

Abstract
Flaviviruses pose a constant threat to human health. These RNA viruses are transmitted by the bite of infected mosquitoes and ticks and regularly cause outbreaks. To identify host factors required for flavivirus infection, we performed full-genome loss of function CRISPR-Cas9 screens. Based on these results, we focused our efforts on characterizing the roles that TMEM41B and VMP1 play in the virus replication cycle. Our mechanistic studies on TMEM41B revealed that all members of the Flaviviridae family that we tested require TMEM41B. We tested 12 additional virus families and found that SARS-CoV-2 of the Coronaviridae also required TMEM41B for infection. Remarkably, single nucleotide polymorphisms present at nearly 20% in East Asian populations reduce flavivirus infection. Based on our mechanistic studies, we propose that TMEM41B is recruited to flavivirus RNA replication complexes to facilitate membrane curvature, which creates a protected environment for viral genome replication.
Keywords: TMEM41B, VMP1, flavivirus, coronavirus, CRISPR, autophagy, flavivirus replication complex, membrane remodeling
Graphical Abstract
Hoffmann et al. determine that the transmembrane protein, TMEM41B, is required for infection by members of the Flaviviridae family of viruses. Loss of TMEM41B reduces viral RNA replication and increases innate immune activation in response to flavivirus infection. Thus, TMEM41B is a potential host target for antiviral therapeutics.
Introduction
Virus outbreaks are a regular yet unpredictable feature of an interconnected world. Flaviviruses are known to cause outbreaks worldwide and are transmitted primarily by the bite of infected mosquitoes and ticks.
Flaviviruses, members of the Flaviviridae family, are positive-sense single-stranded RNA viruses that have caused several notable outbreaks in recent history. For example, West Nile virus (WNV) emerged in New York City in 1999, spread across the continent, and is now endemic in the United States (Kramer et al., 2019; Roehrig et al., 2002). Also noteworthy are the recurring yellow fever virus (YFV) outbreaks that occur in sub-Saharan Africa and South America despite the availability of a highly effective vaccine (Ahmed and Memish, 2017; WHO, 2017). Most recently, the 2016 Zika virus (ZIKV) epidemic swept through South and Central America wreaking havoc on scores of unborn children by causing microcephaly in utero (Hills et al., 2017; Lee and Ng, 2018). In addition to these outbreaks, Aedes albopictus, a vector for ZIKV and dengue virus (DENV) has been found consistently in recent years in central Europe (Müller et al., 2020), and the prevalence of tick-borne diseases is at a record high. These alarming trends correlate with warming temperatures due to climate change, which expand the range of mosquitoes and ticks (Brady and Hay, 2020; Brugueras et al., 2020; Dobler, 2010; McPherson et al., 2017; Medlock et al., 2013). These recent flavivirus outbreaks add to the constant burden of endemic DENV transmission and underscore the devastating impacts that flaviviruses have on human health.
For many viruses with the potential to cause outbreaks, there are no specific treatments. In instances where antiviral therapies do exist, they are often virus specific. One strategy to prepare for and respond to viral outbreaks is to develop drugs that target host factors that viruses require to complete their replication cycles. CRISPR-Cas9 gene disruption has been used to identify host factors required for virus infection and nominate candidate drug targets. We, like others, set out to identify factors required for ZIKV as well as other flaviviruses with a genome-wide loss of function CRISPR-Cas9 screening approach. Here, we present our results and detailed studies focused on two genes, transmembrane protein 41B (TMEM41B) and vacuole membrane protein 1 (VMP1), that were enriched in our screens.
Results
Genome-wide CRISPR-Cas9 Screens for Zika and Yellow Fever Viruses Identify TMEM41B and VMP1 as Required Host Factors
To identify host factors required for ZIKV and YFV, we performed pooled genome-wide CRISPR knockout (KO) screens in B3GALT6-deficient human haploid (HAP1) cells with ZIKV (PRVABC59 strain) and YFV (Asibi strain). The readout for these screens was cell survival. We used B3GALT6 KO cells aiming to decrease cellular heparan sulfate (HS) protein glycosylation, which has been shown to nonspecifically facilitate virus binding and subsequent entry (Cagno et al., 2019). We hypothesized that this alteration in surface protein glycosylation would decrease the enrichment and overabundance of hits related to HS protein glycosylation and nonspecific virus binding. Nevertheless, consistent with a wide range of virus-selected genome-wide loss-of-function screens (Hoffmann et al., 2017; Jae et al., 2014; Realegeno et al., 2017; Riblett et al., 2015), we identified host factors involved in heparan sulfate biosynthesis.
Like other flavivirus host factor screens, we identified genes that are involved in oligosaccharide transfer and genes involved in protein translocation and folding into the endoplasmic reticulum (ER) (Figure 1 A) (Marceau et al., 2016; Savidis et al., 2016; Zhang et al., 2016). Outside of these categories, TMEM41B was enriched in both ZIKV and YFV screens. While several of the abovementioned pathways have been studied in the context of flavivirus infection (Marceau et al., 2016; Ngo et al., 2019; Zhang et al., 2016), little is known about the cellular function of TMEM41B or its role in flavivirus infection. TMEM41B was previously found to be required for synaptic transmission in motor circuit neurons (Imlach et al., 2012; Lotti et al., 2012), and three independent genome-wide CRISPR-Cas9 loss of function screens identified it as a gene that plays an important role in autophagy (Moretti et al., 2018; Morita et al., 2019; Shoemaker et al., 2019). These groups went on to show that TMEM41B is functionally similar to VMP1, which is known to have a role in lipid mobilization and autophagy (Morishita et al., 2019; Ropolo et al., 2007; Zhao et al., 2017).
Figure 1.

Genome-wide CRISPR-Cas9 Screens for Zika and Yellow Fever Viruses Identify TMEM41B and VMP1 as Required Host Factors
(A) Bubble plot of genes significantly enriched in a genome-wide CRISPR KO screen in HAP1 cells challenged with ZIKV (top) and YFV (bottom). Colors indicate association with cellular pathways or protein complexes and domains. Red lines denote Z = ± 2.
(B) Heatmap of Z scores for genes in the autophagy pathway ordered sequentially by functional role: L, lipid mobilization; 1, initiation; 2, nucleation; 3, elongation; 4, sequestration; 5, tethering/fusion. Rows represent replicate screens.
(C) Scatterplot of gene-wise log2 fold change (LFC) from this study (ZIKV) versus Moretti et al. (2018) autophagy screen.
(D) HAP1 WT and (n = 3) individual KO clones for VTT domain-containing proteins infected with ZIKV.
(E) WT and TMEM41B KO HAP1 cells overexpressing individual VTT domain proteins infected with ZIKV.
(F) Same as (E) but in VMP1 KO HAP1 cells.
(G) HAP1 WT and (n = 3–5) individual KO clones for autophagy genes infected with ZIKV.
(H–K) Same as (D–G) but infected with YFV Asibi. Cells were analyzed by flow cytometry and plotted as a percentage of viral antigen-positive cells.
Dots in (D), (G), (H), and (K) represent the average of n = 3 replicates from individual single-cell clones. Error bars in (E), (F), (I), and (J) depict a single KO clone with standard deviation (SD) of n = 3 replicates. See also Figures S1B–S1I.
There are numerous, sometimes conflicting reports, which indicate that autophagy-related genes can promote or restrict Flaviviridae infection. This literature has been recently reviewed by Po-Yuan Ke (Ke, 2018). Our identification of TMEM41B prompted us to interrogate our screen data further for genes involved in autophagy. Of a list of genes with an established role in autophagy, only TMEM41B and VMP1, which act at the early stage of autophagy, scored positively (Figure 1B). This list, however, is not comprehensive, and therefore we next compared our ZIKV screen results to those from a genome-wide CRISPR-Cas9 screen that was designed to identify novel genes required for autophagy (Moretti et al., 2018). As shown in Figure 1C, these two screens showed no overlap with the exception of two genes: TMEM41B and VMP1.
The VTT Domain Proteins TMEM41B and VMP1 Are Required for Flavivirus Infection
TMEM41B and VMP1 are multipass transmembrane proteins that share a stretch of amino acids recently named the VTT domain based on homology among VMP1, TMEM41A/B, and TMEM64 (Morita et al., 2019). Given this homology and the fact that both TMEM41B and VMP1 scored as hits in our flavivirus screens, we tested all four members of the VTT domain family for their requirement in flavivirus infection. Further, in addition to ZIKV and YFV (strain: Asibi) used in our screens, we included three additional flaviviruses: YFV 17D (vaccine strain), DENV-2, and WNV.
To test which of the four VTT domain-containing proteins are required for flavivirus infection, we used CRISPR-Cas9 gene disruption to generate clonal HAP1 cell lines individually lacking VMP1, TMEM41A, TMEM41B, and TMEM64 (Figure S1 A). We then infected three to five independent KO clones for each of these candidate host factors with each of the five flaviviruses listed above or with human parainfluenza virus 3 (hPIV-3) as a non-flavivirus control. Consistent with our screening results, we found that of these four VTT domain-containing proteins, only TMEM41B and VMP1 are required for flavivirus infection (Figures 1D, 1H, S1B, S1D, and S1F). While TMEM41B was not required for hPIV-3 infection, decreased growth of hPIV-3 was observed in the VMP1 KO clones (Figure S1H). VMP1 KO clones appear less viable and grow slower compared to wild-type (WT) cells, which likely accounts for the slight decrease in replication seen for hPIV-3.
Figure S1.

Flavivirus Infection in CRISPR KO Cells for VTT Domain and Autophagy Proteins, Related to Figure 1
(A) Western blots for TMEM41A, TMEM41B, and VMP1 in KO clones lacking individual VTT domain-containing proteins. The expected sizes are indicated by labels on the right. β-actin is shown as loading control. Gene disruption was additionally confirmed by next generation sequencing for each clone.
(B) HAP1 wildtype (WT) and (n = 3) individual knockout (KO) clones for VTT domain-containing proteins infected with YFV 17D (MOI = 0.005 PFU/cell) for 48 h.
(C) HAP1 WT and (n = 3-5) individual KO clones for autophagy genes infected with YFV 17D (MOI = 0.005 PFU/cell) for 48 h.
(D and E) Same as panels B-C but infected with DENV-GFP (MOI = 0.1 PFU/cell) for 96 h.
(F and G) Same as panels B-C but infected with WNV-GFP (MOI = 10 PFU/cell) for 72 h.
(H and I) Same as panels B-C but infected with hPIV-3-GFP (MOI = 0.02 IU/cell) for 48 h.
(J) TMEM41B KO HAP1 cells overexpressing individual VTT domain proteins infected with hPIV-3-GFP (MOI = 0.02 IU/cell) for 48 h.
(K) VMP1 KO HAP1 cells overexpressing individual VTT domain proteins infected with hPIV-3-GFP (MOI = 0.02 IU/cell) for 72 h. Cells were analyzed by flow cytometry and plotted as a percentage of viral antigen positive cells.
Dots in panels B-I represent the average of n = 3-5 replicates from individual single cell clones. Error bars in panels J-K are SD of n = 3 replicates from a single KO clone.
We next expressed each of the four VTT domain-containing proteins individually in TMEM41B KO and VMP1 KO cells to test their ability to restore virus infection. As expected, TMEM41B overexpression completely restored infection in TMEM41B KO cells. Interestingly, we found that VMP1 and TMEM41A overexpression partially compensated for the lack of TMEM41B during flavivirus infection. In contrast, only VMP1 was able to rescue infection in VMP1 KO cells (Figures 1E, 1F, 1I, and 1J). Furthermore, VMP1 expression restored the observed decrease in cell viability in VMP1 KO cells and rescued hPIV-3 replication (Figures S1J and S1K). The capacity of VMP1 to rescue a TMEM41B defect but not vice versa is consistent with what has been shown for TMEM41B and VMP1 in the context of autophagy (Morita et al., 2019).
Classical Autophagy Proteins BECN1, ATG5, and ATG7 Are Not Required for Flavivirus Infection in HAP1 Cells
Since both TMEM41B and VMP1 have been shown to be involved in autophagy, we wondered if canonical autophagy-related genes were essential for flavivirus infection (Moretti et al., 2018; Morita et al., 2019; Ropolo et al., 2007; Shoemaker et al., 2019). To test this, we generated clones lacking BECN1, ATG5, and ATG7, which are known to play a critical role in autophagy (Kang et al., 2011; Mizushima et al., 1998; Tanida et al., 1999). We confirmed KO in these clones by genome sequencing as well as by western blot (for ATG5 and ATG7) (Figure S2 A). ATG5 and ATG7 KO clones lack lipidated LC3 II at baseline levels, indicative of defective autophagy (Tanida et al., 2008; Tanida and Waguri, 2010). We then further tested ATG7 KO clones for their ability to respond to classical autophagy stimuli (e.g., starvation and treatment with Torin 1 in the presence of chloroquine). As expected, ATG7-deficient cells are unable to undergo autophagy. This is shown by western blot in Figure S2B, where autophagy induction leads to LC3 I lipidation and conversion to LC3 II in WT cells but not in ATG7 KO HAP1 clones (Tanida et al., 2008; Tanida and Waguri, 2010). We then tested these autophagy-deficient cells for their ability to support flavivirus infection and found that cells lacking BECN1, ATG5, or ATG7 retained their ability to support flavivirus infection (Figures 1G, 1K, S1C, S1E, S1G, and S1I). These data suggest that in this cell type, canonical autophagy is not required for flavivirus infection. This does not, however, exclude a requirement for non-canonical autophagy mechanisms nor does it suggest TMEM41B and VMP1 are the only autophagy factors required for flavivirus infection. In fact, two other non-classical autophagy genes were enriched in our YFV screen (TBC1D5 and TBC1D20) and in our ZIKV screen (TBC1D20) underscoring the complex interplay between flavivirus replication and the autophagy pathway (Popovic et al., 2012; Popovic and Dikic, 2014; Sidjanin et al., 2016; Sklan et al., 2007) (Figures 1A and 1B).
Figure S2.

Western Blots Confirm Knockout of Autophagy Proteins ATG5 and ATG7 and Autophagy Defects, Related to Figure 1
(A) Western blots for ATG5 and ATG7 in HAP1 WT cells and ATG5 and ATG7 KO clones. The expected sizes are indicated by labels on the right. β-actin is shown as loading control. The anti-ATG5 antibody also recognizes the ATG5/ATG12 conjugate with a higher molecular weight band. Also shown is the differently lipidated LC3 protein (LC3 I/II), indicative of functional autophagy.
(B) ATG7 KO HAP1 clones untreated (left), treated with 250 nM Torin 1 (inducer of autophagy) and 50 μM chloroquine (CQ – block of autophagosome/lysosome fusion and LC3 II turnover) (middle), and serum-starved (induction of autophagy) in Earle’s balanced salt solution (EBSS and treated with CQ) (right) (treatment for 6 h). β-actin is shown as loading control. Functional autophagy and autophagy induction are observed by the appearance of LC3 II, which is absent at baseline in ATG5 and ATG7 KO clones and also upon induction in ATG7 KO clones.
TMEM41B Is a Pan-flavivirus Host Factor
To gain additional insight into the requirement of TMEM41B for flavivirus infection, we extended our studies to include tick-borne flaviviruses, viruses from additional genera within the Flaviviridae family, and a diverse panel of unrelated viruses.
The tick-borne flaviviruses we tested include Powassan virus (POWV), a biosafety level 3 (BSL3) pathogen currently expanding in North America in Ixodes ticks (Dennis et al., 1998; Ebel, 2010; Eisen et al., 2016), and five BSL4 pathogens: two strains of tick-borne encephalitis virus (TBEV) representing the European and Far Eastern clade and three hemorrhagic fever viruses, Omsk hemorrhagic fever virus (OHFV), Kyasanur forest disease virus (KFDV), and Alkhurma hemorrhagic fever virus (AHFV). In addition, we generated TMEM41B KO clones in hepatocellular carcinoma cells (Huh-7.5) and bovine MDBK cells to test additional members in the Flaviviridae: HCV in the hepacivirus genus and BVDV in pestivirus genus. As shown in Figures 2 A–2D, all of these viruses require TMEM41B for infection. This demonstrates that TMEM41B is a pan-flavivirus host factor.
Figure 2.

TMEM41B Is a Pan-flavivirus Host Factor
(A–D) (A) WT and TMEM41B KO HAP1 cells infected with encephalitic or hemorrhagic fever tick-borne flaviviruses. (C) WT and TMEM41B KO Huh-7.5 clones infected with HCV. (D) WT and TMEM41B KO MDBK clones infected with BVDV. For (C) and (D) dots represent the average of n = 3 replicates from individual single cell clones. Virus strain is indicated in the figure. Error bars in (A) and (B) show SD for n = 6 replicates. Error bars in (C) and (D) show SD for n = 3 replicates (WT) and individual KO clones.
(E) Table summarizing results shown in Figure 2 (A–D), Figure S3 (A, B, D, F), and Figure S4 (A–D) from infection experiments with multiple viruses in various mammalian and mosquito cell lines. TBFV, tick-born flaviviruses of (A) and (B); +++, infection comparable to WT cells; +/−, reduced but detectable infection; −, negligible infection.
(F–I) WT, TMEM41B KO, and reconstituted HAP1 cells infected with (−) sense RNA viruses (F); VSV-G-pseudotyped HIV-1 lentivirus (LV-GFP) (G); (+) sense RNA viruses (H); or DNA viruses (I). Cells were analyzed by flow cytometry and plotted as a percentage of viral antigen-positive cells or GFP/RFP-positive cells for reporter viruses expressing a fluorescent protein. Error bars depict SD for n = 3 replicates or the indicated number of clones.
Flaviviruses replicate in a variety of tissue types. Therefore, we generated TMEM41B KO and reconstituted cell clones in additional mammalian cell lines representing lung adenocarcinoma (A549) and placenta carcinoma (JEG3) epithelial cells. In addition, we knocked out and reconstituted the mosquito TMEM41B ortholog in C6/36 cells (Aedes albopictus) and Aag2 cells (Aedes aegypti) to determine if TMEM41B was necessary for flavivirus infection in the mosquito vector. We found that TMEM41B was required in nearly all virus-cell combinations (Figures 2E, S3 A–S3G, and S4A–S4E). However, we observed several virus-cell combinations where TMEM41B appears to be less critical. For example, whereas TMEM41B is critical for YFV infection in HAP1, JEG3, and A549 cells, it is not required in Huh-7.5 cells. Notably we even observed differences in the requirement for TMEM41B among two strains of the same virus: the WT Asibi strain of YFV and the 17D vaccine strain that differ by only 31 amino acids (Figures S3A and S3D) (dos Santos et al., 1995). Given that VMP1 can compensate for TMEM41B deficiency, we hypothesized that differences in TMEM41B and VMP1 expression among cell types could potentially explain this variability. We therefore performed western blots on HAP1, JEG3, A549, and Huh-7.5 cell lysates to compare VMP1 and TMEM41B abundance. We found that full-length TMEM41B is highly expressed in HAP1 and JEG3 cells but is present at lower abundance in A549 and Huh-7.5 cells. Further, the TMEM41B antibody detects several abundant lower molecular weight bands in A549 cells that may or may not be TMEM41B isoforms. VMP1, in contrast, is expressed at lower levels in HAP1 cells compared to JEG3, A549, and Huh-7.5 cells (Figure S4 F). While speculative, we posit that the differential requirement for TMEM41B that we observe in some virus-cell combinations is due to differences in TMEM41B and VMP1 abundance and the extent to which VMP1 can compensate for TMEM41B deficiency.
Figure S3.

Flavivirus Infections in Mammalian TMEM41B KO Cells, Related to Figure 2
(A) WT and TMEM41B KO A549 clones generated with two independent sgRNAs infected with mosquito-borne flaviviruses: YFV 17D, MOI = 0.025 PFU/cell for 48 h; YFV Asibi, MOI = 0.05 PFU/cell for 72 h; ZIKV, MOI = 0.05 PFU/cell for 48 h; DENV-GFP, MOI = 0.1 PFU/cell for 48 h; WNV-GFP, MOI = 1 PFU/cell for 72 h.
(B) WT and TMEM41B KO A549 clones infected for 48 h with tick-borne flaviviruses at MOIs of 0.02 PFU/cell.
(C) WT and TMEM41B KO A549 clones infected for 48 h with hPIV-3-GFP, MOI = 0.02 IU/cell.
(D) WT and TMEM41B KO JEG3 clones generated with two independent sgRNAs infected with mosquito-borne flaviviruses: YFV 17D, MOI = 0.025 PFU/cell for 48 h; YFV Asibi, MOI = 0.05 PFU/cell for 72 h; ZIKV, MOI = 0.025 PFU/cell for 48 h; DENV-GFP, MOI = 0.1 PFU/cell for 72 h; WNV-GFP, MOI = 0.1 PFU/cell for 48 h.
(E) WT and TMEM41B KO JEG3 clones infected for 48 h with hPIV-3-GFP, MOI = 0.02 IU/cell.
(F) WT and TMEM41B KO Huh-7.5 clones generated with two independent sgRNAs infected with mosquito-borne flaviviruses: YFV 17D, MOI = 0.0025 PFU/cell for 48 h; YFV Asibi, MOI = 0.0025 PFU/cell for 48 h; ZIKV, MOI = 0.01 PFU/cell for 48 h; DENV-GFP, MOI = 0.01 PFU/cell for 72 h; WNV-GFP, MOI = 0.01 PFU/cell for 48 h.
(G) WT and TMEM41B KO Huh-7.5 clones infected for 48 h with hPIV-3-GFP, MOI = 0.02 IU/cell. Error bars depict SD for n=3 replicates or the indicated number of clones.
Figure S4.

Flavivirus Infections in Mosquito TMEM41B KO Cells, Related to Figure 2
(A–C) WT and TMEM41B KO and KO reconstituted Aag2 and C6/36 mosquito cell clones generated with independent sgRNAs infected with: ZIKV, MOI = 0.05 PFU/cell for 72 h; DENV, MOI = 0.2 PFU/cell for 96 h; WNV-GFP, MOI = 10 PFU/cell for 72 h.
(D and E) Same as A-C but without TMEM41B reconstitution. YFV 17D, MOI = 10 PFU/cell for 72 h; YFV Asibi, MOI = 0.5 PFU/cell for 72 h; CHIKV, MOI = 0.1 PFU/cell for 72 h (Aag2) and 0.05 PFU/cell for 48 h (C6/36). CHIKV was included as a non-flavivirus control.
(F) Western blot to detect TMEM41B and VMP1 in lysates from indicated cell lines. β-actin is included as a loading control. Cells were analyzed by flow cytometry and plotted as a percentage of viral antigen positive cells or GFP/RFP positive cells for reporter viruses expressing a fluorescent protein. Panels A-E, error bars depict SD for n=3 replicates or the indicated number of clones.
Given that TMEM41B is broadly required among members of the Flaviviridae, we posited that other more distantly related viruses or unrelated viruses may share a similar requirement for TMEM41B. Using the TMEM41B KO and reconstituted HAP1 cells, we queried the TMEM41B requirement for a diverse panel of viruses including negative- and positive-sense RNA viruses, retroviruses, and DNA viruses. Interestingly, we found that Coxsackievirus 3B (CVB-3) infection is enhanced in the absence of TMEM41B, whereas infection with SARS-CoV-2, a member of the Coronaviridae, is impaired similar to the viruses in the Flaviviridae, suggesting that it also requires TMEM41B for infection. Aside from these two viruses, none of the other viruses tested were affected by the lack of TMEM41B (Figures 2F–2I). Our observation that SARS-CoV-2 requires TMEM41B for infection is supported by our recent coronavirus genome-wide CRISPR screening and validation results (Schneider et al., 2020).
Functional TMEM41B Is Conserved across Mammalian and Vector Species
There are four reported TMEM41B isoforms in humans, however, only isoform 1 encodes a fully intact VTT domain. To determine if any of the other three isoforms can support flavivirus infection, we cloned and expressed each isoform in TMEM41B KO cells. Secondary structure predictions indicate that the first ∼47 amino acids of TMEM41B are unstructured (Kelley et al., 2015). Therefore, we also generated a deletion mutant of isoform 1 lacking the first 47 amino acids. A diagram of these TMEM41B constructs is shown in Figure 3 A. We found that only the full-length and N-terminal truncated isoform 1 proteins were able to fully support YFV and ZIKV infection in TMEM41B KO HAP1 cells; however, isoform 4, which contains half of the VTT domain, partially supported YFV infection (Figure 3B). From this we conclude that the TMEM41B VTT domain is required to support flavivirus infection whereas the N terminus is dispensable.
Figure 3.

Functional TMEM41B Is Conserved across Mammalian and Vector Species
(A) Graphical representation of human TMEM41B isoforms and deletion construct. VTT domain is shaded pink. Amino acid length of each isoform is indicated.
(B and C) (B) WT, TMEM41B KO, and TMEM41B KO cells expressing human TMEM41B isoforms or (C) mosquito or tick TMEM41B orthologs, infected with YFV Asibi or ZIKV as indicated.
(D) Table comparing homology between human TMEM41B (isoform 1) and mosquito and tick TMEM41B orthologs. Numbers indicate percent amino acids identity for the full-length protein versus VTT domain only. Protein sequences were aligned using Geneious Software (Geneious 8.1.9. https://www.geneious.com). Cells were analyzed by flow cytometry and plotted as a percentage of viral antigen-positive cells. Error bars depict SD for n = 3 replicates.
It was reported that TMEM41B physically interacts with VMP1, raising the possibility that this interaction may be important for membrane remodeling in the early stages of autophagy (Morita et al., 2019). Based on this we hypothesized that evolutionarily divergent TMEM41B orthologs may not support flavivirus infection in mammalian cells, given that there are extensive amino acid differences among the mosquito, tick, and human TMEM41B orthologs that may prohibit their interaction with human VMP1 (Ishii and Akira, 2005) (Figure 3D). To test if mosquito or tick TMEM41B orthologs could support flavivirus infection, we expressed TMEM41B orthologs from Aedes aegypti mosquitoes, Aedes albopictus mosquitoes, and Ixodes scapularis ticks in TMEM41B KO HAP1 cells and challenged them with YFV and ZIKV. To our surprise, we found that all three TMEM41B orthologs supported virus infection in mammalian cells (Figure 3C). This suggests that despite amino acid differences, evolutionarily divergent TMEM41B orthologs can either functionally interact with human VMP1 or that a direct protein-protein interaction between TMEM41B and VMP1 is not required to support flavivirus infection.
Naturally Occurring TMEM41B SNPs Negatively Impact Flavivirus Infection
Viruses and other pathogens have shaped human genetics over millennia. In the face of strong evolutionary pressure, naturally occurring single nucleotide polymorphisms (SNPs) can be enriched or purged from human populations. We investigated if any missense variants existed in human TMEM41B and found a SNP (rs78813996) at amino acid position 266 that results in an Ile266Val/Leu substitution with Val being the most prevalent variant. This SNP is nearly absent in European and African populations but is present at ∼3.5% frequency in the Latin American population and at a striking ∼20% frequency in the East and South East Asian population (https://www.ncbi.nlm.nih.gov/snp/rs78813996) (Figure 4 A).
Figure 4.

Naturally Occurring TMEM41B SNPs Negatively Impact Flavivirus Infection but Can Maintain Normal Lipid Distribution in Cells
(A) Table shows the frequency of a SNP (rs78813996) in TMEM41B in several human populations.
(B) WT, TMEM41B KO, and TMEM41B KO HAP1 cells expressing WT or TMEM41B SNP variants infected with YFV 17D. Cells were analyzed by flow cytometry and plotted as a percentage of viral antigen-positive cells.
(C) Infected as in (B), and supernatants were collected and titrated by tissue culture infectious dose (TCID50/mL) assay on Huh-7.5 cells. The two-tailed statistical Student’s t test was used to determine statistical significance and is depicted with an asterisk indicating a p value < 0.05.
(D) Same as (B) infected with ZIKV, DENV-GFP, WNV-GFP, and hPIV-3-GFP.
(E) WT and TMEM41B KO Huh-7.5 cell clones stained with Nile red to visualize lipid droplets.
(F) Cumulative frequency of droplets plotted versus droplet area (μm2) for six independent single-cell clones generated with two independent sgRNAs. Inset shows the mean lipid droplet area (μm2) for the six TMEM41B KO clones compared to WT Huh-7.5 cells.
(G) Mean lipid droplet area (μm2) for WT, TMEM41B KO, and TMEM41B KO Huh-7.5 cells (clone 1-1) expressing the indicated TMEM41B variants. Cells were analyzed by flow cytometry and plotted as a percentage of viral antigen positive cells. Error bars depict SD for n = 3 replicates.
We engineered these amino acid variants into a TMEM41B expression construct with a red fluorescent protein (tagRFP) fused to the N terminus. Western blot confirmed that both the tagRFP-TMEM41B WT and SNP I266L and I266V variants were expressed to similar levels in TMEM41B KO HAP1 cells (Figures S5 A and S5B). We then challenged these cells with YFV and found that at 48 h post infection, the percent infection in cells expressing the I266L and I266V variants was reduced by 56% and 62% respectively, and that these cells produced >10-fold less virus as compared to cells expressing WT TMEM41B (Figures 4B and 4C). Consistent with these results, ZIKV, DENV, and WNV infection rates decreased by 41%–67% in cells expressing the SNP variants, whereas the hPIV-3 control was unaffected (Figure 4D). These results suggest that naturally occurring variants in TMEM41B could potentially affect flavivirus pathogenesis in humans.
Figure S5.

Expression of tagRFP-TMEM41B in Reconstituted TMEM41B KO HAP1 Cells, Related to Figure 4
(A) Western blot to detect RFP-TMEM41B in HAP1 cell lysates. Predicted size is indicated by the location of the label. β-actin is included as a loading control.
(B) Flow cytometry analysis to quantify the percentage of RFP-TMEM41B cells.
TMEM41B SNP Constructs Are Able to Maintain Normal Lipid Distribution in Cells
While culturing TMEM41B KO Huh-7.5 cells, we observed a striking visual accumulation of lipids. To confirm that these were lipid droplets, we stained WT and six independent TMEM41B KO Huh-7.5 clones with Nile red and found that in all clones, lipid droplets are considerably larger as compared to WT cells (Figure 4E). We then quantified this observation by measuring the area of individual droplets from a single imaging plane in at least 23 cells per clone and found that the average size of individual lipid droplets was up to 10 times larger among TMEM41B KO clones relative to WT cells (Figure 4F). Moretti et al. (2018) reported a similar observation in TMEM41B KO H4 neuroglioma cells, and it was later shown that these droplets contain neutral and sterol lipids (Kang et al., 2020).
Having found that two naturally occurring TMEM41B variants, I266L and I266V, have a reduced capacity to support flavivirus infection, we next tested if these variants were able to rescue the enlarged lipid droplet phenotype. In addition to the two human variants, we also included the Aedes aegypti TMEM41B ortholog. As shown in Figure 4G, we found that expression of WT human and mosquito TMEM41B as well as the two human TMEM41B variants reduced the mean lipid droplets size to baseline. These results suggest that while these two SNPs negatively impact flavivirus infection, they appear to be fully functional in their ability to maintain normal lipid distribution as measured in this assay.
TMEM41B Co-localizes with Flavivirus NS4A and NS4B Proteins
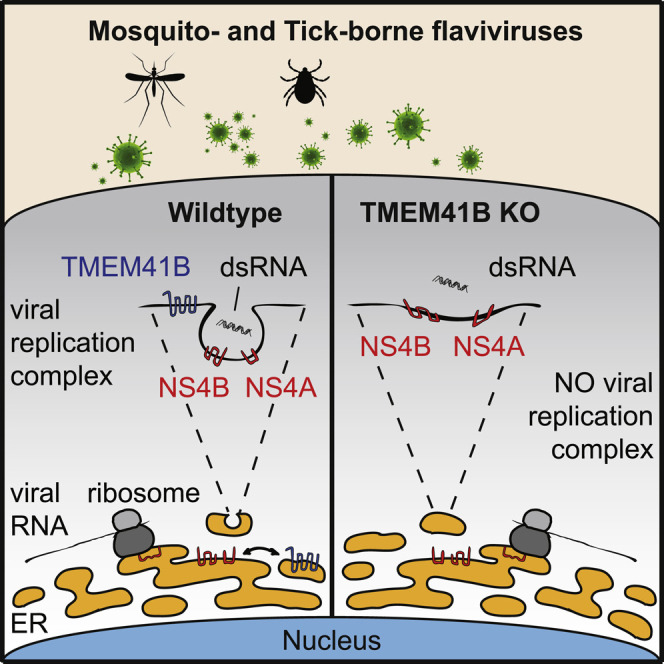
Next, we assayed whether the cellular localization of TMEM41B changed upon virus infection. We utilized the tagRFP-TMEM41B expression construct to monitor TMEM41B cellular localization prior to and during flavivirus infection. Previous imaging and proteomics experiments have shown that TMEM41B localizes to ER membranes (Moretti et al., 2018; Morita et al., 2019). Our observations in TMEM41B KO HAP1 cells expressing tagRFP-TMEM41B are consistent with ER localization. Interestingly, upon infection with either YFV or ZIKV, we observed a striking re-localization of tagRFP-TMEM41B from a diffuse reticular-like pattern to a large cytosolic aggregate that co-localized with viral nonstructural proteins, NS4A (ZIKV) and NS4B (YFV) (Figure 5 A). This is consistent with a previous report that found ZIKV NS4B interacts with TMEM41B (Scaturro et al., 2018). NS4A and NS4B are multipass transmembrane proteins known to induce membrane curvature, which, together with additional nonstructural viral and likely host proteins, form the viral RNA replication complex.
Figure 5.

TMEM41B Co-localizes with Flavivirus NS4A and NS4B Proteins
(A) TMEM41B KO HAP1 cells expressing RFP-TMEM41B visualized in uninfected cells (mock) and YFV- and ZIKV-infected cells 24 h post infection. Anti-NS4A (ZIKV) and anti-NS4B (YFV) antibodies detect viral antigens. Yellow-orange color in the merged column shows the co-localization of TMEM41B with viral antigens. DAPI stains cell nuclei.
(B) Western blot shows that an anti-RFP antibody which recognizes RFP-TMEM41B (but not an IgG antibody control) co-immunoprecipitates ZIKV NS4A in HAP1 cells.
(C) Same as (B) but with YFV infection and visualized with an antibody that detects YFV NS4B.
WCL, whole cell lysate. Of note, the heavy chain of the capture antibodies is detected by the secondary antibody protein A-HRP as indicated ~52 kDa. See also Figure S5.
To further confirm that TMEM41B co-localizes with NS4A and NS4B, we performed co-immunoprecipitation experiments using an anti-RFP antibody to immunoprecipitate tagRFP-TMEM41B. We found that NS4A in lysates from ZIKV-infected cells and NS4B in lysates from YFV-infected cells co-immunoprecipitated with tagRFP-TMEM41B, whereas NS4A or NS4B was not detected in immunoprecipitates from lysates from uninfected cells or infected cell lysates immunoprecipitated with an immunoglobulin G (IgG) isotype control antibody (Figures 5B and 5C). We probed these blots for β-actin to confirm that similar amounts of lysate were used as input, and we probed for RFP to confirm that equal amounts of tagRFP-TMEM41B were immunoprecipitated in both uninfected and infected cells (Figures S6 A and S6B). All together, these results suggest that TMEM41B is recruited to sites of viral RNA replication either indirectly or potentially by direct interaction with NS4A and/or NS4B.
Figure S6.

Co-immunoprecipitation of TMEM41B and Viral NS4A and NS4B Proteins, Related to Figure 5
(A) Shown are western blots from Figure 5 from ZIKV-infected lysates. Left panel was probed with β-actin to show that similar amounts of whole cell lysate were used as input. Right panel was probed with anti-RFP to show that similar amounts of RFP-TMEM41B were immunoprecipitated from mock and infected lysates but not when using an IgG control antibody.
(B) Same as (A) with YFV-infected cells. Of note, the β-actin signal detected in the anti-RFP IgG immunopreciptated fraction is likely an unspecific interaction with the RFP or with TMEM41B as it has been shown that the cytoskeleton (e.g., actins) can interact with organelles (e.g., ER).
Adaptive Mutations in NS4B Restore Flavivirus Infectivity in TMEM41B KO Cells
Viruses exist as diverse populations of variants and can readily adapt to new environments. These genetic adaptations can highlight viral proteins or viral RNA structures that are under selective pressure in a given context. Throughout our experiments, we observed rare YFV and ZIKV antigen-positive cells after infecting clonal TMEM41B KO populations. To gain potential insight into which stage of the virus replication cycle was affected by the absence of TMEM41B, we sought to capitalize on these rare infection events to select for adaptive mutations within the viral genome. We did this by serial passaging of ZIKV-infected cells and culture supernatants to select viral variants capable of replicating in TMEM41B-deficient cells. We performed virus supernatant passaging experiments in human TMEM41B KO A549 and HAP1 cells, and in parallel, we passaged ZIKV-infected TMEM41B KO mosquito Aag2 cells. Serial passages were performed in multiple independent lineages for each cell type, and we similarly passaged virus in WT cells to control for cell culture adaptive mutations that arise, which are unrelated to TMEM41B KO. Figure 6 A shows a graphical outline of these experiments.
Figure 6.

NS4A and NS4B Mutations Bypass TMEM41B Deficiency
(A) Graphical schematic of virus adaptation experiment performed in Aag2 (top) and A549 and HAP1 cells (bottom).
(B) Summary of ZIKV adaptive mutations. Top, graphical representation of the ZIKV genome with mature (proteolytically processed) viral proteins shown as arrows and nucleotide positions listed below. NS4A and NS4B are shown in light blue. Bottom, table missense mutations identified by passaging virus in TMEM41B KO cells. Only mutations that were present in at least three independent passages (irrespective of cell type) and that did not appear in virus populations passaged in WT cells are shown. Boxes show the number of replicates a mutation was identified over the total number of replicates. Of six initial replicates for the A549 TMEM41B KO clones, only five were maintained for 20 passages.
(C) I42T and F91Y mutations were independently engineered in a ZIKV infectious clone to generate virus stocks to infect WT (left) or TMEM41B KO (right) Aag2 cells.
(D) Same as (C) in WT and TMEM41B KO A549 cells.
(E and F) WT and TMEM41B KO A549 cells infected with YFV 17D and Asibi chimeric viruses. (E) 17D backbone with Asibi NS4A and NS4B proteins; (F) Asibi backbone with 17D NS4A and NS4B proteins. Cells were analyzed by flow cytometry and plotted as a percentage of viral antigen positive cells. Error bars depict SD for n = 3 replicates.
After 15–20 rounds of passaging, we deep-sequenced viral cDNA from cell supernatants to identify viral variants that were enriched in virus populations passaged in TMEM41B KO cells relative to virus populations passaged in WT cells. To qualify as a potential adaptive mutation, we applied a stringency filter that required mutations to be present in at least three independent passages (regardless of cell type) and not present in virus that had been passaged in WT cells. Remarkably, all mutations that met these criteria were located in NS4A and NS4B. Further, NS4B mutations at positions Ile42 and Ser236 were selected in both mosquito and mammalian cells (Figure 6B).
To confirm that these mutations we selected enable ZIKV to infect TMEM41B-deficient cells, we chose to introduce two mutations in NS4B, Ile42Thr and Phe91Tyr, into the infectious cDNA clone of the Cambodian ZIKV strain FSS13025 (Shan et al., 2016) and generated viral stocks. Figures 6C and 6D demonstrate that these individual point mutations in NS4B render ZIKV infectious in both mosquito (Aag2) and mammalian (A549) TMEM41B KO cells.
Wild and Vaccine Strain Chimeras Overcome TMEM41B Deficiency
As shown in Figures 2E and S3A, we observed that Asibi and 17D strains of YFV were differentially sensitive to TMEM41B deficiency in A549 cells. The live-attenuated YFV 17D vaccine strain differs from the parental Asibi strain by only 31 amino acids, three of which are located within NS4A (V107I) and NS4B (M95I and Y232H) (dos Santos et al., 1995). Given the strain-specific differences we observed in TMEM41B KO A549 cells, together with the observation that single amino acid changes in NS4B could allow flavivirus replication in TMEM41B KO cells, we next generated 17D:Asibi chimeric viruses by swapping NS4A and NS4B individually or by swapping the region containing both NS4A and NS4B, which includes the 2K protein. Remarkably, we found that in the 17D viral background, NS4B from the Asibi strain significantly increased infection in TMEM41B KO A549 cells, whereas the NS4B from the 17D strain slightly decreased the infectivity of the Asibi strain in TMEM41B KO cells. In contrast, the NS4A changes had no impact on infection. Importantly, parallel infections in WT A549 cells show that the results observed in the KO cells are not due to an overall increase or decrease in viral replication but rather were likely due to a differential requirement for TMEM41B in this cellular context (Figures 6E and 6F).
TMEM41B KO Cells Mount an Exaggerated Innate Immune Response to Flavivirus Inoculum
While studying flavivirus infection in TMEM41B KO cells, we observed an apparent cytotoxicity with high virus inocula (MOI > 5 plaque-forming units [PFU]/cell) that was not seen with similar inocula to WT cells. We hypothesized that this cytotoxicity may have resulted from an exaggerated innate immune response due to increased exposure of viral double-stranded RNA (dsRNA) in TMEM41B-deficient cells. This hypothesis is based on recent evidence supporting a role for TMEM41B in mobilizing lipids and inducing membrane curvature (Moretti et al., 2018; Morita et al., 2019) and our own and other data, which indicate that TMEM41B is likely recruited to flavivirus replication complexes (Scaturro et al., 2018). Flavivirus replication complexes are sites at which viral RNA replication machinery assembles within invaginations into the ER lumen utilizing host curvature-stabilizing proteins (Aktepe and Mackenzie, 2018; Neufeldt et al., 2018; Rajah et al., 2020). Within these structures, dsRNA intermediates that form during viral RNA replication are protected from host innate immune sensors that survey the cytosol for pathogen-associated molecular patterns (PAMPs). In the absence of TMEM41B, viral RNA replication may initiate in aberrant replication complexes that are exposed to the cytosol.
This hypothesis assumes that TMEM41B is required for viral RNA replication, not virus entry. To test this, we transfected WT and TMEM41B KO HAP1 cells with a YFV Renilla reniformis luciferase (Rluc) reporter subgenomic replicon (Jones et al., 2005) to bypass the entry step, and we measured Rluc over a time course. Indeed, results in Figure S7 A confirm that the transfected RNA is translated, but RNA replication is severely impaired. While this does not entirely rule out a role for TMEM41B in virus entry, it nevertheless provides concrete evidence that TMEM41B is required for virus RNA replication.
Figure S7.

TMEM41B’s Role in the Flavivirus Life Cycle, Related to Figure 7
(A) WT and polymerase dead (dDD) YFV Renilla luciferase (Rluc)-reporter replicon RNAs transfected into WT and TMEM41B KO HAP1 cells with Rluc signal plotted as relative light units (RLU) plotted across a time course of 36 h. Rluc signal increases up to 4 h as the transfected RNA is translated. Rluc signal declines between 4 and 12 h as the transfected RNA decays (dDD) or is removed from the translating pool to serve as template for viral RNA replication. Rluc signal increases in WT cells transfected with WT replicon RNA, whereas the signal is dramatically reduced in TMEM41B-deficient cells.
(B and C) WT and TMEM41B KO A549 cells infected with (B) YFV 17D, MOI = 0.025 PFU/cell for 48 h and (C) ZIKV, MOI = 0.05 PFU/cell for 48 h in the presence or absence of innate immune inhibitors. Cells were treated with 500 nM TBK1 and pan-JAK inhibitors or vehicle control (0.05% DMSO) 24 h prior to infection and throughout the experiment. The percentage of infected cells increases in WT cells in the presence of TBK1/JAK inhibitors, but not in TMEM41B KO cells. sg #1 and sg #2 indicates KO clones generated using TMEM41B sgRNA1 and sgRNA2, respectively. Two KO clones were used in each experiment as indicated in the figure. WT and KO clones were assayed in triplicate. Individual dots represent the average of the triplicates and error bars show the standard deviation of results from the average of the KO clones.
We next tested if the cytotoxicity we observed in TMEM41B KO HAP1 cells upon infection with a virus inoculum sufficient to infect most cells during the initial infection was due to innate immune activation. For this, we inoculated WT and TMEM41B KO HAP1 cells with YFV 17D virus (MOI = 0.4 PFU/cell), then quantified viral RNA and mRNA encoding 2′-5′-oligoadenylate synthetase 1 (OAS1) by qPCR at 24 h post infection. OAS1 is a classic interferon (IFN)-stimulated gene (ISG) induced by type I and III IFNs. As shown in Figure 7 A, we found that YFV 17D RNA levels were 100-fold lower in TMEM41B KO cells compared to WT cells, consistent with an inability to replicate; however, OAS1 was highly induced in the KO cells but not induced in WT cells (Figure 7B, left). To determine if OAS1 induction was due to the virus itself or functional viral RNA, we inoculated both WT and TMEM41B KO cells with viral particles inactivated by UV light. UV-inactivated virus failed to induce OAS1 expression in both cell lines suggesting that viral protein production and perhaps initiation of viral RNA replication was required to induce innate immunity (Figure 7B, middle). To control for the possibility that TMEM41B KO cells are hypersensitive to PAMPs in general, we infected both WT and TMEM41B KO cells with a recombinant influenza A virus (IAV) lacking a critical innate immune antagonist protein, NS1 (IAV ΔNS1). This virus is well known for its defect in antagonizing IFN production and ISG expression (García-Sastre et al., 1998; Wang et al., 2000). We found that both WT and TMEM41B KO responded to IAV ΔNS1 as expected, with WT cells expressing even slightly higher amounts of OAS1 mRNA than TMEM41B KO cells (Figure 7B, right).
Figure 7.

TMEM41B KO Cells Mount an Exaggerated Innate Immune Response to Flavivirus Inoculum
(A) Viral RNA quantified by qRT-PCR from WT and TMEM41B KO HAP1 cells infected with YFV 17D.
(B) OAS1 mRNA quantified by qRT-PCR from WT and TMEM41B KO HAP1 cells infected with YFV 17D (with and without UV-inactivation) or with IAV (ΔNS1).
(C) WT and TMEM41B KO HAP1 cells untreated (mock), infected with YFV 17D, or treated with staurosporine (STS) were assayed to detect apoptotic cells via Annexin-V staining. Cells were analyzed by flow cytometry and plotted as a percentage of viral antigen positive cells. Error bars depict SD of n = 3 replicates.
(D) Model for the role of TMEM41B in the flavivirus life cycle. The model is described in the main text.
Upon detection of viral PAMPs, cells secrete IFN to protect neighboring cells from infection then subsequently initiate apoptosis (Drappier and Michiels, 2015; Gusho et al., 2020; Schwartz and Conn, 2019). To assay if apoptosis explained the cytotoxicity we observed in TMEM41B KO cells after virus inoculation, we infected WT and TMEM41B KO cells with YFV 17D (MOI = 4 PFU/cell) and quantified the number of apoptotic cells at 24 h post inoculation. As shown in Figure 7C, we observed an approximate 3-fold increase in the percentage of apoptotic cells in TMEM41B KO cells compared to WT cells. Importantly, at this time point, there was almost no increase in the percentage of apoptotic cells in the infected WT cell population as compared to mock-infected cultures. Further, the positive control, staurosporine (STS), induced apoptosis in approximately 70% of the cells, with no notable difference between WT and TMEM41B KO cells (Figure 7C).
We next sought to test if the enhanced innate immune activation in response to flavivirus infection in TMEM41B-deficient cells could explain the block in virus infection. For this, we utilized inhibitors targeting TBK1 and JAK proteins to block IFN production and signaling, respectively. We found that while the presence of these inhibitors increased infection frequency of YFV-17D and ZIKV in WT cells, they were unable to rescue infection in TMEM41B KO cells (Figures S7B and S7C). Together, these data suggest that sensing dsRNA and innate immune activation is a consequence of TMEM41B-deficiency, but innate immune activation does not fully explain the block in flavivirus replication.
Discussion
Genome-wide CRISPR-Cas9 gene disruption screens are a powerful method of discovery. We applied this method to discover cellular host factors that flaviviruses require to replicate in human cells. As a testament to the robustness of this approach, our results along with CRISPR-Cas9 screens performed by several other research groups identified shared genes and biological pathways. Screens, however, are often only the first step toward understanding new virus-host biology.
In our follow-up studies, we found that all flaviviruses we tested, including both mosquito-borne and tick-borne flaviviruses, required TMEM41B for infection and replication. Further, we found that TMEM41B was required in the mosquito vector as well, supporting a conserved role for TMEM41B in flavivirus infection in distantly related species. Given that TMEM41B is required for flavivirus infection and that TMEM41B is also required for autophagy, the question arose: is autophagy required for flavivirus infection? Our results suggest that under the conditions we tested, the full autophagy pathway is not essential for flavivirus infection. This led us to hypothesize that flaviviruses may hijack TMEM41B and VMP1 for their ability to remodel host cell membranes—a feature required both for autophagy and for the formation of viral replication complexes.
Although every flavivirus we tested required TMEM41B in most cell types, there were several virus-cell combinations we tested where TMEM41B was either not required or played a less essential role in virus infection. This conundrum may be explained by cell-type-specific differences in the abundance of related cellular host proteins such as VMP1 and TMEM41A, which we found can partially support flavivirus infection in the absence of TMEM41B. These findings suggest that TMEM41B may be an important cellular determinant that defines flavivirus tissue tropism and pathogenesis. Along these lines, it is notable that a SNP in TMEM41B present in approximately 20% of East and South East Asian and 3.5% of Latin American populations has a reduced capacity to support flavivirus infection. While it is impossible to know for sure what drove selection of this SNP in these populations, it is conceivable that a pathogenic flavivirus may have been involved.
While variations in the host environment can influence virus tropism, so too can variations in the virus. Our studies uncovered that two highly related YFVs, the virulent Asibi strain and the live attenuated 17D vaccine strain, displayed a differential ability to infect JEG3 and A549 cells lacking TMEM41B. The fact that the YFV Asibi and 17D strains differ by only 31 amino acids provided us with an opportunity to investigate the flavivirus TMEM41B dependency down to the level of an individual viral protein. Our results indicate that amino acid differences in NS4A and NS4B between these two strains may be responsible for their differential TMEM41B requirement. Future studies are needed to better understand how single amino acid changes in NS4B (and possibly NS4A) bypass the strong TMEM41B requirement during flavivirus infection. Together these results highlight plasticity even among “essential” host factors and demonstrate that loss of a critical host factor can be overcome by relatively few genetic changes in the viral genome. This contrasts the notion that resistance barriers may be higher when targeting host factors with antiviral therapeutics as opposed to targeting the virus. When dealing with highly diverse viral populations, multi-target drug cocktails are still likely to be important regardless of whether therapies target host or viral factors.
Our final observation where we demonstrate that TMEM41B cells initiate a strong innate immune response when exposed to a large number of virus particles provides mechanistic insights into the role of TMEM41B in the flavivirus replication cycle. Based on our collective results we propose a model, shown in Figure 7D, where upon flavivirus infection and translation of the viral polyprotein, TMEM41B is recruited to sites on the ER membrane together with NS4A and NS4B where replication complexes are forming (Neufeldt et al., 2018; Paul and Bartenschlager, 2015). This recruitment may be through direct protein:protein interaction or through passive diffusion, where, by mobilizing neutral and sterol lipids, TMEM41B helps lower the local free energy imposed by NS4A- and NS4B-induced membrane curvature. We propose that TMEM41B’s role in facilitating membrane curvature is dependent upon a functional interaction between TMEM41B and VMP1, the latter of which is highly mobile and associates with cell organelles, vesicles, and lipid droplets (Tábara and Escalante, 2016; Zhao et al., 2017). In the absence of TMEM41B, flavivirus NS4A and NS4B proteins (which may or may not be fully processed) assemble and viral RNA replication initiates, but because replication complexes are improperly formed, dsRNA intermediates are exposed to pattern recognition receptors (PRRs). At this point, the infectious replication cycle is aborted because the RNA genome cannot be efficiently copied and the low level of replication that initiates activates innate immune pathways.
Lastly, we and others recently identified TMEM41B as a top scoring hit in genome-wide CRISPR-Cas9 loss-of-function screens designed to identify coronavirus host dependency factors (Baggen et al., 2020; Wang et al., 2020 and accompanying manuscript: Schneider et al., 2020). It will be fascinating to learn if TMEM41B is also required to assemble coronavirus replication complexes. In conclusion, TMEM41B may be a candidate target to inhibit the replication of a broad range of emerging and re-emerging flavivirus and coronavirus pathogens.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| rabbit polyclonal anti-TMEM41A | Proteintech Group, Inc. | Cat.#20768-1-AP; RRID: AB_10693679 |
| rabbit polyclonal anti-TMEM41B | Abnova Corp. | Cat.#PAB20785; RRID: AB_10965049 |
| rabbit monoclonal anti-VMP1 | Cell Signaling Technology, Inc. | Cat.#12929; RRID: AB_2714018 |
| rabbit monoclonal anti-ATG5 | Abcam | Cat.#ab108327; RRID: AB_2650499 |
| rabbit monoclonal anti-ATG7 | Abcam | Cat.#ab52472; RRID: AB_867756 |
| rabbit monoclonal anti-LC3B | Abcam | Cat.#ab192890; RRID: AB_2827794 |
| rabbit polyclonal anti-tagRFP | Evrogen JSC | Cat.#AB233; RRID: AB_2571743 |
| mouse monoclonal anti-β-actin conjugated to HRP | Sigma-Aldrich | Cat.#A3854; RRID: AB_262011 |
| mouse monoclonal anti-YFV | Santa Cruz Biotechnology, Inc. | Cat.#sc-58083; RRID: AB_630447 |
| mouse monoclonal anti-flavivirus group antigen | MilliporeSigma | Cat.#MAB10216; RRID: AB_827205 |
| pan-flavivirus hyperimmune mouse acites fluid (HMAF) | Thomas G. Ksiazek, CDC |
N/A |
| rabbit polyclonal anti-YFV NS4B | GeneTex, Inc. | Cat.#GTX134030 |
| rabbit polyclonal anti-ZIKV NS4A | GeneTex, Inc. | Cat.#GTX133704 |
| rabbit polyclonal anti-SARS-CoV-2 nucleoprotein | GeneTex, Inc. | Cat.#GTX135357; RRID: AB_2868464 |
| mouse monoclonal anti-IAV nucleoprotein | MilliporeSigma | Cat.#MAB8251; RRID: AB_95293 |
| bovine polyclonal anti-BVDV antibody B224 | Kenny V. Brock, Auburn University | N/A |
| normal rabbit polyclonal IgG | Cell Signaling Technology, Inc. | Cat.#2729; RRID: AB_1031062 |
| goat polyclonal anti-rabbit IgG conjugated to HRP | ThermoFisher Scientific, Inc. | Cat.#31462; RRID: AB_228338 |
| goat polyclonal anti-mouse IgG conjugated to HRP | Jackson ImmunoResearch Labs, Inc. | Cat.#115-035-146; RRID: AB_2307392 |
| goat anti-bovine IgG conjugated to FITC | ThermoFisher Scientific, Inc. | Cat.#A18752; RRID: AB_2535529 |
| goat polyclonal anti-mouse IgG conjugated to AF647 | ThermoFisher Scientific, Inc. | Cat.#A-21235; RRID: AB_2535804 |
| goat polyclonal anti-mouse IgG conjugated to AF488 | ThermoFisher Scientific, Inc. | Cat.#A-11001; RRID: AB_2534069 |
| goat polyclonal anti-rabbit IgG conjugated to AF488 | ThermoFisher Scientific, Inc. | Cat.#A-11008; RRID: AB_143165 |
| Protein A conjugated to HRP | ThermoFisher Scientific, Inc. | Cat.#10-1023 |
| Bacterial and Virus Strains | ||
| Endura™ Competent Cells | Lucigen | Cat.#60242 |
| Subcloning Efficiency™ DH5α Competent Cells | ThermoFisher Scientific, Inc. | Cat.#18265017 |
| DENV-2-GFP (based on strain: 16681) | Laboratory of Charles M. Rice | Schoggins et al., 2012 |
| WNV-GFP (based on strain: WNV-TX02) | Laboratory of Ilya V. Frolov | McGee et al., 2010 |
| HCV-RFP (based on chimeric strain: J6/JFH1) | Laboratory of Charles M. Rice | Liu et al., 2015 |
| BVDV (strain: NADL) GenBank: AJ133738.1 |
Laboratory of Charles M. Rice | Mendez et al., 1998 |
| CHIKV-GFP (based on strain: LR2006 OPY1) | Laboratory of Stephen Higgs | Tsetsarkin et al., 2006 |
| hPIV3-GFP (based on strain: JS) | Laboratory of Peter L. Collins | Zhang et al., 2005 |
| BUNV-GFP (based on strain: bunyamwera) | Laboratory of Richard M. Elliott | Shi et al., 2010 |
| VacV-GFP (based on strain: Western Reserve) | Laboratory of Paula Traktman | Schoggins et al., 2014 |
| HSV-1 US11-GFP (based on strain: Patton) | Laboratory of Ian J. Mohr | Benboudjema et al., 2003 |
| AdV-GFP (based on strain: AdV5) | Laboratory of Patrick Hearing | Evans and Hearing, 2003 |
| VSV-GFP (based on strain: Indiana) | Laboratory of John K. Rose | Dalton and Rose, 2001 |
| LCMV-GFP (based on strain: Armstrong) | Laboratory of Juan Carlos de la Torre | Ngo et al., 2015a |
| IAV (strain: A/WSN/33) | Laboratory of Peter Palese | N/A |
| IAV ΔNS1 (based on strain: A/PR/8/34) | Laboratory of Adolfo García-Sastre |
García-Sastre et al., 1998 |
| CVB-3-GFP (based on strain: Nancy) | Laboratory of J. Lindsay Whitton | Feuer et al., 2002 |
| ZIKV (strain: PRVABC59) GenBank: KU501215.1 |
CDC, Ft. Collins | N/A |
| ZIKV (strain: FSS13025) Genebank: KU955593.1 |
Laboratory of Pei-Yong Shi | Shan et al., 2016 |
| YFV (strain: 17D) GenBank: X03700.1 |
Laboratory of Charles M. Rice | N/A |
| YFV (strain: Asibi) GenBank: AY640589.1 |
Laboratory of Charles M. Rice | N/A |
| SARS-CoV-2 (strain: WA1/2020) GenBank: MT246667.1 |
BEI Resources | Cat.#NR-52281 |
| POWV (strain: Byers) GenBank: L06436.1 |
CDC Viral Special Pathogens Branch | VSPB 812027 |
| TBEV (strain: Hypr) GenBank: U39292.1 |
CDC Viral Special Pathogens Branch | VSPB 812222 |
| TBEV (strain: Sofjin) GenBank: JX498940.1 |
CDC Viral Special Pathogens Branch | VSPB 812223 |
| OHFV (strain: Bogoluvovska) GenBank: AY193805.1 |
CDC Viral Special Pathogens Branch | VSPB 812005 |
| KFDV (strain: P9605) GenBank: JF416958.1 |
CDC Viral Special Pathogens Branch | VSPB 811996 |
| AHFV (strain: 200300001) GenBank: JF416954.1 |
CDC Viral Special Pathogens Branch | VSPB 200300001 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Puromycin | MilliporeSigma | Cat.#P8833; CAS: 58-58-2 |
| Blasticidine S | MilliporeSigma | Cat.#203350; CAS: 2079-00-7 |
| Polybrene Infection / Transfection Reagent | MilliporeSigma | Cat.#TR-1003-G |
| doxycycline hyclate | MilliporeSigma | Cat.#D9891; CAS: 24390-14-5 |
| Carbenicillin disodium salt | MilliporeSigma | Cat.#C1389; CAS: 4800-94-6 |
| Polyethylenimine, branched | MilliporeSigma | Cat.#408727; CAS: 9002-98-6 |
| Herculase II Fusion enzyme | Agilent | Cat.#600679 |
| chloroquine diphosphate salt | MilliporeSigma | Cat.#C6628; CAS: 50-63-5 |
| Torin 1 | Selleck Chemicals LLC | Cat.#S2827; CAS: 1222998-36-8 |
| InSolution™ JAK Inhibitor I | MilliporeSigma | Cat.#420097; CAS: 457081-03-7 |
| TBK1 inhibitor BX795 | InvivoGen | Cat.#tlrl-bx7; CAS: 702675-74-9 |
| Hoechst 33342 solution | ThermoFisher Scientific, Inc. | Cat.#62249; CAS: 23491-52-3 |
| NucBlue™ Reagent | ThermoFisher Scientific, Inc. | Cat.#R37605; CAS: 23491-52-3 |
| cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail | MilliporeSigma | Cat.#11836170001 |
| Phenylmethylsulfonyl fluoride (PMFS) | MilliporeSigma | Cat.#10837091001; CAS: 329-98-6 |
| Staurosporine | MilliporeSigma | Cat.#S4400; CAS: 62996-74-1 |
| Critical Commercial Assays | ||
| Nile Red Staining Kit | Abcam | Cat.#ab228553 |
| Pierce™ BCA Protein Assay Kit | ThermoFisher Scientific, Inc. | Cat.#23227 |
| SuperSignal™ West Pico PLUS Chemiluminescent Substrate | ThermoFisher Scientific, Inc. | Cat.#34577 |
| SuperSignal™ West Femto Maximum Sensitivity Substrate | ThermoFisher Scientific, Inc. | Cat.#34095 |
| Dynabeads™ Protein A for Immunoprecipitation | ThermoFisher Scientific, Inc. | Cat.#10001D |
| mMESSAGE mMACHINE™ SP6 Transcription Kit | ThermoFisher Scientific, Inc. | Cat.#AM1340 |
| Pacific Blue Annexin V Apoptosis Detection Kit with 7-AAD | BioLegend, Inc. | Cat.#640926 |
| Applied Biosystems™ PowerUp™ SYBR™ Green Master Mix | ThermoFisher Scientific, Inc. | Cat.#A25742 |
| Maxima H Minus First Strand cDNA Synthesis Kit | ThermoFisher Scientific, Inc. | Cat.#K1681 |
| Deposited Data | ||
| sgRNA sequencing data | This paper | GEO: GSE162040 |
| Experimental Models: Cell Lines | ||
| Human: HAP1 (derived from leukemic cells) | Laboratory of Thijn R. Brummelkamp | RRID: CVCL_Y019 |
| Human: A549 (lung epithelial) | ATCC | Cat.#CCL-185; RRID: CVCL_0023 |
| Human: JEG3 (placenta epithelial) | ATCC | Cat.#HTB-36; RRID: CVCL_0363 |
| Human: Lenti-X™ 293T (embryonic kidney epithelial) | Takara Bio Inc. | Cat.#632180; RRID: CVCL_4401 |
| Human: HEK293T/17 (embryonic kidney epithelial) | ATCC | Cat.#CRL-11268; RRID: CVCL_1926 |
| Human: Huh-7.5 (hepatocyte) | Laboratory of Charles M. Rice | RRID: CVCL_7927 |
| Human: HeLa (cervix epithelial) | ATCC | Cat.#CCL-2; RRID: CVCL_0030 |
| Monkey: VeroE6 (kidney epithelial) | ATCC | Cat.#CRL-1586 RRID: CVCL_0574 |
| Monkey: BSC-40 (kidney fibroblast) | ATCC | Cat.#CRL-2761 RRID: CVCL_3656 |
| Hamster: BHK-21 (kidney fibroblast) | ATCC | Cat.#CCL-10 RRID: CVCL_1914 |
| Bovine: MDBK (kidney epithelial) | ATCC | Cat.#CCL-22; RRID: CVCL_0421 |
| Mosquito: C6/36 (derived from neonate larva of Aedes albopictus) | ATCC | Cat.#CRL-1660 RRID: CVCL_Z230 |
| Mosquito: Aag2 (derived from neonate larva of Aedes aegypti) | Laboratory of Maria-Carla Saleh | RRID: CVCL_Z617 |
| Oligonucleotides | ||
| See Table S1 | N/A | N/A |
| Recombinant DNA | ||
| Plasmid: LentiCas9-Blast | Sanjana et al., 2014 | Addgene: Cat.#52962; RRID: Addgene_52962 |
| Plasmid: LentiGuide-Puro | Sanjana et al., 2014 | Addgene: Cat.#52963; RRID: Addgene_52963 |
| Plasmid: LentiCRISPRv2 | Sanjana et al., 2014 | Addgene: Cat.#52961; RRID: Addgene_52961 |
| Plasmid: pLX304 | Yang et al., 2011 | Addgene: Cat.#25890; RRID: Addgene_25890 |
| Plasmid: pDCC6 | Gokcezade et al., 2014 | Addgene: Cat.#59985; RRID: Addgene_59985 |
| Plasmid: pKRG3-mU6-PUb-hSpCas9-pAc | Rozen-Gagnon et al., 2020 | Addgene: Cat.#162163; RRID: Addgene_162163 |
| Plasmid: pTRIPZ | Horizon Discovery Group | Cat.#RHS4743 |
| Plasmid: pMD2.G | Laboratory of Didier Trono | Addgene: Cat.#12259; RRID: Addgene_12259 |
| Plasmid: psPAX2 | Laboratory of Didier Trono | Addgene: Cat.#12260; RRID: Addgene_12260 |
| Software and Algorithms | ||
| Prism | GraphPad Software, Inc. | https://www.graphpad.com/scientific-software/prism/ |
| FlowJo | FlowJo LLC | https://www.flowjo.com |
| Geneious Prime | Biomatters, Inc. | https://www.geneious.com |
| Phyre2 | Kelley et al., 2015 | http://www.sbg.bio.ic.ac.uk/phyre2 |
| Fiji | Schindelin et al., 2012 | https://imagej.net/Fiji |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Charles M. Rice (ricec@rockefeller.edu).
Materials Availability
Plasmids and cell lines generated in this study are available upon request.
Data and Code Availability
The accession number for the sgRNA sequencing data reported in this paper is GEO: GSE162040. Original data for all virus infection experiments in the paper are available upon request.
Experimental Model and Subject Details
Cell Culture
HAP1 (WT and B3GALT6 KO) (human; sex: male, chronic myeloid leukemia-derived) cells were obtained from Thijn R. Brummelkamp (Netherlands Cancer Institute) and cultured in Iscove’s Modified Dulbecco’s Medium (IMDM, GIBCO) supplemented to contain 10% fetal bovine serum (FBS, Hyclone, GE Healthcare) and 1% non-essential amino acids (NEAA, ThermoFisher Scientific). A549 cells (human; sex: male, lung epithelial), JEG3 cells (human; sex: female, placenta epithelial), HEK293 derivatives HEK293T/17 and Lenti-X 293T cells (human; sex: female, kidney epithelial) obtained from Takara (cat. 632180), Huh-7.5 heptoma cells (human; sex: male, liver epithelial) (Blight et al., 2002), HeLa cells (human; sex: female, cervix epithelial), VeroE6 cells (Chlorocebus sabaeus; sex: female, kidney epithelial), BSC-40 cells (Chlorocebus aethiops; sex: unspecified, kidney epithelial), BHK-21 cells (Mesocricetus auratus; sex: unspecified, kidney fibroblasts) and MDBK cells (bovine; sex: male, kidney epithelial) were cultured in Dulbecco’s Modified Eagle Medium (DMEM, GIBCO) supplemented to contain 10% FBS (or 10% horse serum [ThermoFisher Scientific] in case of MDBK cells) and 1% NEAA. All mammalian cell lines were obtained from the ATCC unless stated otherwise, cultured at 37°C and incubated with 5% CO2. Mosquito C6/36 (Ae. albopictus, sex: unspecified, neonate larva) and Aag2 (Ae. aegypti, sex: unspecified, neonate larva) cells were obtained from ATCC and Dr. Maria-Carla Saleh, respectively. Cells were cultured in Leibovitz’s L-15 Medium, without phenol red (ThermoFisher Scientific), supplemented to contain 10% FBS, 1% NEAA, and ∼0.3 g/L tryptose phosphate broth (Sigma-Aldrich) at 28°C in the absence of CO2. All cell lines tested negative for contamination with mycoplasma and with the exception for Huh-7.5 cells have not been further authenticated.
Method Details
Genome-wide CRISPR-Cas9 Knockout Screen
HAP1 B3GALT6 KO-Cas9 cells were generated by lentiviral transduction of lentiCas9-Blast, a gift from Feng Zhang (Addgene: #52962; http://n2t.net/addgene:52962; RRID: Addgene_52962), followed by selection and expansion in the presence of 1.5 μg/mL blasticidin. The human GeCKO library (A and B) (Shalem et al., 2014) was a gift from Feng Zhang and obtained through Addgene (cat. #1000000049). The plasmid was expanded and lentivirus was produced as previously described (Shalem et al., 2014). Briefly, the library was electroporated into 100 μL of electrocompetent cells and recovered in 10 mL recovery media according to manufacturer’s protocol (Lucigen: 60242) using a GenePulser II (BioRad). Electroporated cells were plated on 20 × 10 cm carbenicillin selective plates and grown overnight at 32°C. Colonies were scraped from all plates in LB media, combined, and centrifuged to obtain a bacterial cell pellet that was used as input material for plasmid maxiprep (QIAGEN: 12662). To produce lentivirus, each A and B half-library was pooled 1:1, then transiently transfected into HEK293T/17 cells in 20 × 15 cm2 dishes. Transfection conditions were 5.4 μg library pool, 3.6 μg psPAX2, 1.8 μg pMD2.G and 21.6 μg branched polyethylenimine (PEI) transfection reagent (MilliporeSigma: 408727) per plate. Media was changed 16 h post-transfection. Lentiviral supernatants were collected, passed through a 0.45 μm Stericup PVDF filter (Millipore), concentrated by ultracentrifugation at 100,000 x g for 2 h at 4°C and stored at −80°C. To deliver the GeCKO (A and B) sgRNA library to cells, 2.03 × 108 HAP1 B3GALT6 KO-Cas9 cells were transduced at a MOI = 0.4 IU/cell to achieve ∼1,600-fold overrepresentation of each sgRNA. Two days later, media was replaced with fresh media containing 1.5 μg/mL puromycin and cells were expanded for six additional days prior to seeding for ZIKV or YFV infection.
We seeded cells at 3.5 × 106 cells per T175 flask and we seeded 15 flasks per replicate (in triplicate) for each virus infection as well as Mock. The following day, the media was removed, and viruses diluted in 10 mL/flask OptiMEM were added to cells. ZIKV was added at MOI = 0.025 PFU/cell and YFV Asibi was added at MOI = 0.5 PFU/cell. After three h on a plate rocker at 37°C, plates were moved to 5% CO2 incubators set to 37°C. Media was changed three days later. Media was changed every 3-4 days to remove dead cells. Mock cells were passaged every 3-4 days and re-seeded at 3.5 × 106 cells/flask to maintain library complexity. Cells were harvested approximately twenty days post infection.
Genomic DNA (gDNA) was isolated via DNeasy Blood & Tissue Kit (QIAGEN: 69504) per the manufacturer’s instructions. The library was amplified from gDNA using a two-step nested PCR approach using the primer sequences listed in Table S1 and described previously (Yau and Rana, 2018). All PCRs were performed using Herculase II Fusion DNA polymerase with manufacturer recommended cycling parameters using 5 μg gDNA per 50 μL reaction volume (Agilent: 600679). After PCR1, PCR products were purified using QIAquick PCR Purification Kit (QIAGEN: 8104) and used as a template for PCR2 to add Illumina sequencing adapters and indexes. PCR products were agarose gel purified, pooled and sequenced on an Illumina NextSeq 500 at the MSKCC Integrated Genomics Operation using standard Illumina sequencing primers and 50 cycles.
Antibodies and Chemicals
Primary antibodies used for western blot include rabbit anti-TMEM41A (Proteintech: 20768-1-AP; RRID: AB_10693679; 1:1,000), rabbit anti-TMEM41B (Abnova: PAB20785; RRID: AB_10965049; 1:1,000), rabbit anti-VMP1 (Cell Signaling Technology: 12929; RRID: AB_2714018; 1:1,000), rabbit anti-ATG5 (Abcam: ab108327; RRID: AB_2650499; 1:5,000), rabbit anti-ATG7 (Abcam: ab52472; RRID: AB_867756; 1:50,000), rabbit anti-LC3B (Abcam: ab192890; RRID: AB_2827794; 1:2,000), rabbit anti-tagRFP (Evrogen: AB233; RRID: AB_2571743; 1:1,000) and mouse anti-β-actin conjugated to HRP (Sigma-Aldrich: A3854; RRID: AB_262011; 1:50,000).
Primary antibodies used for IF, flow cytometry and IP include mouse anti-YFV (Santa Cruz Biotechnology: sc-58083; RRID: AB_630447; 1:1,000), mouse anti-flavivirus group antigen (Millipore: MAB10216; RRID: AB_827205; 1:500), pan-flavivirus hyperimmune mouse acites fluid (HMAF, kindly provided by Thomas G. Ksiazek, CDC; 1:200), rabbit anti-YFV NS4B (GeneTex: GTX134030; 1:1,000), rabbit anti-ZIKV NS4A (GeneTex: GTX133704; 1:1,000), rabbit anti-SARS-CoV-2 nucleoprotein (GeneTex: GTX135357; RRID: AB_2868464; 1:2,000), mouse anti-IAV nucleoprotein (Millipore: MAB8251; RRID: AB_95293; 1:1,000), bovine anti-BVDV polyclonal antibody B224 (kindly provided by Kenny V. Brock, Auburn University; 1:500), rabbit anti-tagRFP (see above) and normal rabbit IgG (Cell Signaling Technology: 2729; RRID: AB_1031062).
Secondary antibodies, proteins and staining solutions used for IF, flow cytometry and IP include goat anti-rabbit IgG conjugated to HRP (ThermoFisher Scientific: 31462; RRID: AB_228338; 1:5,000), goat anti-mouse IgG conjugated to HRP (Jackson ImmunoResearch Labs: 115-035-146; RRID: AB_2307392; 1:10,000), goat anti-bovine IgG conjugated to FITC (ThermoFisher Scientific: A18752; RRID: AB_2535529; 1:1,000), goat anti-mouse IgG conjugated to AF647 (ThermoFisher Scientific: A-21235; RRID: AB_2535804; 1:1,000), goat anti-mouse IgG conjugated to AF488 (ThermoFisher Scientific: A-11001; RRID: AB_2534069; 1:1,000), goat anti-rabbit IgG conjugated to AF488 (ThermoFisher Scientific: A-11008; RRID: AB_143165; 1:2,000) and Protein A-HRP (ThermoFisher Scientific: 10-1023). Hoechst 33342 solution (ThermoFisher Scientific: 62249) was used at 1 μg/mL for nuclear stain. Nile Red Staining Kit (Abcam: ab228553) was used according to the manufacturer’s instructions to stain for intracellular lipid droplets.
Drugs and antibiotics were used at concentrations indicated in figure legends and methods, and include chloroquine diphosphate salt (MilliporeSigma: C6628), Torin 1 (Selleckchem: S2827), doxycycline hyclate (MilliporeSigma: D9891), puromycin dihydrochloride (MilliporeSigma: 8833), blasticidin S (MilliporeSigma), carbenicillin disodium salt (MilliporeSigma: C1389), polybrene (MilliporeSigma: TR-1003-G), Staurosporin (STS – MilliporeSigma: S4400), InSolution JAK Inhibitor I (MilliporeSigma: 420097) and TBK1 inhibitor BX795 (InvivoGen: tlrl-bx7).
Lentivirus Production and Transduction
All ORFs were cloned into a modified pTRIPZ vector for lentivirus delivery to cells. All plasmid sequences are available upon request. Lentivirus stocks were generated in Lenti-X 293T cells by co-transfection of plasmids expressing (1) the ORF of interest (2), HIV gag-pol, and (3) the vesicular stomatitis virus glycoprotein (VSV-G) in a ratio of 0.55:0.35:0.1 using Lipofectamine 2000 Transfection Reagent (ThermoFisher Scientific: 11668030) at a ratio of 2.5 μl reagent to 1 μg DNA. One day prior to transfection, cells were seeded at 4 × 105 cells per well of poly-L-lysine coated 6-well plates. Cells were transfected the following day and six h post transfection media was removed and lentivirus was collected overnight in 2 mL per well. Supernatants were collected at 24 and 48 h, filtered using 0.45-micron syringe filters, and stored at −80°C.
For lentiviral transduction, 3 × 105 cells were transferred to 12-well plates in suspension and transduced with several lentivirus dilutions by spinoculation at 1,000 x g for 60 min at 37°C in medium containing 3% FBS, 20 mM HEPES, and 4 μg/mL polybrene. The following day cells were trypsinized and moved to duplicate wells of 6-well plates. At approximately 48 h post transduction one well of each duplicate was selected with 2 μg/mL puromycin. When selection was complete, the selected and unselected replicates were counted to determine approximate MOI and we proceeded to expand cells that were transduced at MOI < 0.3 to ensure that most cells in the population contained a single integrant. The pTRIPZ constructs are doxycycline inducible and all experiments with these constructs were performed after 24 h of pretreatment and in the continued presence of 2 μg/mL doxycycline.
Generation and Validation of CRISPR KO clones
Mammalian cells
Small guide RNAs (sgRNA) for CRISPR editing were designed using crispor.tefor.net (Haeussler et al., 2016) and cloned into lentiCRISPRv2 plasmid CRISPR KO clones were generated by transfection with the relevant guides, selected with 2 μg/mL puromycin for four days prior to single-cell seeding into 96-well plates in the absence of puromycin at a dilution of 0.7 cells/well to obtain single-cell clonal populations. Clones were expanded and screened for protein knockout by western blot analysis or sequencing analysis. Genomic DNA was extracted using the QIAGEN DNeasy Blood and Tissue Kit (QIAGEN: 69504) and used as a template for amplification of an approximately 500-1,000 bp region flanking the PAM site. Discrete bands were gel-extracted and gene disruption was confirmed by submitting samples to the CCIB DNA Core Facility, Massachusetts General Hospital (Cambridge, MA, USA) for high throughput amplicon sequencing.
We used the following sgRNAs in these studies. sgRNAs targeting TMEM41B in HAP1, A549, JEG3, and Huh-7.5 cells: sgRNA 1: 5′- GTCGCCGAACGATCGCAGTT −3′ and sgRNA 2: 5′- GCTCACCACACGACCCCCGT −3′. sgRNA targeting TMEM41B in MDBK cells: 5′- ATACTGAGAAATATAGAGCC −3′. sgRNA targeting TMEM64 in HAP1 cells: 5¢- GCTCCAGATAGGCGCCGAGC −3¢. sgRNA targeting TMEM41A in HAP1 cells: 5′- TCGCGTCGACAGCAAGTACA −3′. sgRNA targeting VMP1 in HAP1 cells: 5′- GCTCTGGCCATGAAATATGG −3′. sgRNA targeting ATG5 in HAP1 cells: 5′- TTCCATGAGTTTCCGATTGA −3′. sgRNA targeting ATG7 in HAP1 cells: 5′- CCTAGCTACATTGCAACCCA −3′. sgRNA targeting BECN1 in HAP1 cells: 5′- CCTGGATGGTGACACGGTCC −3′. All oligos were purchased from IDT.
Mosquito cells
CRISPR/Cas9 plasmids were generated from the pDDC6 vector, which encodes the human codon-optimized Streptococcus pyogenes Cas9 (hSpCas9; a gift from Peter Duchek) (Addgene plasmid: #59985; http://n2t.net/addgene:59985; RRID: Addgene_59985). For expression in mosquito cells, we replaced the dme phsp70 promoter (hsp70Bb) in pDCC6 with the Ae. aegypti polyubiquitin promoter (aae PUb) and we replaced the dme U6-2 promoter with the Ae. aegypti U6 promoter (aae U6; AAEL017774). We also added a selectable puromycin resistance cassette to the hSpCas9. Plasmid sequence is available from Addgene (plasmid #162163). The following sgRNAs were cloned into the abovementioned plasmid. sgRNAs targeting TMEM41B ortholog (AAEL022930-RB) in Aag2 cells: 5′- CAAAGATCTCTACTACCTGG −3′ and 5′- TATGTAGACGAGGACCACGC −3′. sgRNAs targeting TMEM41B ortholog (AALF002881-RA) in C6/36 cells: 5'- CAAACAGTTGGGACGAGTGC −3′ and 5′- TGTAGCTGAGGAGGTCTTCC −3′. All oligos were purchased from IDT. Cells were transfected with the appropriate plasmids using Fugene HD Transfection Reagent (Promega: E2311) according to the manufacturer’s protocol. Cells were seeded at ∼50% confluency and complexes were formed using a ratio of 3:1 transfection reagent to plasmid DNA. To generate single cell clones, cells were seeded in 96-well plates (∼0.7 cells/well) in 50% fresh media, and 50% conditioned media and expanded. Gene disruption was confirmed by high throughput amplicon sequencing and described above. For additional details please see (Rozen-Gagnon et al., 2020).
Viruses and Infections
Virus stocks used in this study include: DENV-GFP (Schoggins et al., 2012) (derived from IC30P-A, a full-length infectious clone of strain 16681) grown in Huh-7.5 cells, WNV-GFP (McGee et al., 2010) (derived from pBELO-WNV-GFP-RZ ic) grown in Huh-7.5 cells, HCV-RFP (Liu et al., 2015) (based on Jc1-378-1) grown in Huh-7.5 cells, BVDV-NADL (Mendez et al., 1998) grown in MDBK cells, CHIKV-GFP (Tsetsarkin et al., 2006) (derived from pCHIKV-LR 5′GFP) grown in BHK-21 cells, hPIV3-GFP (Zhang et al., 2005) (based on strain JS) grown in VeroE6 cells, BUNV-GFP (Shi et al., 2010) (based on rBUN-del7GFP) grown in BHK-21 cells, VacV-GFP (Schoggins et al., 2014) (based on strain Western Reserve) grown in BSC-40 cells, HSV-1 US11-GFP (Benboudjema et al., 2003) grown in VeroE6 cells, AdV5-GFP (Evans and Hearing, 2003) (generously provided by Patrick Hearing, Stony Brook University), VSV-GFP (Dalton and Rose, 2001) grown in BHK-21 cells, LCMV-GFP (Ngo et al., 2015a) (generously provided by J.C. de la Torre, Scripps Research), influenza viruses A/WSN/33 and A/PR/8/34 ΔNS1 (generously provided by Peter Palese and Adolfo García-Sastre, Mount Sinai School of Medicine). LV-GFP was generated in Lenti-X 293T as described above. CVB-3-GFP (Feuer et al., 2002) (derived from infectious clone pMKS1-GFP) was amplified in HeLa cells and titrated by TCID50. SARS-CoV-2 (WA1/2020 obtained from BEI Resources) and ZIKV (PRVABC59 obtained from the CDC, Ft. Collins) were amplified in Huh-7.5 and HAP1 cells respectively and titrated by standard plaque assay on Huh-7.5 cells. YF viruses (17D and Asibi) and the Cambodian ZIKV (Shan et al., 2016) (strain: FSS13025, generously provided by Pei-Yong Shi) were generated from infectious clones as described below. Experiments with YFV (strain: Asibi), WNV-GFP, CHIKV-GFP and SARS-CoV-2 were carried out in biosafety level 3 (BSL3) containment in compliance with institutional and federal guidelines. The tick-borne viruses POWV (strain: Byers, VSPB 812027), TBEV (strain: Hypr, VSPB 812222 and Sofjin, VSPB 812223), OHFV (strain: Bogoluvovska, VSPB 812005), KFDV (strain: P9605, VSPB 811996) and AHFV (strain: 200300001, VSPB 200300001) were generated at the CDC and experiments were carried out in BSL4 containment in compliance with institutional and federal guidelines (Flint et al., 2014; Zaki, 1997).
Generation of recombinant YFV and ZIKV
Recombinant viruses for the Cambodian ZIKV (strain: FSS13025), the YFV vaccine strain 17D and wildtype Asibi strain, as well as the YF chimeric 17D and Asibi viruses were generated using plasmids encoding the infectious clones. To generate mutant and chimeric viruses, overlapping oligos were created to either introduce point mutations or to allow the amplification of specific coding sequences. PCR reactions were run under standard conditions using KOD Hot Start Master Mix (Sigma-Aldrich: 71842). After subsequent ligation with T4 ligase (NEB: M0202), the plasmid DNA was amplified in competent DH5-ɑ E.coli cells (ThermoFisher Scientific: 18265017). All plasmids encoding WT and mutant/chimeric viruses were sequenced for the absence of unwanted mutations within the viral coding region. For in vitro transcription, 2 μg linearized plasmid DNA was used in a 30 μl reaction with the mMessage mMachine SP6 Transcription Kit (ThermoFisher Scientific: AM1340). RNA integrity was examined by electrophoresis on a 1% agarose gel. Huh-7.5 cells were subsequently electroporated with 4 μg of YFV RNA mixed into 6 × 106 cells, while C6/36 cells were used for electroporating ZIKV RNA under similar conditions. Electroporated cells were incubated for 48-72 h before collecting and titrating supernatants by standard plaque assay in Huh-7.5 cells.
All virus infections and quantification were performed using the doses and time points reported in the figure legends and below for quantification by flow cytometry or microscopy. For infections, cells were seeded at a density of 1 × 104 cells/well into black, glass-bottom 96-well plates (PerkinElmer: 6055302) and at 2.5 × 104 - 5 × 104 cells/well into regular tissue culture 24-well plates. The next day, cells were washed 1x with Opti-MEM (GIBCO) and adsorbed with virus inoculum (50 μl for 96-well and 200 μl for 24-well plates) prepared in Opti-MEM at 37°C. After 1-2 h inoculum was removed, cells were washed with Opti-MEM, fresh media was added, and cells were incubated at 37°C. At the final time point dependent on viral replication kinetics, cells in 24-well plates were lifted with Accumax cell dissociation medium (Sigma: A7089) and fixed with 2% paraformaldehyde (PFA). Cells infected with GFP/RFP reporter viruses were pelleted at 930 x g for 5 min at 4°C, resuspended in cold phosphate-buffered saline (PBS) containing 3% FBS and stored at 4°C until analysis using a LSRII flow cytometer (BD Biosciences). FlowJo software v10 (Treestar) was used to obtain the percentage of GFP/RFP-expressing cells, which were graphed using Prism8 (Graphpad).
For non-fluorescent reporter viruses, cells were washed 1x in PBS (+ 2% FBS), permeabilized in 1x BD perm (BD Biosciences: 554723) for 1 h at RT followed by overnight incubation with primary antibody. Cells were then washed three times with BD perm, incubated with secondary antibody for 1 h at RT, washed an additional three times with BD perm, and once with PBS (+ 2% FBS) before being transferred to FACS tubes for flow-cytometry analysis similar as for aforementioned reporter viruses.
Cells infected in 96-well plates were fixed with 10% neutral-buffered formalin, permeabilized with 0.1% Triton X-100 in PBS for 10 min at RT, washed in PBS with 0.01% Tween20 (PBS-T) and incubated with primary antibody (pan-flavivirus HMAF) overnight. The secondary antibody was used at RT for 1 h together with NucBlue (ThermoFisher Scientific: R37605) to stain for nuclei. Plates were subsequently read using the Perkin Elmer Operetta confocal high content imager with a 10x objective. Images were analyzed with a cell segmentation workflow using Harmony software v4.1 Perkin Elmer, Waltham, MA.
Details of virus infections related to Figures 1, 2, 3, 4, 5, 6, and 7
Related toFigure 1: For ZIKV infections in HAP1 cells comparing WT versus VTT domain protein KO clones and BECN1, ATG5, and ATG7 KO clones, cells were infected for 48 h at MOI = 0.5 PFU/cell. For ZIKV infections in HAP1 cells comparing WT versus TMEM41B KO or VMP1 KO cells expressing VTT domain proteins, cells were infected for 48 h at MOI = 0.25 PFU/cell. Similar experiments with YFV Asibi were performed with MOI = 0.1 PFU/cell for 72 h.
Related toFigure 2: WT and TMEM41B KO HAP1 cells were infected with POWV for 48 h at MOI = 0.02 PFU/cell. All other tick-borne flaviviruses, including TBEV strains Hypr and Sofjin, OHFV, KFDV, and AHFV were inoculated at MOI = 0.02 PFU/cell and harvested at 24 h post infection. WT and TMEM41B KO Huh-7.5 clones were infected with HCV-RFP for 72 h at MOI = 0.05 PFU/cell. WT and TMEM41B KO MDBK clones were infected with BVDV for 48 h at MOI = 0.01 PFU/cell. WT, TMEM41B KO and reconstituted HAP1 cells were infected with influenza A virus (IAV) at MOI = 0.1 PFU/cell for 24 h; bunyamwera virus (BUNV-GFP) at MOI = 0.025 IU/cell for 48 h; human parainfluenza virus 3 (h-PIV3-GFP) at MOI = 0.02 IU/cell for 48 h; lymphocytic choriomeningitis virus (LCMV-GFP) at MOI = 0.01 IU/cell for 48 h; vesicular stomatitis virus (VSV-GFP) at MOI = 0.01 PFU/cell for 24 h; VSV-G-pseudotyped HIV-1 lentivirus (LV-GFP) at MOI = 1:25 dilution of virus stock for 48 h; Coxsackie virus B3 (CVB-3-GFP) at MOI = 1:100 dilution of virus stock for 8 h; chikungunya virus (CHIKV-GFP) at MOI = 0.025 PFU/cell for 24 h; adenovirus 5 (AdV5-GFP) at MOI = 200 particles/cell for 24 h; herpes simplex virus 1 (HSV-1-GFP) at MOI = 2.5 PFU/cell for 8 h; vaccinia virus (VacV-GFP) at MOI = 0.005 PFU/cell for 48 h. For SARS-CoV-2 infection, WT, TMEM41B KO and reconstituted HAP1 cells were modified to stably express ACE2 and TMPRSS2 and infected at MOI = 1 PFU/cell for 24 h. Cells were analyzed by flow cytometry and plotted as a percentage of viral antigen positive cells or GFP/RFP positive cells for reporter viruses expressing a fluorescent protein.
Related toFigure 3: WT, TMEM41B KO, and TMEM41B KO cells expressing human TMEM41B isoforms or mosquito and tick TMEM41B orthologs were infected with YFV Asibi at MOI = 0.1 PFU/cell for 72 h, or ZIKV at MOI = 0.25 PFU/cell for 48 h. TMEM41B isoforms are as follows: Isoform 1, UniProt ID: Q5BJD5-1; isoform 2, UniProt ID: Q5BJD5-2; isoform 3, UniProt ID: Q5BJD5-3; isoform 4, UniProt ID: E9PJ42.
Related toFigure 4: WT, TMEM41B KO, and TMEM41B KO HAP1 cells expressing WT or TMEM41B SNP variants were infected with YFV 17D at MOI = 0.005 PFU/cell for 48 h; ZIKV at MOI = 0.25 PFU/cell for 48 h; DENV-GFP at MOI = 0.1 PFU/cell for 96 h; WNV-GFP at MOI = 1 PFU/cell for 72 h; and hPIV-3-GFP at MOI = 0.02 IU/cell for 48 h.
Related toFigure 5: TMEM41B KO HAP1 cells expressing RFP-TMEM41B were infected with YFV 17D at MOI = 1 and ZIKV at MOI = 2.5 PFU/cell for 24 h.
Related toFigure 6: WT Aag2 (n = 1) and TMEM41B KO clones (n = 2) were infected with ZIKV (strain: PRVABC59) at MOI = 0.5 PFU/cell in triplicate. Cells were passaged every 2-3 days for 15 passages after which supernatants were collected and viral RNA was reverse transcribed and sequenced with next generation sequencing (NGS). WT (n = 1) and TMEM41B KO A549 clones (n = 2) and WT (n = 1) and TMEM41B KO HAP1 clones (n = 2), were infected with ZIKV (strain: FSS13025) at MOI = 5 PFU/cell in triplicate. Supernatants were diluted 1:2 and used to inoculate naive cells every 3-4 days for 20 passages after which supernatants were collected and viral RNA was reverse transcribed and sequenced with next generation sequencing (NGS). Missense mutations I42T and F91Y were independently engineered in a ZIKV infectious clone and used to generate virus stocks to infect WT or TMEM41B KO Aag2 cells at MOI = 0.001 PFU/cell for 72 h. WT and TMEM41B KO A549 cells were infected with WT and mutant viruses at MOI = 0.05 PFU/cell for 48 h. YFV 17D and Asibi chimeric viruses were generated and used to infect WT and TMEM41B KO A549 cells as follows: 17D backbone with Asibi NS4A and NS4B proteins were inoculated at MOI = 0.1 PFU/cell for 48 h; Asibi backbone with 17D NS4A and NS4B proteins were inoculated at MOI = 0.1 PFU/cell for 48 h.
Related toFigure 7: To quantify viral RNA and OAS1 by qRT-PCR, WT and TMEM41B KO HAP1 cells were infected with YFV 17D (with and without UV-inactivation) at MOI = 0.4 PFU/cell for 24 h. Cells were infected with IAV (ΔNS1) at MOI = 0.1 PFU/cell for 24 h. For apoptosis assay, WT and TMEM41B KO HAP1 cells were infected YFV 17D at MOI = 4 PFU/cell for 24 h or treated with 250 nM staurosporine (STS) and assayed to detect apoptotic cells via Annexin-V staining.
Sequence Analysis of Adapted ZIKVs
To determine sequences of ZIKVs passaged in mosquito (ZIKV strain: PRVABC59) and in mammalian WT and TMEM41B KO cells (ZIKV strain: FSS13025), RNA from 200 μl supernatants of each individual passage was purified by adding 800 μl TRIzol Reagent (ThermoFisher Scientific: 15596026) plus 200 μl chloroform then centrifuged at 12,000 g x 5 min. The upper aqueous phase was moved to a new tube and an equal volume of isopropanol was added. This was then added to an RNeasy mini kit column (QIAGEN: 74014) and further purified following the manufacturer’s instructions. Viral RNA was reverse transcribed into cDNA using the Maxima H Minus First Strand cDNA Synthesis Kit (ThermoFisher Scientific: K1681) according to the manufacturer’s protocol, amplified using ZIKV-specific primers (available upon request) and submitted to the CCIB DNA Core Facility, Massachusetts General Hospital (Cambridge, MA, USA) for high throughput amplicon sequencing. Briefly, samples were sheared, ligated to Illumina compatible adapters with unique barcodes, and sequenced from both ends on the MiSeq platform with 2x150 run parameters.
Immunofluorescence
Unless otherwise described, cells were fixed in 2% PFA prepared in PBS at RT for 10 min. Cells were then directly stained for intracellular lipid droplets using the Nile Red Staining Kit (Abcam: ab228553) according to the manufacturer’s instructions, or were processed for antigen staining by blocking with PBTG (PBS containing 10% normal goat serum, 1% bovine serum albumin (BSA), 0.1% Triton X-100) at RT for 1-2 h. Cells were then incubated with primary antibodies diluted in PBTG (concentrations above) at 4°C overnight. Following four washes with PBS-T, cells were stained with AlexaFluor-conjugated secondary antibodies in PBTG at RT for 1 h. Following one wash with PBS-T, cells were incubated with Hoechst nuclear stain diluted in PBS at RT for 15 min, followed by three additional washes with PBS-T and a final wash with PBS. Images were acquired using a 40x oil immersion objective on an inverted Zeiss Axiovert 200 spinning disk confocal microscope using solid-state 491, 561 and 644 nm lasers (Spectral Applied) for excitation for collection with an Andor iXon 512x512 EMCCD camera using MetaMorph software. Images acquired staining for antigen and intracellular lipid droplets were analyzed using Fiji software (Schindelin et al., 2012).
Western Blot
Cell lysates were prepared in RIPA buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 0.1% sodium dodecyl sulfate [SDS], 0.5% SDC, 1% NP-40 with addition of cOmplete Mini EDTA-free protease inhibitor tablet [Millipore/Sigma: 11836170001]) and incubated on ice for 30 min with frequent vortexing and then clarified at 14,000 x g for 20 min at 4°C. Protein concentration was determined by bicinchoninic (BCA) protein assay (Pierce BCA Protein Assay Kit, ThermoFisher Scientific: 23227) and samples were resolved on NuPAGE 4%–12% Bis-Tris gels and Novex 4%–20% Tris-Glycine gels (Invitrogen) followed by transfer onto polyvinylidene fluoride (PVDF) membrane (Millipore/Sigma: IPVH00010). Membranes were blocked with 5% milk in PBS-T and incubated with primary antibody at 4°C overnight in 5% milk PBST. Membranes were washed 3x with PBST and incubated with secondary antibodies. For chemiluminescent readout, the membranes were incubated with HRP-conjugated secondary antibody and exposed using SuperSignal West Pico PLUS Chemiluminescent Substrate or SuperSignal West Femto Maximum Sensitivity Substrate (ThermoFisher Scientific: 34577 and 34095) by film in a dark room.
Immunoprecipitation
TMEM41B KO HAP1 cells reconstituted with full length TMEM41B fused to tagRFP (infected and uninfected) were collected and lysed in nonyl phenoxypolyethoxylethanol (NP-40) buffer (10 mM HEPES, pH 7.5, 150 mM KCl, 3 mM MgCl2, 0.5% NP-40) supplemented with cOmplete Mini EDTA-free protease inhibitor tablet (Millipore/Sigma: 11836170001) and 0.1 mM phenylmethylsulfonyl fluoride (PMSF) (Millipore/Sigma: 10837091001). Protein concentrations were determined by BCA assay as described above. Per sample, 50 μL (1.5 mg) Dynabeads Protein A for Immunoprecipitation (ThermoFisher Scientific: 10001D) were prepared and linked to 5 μg antibodies (rabbit anti-tagRFP and normal rabbit IgG control) according to the manufacture’s protocol. 100 μg of whole cell lysate (WCL) were incubated with beads-antibody complexes for 70 min at RT followed by wash steps according to the manufacture’s protocol. The resulting beads-antibody-antigen complexes were subsequently treated with elution buffer, mixed with NuPAGE® LDS Sample Buffer (ThermoFisher Scientific: NP0008) and NuPAGE® Sample Reducing Agent (ThermoFisher Scientific: NP0009) as per manufacturer’s instructions and denatured for 15 min at 70°C. The precipitated proteins within the eluted fractions were resolved by western blot as described above.
YFV Subgenomic Reporter Replicon
For in vitro transcription of a Renilla reniformis luciferase encoding YFV subgenomic reporter replicon, 1 μg of linearized DNA template was used in a 20 μl reaction of the mMessage mMachine SP6 Transcription Kit (ThermoFisher Scientific: AM1340) following the manufacturer’s protocol and adding 1 μl of additional GTP. In vitro transcribed RNA was purified using the RNeasy Mini Kit (QIAGEN: 74104), examined for integrity by electrophoresis on a 1% agarose gel and quantified using a NanoDrop spectrophotometer. Next, HAP1 cells (WT, TMEM41B KO and tagRFP-TMEM41B reconstituted KO) were seeded into 24-well plates at 5 × 104 cells/well and incubated overnight at 37°C. The next day, 250 ng RNA/well of either WT or the polymerase dead (dDD) YFV replicon were transfected using Lipofectamine 2000 Transfection Reagent (ThermoFisher Scientific: 11668030) at a ratio of 2.5:1 μl transfection reagent to μg RNA. Cells were harvested at indicated time points by washing 1x with 250 μl PBS, adding 150 μl of diluted 5x Passive Lysis Buffer (Promega: E1941), incubating for 20 min at RT and storing lysates at −20°C. Samples harvested at different time points were processed together using the Dual-Luciferase® Reporter Assay System (Promega: E1910) and a FLUOstar Omega multimode microplate reader (BMG LABTECH). Raw data was analyzed, normalized and graphed using Excel (Microsoft) and Prism8 (Graphpad).
Apoptosis Assay
Hap1 WT and TMEM41B KO cells were seeded into 24-well plates at 5 × 104 cells/well. The next day, cells were infected with the YFV vaccine strain 17D, UV-inactivated YFV 17D and IAV (ΔNS1) diluted in Opti-MEM for 2 h at 37°C. Staurosporine (STS) diluted in the culture medium was added to the cells simultaneously. After 2 h cells were washed with Opti-MEM and cultured in fresh medium. After 24 h, detached cells (from supernatant and PBS wash) together with the trypsinized cells were harvested into tubes and samples were prepared on ice for flow cytometry using the Pacific Blue Annexin V Apoptosis Detection Kit with 7-AAD (Biolegend: 640926) according to the manufacturer’s protocol. Cells were subsequently washed twice in binding buffer, followed by fixation in 2% PFA in PBS before subjecting them to flow cytometry using the LSR-II (BD Biosciences). Data was analyzed by FlowJo v10 and graphed by Prism 8 (GraphPad).
ISG response
HAP1 WT and TMEM41B KO cells were seeded into 24-well plates at 7.5 × 104 cells/well. The next day, cells were infected with the YFV vaccine strain 17D, UV-inactivated YFV 17D and IAV (ΔNS1) diluted in Opti-MEM for 2 h at 37°C. After 2 h cells were washed with Opti-MEM before adding fresh culture medium. After 24 h at 37°C, the cells were washed once with PBS, trypsinized, and pelleted before extracting total RNA by using RNeasy Mini Kit (QIAGEN: 74104) following the manufacturer’s instructions. Next, 0.2 μg RNA were reverse transcribed into cDNA using random hexamer primers with the Superscript III First-Strand Synthesis System Kit (Invitrogen: 18080051) following the manufacturer’s instructions. Gene expression was quantified by qRT-PCR using PowerUp SYBR Green Master Mix (Applied Biosystems: A25742) and gene-specific primers: RPS11: forward: 5′- GCCGAGACTATCTGCACTAC −3′ and reverse: 5′- ATGTCCAGCCTCAGAACTTC- 3′; OAS1: forward: 5′- AGAAATACCCCAGCCAAATCTCT −3′ and reverse: 5′- TGAGGAGCCACCCTTTACCA −3′. The following PCR conditions were used: 56°C for 2 min and 95°C for 10 min (initial denaturation); 45 cycles 95°C for 15 s, 56°C for 15 s and 72°C for 20 s (PCR); followed by 95°C for 10 s, 65°C for 10 s, a slow increase to 95°C (0.07°C/sec) and a final cooling to 50°C (melt curve). The data were analyzed by melt curve analysis for product specificity as well as ΔΔCT analysis for fold changes (after normalization to housekeeping genes) and graphed using Prism 8 (GraphPad).
Quantification and Statistical Analysis
Statistical details of experiments can be found in the figure legends, including the statistical tests used, exact value of n, what n represents (e.g., number of replicates or number of cell clones) and the definition of error bars (e.g., standard deviation).
Analysis of CRISPR-Cas9 Screen Data
FASTQ files were processed and trimmed to retrieve sgRNA target sequences followed by enumeration of sgRNAs in the reference sgRNA library file using MAGeCK (Li et al., 2014). Z-scores were computed using the following approach: for each condition, the log2 fold change with respect to the initial condition was computed. A natural cubic spline with 4 degrees of freedom was fit to each pair of infected and control cells and residuals were extracted. To obtain gene-wise data, the mean residuals for each group of sgRNAs was calculated, a z-score was computed, and a p value was determined using a 2-sided normal distribution test. P values were combined across biological replicates using Fisher’s sumlog approach and corrected for multiple testing using the method of Benjamini & Hochberg.
Analysis of lipid droplets in WT versus TMEM41B KO cells
Upon staining intracellular lipid droplets using Nile red, images were analyzed using Fiji software (Schindelin et al., 2012) as follows: A maximum z-projection was made from the acquired image stacks, and the particles were identified through a fixed threshold based on control images. After an applied watershed, particles under 5 pixels were excluded as they could not be distinguished from background/noise in control images, and those greater than 5 pixels were counted.
Analysis of virus infection in cells expressing WT versus TMEM41B SNP variants
Statistical analysis was performed in Prism (GraphPad Software, v8.0, 2018). The two-tailed Student’s t test was used comparing two conditions. All conditions met the normality assumption by the Shapiro-Wilk test. Statistical significance was defined as a p value < 0.05.
Acknowledgments
We thank the following investigators for contributing viral molecular clones and viral stocks: P. Hearing (AdV5-GFP), I.J. Mohr (HSV-1-GFP), P.L. Collins (hPIV-3-GFP), J.K. Rose (VSV-GFP), J.C. de la Torre (LCMV-GFP), P. Palese (IAV), A. García-Sastre (IAV ΔNS1), J.L. Whitton (CVB-3-GFP), P. Traktman (VacV-GFP), I.V. Frolov (WNV-GFP), S. Higgs (CHIKV-GFP), R.M. Elliott (BUNV-GFP), P.Y. Shi (ZIKV: FSS13025), Centers for Disease Control and Prevention (ZIKV: PRVABC59), and BEI Resources (SARS-CoV-2). We thank the Rockefeller University Flow Cytometry and Bio-Imaging Resource Centers for their training and advice. We thank Joe Luna and Francisco J. Sánchez-Rivera for their boundless enthusiasm and for stepping up in time of crisis to allow this manuscript to be written. We thank Yingpu Yu for engaging discussion and generosity. We thank Alison W. Ashbrook and Lauren C. Aguado for critical feedback on short notice. We thank Aileen O’Connell, Santa Maria Pecoraro Di Vittorio, Glen Santiago, Mary Ellen Castillo, Arnella Webson, and Sonia Shirley for outstanding administrative or technical support. The research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award number R01AI124690 (to C.M. Rice). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health nor does it represent the official position of the Centers for Disease Control and Prevention. The project was supported in part by NIH grant P30 CA008748 (to C.M. Rudin). F.S. received a MD fellowship from Boehringer Ingelheim Fonds. This work was also supported by the generosity of the BAWD Foundation and the Robertson Foundation.
Author Contributions
Conceptualization, H.-H.H., W.M.S., K.R.-G., L.A.M., F.S., M.R.M., J.T.P., and C.M. Rice; Methodology, H.-H.H., W.M.S., and K.R.-G.; Formal Analysis, J.T.P.; Investigation, H.-H.H., W.M.S., K.R.-G., L.A.M., F.S., B.R., E.J., X.W., S.Y., and L.K.M.; Supervision, M.R.M., J.T.P., and C.M. Rice; Visualization, H.-H.H., W.M.S., and J.T.P.; Writing – Original Draft, H.-H.H. and W.M.S.; Writing – Review & Editing, H.-H.H., W.M.S., L.A.M., F.S., B.R., M.R.M., L.K.M., J.T.P., and C.M. Rice; Funding Acquisition, H.-H.H., W.M.S., C.M. Rudin, M.R.M., J.T.P., and C.M. Rice.
Declaration of Interests
C.M. Rice is a founder of Apath LLC; a Scientific Advisory Board member of Imvaq Therapeutics, Vir Biotechnology, and Arbutus Biopharma; and an advisor for Regulus Therapeutics and Pfizer. The remaining authors declare no competing interests. C.M. Rudin serves on the Scientific Advisory Boards of Bridge Medicines, Earli, and Harpoon Therapeutics.
Published: December 9, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.cell.2020.12.005.
Supplemental Information
References
- Ahmed Q.A., Memish Z.A. Yellow fever from Angola and Congo: a storm gathers. Trop. Doct. 2017;47:92–96. doi: 10.1177/0049475517699726. [DOI] [PubMed] [Google Scholar]
- Aktepe T.E., Mackenzie J.M. Shaping the flavivirus replication complex: It is curvaceous! Cell. Microbiol. 2018;20:e12884. doi: 10.1111/cmi.12884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baggen J., Persoons L., Jansen S., Vanstreels E., Jacquemyn M., Jochmans D., Neyts J., Dallmeier K., Maes P., Daelemans D. Identification of TMEM106B as proviral host factor for SARS-CoV-2. bioRxiv. 2020 doi: 10.1101/2020.09.28.316281. [DOI] [Google Scholar]
- Benboudjema L., Mulvey M., Gao Y., Pimplikar S.W., Mohr I. Association of the herpes simplex virus type 1 Us11 gene product with the cellular kinesin light-chain-related protein PAT1 results in the redistribution of both polypeptides. J. Virol. 2003;77:9192–9203. doi: 10.1128/JVI.77.17.9192-9203.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blight K.J., McKeating J.A., Rice C.M. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 2002;76:13001–13014. doi: 10.1128/JVI.76.24.13001-13014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady O.J., Hay S.I. The Global Expansion of Dengue: How Aedes aegypti Mosquitoes Enabled the First Pandemic Arbovirus. Annu. Rev. Entomol. 2020;65:191–208. doi: 10.1146/annurev-ento-011019-024918. [DOI] [PubMed] [Google Scholar]
- Brugueras S., Fernández-Martínez B., Martínez-de la Puente J., Figuerola J., Porro T.M., Rius C., Larrauri A., Gómez-Barroso D. Environmental drivers, climate change and emergent diseases transmitted by mosquitoes and their vectors in southern Europe: A systematic review. Environ. Res. 2020;191:110038. doi: 10.1016/j.envres.2020.110038. [DOI] [PubMed] [Google Scholar]
- Cagno V., Tseligka E.D., Jones S.T., Tapparel C. Heparan Sulfate Proteoglycans and Viral Attachment: True Receptors or Adaptation Bias? Viruses. 2019;11:596. doi: 10.3390/v11070596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton K.P., Rose J.K. Vesicular stomatitis virus glycoprotein containing the entire green fluorescent protein on its cytoplasmic domain is incorporated efficiently into virus particles. Virology. 2001;279:414–421. doi: 10.1006/viro.2000.0736. [DOI] [PubMed] [Google Scholar]
- Dennis D.T., Nekomoto T.S., Victor J.C., Paul W.S., Piesman J. Reported distribution of Ixodes scapularis and Ixodes pacificus (Acari: Ixodidae) in the United States. J. Med. Entomol. 1998;35:629–638. doi: 10.1093/jmedent/35.5.629. [DOI] [PubMed] [Google Scholar]
- Dobler G. Zoonotic tick-borne flaviviruses. Vet. Microbiol. 2010;140:221–228. doi: 10.1016/j.vetmic.2009.08.024. [DOI] [PubMed] [Google Scholar]
- dos Santos C.N., Post P.R., Carvalho R., Ferreira I.I., Rice C.M., Galler R. Complete nucleotide sequence of yellow fever virus vaccine strains 17DD and 17D-213. Virus Res. 1995;35:35–41. doi: 10.1016/0168-1702(94)00076-o. [DOI] [PubMed] [Google Scholar]
- Drappier M., Michiels T. Inhibition of the OAS/RNase L pathway by viruses. Curr. Opin. Virol. 2015;15:19–26. doi: 10.1016/j.coviro.2015.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebel G.D. Update on Powassan virus: emergence of a North American tick-borne flavivirus. Annu. Rev. Entomol. 2010;55:95–110. doi: 10.1146/annurev-ento-112408-085446. [DOI] [PubMed] [Google Scholar]
- Eisen R.J., Eisen L., Beard C.B. County-Scale Distribution of Ixodes scapularis and Ixodes pacificus (Acari: Ixodidae) in the Continental United States. J. Med. Entomol. 2016;53:349–386. doi: 10.1093/jme/tjv237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans J.D., Hearing P. Distinct roles of the Adenovirus E4 ORF3 protein in viral DNA replication and inhibition of genome concatenation. J. Virol. 2003;77:5295–5304. doi: 10.1128/JVI.77.9.5295-5304.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuer R., Mena I., Pagarigan R., Slifka M.K., Whitton J.L. Cell cycle status affects coxsackievirus replication, persistence, and reactivation in vitro. J. Virol. 2002;76:4430–4440. doi: 10.1128/JVI.76.9.4430-4440.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint M., McMullan L.K., Dodd K.A., Bird B.H., Khristova M.L., Nichol S.T., Spiropoulou C.F. Inhibitors of the tick-borne, hemorrhagic fever-associated flaviviruses. Antimicrob. Agents Chemother. 2014;58:3206–3216. doi: 10.1128/AAC.02393-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Sastre A., Egorov A., Matassov D., Brandt S., Levy D.E., Durbin J.E., Palese P., Muster T. Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology. 1998;252:324–330. doi: 10.1006/viro.1998.9508. [DOI] [PubMed] [Google Scholar]
- Gokcezade J., Sienski G., Duchek P. Efficient CRISPR/Cas9 plasmids for rapid and versatile genome editing in Drosophila. G3 (Bethesda) 2014;4:2279–2282. doi: 10.1534/g3.114.014126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusho E., Baskar D., Banerjee S. New advances in our understanding of the “unique” RNase L in host pathogen interaction and immune signaling. Cytokine. 2020;133:153847. doi: 10.1016/j.cyto.2016.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeussler M., Schönig K., Eckert H., Eschstruth A., Mianné J., Renaud J.B., Schneider-Maunoury S., Shkumatava A., Teboul L., Kent J., et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016;17:148. doi: 10.1186/s13059-016-1012-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hills S.L., Fischer M., Petersen L.R. Epidemiology of Zika Virus Infection. J. Infect. Dis. 2017;216(suppl_10):S868–S874. doi: 10.1093/infdis/jix434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann H.H., Schneider W.M., Blomen V.A., Scull M.A., Hovnanian A., Brummelkamp T.R., Rice C.M. Diverse Viruses Require the Calcium Transporter SPCA1 for Maturation and Spread. Cell Host & Microbe. 2017;22:460–470.e5. doi: 10.1016/j.chom.2017.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imlach W.L., Beck E.S., Choi B.J., Lotti F., Pellizzoni L., McCabe B.D. SMN is required for sensory-motor circuit function in Drosophila. Cell. 2012;151:427–439. doi: 10.1016/j.cell.2012.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii K.J., Akira S. Innate immune recognition of nucleic acids: beyond toll-like receptors. Int. J. Cancer. 2005;117:517–523. doi: 10.1002/ijc.21402. [DOI] [PubMed] [Google Scholar]
- Jae L.T., Raaben M., Herbert A.S., Kuehne A.I., Wirchnianski A.S., Soh T.K., Stubbs S.H., Janssen H., Damme M., Saftig P., et al. Virus entry. Lassa virus entry requires a trigger-induced receptor switch. Science. 2014;344:1506–1510. doi: 10.1126/science.1252480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones C.T., Patkar C.G., Kuhn R.J. Construction and applications of yellow fever virus replicons. Virology. 2005;331:247–259. doi: 10.1016/j.virol.2004.10.034. [DOI] [PubMed] [Google Scholar]
- Kang R., Zeh H.J., Lotze M.T., Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18:571–580. doi: 10.1038/cdd.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Z.B., Moutsatsos I., Moretti F., Bergman P., Zhang X., Nyfeler B., Antczak C. Fluopack screening platform for unbiased cellular phenotype profiling. Sci. Rep. 2020;10:2097. doi: 10.1038/s41598-020-58861-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke P.Y. The Multifaceted Roles of Autophagy in Flavivirus-Host Interactions. Int. J. Mol. Sci. 2018;19:3940. doi: 10.3390/ijms19123940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley L.A., Mezulis S., Yates C.M., Wass M.N., Sternberg M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015;10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer L.D., Ciota A.T., Kilpatrick A.M. Introduction, Spread, and Establishment of West Nile Virus in the Americas. J. Med. Entomol. 2019;56:1448–1455. doi: 10.1093/jme/tjz151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.Y., Ng L.F.P. Zika virus: from an obscurity to a priority. Microbes Infect. 2018;20:635–645. doi: 10.1016/j.micinf.2018.02.009. [DOI] [PubMed] [Google Scholar]
- Li W., Xu H., Xiao T., Cong L., Love M.I., Zhang F., Irizarry R.A., Liu J.S., Brown M., Liu X.S. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 2014;15:554. doi: 10.1186/s13059-014-0554-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D., Ji J., Ndongwe T.P., Michailidis E., Rice C.M., Ralston R., Sarafianos S.G. Fast hepatitis C virus RNA elimination and NS5A redistribution by NS5A inhibitors studied by a multiplex assay approach. Antimicrob. Agents Chemother. 2015;59:3482–3492. doi: 10.1128/AAC.00223-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotti F., Imlach W.L., Saieva L., Beck E.S., Hao le T., Li D.K., Jiao W., Mentis G.Z., Beattie C.E., McCabe B.D., Pellizzoni L. An SMN-dependent U12 splicing event essential for motor circuit function. Cell. 2012;151:440–454. doi: 10.1016/j.cell.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marceau C.D., Puschnik A.S., Majzoub K., Ooi Y.S., Brewer S.M., Fuchs G., Swaminathan K., Mata M.A., Elias J.E., Sarnow P., Carette J.E. Genetic dissection of Flaviviridae host factors through genome-scale CRISPR screens. Nature. 2016;535:159–163. doi: 10.1038/nature18631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGee C.E., Shustov A.V., Tsetsarkin K., Frolov I.V., Mason P.W., Vanlandingham D.L., Higgs S. Infection, dissemination, and transmission of a West Nile virus green fluorescent protein infectious clone by Culex pipiens quinquefasciatus mosquitoes. Vector Borne Zoonotic Dis. 2010;10:267–274. doi: 10.1089/vbz.2009.0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherson M., García-García A., Cuesta-Valero F.J., Beltrami H., Hansen-Ketchum P., MacDougall D., Ogden N.H. Expansion of the Lyme Disease Vector Ixodes Scapularis in Canada Inferred from CMIP5 Climate Projections. Environ. Health Perspect. 2017;125:057008. doi: 10.1289/EHP57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medlock J.M., Hansford K.M., Bormane A., Derdakova M., Estrada-Peña A., George J.C., Golovljova I., Jaenson T.G., Jensen J.K., Jensen P.M., et al. Driving forces for changes in geographical distribution of Ixodes ricinus ticks in Europe. Parasit. Vectors. 2013;6:1. doi: 10.1186/1756-3305-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez E., Ruggli N., Collett M.S., Rice C.M. Infectious bovine viral diarrhea virus (strain NADL) RNA from stable cDNA clones: a cellular insert determines NS3 production and viral cytopathogenicity. J. Virol. 1998;72:4737–4745. doi: 10.1128/jvi.72.6.4737-4745.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N., Noda T., Yoshimori T., Tanaka Y., Ishii T., George M.D., Klionsky D.J., Ohsumi M., Ohsumi Y. A protein conjugation system essential for autophagy. Nature. 1998;395:395–398. doi: 10.1038/26506. [DOI] [PubMed] [Google Scholar]
- Moretti F., Bergman P., Dodgson S., Marcellin D., Claerr I., Goodwin J.M., DeJesus R., Kang Z., Antczak C., Begue D., et al. TMEM41B is a novel regulator of autophagy and lipid mobilization. EMBO Rep. 2018;19:e45889. doi: 10.15252/embr.201845889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishita H., Zhao Y.G., Tamura N., Nishimura T., Kanda Y., Sakamaki Y., Okazaki M., Li D., Mizushima N. A critical role of VMP1 in lipoprotein secretion. eLife. 2019;8:e48834. doi: 10.7554/eLife.48834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita K., Hama Y., Mizushima N. TMEM41B functions with VMP1 in autophagosome formation. Autophagy. 2019;15:922–923. doi: 10.1080/15548627.2019.1582952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller P., Engeler L., Vavassori L., Suter T., Guidi V., Gschwind M., Tonolla M., Flacio E. Surveillance of invasive Aedes mosquitoes along Swiss traffic axes reveals different dispersal modes for Aedes albopictus and Ae. japonicus. PLoS Negl. Trop. Dis. 2020;14:e0008705. doi: 10.1371/journal.pntd.0008705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neufeldt C.J., Cortese M., Acosta E.G., Bartenschlager R. Rewiring cellular networks by members of the Flaviviridae family. Nat. Rev. Microbiol. 2018;16:125–142. doi: 10.1038/nrmicro.2017.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo N., Henthorn K.S., Cisneros M.I., Cubitt B., Iwasaki M., de la Torre J.C., Lama J. Identification and Mechanism of Action of a Novel Small-Molecule Inhibitor of Arenavirus Multiplication. J. Virol. 2015;89:10924–10933. doi: 10.1128/JVI.01587-15. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Ngo A.M., Shurtleff M.J., Popova K.D., Kulsuptrakul J., Weissman J.S., Puschnik A.S. The ER membrane protein complex is required to ensure correct topology and stable expression of flavivirus polyproteins. eLife. 2019;8:e48469. doi: 10.7554/eLife.48469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul D., Bartenschlager R. Flaviviridae Replication Organelles: Oh, What a Tangled Web We Weave. Annu. Rev. Virol. 2015;2:289–310. doi: 10.1146/annurev-virology-100114-055007. [DOI] [PubMed] [Google Scholar]
- Popovic D., Dikic I. TBC1D5 and the AP2 complex regulate ATG9 trafficking and initiation of autophagy. EMBO Rep. 2014;15:392–401. doi: 10.1002/embr.201337995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovic D., Akutsu M., Novak I., Harper J.W., Behrends C., Dikic I. Rab GTPase-activating proteins in autophagy: regulation of endocytic and autophagy pathways by direct binding to human ATG8 modifiers. Mol. Cell. Biol. 2012;32:1733–1744. doi: 10.1128/MCB.06717-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajah M.M., Monel B., Schwartz O. The entanglement between flaviviruses and ER-shaping proteins. PLoS Pathog. 2020;16:e1008389. doi: 10.1371/journal.ppat.1008389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Realegeno S., Puschnik A.S., Kumar A., Goldsmith C., Burgado J., Sambhara S., Olson V.A., Carroll D., Damon I., Hirata T., et al. Monkeypox Virus Host Factor Screen Using Haploid Cells Identifies Essential Role of GARP Complex in Extracellular Virus Formation. J. Virol. 2017;91:e00011-17. doi: 10.1128/JVI.00011-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riblett A.M., Blomen V.A., Jae L.T., Altamura L.A., Doms R.W., Brummelkamp T.R., Wojcechowskyj J.A. A Haploid Genetic Screen Identifies Heparan Sulfate Proteoglycans Supporting Rift Valley Fever Virus Infection. J. Virol. 2015;90:1414–1423. doi: 10.1128/JVI.02055-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roehrig J.T., Layton M., Smith P., Campbell G.L., Nasci R., Lanciotti R.S. The emergence of West Nile virus in North America: ecology, epidemiology, and surveillance. Curr. Top. Microbiol. Immunol. 2002;267:223–240. doi: 10.1007/978-3-642-59403-8_11. [DOI] [PubMed] [Google Scholar]
- Ropolo A., Grasso D., Pardo R., Sacchetti M.L., Archange C., Lo Re A., Seux M., Nowak J., Gonzalez C.D., Iovanna J.L., Vaccaro M.I. The pancreatitis-induced vacuole membrane protein 1 triggers autophagy in mammalian cells. J. Biol. Chem. 2007;282:37124–37133. doi: 10.1074/jbc.M706956200. [DOI] [PubMed] [Google Scholar]
- Rozen-Gagnon K., Yi S., Jacobson E., Novack S., Rice C.M. A selectable, plasmid-based system to generate CRISPR/Cas9 gene edited and knock-in mosquito cell lines. bioRxiv. 2020 doi: 10.1101/2020.10.09.333641. [DOI] [Google Scholar]
- Sanjana N.E., Shalem O., Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savidis G., McDougall W.M., Meraner P., Perreira J.M., Portmann J.M., Trincucci G., John S.P., Aker A.M., Renzette N., Robbins D.R., et al. Identification of Zika Virus and Dengue Virus Dependency Factors using Functional Genomics. Cell Rep. 2016;16:232–246. doi: 10.1016/j.celrep.2016.06.028. [DOI] [PubMed] [Google Scholar]
- Scaturro P., Stukalov A., Haas D.A., Cortese M., Draganova K., Płaszczyca A., Bartenschlager R., Götz M., Pichlmair A. An orthogonal proteomic survey uncovers novel Zika virus host factors. Nature. 2018;561:253–257. doi: 10.1038/s41586-018-0484-5. [DOI] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider W.M., Luna J.M., Hoffmann H.-H., Sánchez-Rivera F.J., Leal A.A., Ashbrook A.W., Le Pen J., Ricardo-Lax I., Michailidis E., Peace A., et al. Genome-scale identification of SARS-CoV-2 and pan-coronavirus host factor networks. Cell. 2020;184 doi: 10.1016/j.cell.2020.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins J.W., Dorner M., Feulner M., Imanaka N., Murphy M.Y., Ploss A., Rice C.M. Dengue reporter viruses reveal viral dynamics in interferon receptor-deficient mice and sensitivity to interferon effectors in vitro. Proc. Natl. Acad. Sci. USA. 2012;109:14610–14615. doi: 10.1073/pnas.1212379109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins J.W., MacDuff D.A., Imanaka N., Gainey M.D., Shrestha B., Eitson J.L., Mar K.B., Richardson R.B., Ratushny A.V., Litvak V., et al. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature. 2014;505:691–695. doi: 10.1038/nature12862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S.L., Conn G.L. RNA regulation of the antiviral protein 2′-5′-oligoadenylate synthetase. Wiley Interdiscip. Rev. RNA. 2019;10:e1534. doi: 10.1002/wrna.1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O., Sanjana N.E., Hartenian E., Shi X., Scott D.A., Mikkelson T., Heckl D., Ebert B.L., Root D.E., Doench J.G., Zhang F. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan C., Xie X., Muruato A.E., Rossi S.L., Roundy C.M., Azar S.R., Yang Y., Tesh R.B., Bourne N., Barrett A.D., et al. An Infectious cDNA Clone of Zika Virus to Study Viral Virulence, Mosquito Transmission, and Antiviral Inhibitors. Cell Host Microbe. 2016;19:891–900. doi: 10.1016/j.chom.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X., van Mierlo J.T., French A., Elliott R.M. Visualizing the replication cycle of bunyamwera orthobunyavirus expressing fluorescent protein-tagged Gc glycoprotein. J. Virol. 2010;84:8460–8469. doi: 10.1128/JVI.00902-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker C.J., Huang T.Q., Weir N.R., Polyakov N.J., Schultz S.W., Denic V. CRISPR screening using an expanded toolkit of autophagy reporters identifies TMEM41B as a novel autophagy factor. PLoS Biol. 2019;17:e2007044. doi: 10.1371/journal.pbio.2007044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidjanin D.J., Park A.K., Ronchetti A., Martins J., Jackson W.T. TBC1D20 mediates autophagy as a key regulator of autophagosome maturation. Autophagy. 2016;12:1759–1775. doi: 10.1080/15548627.2016.1199300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sklan E.H., Serrano R.L., Einav S., Pfeffer S.R., Lambright D.G., Glenn J.S. TBC1D20 is a Rab1 GTPase-activating protein that mediates hepatitis C virus replication. J. Biol. Chem. 2007;282:36354–36361. doi: 10.1074/jbc.M705221200. [DOI] [PubMed] [Google Scholar]
- Tábara L.C., Escalante R. VMP1 Establishes ER-Microdomains that Regulate Membrane Contact Sites and Autophagy. PLoS ONE. 2016;11:e0166499. doi: 10.1371/journal.pone.0166499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanida I., Waguri S. Measurement of autophagy in cells and tissues. Methods Mol. Biol. 2010;648:193–214. doi: 10.1007/978-1-60761-756-3_13. [DOI] [PubMed] [Google Scholar]
- Tanida I., Mizushima N., Kiyooka M., Ohsumi M., Ueno T., Ohsumi Y., Kominami E. Apg7p/Cvt2p: A novel protein-activating enzyme essential for autophagy. Mol. Biol. Cell. 1999;10:1367–1379. doi: 10.1091/mbc.10.5.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanida I., Ueno T., Kominami E. LC3 and Autophagy. Methods Mol. Biol. 2008;445:77–88. doi: 10.1007/978-1-59745-157-4_4. [DOI] [PubMed] [Google Scholar]
- Tsetsarkin K., Higgs S., McGee C.E., De Lamballerie X., Charrel R.N., Vanlandingham D.L. Infectious clones of Chikungunya virus (La Réunion isolate) for vector competence studies. Vector Borne Zoonotic Dis. 2006;6:325–337. doi: 10.1089/vbz.2006.6.325. [DOI] [PubMed] [Google Scholar]
- Wang X., Li M., Zheng H., Muster T., Palese P., Beg A.A., García-Sastre A. Influenza A virus NS1 protein prevents activation of NF-kappaB and induction of alpha/beta interferon. J. Virol. 2000;74:11566–11573. doi: 10.1128/jvi.74.24.11566-11573.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X., Boehm J.S., Yang X., Salehi-Ashtiani K., Hao T., Shen Y., Lubonja R., Thomas S.R., Alkan O., Bhimdi T., et al. A public genome-scale lentiviral expression library of human ORFs. Nat. Methods. 2011;8:659–661. doi: 10.1038/nmeth.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau E.H., Rana T.M. Next-Generation Sequencing of Genome-Wide CRISPR Screens. Methods Mol. Biol. 2018;1712:203–216. doi: 10.1007/978-1-4939-7514-3_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R., Simoneau C.R., Kulsuptrakul J., Bouhaddou M., Travisano K.A., Hayashi J.M., Carlson-Stevermer J., Zengel J.R., Richards C.M., Fozouni P., et al. Genetic Screens Identify Host Factors for SARS-CoV-2 and Common Cold Coronaviruses. Cell. 2020;184 doi: 10.1016/j.cell.2020.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO Yellow fever in Africa and the Americas, 2016. Wkly Epidemiol Rec. 2017;92:442–452. [PubMed] [Google Scholar]
- Zaki A.M. Isolation of a flavivirus related to the tick-borne encephalitis complex from human cases in Saudi Arabia. Trans. R. Soc. Trop. Med. Hyg. 1997;91:179–181. doi: 10.1016/s0035-9203(97)90215-7. [DOI] [PubMed] [Google Scholar]
- Zhang L., Bukreyev A., Thompson C.I., Watson B., Peeples M.E., Collins P.L., Pickles R.J. Infection of ciliated cells by human parainfluenza virus type 3 in an in vitro model of human airway epithelium. J. Virol. 2005;79:1113–1124. doi: 10.1128/JVI.79.2.1113-1124.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R., Miner J.J., Gorman M.J., Rausch K., Ramage H., White J.P., Zuiani A., Zhang P., Fernandez E., Zhang Q., et al. A CRISPR screen defines a signal peptide processing pathway required by flaviviruses. Nature. 2016;535:164–168. doi: 10.1038/nature18625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.G., Chen Y., Miao G., Zhao H., Qu W., Li D., Wang Z., Liu N., Li L., Chen S., et al. The ER-Localized Transmembrane Protein EPG-3/VMP1 Regulates SERCA Activity to Control ER-Isolation Membrane Contacts for Autophagosome Formation. Mol Cell. 2017;67:974–989.e6. doi: 10.1016/j.molcel.2017.08.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the sgRNA sequencing data reported in this paper is GEO: GSE162040. Original data for all virus infection experiments in the paper are available upon request.