Shukla AK; Petyuk VA; Campbell-Thompson M; Mathews CE; Smith RD; Qian WJ; Kelly RT Nanodroplet processing platform for deep and quantitative proteome profiling of 10–100 mammalian cells. Nat. Commun 2018, 9, 882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Lizotte PH; Jones RE; Keogh L; Ivanova E; Liu H; Awad MM; Hammerman PS; Gill RR; Richards WG; Barbie DA; Bass AJ; Bueno R; English JM; Bittinger M; Wong KK Fine needle aspirate flow cytometric phenotyping characterizes immunosuppressive nature of the mesothelioma microenvironment. Sci. Rep 2016, 6, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Lin P; Yao Z; Sun Y; Li W; Liu Y; Liang K; Liu Y; Qin J; Hou X; Chen L Deciphering novel biomarkers of lymph node metastasis of thyroid papillary microcarcinoma using proteomic analysis of ultrasound-guided fine-needle aspiration biopsy samples. J. Proteomics 2019, 204, 103414. [DOI] [PubMed] [Google Scholar]
- (10).Li S; Plouffe BD; Belov AM; Ray S; Wang X; Murthy SK; Karger BL; Ivanov AR An Integrated Platform for Isolation, Processing, and Mass Spectrometry-based Proteomic Profiling of Rare Cells in Whole Blood. Mol. Cell. Proteomics 2015, 14, 1672–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Shen Y; Tolić N; Masselon C; Paša-Tolić L; Camp DG; Hixson KK; Zhao R; Anderson GA; Smith RS Ultrasensitive Proteomics Using High-Efficiency On-Line Micro-SPE-NanoLC-NanoESI MS and MS/MS. Anal. Chem 2004, 76, 144–154. [DOI] [PubMed] [Google Scholar]
- (12).Cifani P; Kentsis A High Sensitivity Quantitative Proteomics Using Automated Multidimensional Nano-flow Chromatography and Accumulated Ion Monitoring on Quadrupole-Orbitrap-Linear Ion Trap Mass Spectrometer. Mol. Cell. Proteomics 2017, 16, 2006–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Greguš M; Kostas JC; Ray S; Abbatiello SE; Ivanov AR Improved Sensitivity of Ultralow Flow LC-MS-Based Proteomic Profiling of Limited Samples Using Monolithic Capillary Columns and FAIMS Technology. Anal. Chem 2020, 92, 14702–14712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Budnik B; Levy E; Harmange G; Slavov N SCoPE-MS: mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biol. 2018, 19, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Cong Y; Liang Y; Motamedchaboki K; Huguet R; Truong T; Zhao R; Shen Y; Lopez-Ferrer D; Zhu Y; Kelly RT Improved Single-Cell Proteome Coverage Using Narrow-Bore Packed NanoLC Columns and Ultrasensitive Mass Spectrometry. Anal. Chem 2020, 92, 2665–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Hata K; Izumi Y; Hara T; Matsumoto M; Bamba T In-Line Sample Processing System with an Immobilized Trypsin-Packed Fused-Silica Capillary Tube for the Proteomic Analysis of a Small Number of Mammalian Cells. Anal. Chem 2020, 92, 2997–3005. [DOI] [PubMed] [Google Scholar]
- (17).Shao X; Wang X; Guan S; Lin H; Yan G; Gao M; Deng C; Zhang X Integrated Proteome Analysis Device for Fast Single-Cell Protein Profiling. Anal. Chem 2018, 90, 14003–14010. [DOI] [PubMed] [Google Scholar]
- (18).Lombard-Banek C; Moody SA; Manzini MC; Nemes P Microsampling Capillary Electrophoresis Mass Spectrometry Enables Single-Cell Proteomics in Complex Tissues: Developing Cell Clones in Live Xenopus laevis and Zebrafish Embryos. Anal. Chem 2019, 91, 4797–4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Specht H; Emmott E; Petelski AA; Huffman RG; Perlman DH; Serra M; Kharchenko P; Koller A; Slavov N Single-cell proteomic and transcriptomic analysis of macrophage heterogeneity using SCoPE2. Genome Biol. 2021, 22, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Cho HR; Park JS; Wood TD; Choi YS Longitudinal Assessment of Peptide Recoveries from a Sample Solution in an Autosampler Vial for Proteomics. Bull. Korean Chem. Soc 2015, 36, 312–321. [Google Scholar]
- (21).Feist P; Hummon AB Proteomic challenges: sample preparation techniques for microgram-quantity protein analysis from biological samples. Int. J. Mol. Sci 2015, 16, 3537–3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Kasuga K; Katoh Y; Nagase K; Igarashi K Microproteomics with microfluidic-based cell sorting: Application to 1000 and 100 immune cells. Proteomics 2017, 17, 1600420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Myers SA; Rhoads A; Cocco AR; Peckner R; Haber AL; Schweitzer LD; Krug K; Mani DR; Clauser KR; Rozenblatt-Rosen O; Hacohen N; Regev A; Carr SA Streamlined protocol for deep proteomic profiling of FAC-sorted cells and its application to freshly isolated murine immune cells. Mol. Cell. Proteomics 2019, 18, 995–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Xu K; Liang Y; Piehowski PD; Dou M; Schwarz KC; Zhao R; Sontag RL; Moore RJ; Zhu Y; Kelly RT Benchtop-compatible sample processing workflow for proteome profiling of < 100 mammalian cells. Anal. Bioanal. Chem 2019, 411, 4587–4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Chen Q; Yan G; Gao M; Zhang X Ultrasensitive Proteome Profiling for 100 Living Cells by Direct Cell Injection, Online Digestion and Nano-LC-MS/MS Analysis. Anal. Chem 2015, 87, 6674–6680. [DOI] [PubMed] [Google Scholar]
- (26).Ethier M; Hou W; Duewel HS; Figeys D The Proteomic Reactor: A Microfluidic Device for Processing Minute Amounts of Protein Prior to Mass Spectrometry Analysis. J. Proteome Res 2006, 5, 2754–2759. [DOI] [PubMed] [Google Scholar]
- (27).Zhang Z; Dubiak KM; Huber PW; Dovichi NJ Miniaturized Filter-Aided Sample Preparation (MICRO-FASP) Method for High Throughput, Ultrasensitive Proteomics Sample Preparation Reveals Proteome Asymmetry in Xenopus laevis Embryos. Anal. Chem 2020, 92, 5554–5560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Kulak NA; Pichler G; Paron I; Nagaraj N; Mann M Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 2014, 11, 319–324. [DOI] [PubMed] [Google Scholar]
- (29).Wiśniewski JR; Zougman A; Nagaraj N; Mann M Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [DOI] [PubMed] [Google Scholar]
- (30).Hughes CS; Foehr S; Garfield DA; Furlong EE; Steinmetz LM; Krijgsveld J Ultrasensitive proteome analysis using paramagnetic bead technology. Mol. Syst. Biol 2014, 10, 757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Zougman A; Selby PJ; Banks RE Suspension trapping (STrap) sample preparation method for bottom-up proteomics analysis. Proteomics 2014, 14, 1006–1000. [DOI] [PubMed] [Google Scholar]
- (32).Leipert J; Tholey A Miniaturized sample preparation on a digital microfluidics device for sensitive bottom-up microproteomics of mammalian cells using magnetic beads and mass spectrometry-compatible surfactants. Lab Chip 2019, 19, 3490–3498. [DOI] [PubMed] [Google Scholar]
- (33).Li ZY; Huang M; Wang XK; Zhu Y; Li JS; Wong CCL; Fang Q Nanoliter-Scale Oil-Air-Droplet Chip-Based Single Cell Proteomic Analysis. Anal. Chem 2018, 90, 5430–5438. [DOI] [PubMed] [Google Scholar]
- (34).Rappsilber J; Mann M; Ishihama Y Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc 2007, 2, 1896–1906. [DOI] [PubMed] [Google Scholar]
- (35).Huffman RG; Chen A; Specht H; Slavov N DO-MS: Data-Driven Optimization of Mass Spectrometry Methods. J. Proteome Res 2019, 18, 2493–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Mellacheruvu D; Wright Z; Couzens AL; Lambert JP; St-Denis NA; Li T; Miteva YV; Hauri S; Sardiu ME; Low TY; Halim VA; Bagshaw RD; Hubner NC; Al-Hakim A; Bouchard A; Faubert D; Fermin D; Dunham WH; Goudreault M; Lin ZY; Badillo BG; Pawson T; Durocher D; Coulombe B; Aebersold R; Superti-Furga G; Colinge J; Heck AJ; Choi H; Gstaiger M; Mohammed S; Cristea IM; Bennett KL; Washburn MP; Raught B; Ewing RM; Gingras AC; Nesvizhskii AI The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat. Methods 2013, 10, 730–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Pathan M; Keerthikumar S; Ang C-S; Gangoda L; Quek CYJ; Williamson NA; Mouradov D; Sieber OM; Simpson RJ; Salim A; Bacic A; Hill AF; Stroud DA; Ryan MT; Agbinya JI; Mariadason JM; Burgess AW; Mathivanan S FunRich: An open access standalone functional enrichment and interaction network analysis tool. Proteomics 2015, 15, 2597–2601. [DOI] [PubMed] [Google Scholar]
- (38).Hulsen T; de Vlieg J; Alkema W BioVenn - a web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genomics 2008, 9, 488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Spitzer M; Wildenhain J; Rappsilber J; Tyers M BoxPlotR: a web tool for generation of box plots. Nat. Methods 2014, 11, 121–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Rappsilber J; Ishihama Y; Mann M Stop and Go Extraction Tips for Matrix-Assisted Laser Desorption-Ionization, Nanoelectrospray, and LC/MS Sample Pretreatment in Proteomics. Anal. Chem 2003, 75, 663–670. [DOI] [PubMed] [Google Scholar]
- (41).Smejkal GB; Li C; Robinson MH; Lazarev AV; Lawrence NP; Chernokalskaya E Simultaneous Reduction and Alkylation of Protein Disulfides in a Centrifugal Ultrafiltration Device Prior to Two-Dimensional Gel Electrophoresis. J. Proteome Res 2006, 5, 983–987. [DOI] [PubMed] [Google Scholar]
- (42).Herbert BR; Molloy MP; Gooley AA; Walsh BJ; Bryson WG; Williams KL Improved Protein Solubility in Two-dimensional Electrophoresis Using Tributyl Phosphine as Reducing Agent. Electrophoresis 1998, 19, 845–851. [DOI] [PubMed] [Google Scholar]
- (43).Glatter T; Ludwig C; Ahrné E; Aebersold R; Heck AJR; Schmidt A Large-scale quantitative assessment of different in-solution protein digestion protocols reveals superior cleavage efficiency of tandem Lys-C/trypsin proteolysis over trypsin digestion. J. Proteome Res 2012, 11, 5145–5156. [DOI] [PubMed] [Google Scholar]
- (44).Yurinskaya V; Aksenov N; Moshkov A; Model M; Goryachaya T; Vereninov A A comparative study of U937 cell size changes during apoptosis initiation by flow cytometry, light scattering, water assay and electronic sizing. Apoptosis 2017, 22, 1287–1295. [DOI] [PubMed] [Google Scholar]
- (45).Kyte J; Doolittle RF A Simple Method for Displaying the Hydropathic Character of a Protein. J. Mol. Biol 1982, 157, 105–132. [DOI] [PubMed] [Google Scholar]
- (46).Klont F; Bras L; Wolters JC; Ongay S; Bischoff R; Halmos GB; Horvatovich P Assessment of Sample Preparation Bias in Mass Spectrometry-Based Proteomics. Anal. Chem 2018, 90, 5405–5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Giansanti P; Tsiatsiani L; Low TY; Heck AJR Six alternative proteases for mass spectrometry-based proteomics beyond trypsin. Nat. Protoc 2016, 11, 993–1006. [DOI] [PubMed] [Google Scholar]
- (48).Xiang P; Zhu Y; Yang Y; Zhao Z; Williams SM; Moore RJ; Kelly RT; Smith RD; Liu S Picoflow Liquid Chromatography-Mass Spectrometry for Ultrasensitive Bottom-Up Proteomics Using 2-μm-i.d. Open Tubular Columns. Anal. Chem 2020, 92, 4711–4715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Zhu Y; Zhao R; Piehowski PD; Moore RJ; Lim S; Orphan VJ; Paša-Tolić L; Qian WJ; Smith RD; Kelly RT Subnanogram proteomics: impact of LC column selection, MS instrumentation and data analysis strategy on proteome coverage for trace samples. Int. J. Mass Spectrom 2018, 427, 4–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.