

Abstract
Introduction
While monophasic and relapsing forms of myelin oligodendrocyte glycoprotein antibody associated disorders (MOGAD) are increasingly diagnosed world-wide, consensus on management is yet to be developed.
Objective
To survey the current global clinical practice of clinicians treating MOGAD.
Method
Neurologists worldwide with expertise in treating MOGAD participated in an online survey (February–April 2019).
Results
Fifty-two responses were received (response rate 60.5%) from 86 invited experts, comprising adult (78.8%, 41/52) and pædiatric (21.2%, 11/52) neurologists in 22 countries. All treat acute attacks with high dose corticosteroids. If recovery is incomplete, 71.2% (37/52) proceed next to plasma exchange (PE). 45.5% (5/11) of paediatric neurologists use IV immunoglobulin (IVIg) in preference to PE. Following an acute attack, 55.8% (29/52) of respondents typically continue corticosteroids for ≤ 3 months; though less commonly when treating children. After an index event, 60% (31/51) usually start steroid-sparing maintenance therapy (MT); after ≤ 2 attacks 92.3% (48/52) would start MT. Repeat MOG antibody status is used by 52.9% (27/51) to help decide on MT initiation. Commonly used first line MTs in adults are azathioprine (30.8%, 16/52), mycophenolate mofetil (25.0%, 13/52) and rituximab (17.3%, 9/52). In children, IVIg is the preferred first line MT (54.5%; 6/11). Treatment response is monitored by MRI (53.8%; 28/52), optical coherence tomography (23.1%; 12/52) and MOG antibody titres (36.5%; 19/52). Regardless of monitoring results, 25.0% (13/52) would not stop MT.
Conclusion
Current treatment of MOGAD is highly variable, indicating a need for consensus-based treatment guidelines, while awaiting definitive clinical trials.
Keywords: Myelin oligodendrocyte glycoprotein, MOG, MOGAD, Survey
Introduction
Myelin oligodendrocyte glycoprotein antibody associated disorders (MOGAD) have been widely recognised as a distinct clinical entity only in the last decade, following the development of reliable cell-based assays using full-length human MOG as the target antigen [1, 2]. They encompass monophasic and relapsing presentations of central demyelination. Within the ‘neuromyelitis optica’ phenotype, optic neuritis is more common than transverse myelitis [3–8]. The clinical spectrum has since expanded to include brainstem and cortical encephalitis [9–12]. The most frequent presentation in young children is acute disseminated encephalomyelitis (ADEM) [8, 12, 13].
In comparison to aquaporin-4 antibody positive neuromyelitis optica spectrum disorders (AQP4-Ab NMOSD), relapse is less common. Approximately half of MOGAD patients may have monophasic disease, but some experience frequent relapses despite immunosuppressive therapy [14–17]. The value of antibody titres in predicting relapse is not yet fully understood. Overall, motor and visual disability outcomes seem better in MOGAD than in AQP4-Ab NMOSD [5, 14, 17], but the impact of relapses on long-term disability is unclear.
The unpredictability of MOGAD presents a challenge when developing treatment paradigms. Retrospective studies suggest that both acute and maintenance immunotherapy improve outcomes. However, there are no randomised controlled trials (RCTs) in MOGAD and an international, evidence-based consensus on management is yet to be developed. The objective of this survey is to describe the current clinical practice of neurologists treating adults and children with MOGAD internationally, to identify common themes, divergent practices and unanswered questions, which could inform the planning of collaborative studies and clinical trials.
Methods
The survey was created with the ‘Survey Monkey’ web-based tool (https://www.surveymonkey.co.uk). It comprised a mix of 34 multiple choice, ranking, or free-text questions (see online supplementary material).
Eighty-six neurologists were invited to participate via email. Invites were sent out to prospective attendees at the 7th Focused Workshop of The European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS) on MOGAD, which took place on 7th–8th March 2019, in Athens, Greece. The survey was closed to meeting attendees on 6th March to avoid obtaining biased responses following the meeting. Additional neurologists blinded to the workshop discussions and conclusions were invited to complete the survey, with the aim of creating a diverse global representation.
All responses were obtained throughout February to April 2019.
Results
A. Respondent details and scope of practice
Fifty-two responses were received (response rate 60.5%) from neurologists practising in 22 countries—Argentina (1), Australia (4), Brazil (1), Canada (2), China (1), Denmark/Hungary (1), France (3), French Martinique (1), Germany (5), Italy (4), India (1), Japan (2), Malaysia (2), Netherlands (1), Republic of Korea (1), Spain (1), Switzerland (1), Thailand (1), Turkey (3), United Kingdom (7) and United States of America (9).
Respondents were adult (78.8%, 41/52) and paediatric (21.2%, 11/52) neurologists, with 43.9% (18/41) of adult neurologists also involved in the specialist care of children. The median (range) number of MOGAD patients under each neurologist’s care was 20 (3–130), with each seeing a median (range) of 5 (0–50) new patients in the last 12 months.
The majority of neurologists (53.8%, 28/52) indicated that their management of MOGAD did not follow a published consensus, guideline or policy. International publications were cited by 32.7% (17/52) and included review or opinion articles [18–23] and observational studies [4, 8, 13, 24, 25]. National or regional/hospital policies were followed by 9.6% (5/52) and 15.4% (8/52) respectively.
For ease of review, the rest of the survey results have been condensed into 12 core questions.
B. Acute attack therapy
Question 1: Please state your usual dose and duration of high dose corticosteroid (HDCS) therapy for acute attacks of MOGAD (response rate 88.5%, 46/52)
For adult patients, 100% (36/36) used intravenous (IV) methylprednisolone at a dose of 1000 mg daily; 11.1% (4/36) substituted oral methylprednisolone 500 mg daily for milder attacks. Duration of IV therapy, if specified, was 3–5 days for 87.8% (29/33), with 12.1% (4/33) extending up to 10 days. For paediatric patients, 100% (10/10) used IV methylprednisolone at an actual body weight adjusted dose of 20–30 mg/kg daily (maximum 1 g daily) for 3–5 days.
Question 2: For severe attacks or if recovery is incomplete after HDCS therapy, what is your next choice of acute therapy? (response rate 100%, 52/52)
Respondents ranked up to six options in order of preference: plasma exchange/immunoadsorption (PE), intravenous immunoglobulin (IVIg), rituximab, cyclophosphamide, repeat HDCS, other (free text). They were advised to consider local availability, restrictions and cost. The most popular first choice among adult neurologists was PE in 80.5% (33/41); the remaining 19.5% (8/41) repeated HDCS initially. Paediatric neurologists were divided between PE (36.4%, 4/11), IVIg (36.4%, 4/11) and repeat HDCS (27.3%, 3/11).
We also calculated the mean preference score (between 0 and 6) for each acute therapy, with higher scores denoting earlier use by more respondents (Fig. 1). PE (5.60) was the preferred choice, followed by repeat HDCS (3.42), IVIg (3.40), rituximab (2.52), cyclophosphamide (1.42) and other therapies (0.27). The order was unchanged when analysing only adult neurologist responses. Paediatric neurologists preferred IVIg (5.18) over PE (4.91). When respondents were asked to disregard local restrictions and treatment costs (response rate 94.2%, 49/52), preference increased for IVIg (3.78), reduced for repeat HDCS (3.06), and were otherwise unchanged.
Fig. 1.
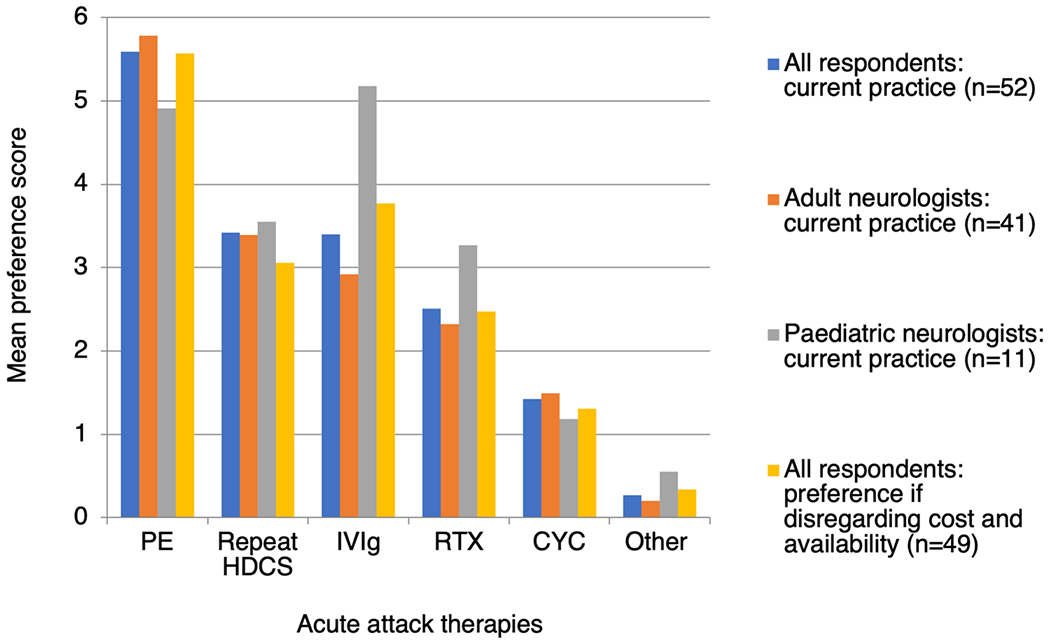
Neurologists’ preferences for escalation of acute attack therapies in MOGAD. Mean preference scores were calculated as follows: 6 points were given if ranked 1st, 5 points if ranked 2nd, and so on, with 0 points if not ranked at all. The total number of points for each therapy was then divided by the total number of respondents (52). Higher scores therefore indicate earlier use by more respondents. PE plasma exchange or immunoadsorption, HDCS high dose corticosteroids, IVIg intravenous immunoglobulin, RTX rituximab, CYC cyclophosphamide
The ‘other’ acute therapies specified were mitoxantrone (1), tocilizumab (1), azathioprine (2) and further courses of IVIg (1). Additionally, 7.7% (4/52) indicated that they initiate PE with HDCS in severe attacks, rather than waiting to assess response.
Question 3: After a recent first attack, do you give a prolonged (> 3 months) course of oral corticosteroid therapy? (response rate 100%, 52/52)
59.7% (31/52) of respondents answered ‘usually’ or ‘always’, 23.1% (12/52) answered ‘sometimes’ and 17.3% (9/52) answered ‘rarely’ or ‘never’ (Fig. 2). A greater proportion of paediatric than adult neurologists answered ‘never’ (27.3% versus 4.9%). Factors reported to influence this decision were attack severity (6), speed/extent of recovery (4), patient preference/co-morbidities (2) and attack topography (1) i.e. less likely to treat an ADEM-like attack than optic neuritis for greater than 3 months.
Fig. 2.
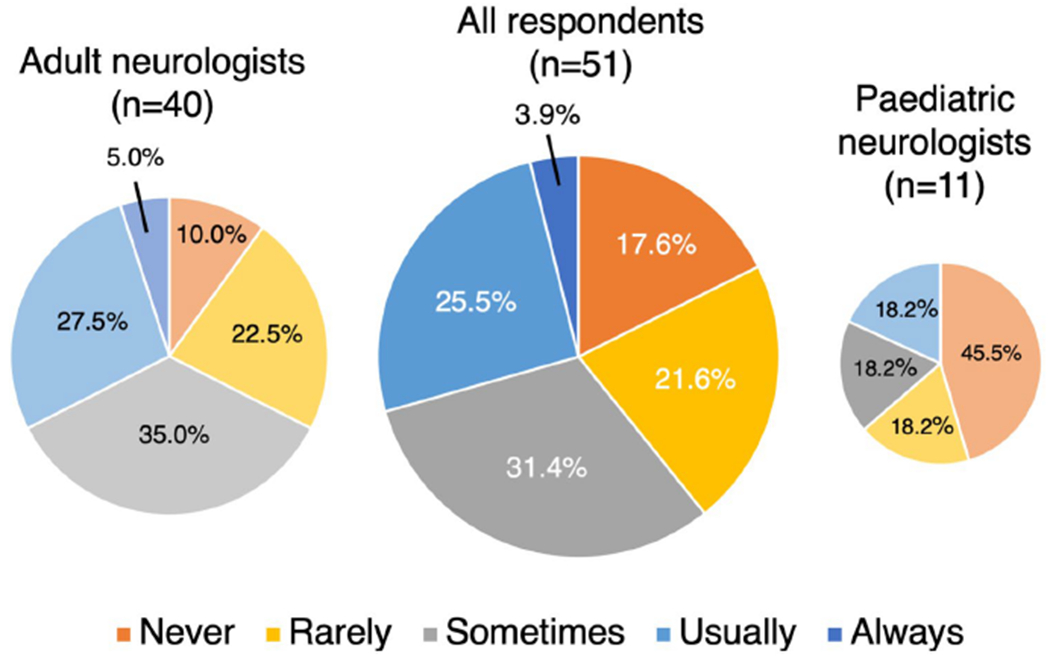
How frequently do neurologists’ treat a first attack of MOGAD with oral corticosteroid therapy for greater than 3 months?
88.5% (46/52) stated their preferred minimum duration of oral corticosteroid treatment: < 3 months 15.2% (7/46), ≥ 3 months 41.3% (19/46), ≥ 6 months 39.1% (18/46), ≥ 9 months 2.2% (1/46), and ≥ 18 months 2.2% (1/46). Dose tapering strategies were individualised. A minority of respondents (16.7%, 8/48) used repeat MOG-Ab titres to help determine the duration of corticosteroid treatment. Repeat testing was timed at 3 months (25.0%, 2/8), 6 months (37.5%, 3/8) or unspecified (37.5%, 3/8).
C. Starting steroid-sparing maintenance therapy
Question 4: Would you recommend starting maintenance therapy after a first attack of confirmed MOGAD? (response rate 98.1%, 51/52)
39.2% (20/51) of respondents answered ‘usually’ or ‘always’, 31.4% (16/51) answered ‘sometimes’ and 29.4% (15/51) answered ‘rarely’ or ‘never’ (Fig. 3). A greater proportion of paediatric than adult neurologists answered ‘never’ (45.5% versus 10.0%). Respondents justified their answers as follows: Relapse risk was perceived variably as low (16), high (5), or ‘impossible to predict’ (1). Other factors considered included the onset attack severity or recovery (13), the onset attack topography (8), the titre and persistence of MOG-Ab (7), and patient or clinician concerns about steroid-related adverse effects (6).
Fig. 3.
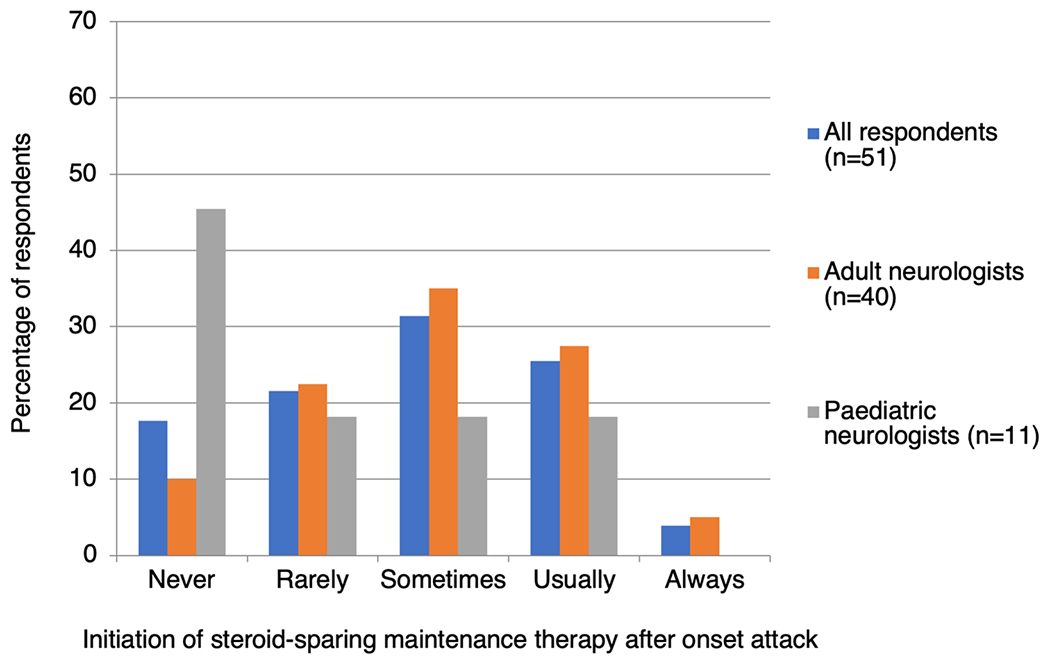
How frequently do neurologists start steroid-sparing maintenance therapy after an onset attack of MOGAD?
Question 5: Would you recommend starting maintenance therapy after two or more confirmed attacks of MOGAD? (response rate 100%, 52/52)
92.3% (48/52) respondents answered ‘usually’ or ‘always’, 5.8% (3/52) answered ‘sometimes’ and 1.9% (1/52) answered ‘rarely’ (Fig. 4). There was general agreement between adult and paediatric neurologists, though fewer paediatric neurologists answered ‘always’ (36.5% versus 68.5%). Repeat MOG-Ab testing is used by 47.1% (24/51) to inform this decision, but the timing and interpretation of testing is highly variable. Additional factors that influence the decision to start maintenance therapy after two or more attacks included the attack interval (5), attack severity and recovery (4), patient preference (3), the patient’s tolerance of corticosteroids (3), and patient age (2).
Fig. 4.
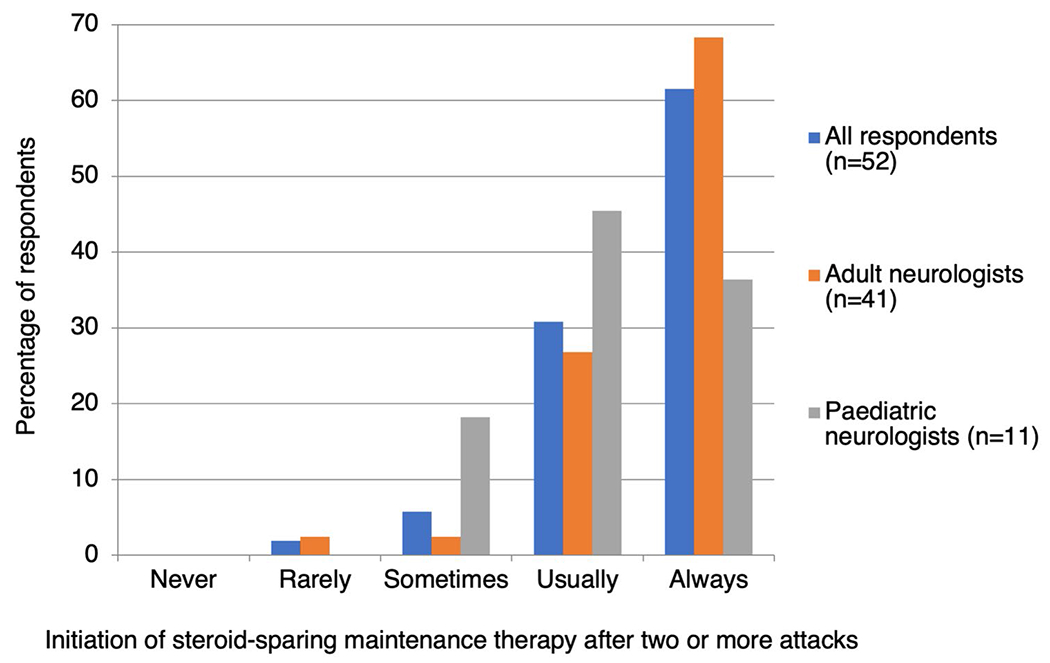
How frequently do neurologists start steroid-sparing maintenance therapy after two or more attacks of MOGAD?
Respondents also differed on the minimum interval between symptomatic flares that they use to define a second attack: 40.4% (21/52) answered 1 month, 36.5% (19/52) 3 months, and 1.9% (1/52) 6 months, whereas 21.2% (11/52) felt it was important to be flexible on this interval, depending on the disease topography or treatment history.
D. Choosing and switching maintenance therapies
Question 6 (adult neurologists only): Taking account of local availability, restrictions and cost, what would be your first-choice maintenance therapy for treating an otherwise healthy 38-year-old male with relapsing MOGAD? (response rate 100%, 41/41)
The most common first choice therapy was azathioprine (39.0%, 16/41), followed by mycophenolate mofetil (31.7%, 13/41), rituximab (22.0%, 9/41), tacrolimus (4.9%, 2/41) and mitoxantrone (2.4%, 1/41). One of these agents may be combined with low-dose oral corticosteroid ‘always’ (20.0%, 8/40), ‘usually’ (10.0%, 4/40), ‘sometimes’ (30.0%, 12/40), ‘rarely’ (22.5%, 9/40) or ‘never’ (17.5%, 7/40).
For patients relapsing on first-line azathioprine, 68.7% (11/16) escalate therapy to rituximab and 31.3% (5/16) escalate to mycophenolate mofetil. For this clinical scenario, Fig. 5 shows the popularity of each drug as a first-, second- or third-line treatment following breakthrough relapses.
Fig. 5.
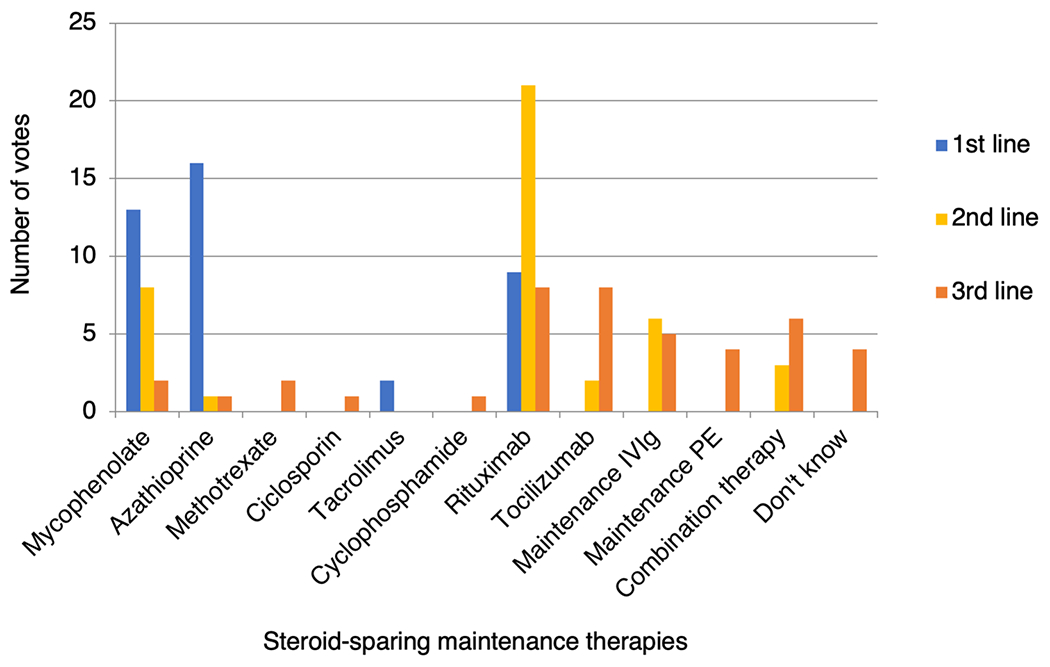
Popularity of individual steroid-sparing maintenance therapies as first-, second- and third-line treatments for a 38-year-old male with relapsing MOGAD. PE plasma exchange or immunoadsorption, IVIg intravenous immunoglobulin
Question 7 (paediatric neurologists only): Taking account of local availability, restrictions and cost, what would be your first-choice maintenance therapy for treating an otherwise healthy 6-year-old female with relapsing MOGAD? (response rate 100%, 11/11)
The most common first choice therapy was IVIg (45.5%, 5/11), followed by azathioprine (18.2%, 2/11) or rituximab (18.2%, 2/11). One of these agents may be combined with low-dose oral corticosteroid ‘always’ (9.1%, 1/11), ‘usually’ (18.2%, 2/11), ‘sometimes’ (36.4%, 4/11), ‘rarely’ (27.3%, 3/11) or ‘never’ (9.1%, 1/11). Figure 6 shows the popularity of each drug as a first-, second- or third-line treatment following breakthrough relapses.
Fig. 6.
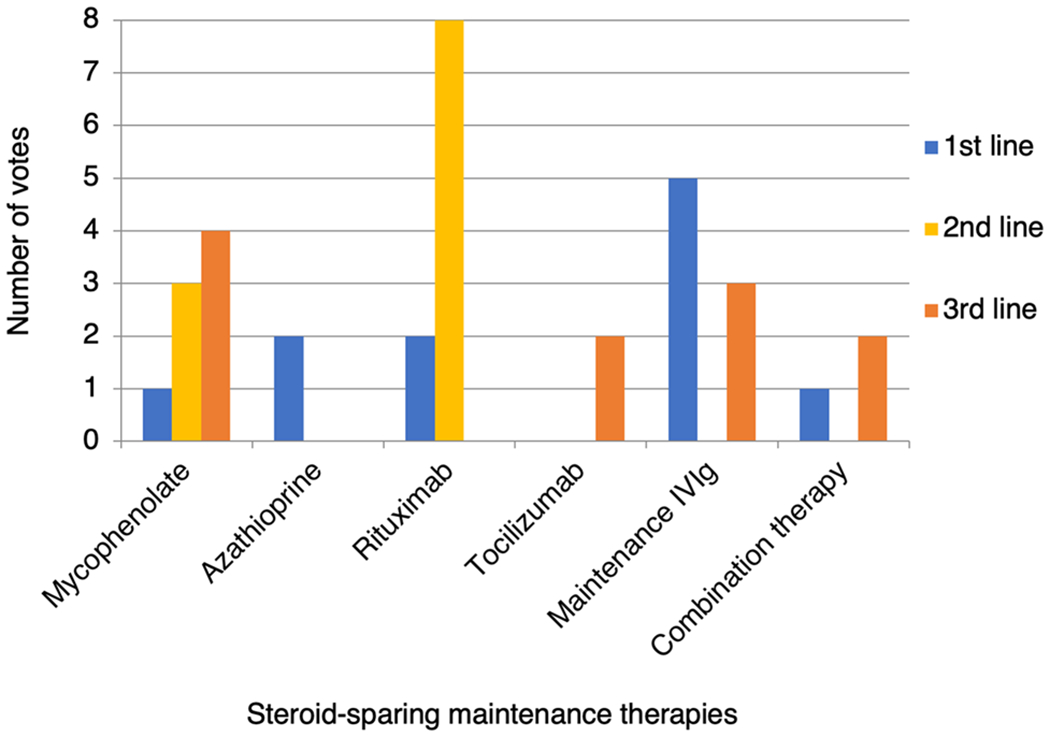
Popularity of individual steroid-sparing maintenance therapies as first-, second- and third-line treatments for a 6-year-old female with relapsing MOGAD. IVIg intravenous immunoglobulin
Question 8: Taking account of local availability, restrictions and cost, what would be your first-choice maintenance therapy for treating an otherwise healthy 16-year-old female with relapsing MOGAD? (response rate 100%, 52/52)
Respondents ranked up to eight options in order of preference. The most common first choice therapy was azathioprine (42.3%, 22/52), followed by rituximab (25.0%, 13/52), IVIg (13.5%, 10/52) and mycophenolate mofetil (13/5%, 7/52). The most common first choice amongst paediatric neurologists was IVIg (45.5%, 5/11).
We then generated overall preference scores for each treatment (as for question 2). Overall, rituximab was the preferred treatment choice (6.33), followed by azathioprine (5.54), mycophenolate mofetil (4.49), IVIg (4.12), PE (2.27), tocilizumab (1.96), methotrexate (1.54) and ciclosporin (0.81) (Fig. 7). Compared to adult neurologists, paediatric neurologists expressed greater preference for IVIg (5.64 versus 3.71) and tocilizumab (3.27 versus 1.96), and less preference for azathioprine (3.91 versus 5.98) and mycophenolate mofetil (4.00 versus 5.00).
Fig. 7.
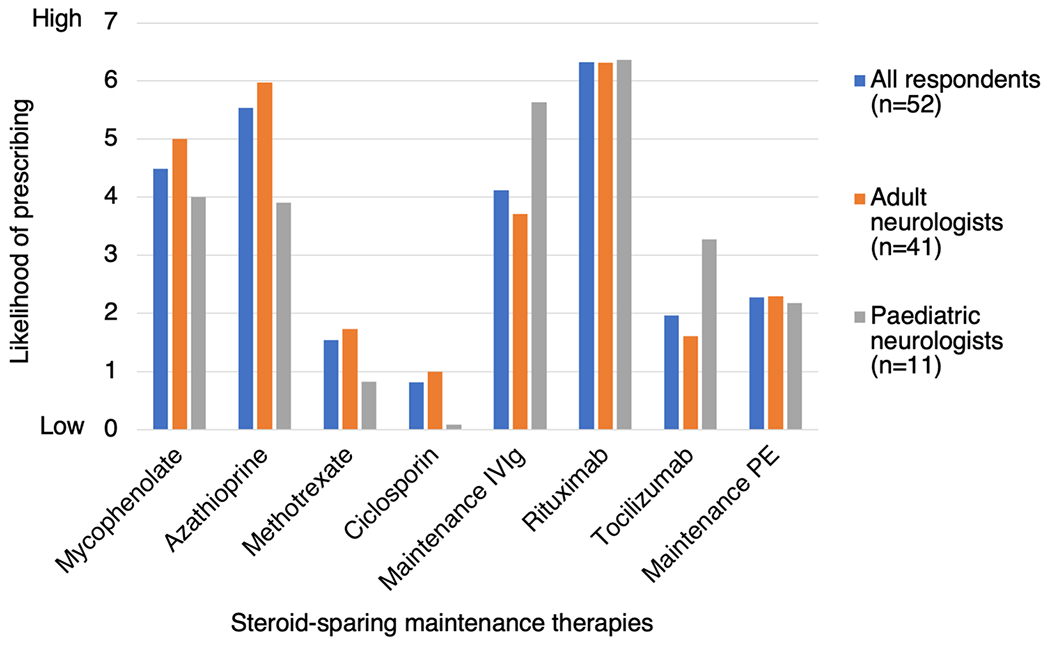
Neurologists’ preferences for individual steroid-sparing maintenance therapies to treat a 16-year-old female with relapsing MOGAD. Respondents were asked to rank eight different maintenance therapies in order of preference. No rank was given if the respondent would not consider using that therapy. Rankings were then converted to mean scores. PE plasma exchange or immunoadsorption, IVIg intravenous immunoglobulin
Question 9: Disregarding cost, availability and safety profiles, how would you rank the following treatment options solely on their effectiveness at maintaining remission? (response rate 100%, 52/52)
Respondents ranked up to ten treatment options in order of perceived effectiveness. They were advised to only rank treatments that they had experience administering to MOGAD patients. Mean scores were then calculated for each treatment (Fig. 8). The majority of neurologists had experience using rituximab (92.3%, 48/52), azathioprine (84.6%, 44/52), mycophenolate mofetil (78.8%, 41/52), IVIg (76.9%, 40/52), prednisolone up to 0.5 mg/kg/day (73.1%, 38/52) or PE (53.9%, 28/52). Of these, rituximab (8.23) was perceived to be most effective, followed by prednisolone 0.5 mg/kg/day (7.97), mycophenolate mofetil (7.68), azathioprine (7.43), IVIg (7.30) and PE (5.46). Fewer neurologists had experience giving tocilizumab (40.4%, 21/52), methotrexate (34.6%, 18/52), eculizumab (21.2%, 11/52) or ciclosporin (13.5%, 7/52). Of these lesser used therapies, tocilizumab (6.86) was perceived to be most effective, followed by eculizumab (6.45), ciclosporin (5.57) and methotrexate (4.83).
Fig. 8.
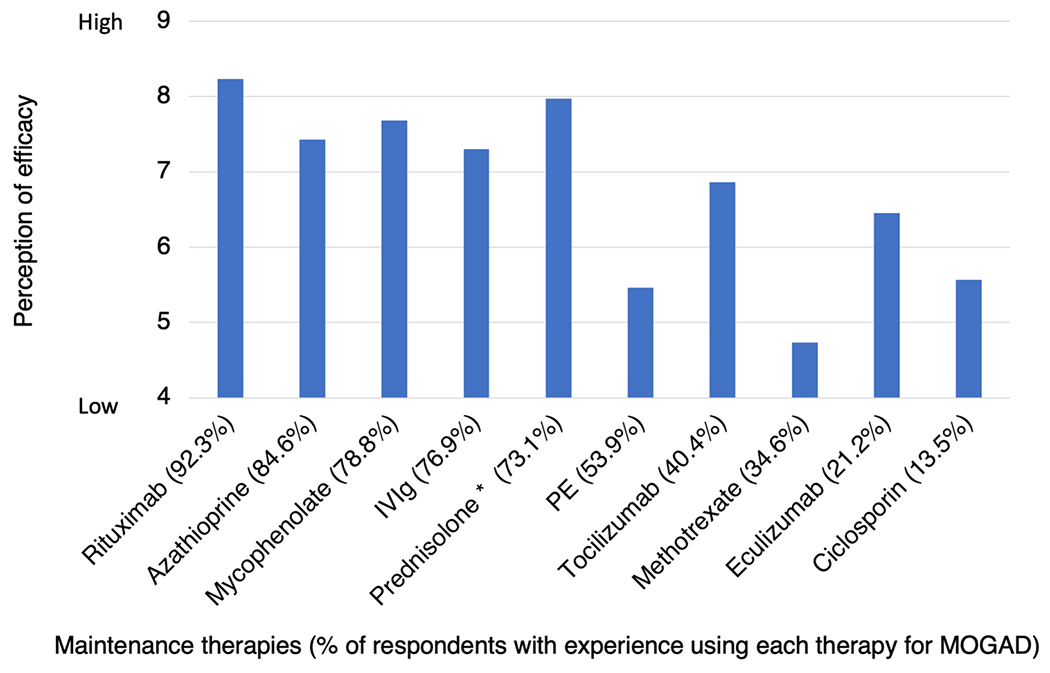
Neurologists’ perceptions of the effectiveness of individual maintenance therapies at preventing relapses of MOGAD. Respondents were asked to rank ten different maintenance therapies in order of their perceived efficacy. No rank was given if the respondent had no experience of using that therapy in MOGAD. Rankings were then converted to mean scores. The percentage of respondents with experience of using each therapy is given in parentheses. PE plasma exchange or immunoadsorption, IVIg intravenous immunoglobulin; *Prednisolone dose up to 0.5 mg/kg/day
Question 10: Which MS disease modifying therapies (MS-DMTs) do you think could be beneficial for treating MOGAD? (response rate 98.1%, 51/52)
Respondents could either select from up to ten licensed MS-DMTs (82.4%, 42/51), or choose ‘none’ (17.6%, 9/51). Ocrelizumab was chosen by 72.5% (37/51). Other therapies with a minority of votes included haematopoietic stem cell transplant (17.6%, 9/51), cladribine (15.7%, 8/51), teriflonumide (13.7, 7/51), natalizumab (9.8%, 5/51), alemtuzumab (7.8%, 4/51), fingolimod (7.8%, 4/51) and dimethyl fumarate (5.9%, 3/51). Beta-interferon and glatiramer acetate received no votes.
E. Assessing treatment response and stopping immunotherapy
Question 11: How do you routinely monitor the efficacy of maintenance therapy? (response rate 100%, 52/52)
All respondents (100%, 52/52) use prevention of relapses as an indicator of drug efficacy. In addition, 59.6% (31/52) routinely assess disability (e.g. extended disability status scale [EDSS] score, timed 25-foot walk or visual assessments). Some regularly monitor asymptomatic patients using MRI (53.8%, 28/52), optical coherence tomography (OCT) (23.1%, 12/52), and repeated MOG-Ab titres (36.5%, 19/52). Frequency of monitoring is variable and often individualised.
Question 12: For patients with relapsing MOGAD, in which circumstances would you recommend stopping maintenance immunotherapy? (response rate 100%, 52/52)
Twenty-five percent (13/52) of respondents would not stop maintenance immunotherapy in MOGAD. Others may recommend stopping treatment if the patient remains relapse-free after 1 year (5.8%, 3/52), 2 years (23.1%, 12/52) or 5 years (44.2%, 23/52). Alternatively, 17.3% (9/52) contemplate stopping treatment if the patient becomes MOG-Ab negative, irrespective of the time in clinical remission. Finally, 40.4% (21/52) described an individualised approach to stopping immunotherapy, considering not only time in remission and serostatus, but also the frequency and severity of prior attacks and the patient’s level of disability.
Discussion
This survey summarised the current global expert approach to the treatment of MOGAD. There was consensus on use of high dose corticosteroids and plasma exchange in acute attacks, and of purine synthesis inhibitors (azathioprine and mycophenolate mofetil) or rituximab as maintenance therapies for relapse prevention. This mirrors treatment of AQP4-Ab NMOSD, where the high risk of permanent disability mandates aggressive acute attack therapy and lifelong immunosuppression in most cases [26–28].
The survey also highlighted areas of divergent practice, such as the duration of oral corticosteroid therapy administered after a single attack, indications for starting and stopping steroid-sparing immunotherapy, individual drug choices, and the use of paraclinical tools (MRI, OCT and MOG-Ab titres) to monitor treatment response. Paediatric neurologists were less likely than adult neurologists to use PE for acute attacks, less likely to use prolonged oral corticosteroid therapy, and more likely to use IVIg as an acute or maintenance therapy. Notably, many responses indicated a complex and individualised approach to treating MOGAD, largely due to a lack of adequate data.
The uncertainty of relapse risk in MOGAD is probably a major reason for the variability in maintenance immunotherapy prescribing. The largest studies of incident cohorts (patients diagnosed as MOG-Ab seropositive at the time of their first attack) have indicated a relapse risk of 36% at 16 months or 43% at 2 years [16, 17]. However, more patients may be at risk of a second attack occurring beyond 2 years. An earlier study with a mean of 6.3 years follow-up quoted a relapse risk of 80%, increasing to 93% in cases with over 8 years follow-up [4]. However, these estimates may have been inflated by ascertainment bias, due to inclusion of non-incident cases who were diagnosed only when relapse occurred; many monophasic cases may have gone untested and been missed in this study. Variable use of immunotherapies has also biased relapse risk in these retrospective studies.
Age at first attack, attack topography, and MOG-Ab titres seem to influence relapse risk, but no one factor accurately predicts the disease course. Patients presenting with optic neuritis appear more likely to relapse early than those with transverse myelitis or ADEM [4, 7, 16]. Numerous studies have identified correlation between MOG-Ab titres and disease activity at a population level [16, 17, 29, 30], but as with AQP4-Ab NMOSD, MOG-Ab titres do not reliably predict relapses in individual patients. Many persistently seropositive patients do not relapse, and seronegative patients can relapse, with or without a return of detectable MOG-Ab [14, 31]. In adults, high titres at onset are associated with more severe presentations, but do not predict future disease course [32]. In children, one study reported that a high MOG-Ab titre (≥ 1:1280) at onset attack predicted relapse with 46% sensitivity and 86% specificity [33]. A prospective cohort study with a median of 4 years follow-up found that 57% of children become seronegative with a median time from first attack to seronegative conversion of 1 year [34]. Relapse occurred in 38% of persistently seropositive children and in 15% of those who became seronegative. Children with MOG-Ab positive relapsing ADEM have also been reported to become transiently seronegative between attacks [35]. This inability of serostatus to accurately predict relapses may explain why the majority of neurologists surveyed did not routinely use longitudinal MOG-Ab testing to aid treatment decisions.
Another area of controversy is the impact of relapses on long-term disability. MOGAD attacks are usually milder and more steroid-responsive than in AQP4-Ab NMOSD. Earlier diagnosis and treatment of relapses may improve recovery versus index attacks. This may explain why one study found no difference between relapsing and monophasic patients in the proportion of patients with major disability (EDSS score ≥ 3.0 or visual acuity of 20/100) [17]. However, other studies identified cumulative disability with repeated attacks, and recognised that patients with good recovery from their onset attack were at risk of disabling relapses, suggesting a role for maintenance immunotherapy [16, 36].
The level of evidence to support selection amongst maintenance therapies in MOGAD is poor. No treatments have been evaluated in RCTs, but several observational studies have examined treatment responses. MOGAD is clearly steroid-responsive, with several studies noting that early relapses frequently occur on withdrawal of corticosteroids [4, 8, 16]. Treatment for longer than 3 months following onset attack was associated with a lower relapse risk in a large British cohort [16], which may explain why 59.7% of neurologists surveyed usually or always treat with oral corticosteroids for at least 3 months. One paediatric study reported relapses on all maintenance therapies, except for patients receiving prednisolone ≥ 10 mg daily [37]. However, the adverse metabolic effects of exogenous corticosteroids limit their use, particularly in children.
Standard first-line NMOSD therapies, including azathioprine, mycophenolate, methotrexate, rituximab and IVIg, are associated with reduced relapse rates in observational studies of MOGAD [4, 8, 13, 24, 36, 37]. However, small numbers of patients on each individual therapy render comparison difficult. Drug choice is therefore likely to depend on availability, cost, and individual patient factors, as reflected by the heterogeneity of responses in this survey. The overall perception in this survey was that rituximab may be the most effective of the commonly used therapies. Interestingly, some studies have reported relatively frequent relapses in small numbers of RTX-treated MOGAD patients [8, 13], and a larger retrospective study found a modest 43% decline in relapse rate following initiation of rituximab, albeit in a relatively selected population with high disease activity [38]. One paediatric study suggested superior efficacy of IVIg over rituximab, azathioprine and mycophenolate mofetil, though only 12/102 patients received this treatment [13]. This may in part explain, together with its favourable safety profile, why IVIg was favoured by paediatric neurologists in this survey.
Only a minority of survey respondents had experience treating MOGAD with tocilizumab (interleukin-6 [IL-6] blockade) or eculizumab (terminal complement inhibitor). IL-6 plays a crucial role in the induction of experimental autoimmune encephalitis (EAE), the murine model of MOGAD [39], and CSF IL-6 is elevated in MOGAD patients during acute attacks [40, 41]. Tocilizumab has been reported to induce remission in rituximab-refractory MOGAD [42–44]. The exact role of complement in MOGAD is less established than in AQP4-Ab NMOSD, but human MOG-Ab can initiate complement-dependent demyelination in animal models [45–48]. Further pre-clinical and clinical studies are therefore needed to better define the role of novel therapies in MOGAD.
As an anti-CD20 B-cell depleting therapy, like rituximab, it is unsurprising that 72.5% of respondents felt that ocrelizumab may be effective in treating MOGAD. The effect of other MS-DMTs on MOGAD is unknown, but some studies have suggested poor efficacy, particularly of beta-interferon [4, 13, 35, 49]. The detrimental effect of some MS-DMTs in AQP4-Ab NMOSD has not been observed in MOGAD thus far.
In summary, treating MOGAD is currently complicated due to the heterogenous clinical spectrum and the lack of data. Thus, neurologists appear to be individualizing therapy based on patient-specific factors.
There are limitations to this survey. Availability of diagnostic assays varies globally and consequently MOGAD is not diagnosed in many regions of the world, which are underrepresented. Racial differences in MOGAD phenotypes or the efficacy and tolerability of immunotherapies may affect regional treatment paradigms. Furthermore, neurologists inevitably acquire unconscious biases due to differing clinical exposures, health care systems, drug costs and availabilities. Prescribing practices do not necessarily reflect optimal treatment, though we have tried to address these issues.
This survey provides a current cross-sectional view of how ‘MOGAD experts’ treat patients in the face of limited evidence and should serve as rough map to prevent nonexperts from ‘straying too far from the path’. It also highlights the need for prospective observational studies with long-term follow-up of incident cohorts and systematic testing of MOG-Ab titres to establish the natural history of MOGAD. RCTs should follow and will best establish the role of individual immunotherapies.
The more favourable outcomes of MOGAD create genuine equipoise and should make it more suited to placebo-controlled trials than AQP4-Ab NMOSD. Ideally, studies should examine different patient groups and attack types separately to generate the most clinically meaningful data. In the meantime, the results of this survey emphasise the importance of taking an individualised approach to treating MOGAD, in which patients make informed treatment decisions and are actively encouraged to participate in research.
Acknowledgments
Funding The UK Neuromyelitis Optica Diagnostic and Advisory Service is funded by the Highly Specialised Commissioning Division of NHS England. There was no formal sponsorship for this study.
Conflicts of interest D.H. Whittam, E.Gibbons, V. Karthikeayan, R. Kneen, S. Chandratre, J. de Seze, K. Deiva, R.Q. Hintzen, I. Kleiter, K. Rostasy, P. Huppke, F. Paul, A.K. Pröbstel, M.P. Amato, M. Nosadini, M.M. Mancardi, Z. Illes, A. Siva, G. Akman-Demir, L. Pandit, M. Apiwattankul, J.Y. Hor, S. Viswanathan, W. Qiu, H.J. Kim, I. Nakashima, R.C. Dale, M. Boggild, S. Broadley, M.A. Lana-Peixoto, P. Cabre, B.G. Weinshenker, B. Greenberg, M. Matiello, E.C. Klawiter, J.L. Bennett, A.I. Wallach, I. Kister, B.L. Banwell, D. Pohl, M.Levy, M.I. Leite, T. Solomon: nothing to disclose. O. Ciccarelli is a consultant for Roche, Novartis, Teva, Biogen and Merck. B Wildemann has received research grants and/or honoria from Merck Serono, Biogen, Teva, Novartis, Sanofi Genzyme, Bayer Healthcare, and research grants from Bundesministerium für Bildung und Forschung, Deutsche Forschungsgemeinschaft, Dietmar Hopp Foundation and the Klaus Tschira Foundation. S. Jarius’s work was indirectly supported by research grants from Dietmar Hopp Stiftung and from Merck Serono. I. Kleiter has received speaker honoraria and travel funding from Bayer, Biogen, Novartis, Merck, Sanofi Genzyme, Roche; speaker honoraria from Mylan; travel funding from the Guthy-Jackson Charitable Foundation; consulted for Alexion, Bayer, Biogen, Celgene, Chugai, IQVIA, Novartis, Merck, Roche; and research support from Chugai, Diamed. B. Hemmer has served on scientific advisory boards for F. Hoffmann-La Roche Ltd, Novartis, and Bayer AG; he has served as DMSC member for AllergyCare and TG Therapeutics; he or his institution have received speaker honoraria from Medimmune, Novartis, Desitin, and F. Hoffmann-La Roche Ltd; his institution has received research support from Chugai Pharmaceuticals; holds part of two patents; one for the detection of antibodies and T cells against KIR4.1 in a subpopulation of MS patients and one for genetic determinants of neutralizing antibodies to interferon β. O. Aktas reports grants from the German Research Foundation (DFG) and the German Ministry of Education and Research (BMBF), grants and personal fees from Bayer HealthCare, Biogen, Genzyme, Novartis, Teva and Viela Bio, and personal fees from Almirall, MedImmune, Merck Serono and Roche. G. Arrambide has received compensation for consulting services or participation in advisory boards from Sanofi, Merck, and Roche; research support from Novartis; travel expenses for scientific meetings from Novartis, Roche, Stendhal, and ECTRIMS; and speaking honoraria from Sanofi, Merck, and Novartis. M. Tintore has received compensation for consulting services and speaking honoraria from Almirall, Bayer Schering Pharma, Biogen-Idec, Genzyme, Merck-Serono, Novartis, Roche, Sanofi-Aventis, and Teva Pharmaceuticals. MT is co-editor of Multiple Sclerosis Journal-ETC. M. Capobianco received personal honoraria for speaking at meeting or participating in advisory boards from Biogen, Merck, Novartis, Roche, Sanofi, Teva. A. Altintas received travel grants and/or speaker honoraria from Merck, Generica and Novartis. H.J. Kim received research support from the Ministry of Science and ICT, Genzyme, Merck Serono, Teva-Handok, and UCB; received consultancy/speaker fees from Celltrion, Eisai, HanAll BioPharma, MedImmune, Merck Serono, Novartis, Sanofi Genzyme, Teva-Handok, and UCB; serves on a steering committee for MedImmune/VielaBio; is a co-editor for the Multiple Sclerosis Journal—Experimental, Translational, and Clinical, and an associated editor for the Journal of Clinical Neurology. K. Fujihara received consultancy/speaker fees: Alexion Pharmaceuticals, Chugai, Asahi Kasei Medical, Biogen, Eisai, Mitsubishi-Tanabe Pharma, Nihon, Novartis Pharmaceuticals, ONO Pharmaceutical, Takeda, and Teijin. S. Ramanathan has received an Early Career Fellowship from the National Health and Medical Research Council (Australia). D.K. Sato has received a Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (KAKENHI 15K19472); research support from CNPq/Brasil (425331/2016-4), FAPERGS/MS/CNPq/SESRS (17/2551-0001391-3) PPSUS/Brazil, TEVA (research grant for EMOCEMP Investigator Initiated Study), and Euroimmun AG (Neuroimmunological Complications associated with Arboviruses); and speaker honoraria from Biogen, Novartis, Genzyme, TEVA, Merck-Serono, Roche, and Bayer and has participated in advisory boards for Shire, Roche, TEVA, Merck-Serono and Quest/Athena Diagnostics. S. Tenembaum serves as a non-remunerated editorial board member of Neurology: Neuroimmunology & Neuroinflammation. She has received speaker and consulting fees from Biogen-Idec Argentina, Merck Serono LATAM, Genzyme-Sanofi, Novartis, and Teva Neuroscience during the last 3 years. D.M. Wingerchuk received grant support paid to Mayo Clinic from Alexion and TerumoBCT, consultant fees from MedImmune, Celgene, Novartis, and ONO Pharmaceuticals. A. Traboulsee: has received research funding from Chugai, Roche, and Sanofi Genzyme; received honoraria or travel support from Consortium of MS Centers, MS Society of Canada, Biogen, Teva, Roche, Merck/EMD Serono, Sanofi Genzyme, Chugai. J. Palace is partly funded by highly specialised services to run a national congenital myasthenia service and a neuromyelitis optica service. She has received support for scientific meetings and honorariums for advisory work from Merck Serono, Biogen Idec, Novartis, Teva, Chugai Pharma and Bayer Schering, Alexion, Roche, Genzyme, MedImmune, EuroImmun, MedDay, Abide ARGENX, UCB and Viela Bio and grants from Merck Serono, Novartis, Biogen Idec, Teva, Abide, MedImmune, Bayer Schering, Genzyme, Chugai and Alexion. She has received grants from the MS society, Guthrie Jackson Foundation, NIHR, Oxford Health Services Research Committee, EDEN, MRC, GMSI, John Fell and Myaware for research studies. R. Marignier serves on the scientific advisory board for Novartis and Medimmune; received speaker honoraria and travel funding from Novartis, Biogen, Teva, Sanofi-Aventis/Genzyme, Merck. M. Lim has received consultation fees from CSL Behring; received travel grants from Merck Serono; and was awarded educational grants to organize meetings by Novartis, Biogen Idec, Merck Serono and Bayer. S. Huda has received research support from the Neuromyelitis Optica UK charity. A. Jacob served on the scientific advisory board for Shire Pharmaceuticals; received travel funding and/or speaker honoraria from Biogen Idec, Shire, and Terumo BCT; consulted for Shire Pharmaceuticals; and received research support from Biogen, Alexion Pharmaceuticals, NHS, and University of Liverpool.
Footnotes
Availability of data and material The survey can be viewed in its original format in the online supplementary material. Complete results are available upon reasonable request to the corresponding author.
References
- 1.O’Connor KC, McLaughlin KA, De Jager PL et al. (2007) Self-antigen tetramers discriminate between myelin autoantibodies to native or denatured protein. Nat Med 13(2):211–217. 10.1038/nm1488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Waters P, Woodhall M, O’Connor KC et al. (2015) MOG cell-based assay detects non-MS patients with inflammatory neurologic disease. Neurol Neuroimmunol Neuroinflamm 2(3):e89. 10.1212/NXI.0000000000000089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kitley J, Woodhall M, Waters P et al. (2012) Myelin-oligodendrocyte glycoprotein antibodies in adults with a neuromyelitis optica phenotype. Neurology 79(12):1273–1277. 10.1212/WNL.0b013e31826aac4e [DOI] [PubMed] [Google Scholar]
- 4.Jarius S, Ruprecht K, Kleiter I et al. (2016) MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 2: Epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. J Neuroinflamm 13(1):280. 10.1186/s12974-016-0718-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kitley J, Waters P, Woodhall M et al. (2014) Neuromyelitis optica spectrum disorders with aquaporin-4 and myelin-oligodendrocyte glycoprotein antibodies: a comparative study. JAMA Neurol 71(3):276–283. 10.1001/jamaneurol.2013.5857 [DOI] [PubMed] [Google Scholar]
- 6.Sato DK, Callegaro D, Lana-Peixoto MA et al. (2014) Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders. Neurology 82(6):474–481. 10.1212/WNL.0000000000000101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pandit L, Sato DK, Mustafa S et al. (2016) Relapsing optic neuritis and isolated transverse myelitis are the predominant clinical phenotypes for patients with antibodies to myelin oligodendrocyte glycoprotein in India. Mult Scler J Exp Transl Clin 2:2055217316675634. 10.1177/2055217316675634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ramanathan S, Mohammad S, Tantsis E et al. (2018) Clinical course, therapeutic responses and outcomes in relapsing MOG antibody-associated demyelination. J Neurol Neurosurg Psychiatry 89(2):127–137. 10.1136/jnnp-2017-316880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ogawa R, Nakashima I, Takahashi T et al. (2017) MOG antibody-positive, benign, unilateral, cerebral cortical encephalitis with epilepsy. Neurol Neuroimmunol Neuroinflamm 4:e322. 10.1212/NXI.0000000000000322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hamid SHM, Whittam D, Saviour M et al. (2018) Seizures and encephalitis in myelin oligodendrocyte glycoprotein IgG disease vs aquaporin 4 IgG disease. JAMA Neurol 75(1):65–71. 10.1001/jamaneurol.2017.3196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fujimori J, Takai Y, Nakashima I et al. (2017) Bilateral frontal cortex encephalitis and paraparesis in a patient with anti-MOG antibodies. J Neurol Neurosurg Psychiatry 88(6):534–536. 10.1136/jnnp-2016-315094 [DOI] [PubMed] [Google Scholar]
- 12.Cobo-Calvo A, Ruiz A, D’Indy H et al. (2017) MOG antibody-related disorders: common features and uncommon presentations. J Neurol 264(7):1945–1955. 10.1007/s00415-017-8583-z [DOI] [PubMed] [Google Scholar]
- 13.Hacohen Y, Wong YY, Lechner C et al. (2018) Disease course and treatment responses in children with relapsing myelin oligodendrocyte glycoprotein antibody-associated disease. JAMA Neurol 75(4):478–487. 10.1001/jamaneurol.2017.4601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Höftberger R, Sepúlveda M, Armangue T et al. (2015) Antibodies to MOG and AQP4 in adults with neuromyelitis optica and suspected limited forms of the disease. Mult Scler 21(7):866–874. 10.1177/1352458514555785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mariotto S, Ferrari S, Monaco S et al. (2017) Clinical spectrum and IgG subclass analysis of anti-myelin oligodendrocyte glycoprotein antibody-associated syndromes: a multicenter study. J Neurol 264(12):2420–2430. 10.1007/s00415-017-8635-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jurynczyk M, Messina S, Woodhall MR et al. (2017) Clinical presentation and prognosis in MOG-antibody disease: a UK study. Brain 140(2):3128–3138. 10.1093/brain/awx276 [DOI] [PubMed] [Google Scholar]
- 17.Cobo-Calvo A, Ruiz A, Maillart E et al. (2018) Clinical spectrum and prognostic value of CNS MOG autoimmunity in adults: the MOGADOR study. Neurology 90(21):e1858–e1869. 10.1212/WNL.0000000000005560 [DOI] [PubMed] [Google Scholar]
- 18.Juryńczyk M, Jacob A, Fujihara K, Palace J (2019) Myelin oligodendrocyte glycoprotein (MOG) antibody-associated disease: practical considerations. Pract Neurol 19(3):187–195. 10.1136/practneurol-2017-001787 [DOI] [PubMed] [Google Scholar]
- 19.Hacohen Y, Banwell B (2019) Treatment approaches for MOG-Ab-associated demyelination in children. Curr Treat Options Neurol 21(1):2. 10.1007/s11940-019-0541-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wynford-Thomas R, Jacob A, Tomassini V (2019) Neurological update: MOG antibody disease. J Neurol 266(5):1280–1286. 10.1007/s00415-018-9122-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jarius S, Paul F, Asgari N et al. (2018) MOG encephalomyelitis: international recommendations on diagnosis and antibody testing. J Neuroinflamm 15(1):134. 10.1186/s12974-018-1144-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dos Passos GR, Oliveira LM, da Costa BK et al. (2018) MOG-IgG-Associated Optic neuritis, encephalitis, and myelitis: lessons learned from neuromyelitis optica spectrum disorder. Front Neurol 9:217. 10.3389/fneur.2018.00217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Borisow N, Mori M, Kuwabara S, Scheel M, Paul F (2018) Diagnosis and treatment of NMO spectrum disorder and MOG-encephalomyelitis. Front Neurol 9:888. 10.3389/fneur.2018.00888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pandit L, Mustafa S, Nakashima I, Takahashi T, Kaneko K (2018) MOG-IgG-associated disease has a stereotypical clinical course, asymptomatic visual impairment and good treatment response. Mult Scler J Exp Transl Clin 4(3):2055217318787829. 10.1177/2055217318787829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Montcuquet A, Collongues N, Papeix C et al. (2017) Effectiveness of mycophenolate mofetil as first-line therapy in AQP4-IgG, MOG-IgG, and seronegative neuromyelitis optica spectrum disorders. Mult Scler 23(10):1377–1384. 10.1177/1352458516678474 [DOI] [PubMed] [Google Scholar]
- 26.Sellner J, Boggild M, Clanet M et al. (2010) EFNS guide-lines on diagnosis and management of neuromyelitis optica. Eur J Neurol 17(8): 1019–1032. 10.1111/j.1468-1331.2010.03066.x [DOI] [PubMed] [Google Scholar]
- 27.Palace J, Leite MI, Jacob A (2012) A practical guide to the treatment of neuromyelitis optica. Pract Neurol 12(4):209–214. 10.1136/practneurol-2012-000237 [DOI] [PubMed] [Google Scholar]
- 28.Kimbrough DJ, Fujihara K, Jacob A et al. (2012) Treatment of neuromyelitis optica: review and recommendations. Mult Scler Relat Disord 1(4):180–187. 10.1016/j.msard.2012.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jarius S, Ruprecht K, Kleiter I et al. (2016) MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 1: Frequency, syndrome specificity, influence of disease activity, long-term course, association with AQP4-IgG, and origin. J Neuroinflamm 13(1):279. 10.1186/s12974-016-0717-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hyun JW, Woodhall MR, Kim SH et al. (2017) Longitudinal analysis of myelin oligodendrocyte glycoprotein antibodies in CNS inflammatory diseases. J Neurol Neurosurg Psychiatry 88(10):811–817. 10.1136/jnnp-2017-315998 [DOI] [PubMed] [Google Scholar]
- 31.Spadaro M, Gerdes LA, Krumbholz M et al. (2016) Autoantibodies to MOG in a distinct subgroup of adult multiple sclerosis. Neurol Neuroimmunol Neuroinflamm 3(5):e257. 10.1212/NXI.0000000000000257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cobo-Calvo A, Sepúlveda M, D’Indy H et al. (2019) Usefulness of MOG-antibody titres at first episode to predict the future clinical course in adults. J Neurol 266(4):800–815. 10.1007/s00415-018-9160-9 [DOI] [PubMed] [Google Scholar]
- 33.Hennes E, Baumann M, Schanda K et al. (2017) Prognostic relevance of MOG antibodies in children with an acquired demyelinating syndrome. Neurology 89(9):900–908. 10.1212/WNL.0000000000004312 [DOI] [PubMed] [Google Scholar]
- 34.Waters P, Fadda G, Woodhall M et al. (2019) Serial anti-myelin oligodendrocyte glycoprotein antibody analysis and outcomes in children with demyelinating syndromes. JAMA Neurol. 10.1001/jamaneurol.2019.2940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Duignan S, Wright S, Rossor T et al. (2018) Myelin oligodendrocyte glycoprotein and aquaporin-4 antibodies are highly specific in children with acquired demyelinating syndromes. Dev Med Child Neurol 60:958–962. 10.1111/dmcn.13703 [DOI] [PubMed] [Google Scholar]
- 36.Cobo-Calvo A, Sepúlveda M, Rollot F et al. (2019) Evaluation of treatment response in adults with relapsing MOG-Ab-associated disease. J Neuroinflamm 16(1): 134. 10.1186/s12974-019-1525-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wong YYM, Hacohen Y, Armangue T et al. (2018) Paediatric acute disseminated encephalomyelitis followed by optic neuritis: disease course, treatment response and outcome. Eur J Neurol 25(5):782–786. 10.1111/ene.13602 [DOI] [PubMed] [Google Scholar]
- 38.Whittam DH, Cobo-Calvo A, Lopez-Chiriboga AS et al. (2018) Treatment of MOG-IgG-associated demyelination with Rituximab: a multinational study of 98 patients (S13.003). Talk presented at: 70th American Academy of Neurology Annual Meeting, Los Angeles, USA, 21–27 April, 2018 [Google Scholar]
- 39.Okuda Y, Sakoda S, Fujimura H, Saeki Y, Kishimoto T, Yanagihara T (1999) IL-6 plays a crucial role in the induction phase of myelin oligodendrocyte glycoprotein 35–55 induced experimental autoimmune encephalomyelitis. J Neuroimmunol 101(2):188–196. 10.1016/s0165-5728(99)00139-3 [DOI] [PubMed] [Google Scholar]
- 40.Kaneko K, Sato DK, Nakashima I et al. (2018) CSF cytokine pro-file in MOG-IgG+ neurological disease is similar to AQP4-IgG+ NMOSD but distinct from MS: a cross-sectional study and potential implications. J Neurol Neurosurg Psychiatry 89(9):927–936. 10.1136/jnnp-2018-317969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kothur K, Wienholt L, Tantsis EM et al. (2016) B cell, Th17, and neutrophil related cerebrospinal fluid cytokine/chemokines are elevated in MOG antibody associated demyelination. PLoS ONE 11(2):e0149411. 10.1371/journal.pone.0149411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Novi G, Gastaldi M, Franciotta D, Pesce G, Benedetti L, Uccelli A (2019) Tocilizumab in MOG-antibody spectrum disorder: a case report. Mult Scler Relat Disord 27:312–314. 10.1016/j.msard.2018.11.012 [DOI] [PubMed] [Google Scholar]
- 43.Hayward-Könnecke H, Reindl M, Martin R, Schippling S (2019) Tocilizumab in severe recurrent anti-MOG-associated optic neuritis. Neurology 92(16):765–767. 10.1212/WNL.0000000000007312 [DOI] [PubMed] [Google Scholar]
- 44.Ringelstein M, Kleiter I, Rommer P et al. (2019) Long-term interleukin-6-receptor blockade in neuromyelitis optica spectrum disorder and MOG associated encephalomyelitis (P1344). Poster presented at: 35th Congress of the European Committee for Treatment and Research in Multiple Sclerosis, Stockholm, Sweden, 11–13 September, 2019 [Google Scholar]
- 45.Mader S, Gredler V, Schanda K et al. (2011) Complement activating antibodies to myelin oligodendrocyte glycoprotein in neuromyelitis optica and related disorders. J Neuroinflamm 8:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Peschl P, Schanda K, Zeka B et al. (2017) Human antibodies against the myelin oligodendrocyte glycoprotein can cause complement-dependent demyelination. J Neuroinflamm 14(1):208. 10.1186/s12974-017-0984-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fang L, Kang X, Wang Z et al. (2019) Myelin oligodendrocyte glycoprotein-IgG contributes to oligodendrocytopathy in the presence of complement, distinct from astrocytopathy induced by AQP4-IgG. Neurosci Bull 35(5):853–866. 10.1007/s12264-019-00375-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chamberlain JL, Huda S, Whittam DH, Matiello M, Morgan BP, Jacob A (2019) Role of complement and potential of complement inhibitors in myasthenia gravis and neuromyelitis optica spectrum disorders: a brief review. J Neurol. 10.1007/s00415-019-09498-4 [DOI] [PubMed] [Google Scholar]
- 49.Wildemann B, Jarius S, Schwarz A et al. (2017) Failure of alemtuzumab therapy to control MOG encephalomyelitis. Neurology 89(2):207–209. 10.1212/WNL.0000000000004087 [DOI] [PubMed] [Google Scholar]