

Abstract
Though significant efforts are in progress for developing drugs and vaccines against COVID-19, limited therapeutic agents are available currently. Thus, it is essential to undertake COVID-19 research and to identify therapeutic interventions in which computational modeling and virtual screening of lead molecules provide significant insights. The present study aimed to predict the interaction potential of natural lead molecules against prospective protein targets of SARS-CoV-2 by molecular modeling, docking, and dynamic simulation. Based on the literature survey and database search, fourteen molecular targets were selected and the three targets which lack the native structures were computationally modeled. The drug-likeliness and pharmacokinetic features of ninety-two natural molecules were predicted. Four lead molecules with ideal drug-likeliness and pharmacokinetic properties were selected and docked against fourteen targets, and their binding energies were compared with the binding energy of the interaction between Chloroquine and Hydroxychloroquine to their usual targets. The stabilities of selected docked complexes were confirmed by MD simulation and energy calculations. Four natural molecules demonstrated profound binding to most of the prioritized targets, especially, Hyoscyamine and Tamaridone to spike glycoprotein and Rotiorinol-C and Scutifoliamide-A to replicase polyprotein-1ab main protease of SARS-CoV-2 showed better binding energy, conformational and dynamic stabilities compared to the binding energy of Chloroquine and its usual target glutathione-S-transferase. The aforementioned lead molecules can be used to develop novel therapeutic agents towards the protein targets of SARS-CoV-2, and the study provides significant insight for structure-based drug development against COVID-19.
Keywords: COVID-19, SARS-CoV-2, Computational virtual screening, Hyoscyamine, Rotiorinol-C, Scutifoliamide-A, Tamaridone, Molecular dynamic simulation
1. Introduction
Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV-2) is responsible for Coronavirus disease-2019 (COVID-19), formerly known as nCoV-2019, which was first reported in the city of Wuhan, China in December 2019 [1]. Globally, 116 million confirmed cases and 2.58 million deaths have been reported as of the first week of March 2021 as per the latest reports of the World Health Organization (WHO) [2]. The WHO declared COVID-19 as a pandemic due to the impact of a global outbreak, and the severity of the infection caused by SARS-CoV-2 [3]. It is one of the challenging outbreaks the world has ever experienced, and most of the developed countries are at the risk of community outbreaks [[4], [5], [6]]. While there were several asymptomatic cases reported, the infection mainly characterized by the symptoms such as fever, muscle and body aches, cough, sore throat, shortness of breath for about 5–6 days after the infection, whereas, it causes a severe form of pneumonia in the elderly and immune-compromised people that resulted in worldwide panic leading to an international concern [[7], [8], [9], [10]]. Presently, more than 90% of the deaths are reported due to COVID-19, and the major countries with high mortality rates reported include the United States of America, India, Brazil, Russian Federation, The United Kingdom, France, Spain, Italy, Turkey, Germany, Colombia and Argentina [2]. Currently, there are limited therapeutic agents available for the treatment of COVID-19, which demonstrates the severity of the infection. Thus, it is essential to undertake COVID-19 research and to screen putative drug targets and lead molecules for the development of therapeutic strategies towards COVID-19.
The surface spike proteins (S), membrane proteins (M), envelope proteins (E), main protease-replicase polyproteins (rep and protein 1a), and few non-structural proteins (nsp6, nsp8, and nsp10) are responsible for the pathogenesis mechanism of coronavirus [10]. Spike glycoproteins protruding on the surface of the virus mediates the viral entry into the host cells [11,12]. Five regions were identified in spike proteins (residues 274–306, 510–586, 587–628, 784–803, and 870–893) that represent the regions associated with the high rate of an immune response [13]. The nucleoprotein such as replicase polyprotein and RNA-dependent RNA polymerase (RdRp) played a pivotal role in the virus replication and form a ribonucleoprotein complex with the viral RNA through the N-terminal domain (N-NTD) of protein N. The membrane protein involved in the protein-protein interactions required for assembly of coronaviruses, and determined as a protective antigen in humoral responses which functions as an ion channel [14]. Replicase polyproteins such as main protease (Mpro) and papain-like protease (PLpro) are involved in the cleavage of polyproteins, and non-structural proteins (nsp) are involved in the assembly of replicase-transcriptase complex (RTC) [[14], [15], [16]]. Therefore, these proteins can be considered as potential targets to develop therapeutic agents against SARS-CoV-2.
The drugs that were suggested in the treatment of COVID-19 include Hydroxychloroquine, Chloroquine, Ritonavir, Lopinavir, and Remdesvir, which are used for the treatment of Malaria, AIDS, Ebola, and Influenza [17,18]. Hence, there is a need to screen more reliable approaches to combat the infections caused by SARS-CoV-2. Computational biology studies are one of the promising approaches to reduce the time and cost of various stages of drug discovery when compared to the conventional methods of drug development. The major approaches of computational biology in drug discovery include molecular modeling of potential targets, computer-aided virtual screening of novel lead molecules, and molecular dynamic simulation studies of the target-lead complexes. These methods can be used to screen potential lead molecules against SARS-CoV-2. Several studies have demonstrated that the bioactive molecules from natural sources can be used to treat several viral infections because the natural compounds possess fewer side effects, ideal drug likeliness, pharmacokinetic and toxicity properties, and their binding potential to the molecular targets [19,20].
The present study aimed to predict the binding potential of selected natural leads against multiple protein targets of SARS-CoV-2 when compared to the binding of two currently suggested drugs to their usual targets by computational virtual screening, molecular modeling, and dynamic simulation studies.
2. Materials and methods
2.1. Identification of probable molecular targets
The drug targets of SARS CoV – 2 were identified based on the literature survey, database search, and knowledge on their role in pathogenicity especially mediating host cell and replication of RNA. The major targets involved in the virulence mechanism of SARS-CoV-2 are spike glycoproteins, replicase, membrane proteins, envelope proteins, and non-structural proteins. A total of fourteen probable drug targets, namely protein S, E, M5, rep, 1a, 7a, 3a, nsp6, nsp7, nsp8, 9b, nsp10, nsp12, and receptor-binding domain (RBD) were identified as potential molecular targets. Out of fourteen targets identified, the three-dimensional (3D) structures of S, E, M5, rep, 1a, Protein7a, Protein9b, nsp7, nsp8, nsp12, and RBD are available in their native forms. The structural details of the selected targets are shown in Table 1 . The 3D structures of spike glycoprotein available in post-fusion (PDB: 1WYY; resolution: 2.2 Å) [22], open state (PDB: 6VYB; resolution 3.2 Å) [10] and closed state (PDB: 6VXX; resolution 2.8 Å) [10] were retrieved from protein data bank (PDB) [21]. Similarly, the structures of the main protease (Mpro) (PDB: 1Q2W; resolution 1.8 Å) [23], Mpro (PDB: 5RE4; resolution 1.88 Å) [24], membrane protein (chain C, F) (PDB: 3I6G; resolution 2.201 Å) [25], envelope protein (PDB: 2MM4) [26], Protein7a (PDB: 1XAK; resolution 1.8 Å) [27], Protein9b (PDB: 2CME; resolution 2.8 Å) [28], ribosome binding protein (RBD) (PDB: 6M17; resolution 2.9 Å) [29], RNA dependent RNA polymerase (RdRp) (PDB: 6M71; resolution 2.9 Å) [30] with chain A (non-structural protein 12; nsp12), chain B and D (non-structural protein 7; nsp7), and C (non-structural protein 8; nsp8) were selected as a drug target.
Table 1.
The structural description of eleven probable drug targets (with known 3D structures) that are involved in the virulence and pathogenesis of SARS-CoV-2.
| PDB ID | Name of the protein | Chain | Resolution | R-Value Free | R-Value Work | Experimental method | Secondary structure | References |
|---|---|---|---|---|---|---|---|---|
| 1WYY | Post-fusion hairpin conformation of the spike glycoprotein | A, B | 2.2 Å | 0.249 | 0.210 | X-ray diffraction | 66% helical | Duquerroy et al., 2005 |
| 6VYB | SARS-CoV-2 spike ectodomain structure (open state) | A, B, C | 3.2 Å | Aggregation state: Particle | Reconstruction method: Single particle | Electron Microscopy | 16% helical, 25% beta sheet | Walls et al., 2020 |
| 6VXX | Structure of the SARS-CoV-2 spike glycoprotein (closed state) | A, B, C | 2.8 Å | Aggregation state: Particle | Reconstruction method: Single particle | Electron Microscopy | 16% helical, 25% beta sheet | Walls et al., 2020 |
| 1Q2W | X-Ray Crystal Structure of the SARS Coronavirus Main Protease | A, B | 1.86 Å | 0.249 | 0.194 | X-ray diffraction | 24% helical, 28% beta sheet | Bonanno et al., 2003 |
| 5RE4 | Crystal Structure of SARS-CoV-2 main protease in complex with Z1129283193 | A | 1.88 Å | 0.266 | 0.199 | X-ray diffraction | 26% helical, 28% beta sheet | Fearon et al., to be published |
| 3I6G | Newly identified epitope Mn2 from SARS-CoV M protein complexed withHLA-A*0201 | C, F | 2.201 Å | 0.246 | 0.205 | X-ray diffraction | 26% helical, 39% beta sheet | Liu et al., 2010 |
| 2MM4 | Structure of a Conserved Golgi Complex-targeting Signal in Coronavirus Envelope Proteins | A | – | Conformers Calculated: 200 | Conformers Submitted: 15 | NMR | 65% helical | Li et al., 2014 |
| 1XAK | Structure of the sars-coronavirus orf7a accessory protein | A | 1.8 Å | 0.275 | 0.223 | X-ray diffraction | 48% beta sheet | Nelson et al., 2005 |
| 2CME | The crystal structure of SARS coronavirus ORF-9b protein | A | 2.8 Å | 0.289 | 0.266 | X-ray diffraction | 3% helical, 44% beta sheet | Meier et al., 2006 |
| 6M17 | The 2019-nCoV RBD/ACE2-B0AT1 complex | E, F | 2.9 Å | Aggregation state: Particle | Reconstruction method: Single particle | Electron Microscopy | 3% helical, 23% beta sheet | Yan et al., 2020 |
| 6M71 | SARS-Cov-2 RNA-dependent RNA polymerase in complex with cofactors | A, B. C, D | 2.9 Å | Aggregation state: Particle | Reconstruction method: Single particle | Electron Microscopy | 47% helical, 17% beta sheet | Gao et al., 2020 |
2.2. Model building and validation
Out of fourteen identified targets, the 3D structures of three proteins namely Protein3a, nsp6, and nsp10 were not reported in their native forms. Thus, the amino acid sequences of Protein3a (UniProt ID: P59632), nsp6 (UniProt ID: P59634), and nsp10 (UniProt ID: P0C6U8) were retrieved from UniProt KB [31], and the 3D structures of these proteins were predicted from the basic amino acid sequences by ab initio (de novo) modelling by C–I-Tasser [32]. The 3D models of three targets were energy minimized by ModRefiner [33]. The secondary structures of the hypothetical models were assessed by STRIDE [34]. The hypothetical models were analyzed and computationally validated by ProSa [35], Verify3D [36], ProCheck [37], ERRAT [38], and ANOLEA [39] in terms of the reliability, model quality, stereochemical quality (Ramachandran plot), quality factor (error function), structural and force field parameters, respectively.
2.3. Identification and virtual screening of natural lead molecules
Based on the relevance of natural compounds in medicinal chemistry research, the lead molecules present in various natural sources like plants and microorganisms with antiviral properties were identified by literature survey and database search. The details of these compounds are shown in Supplementary Material, Table S1. A total of ninety-two natural compounds were identified, and their 3D structures were retrieved from PubChem [40] and Chemspider [41] databases in.sdf and. mol formats, respectively (Supplementary Materials, Table S1). The retrieved sdf files were converted into.pdb by Open Babel [42]. The drug likeliness, pharmacokinetics (adsorption, distribution, metabolism, excretion -ADME), and toxicity properties were predicted using the molecular descriptors, filters, and statistical models available at PreADMET [43] and SwissADME [44]. The molecular filters used to predict drug likeliness included Lipinski's rule of five [45], Comprehensive Medicinal Chemistry (CMC) rule, Egan's rule [46], MDL Drug Data Report (MDDR) like rule [47], World Drug Index (WDI) like rule, Ghose filter [48], lead-like rule [49] and bioavailability score [50]. The statistical models such as blood-brain barrier (BBB) [51], caco2 cell permeability [52], human intestinal absorption (HIA) [53], lipophilicity [54], and water solubility [55] were predicted by PreADMET and SwissADME. The statistical models such as daphnia toxicity, Ames test [56], algae test, hERG inhibition, carcinogenicity in rats and mice, Minnow and Medaka fish toxicity and mutagenicity in terms of TA100_NA (-S9), TA1535_10RLI (-S9), TA100_10RLI (þS9), and TA1535_NA (þS9) strain of Salmonella typhimurium were used to predict the toxicity features. The molecules that qualified the features required for drug likeliness, pharmacokinetics, and toxicity were selected.
2.4. Molecular docking studies
Four potential molecules that qualified the drug-likeliness, ADME, and toxicity properties were selected, and the binding potentials of these molecules to fourteen selected targets of SARS CoV-2 were predicted using molecular docking by AutoDock tools and AutoDock Vina [57]. The binding sites of selected target proteins were predicted by Depth [58] and CastP [59]. The protein targets were prepared by adding polar hydrogen atoms and Gasteiger partial charges. The ligands were prepared by setting the number of torsions and detecting the root atoms. The ligand and receptor files were written in. PDBQT format. The grid box for each target was prepared based on the dimensions of x, y, z coordinates of the binding pocket, and the configuration file was prepared. Each ligand molecule was docked against the selected target, and the output log files were prepared. Out of various docked conformations, the best-docked poses of the complexes were selected based on the binding energy (kcal/mol), cluster RMSD, number of hydrogen bonds, and other weak interactions, which stabilized the docked complexes. The binding energies associated with the interaction between natural molecules and selected targets were compared with the binding energy of the interaction between Chloroquine and Hydroxychloroquine towards their usual targets namely glutathione S transferase (PDB: 1OKT) of Plasmodium falciparum and human angiotensin-converting enzyme (ACE)-2 (PDB: 1R42), respectively. The results of these docked complexes were considered as the control for the comparison of the binding potential of natural molecules and the selected targets.
2.5. Molecular dynamic simulation studies
The molecular dynamic (MD) simulation studies were performed to confirm the stabilities of the best-docked complexes of ligands and their targets along with the interaction of Chloroquine and glutathione S transferase. The MD simulation was performed by the Desmond module of Schrödinger's suite [60]. The protein-ligand complexes were subjected to pre-processing and hydrogen bond assignment was performed with standard parameters. The simulation system was prepared by utilizing the system builder. TIP3P was selected as the solvent model, and the boundary conditions were defined by an orthorhombic box with minimized volume encapsulating the complex. OPLS3e was used as a force field, and the system neutralized by adding Cl− or Na+ ions based on the total charges of the system. The MD simulations were performed at 300 K using the constant number of particles, pressure, and temperature (NPT) ensembles for 100 ns at an interval of 1 ns? The model was unfitted for 10ns before performing the MD simulation. The time-step adjusted to 2 fs (fs), a short-range cut-off with a radius of 9.0 Å optimized, and no restraints were pre-defined. The simulation showed a temperature increase of 10K per time-step after the solvation of the binding pocket. The changes in RMS and protein-ligand contacts during simulation were generated by the simulation interaction diagram tool.
The trajectories of RMSD and RMSF of proteins and ligands were estimated using the following formula:
Where N is the number of atoms in the atom selection; tref is the reference time, (first frame used as reference, time t = 0); and r’ is the position of the selected atoms in frame x after superimposing on the reference frame, where frame x is recorded at time tx.
Where, T is the trajectory time over which the RMSF calculated, tref is the reference time (time t = 0); r is the position of an atom in the reference at time tref, and r’ is the position of atom at time t after superimposing on the reference frame.
The protein-ligand RMSD, protein and ligand RMSF, protein-ligand contacts, ligand properties, and ligand torsion profile trajectories were analyzed using the simulation interaction diagram tool.
2.6. Molecular mechanics Poisson-Boltzmann surface area (MM-PBSA) calculations
The calculation of model affinities of each docked complex (protein-ligand interactions) after a minimal simulation period of 1ns performed by GROMACS [[61], [62], [63]] using CHARMM36 all-atom force field. CGenFF (Force field generator) was used to assign the parameters and charges to ligand to generate an index file. A solvation dodecahedron box generated with TIP3P solvent, and ions were added to neutralize the system charges. The energy minimization was carried out at 0.5 ns? Equilibration of the number of particles, volume, and temperature (NVT) ensemble was carried out before producing molecular dynamics of 1 ns? The trajectory files generated were used as the input files. The calculation of energy by MMPBSA carried out using the following command:
g_mmpbsa -f traj.xtc -s topol.tpr -n index.ndx -mme -mm energy_MM.xvg
The output was reported in terms of binding energy (kcal/mol).
3. Results and discussion
3.1. Prospective molecular targets
Fourteen target proteins namely protein S, M, E, rep, 1a, 3a, 7a, 9b, nsp6, nsp7, nsp8, nsp10, nsp12, and RBD were identified based on an extensive literature survey and database search. Eleven targets possessed native structures (Table 1), and three targets, which lack the 3D structures were computationally modeled (Table 2 ). Spike glycoprotein S (PDB: 1WYY, 6VXX, 6VYB) was identified to be one of the most important targets due to its involvement in mediating host transfer by attaching the virion to the cell surface, and interacting with the host, and promote the infection [64,65]. The post-fusion conformation of the spike glycoprotein is a hairpin structure comprised of two heptad repeats that are stabilized by Asparagine and Glutamine residues, and form hydrogen bonds between the two heptad domains. The open and closed conformations of the surface spike glycoprotein act as an important part of interaction with the host cell surface and, three human Angiotensin-Converting Enzyme 2 (hACE2) motifs were observed in the closed conformation, thus, the conformation is important for the interaction with hACE2 at host cell surface initiating the viral entry [10]. The receptor-binding domain of the membrane protein interacted with the human cellular receptor hACE2 that recognizes SARS CoV-2 [29]. The membrane protein M is involved in morphogenesis, envelope membrane protein E induces apoptosis by activating host inflammasome, and replicase protein rep involved in transcription and replication of viral RNA, which contain proteinases that cleave the polyprotein [66,67]. Replicase polyprotein 1a is a proteinase that cleaves the N and C- terminal of replicase polyprotein and involved in suppressing the host gene expression [68]. RNA-dependent RNA polymerase consists of three non-structural proteins namely nsp7, nsp8, and nsp12 [30]. Protein3a induces apoptosis [69], protein 7a involved in the viral replication in cell culture [70], Protein9b is a lipid-binding protein, non-structural protein6 (nsp6) initiates induction of autophagosomes, non-structural protein8 (nsp8) acts as primase in the viral replication and non-structural protein10 (nsp10) involved in viral mRNA cap methylation [[71], [72], [73]]. Therefore, based on the functional role and virulence mechanisms, these proteins were considered to be putative molecular targets for drug screening in the present study.
Table 2.
The description of computational validation of three probable drug targets of SARS-CoV-2 that are involved in the pathogenesis mechanism of coronavirus modeled by ab initio approach using the C–I Tasser server.
| UniProt ID | Organism | Entry names | Protein names | Gene | Function | Length | ProSA (z- score) | ERRAT (overall quality factor) | WHATIF Ramachandran (Z-score) | Verify3D (residues having averaged 3D-1D score ≥ 0.2) | Anolea (e/kt) | ProCheck |
||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Favored region | Allowed region | Outlier region | ||||||||||||
| P59632 | Human SARS coronavirus (SARS-CoV) | AP3A_CVHSA | Protein 3a | 3a | induces apoptosis | 274 | −3.33 | 39.62 | −5.763 | 50.82% | 359 | 69% | 35.8% | 5.2% |
| P0C6U8 | Human SARS coronavirus (SARS-CoV) | R1A_CVHSA | Non-structural protein 10 | Nsp10 | essential for viral mRNA cap methylation | 139 | −4.88 | 30.71 | −6.042 | 63.64% | 871 | 68.8% | 27.9% | 3.2% |
| P59634 | Human SARS coronavirus (SARS-CoV) | NS6_CVHSA | Non-structural protein 6 | Nsp6 | induction of autophagosomes | 63 | −5.67 | 86.84 | 0.783 | 64.65% | −528 | 89.3% | 9.1% | 1.6% |
3.2. Hypothetical model building and validation of selected targets
The 3D structures of target proteins such as Protein3a, non-structural protein6, and non-structural protein10, which lack native structures were computationally predicted. The sequence size of Protein3a, 6, and 10 were 274, 63, and 139 amino acids, respectively. The 3D structures of these sequences were predicted by C–I-Tasser, which utilizes deep-convolutional neural-network-based contact-map prediction [32].
The theoretical model of Protein 3a is shown in Fig. 1 a. The energy of the modeled protein predicted by Anolea was found to be 1716e/kt, and energy was minimized to 879e/kt by ModRefiner (Table 2). The secondary structure of modeled protein comprised of 26.18, 29.82, 10.18, 33.82 alpha-helices, beta-sheet, beta turns, and coils, respectively (Fig. 1b). Ramachandran plot of the model suggested that 69%, 35.8%, and 5.2% residues were located in the most favored region, additionally allowed region, and outlier region of the plot, respectively (Fig. 1c). The stereo-chemical validation in terms of z-score obtained from ProSA was found to be −4.88, which is comparable to the z-score of experimental structures (Fig. 1d). The VERIFY3D plot indicated that 63.64% of the residues possessed a 3D-1D score of ≥ 0.2, suggested a good quality model (Fig. 1e). The Z-score predicted by WHATIF showed a value of −6.042 (Table 2). The overall quality of the model is predicted to be 30.71 (Fig. 1f). Thus, based on the computational validation, it is suggested that the hypothetical model of Protein 3a showed good stereochemical validity, secondary structural alignment, and potential energy, which can be used as a molecular target.
Fig. 1.

Computational modeling and model validation of Protein3a (a) Theoretical model of the target protein modeled using C–I-Tasser, (b) Secondary structure prediction of the theoretical model by STRIDE (c) Ramachandran plot assessment of the modeled residues of the target protein obtained by ProCheck (d) Stereo-chemical validation of the theoretical model in terms of z-score obtained from ProSA (e) The residues qualifying the 3D-1D score predicted by Verify 3D (f) The quality factor of the model obtained by the output plot of error function predicted by ERRAT.
The hypothetical model of nsp6 visualized by PyMol is illustrated in Fig. 2 a. The energy of the protein model was minimized from 366e/kt to 359e/kt by ModRefiner. The secondary structure of the model comprised 70.4, 9.84, 8.20, and 11.48% alpha-helices, beta-sheet, beta turns, and coils, respectively (Fig. 2b). A Ramachandran plot of the model is shown in Fig. 2c, which revealed that 89.3%, 9.1%, 1.6% of the residues were present in the most favored region, additionally allowed regions, and outlier regions of the plot, respectively. The stereo-chemical validation by ProSA in terms of Z-score was found to be −3.33, a score closer to the z-score of native structures (Fig. 2d). The plot obtained by VERIFY3D indicated that 50.82% of the residues showed a 3D-1D score of ≥ 0.2 (Fig. 2e). The Z-score predicted by WHATIF was found to be −5.67 (Table 2). The overall model quality was found to be 39.62 (Fig. 2f). From the computational prediction, it was evident that the hypothetical model demonstrated structural and stereochemical qualities, thus, the model can consider as a potential molecular target.
Fig. 2.

Computational modeling and model validation of Protein6 (a) Theoretical model of the target protein modeled using C–I-Tasser, (b) Secondary structure prediction of the theoretical model by STRIDE (c) Ramachandran plot assessment of the modeled residues of the target protein obtained by ProCheck (d) Stereo-chemical validation of the theoretical model in terms of z-score obtained from ProSA (e) The residues qualifying the 3D-1D score predicted by Verify 3D (f) The quality factor of the model obtained by the output plot of error function predicted by ERRAT.
Similarly, the hypothetical model of nsp10 is shown in Fig. 3 a. The energy of the protein model was minimized from −636e/kt to −740e/kt (Table 2). The secondary structure of the modeled protein comprised 19.4, 25.9, 11.51, and 43.17% of alpha-helices, beta-sheet, beta turns, and coils, respectively (Fig. 3b). Ramachandran plot of the model predicted by ProCheck showed that 68.8, 27.9, and 3.2 residues were located in the favored, allowed, and outlier regions, respectively (Fig. 3c). The stereo-chemical quality obtained from ProSA was found to be −4.05 in terms of Z-score, which was in the range of the z-score of the experimental structures (Fig. 3d). The plot of the theoretical model obtained by VERIFY3D indicated that 70.5% of the residues were found to be a 3D-1D score of ≥ 0.2 (Fig. 3e). The Z-score predicted by WHATIF showed a value of 0.334 (Table 2). The quality of the model validated by ERRAT showed that a quality factor of 93.6 (Fig. 3f). Therefore, the computational validation showed that the 3D model of nsp10 was ideal in terms of stereochemical and structural stabilities, and the model can be considered as a target for the lead screening.
Fig. 3.

Computational modeling and model validation of Protein10 (a) Theoretical model of the target protein modeled using C–I-Tasser, (b) Secondary structure prediction of the theoretical model by STRIDE (c) Ramachandran plot assessment of the modeled residues of the target protein obtained by ProCheck (d) Stereo-chemical validation of the theoretical model in terms of z-score obtained from ProSA (e) The residues qualifying the 3D-1D score predicted by Verify 3D (f) The quality factor of the model obtained by the output plot of error function predicted by ERRAT.
3.3. Computational screening of natural lead molecules
The virtual screening of 92 natural compounds from PubChem and ChemSpider databases showed that 48 of them were qualified for the drug-likeliness predicted by various filters available at PreADMET and SwissADME. The predicted drug-likeliness of natural lead molecules is shown in Supplementary Material, Table S2. When 48 lead molecules were subjected to ADME prediction, 16 compounds demonstrated ideal skin and caco2 cell permeabilities, human intestinal absorption, blood-brain barrier penetration, gastrointestinal absorption, lipophilicity, and water-solubility. The ADME features of the selected molecules are shown in Supplementary Material, Table S3. When sixteen molecules that qualified the ADME properties were screened for toxicity prediction, four molecules were qualified the toxicity features such as carcinogenicity, mutagenicity, algae and fish toxicity, and hERG inhibition. Thus, out of 92 molecules screened by in silico approach, four molecules were qualified for drug-likeliness, ADME, and toxicity features. The selected molecules were Hyoscyamine, Rotiorinol-C, Scutifoliamide-A, and Tamaridone, which are present in Datura stramonium, Chaetorium cupreum, Piper scutifolium, and Tamarix dioica, respectively. These natural compounds were prioritized as the lead molecules, and their interactions towards fourteen molecular targets of SARS-CoV-2 were computationally modeled and validated.
3.4. Molecular docking studies
The binding potential of selected natural lead molecules namely Hyoscyamine, Rotiorinol-C, Scutifoliamide-A, and Tamaridone towards fourteen molecular targets (S, M, E, rep, 1a, 3a, 7a, 9b, nsp6, nsp7, nsp8, nsp10, nsp12, and RBD) of SARS-CoV-2 were predicted by molecular docking. The best-docked conformations generated were analyzed and scrutinized based on binding energy (kcal/mol), the number of hydrogen bonds and other stabilizing interactions, and cluster RMSD. The binding potentials of four selected natural molecules to the prioritized targets of SARS-CoV-2 were predicted by molecular docking studies are shown in Table 3 .
Table 3.
The binding potential of selected natural lead molecules towards the probable drug targets of human SARS-CoV-2 predicted by molecular docking studies.
| Protein name | PDB ID | Ligand names | Ligand structure | Binding affinity (Kcal/mol) | RMSD (Å) | Interacting residues | Hydrogen bonds |
|---|---|---|---|---|---|---|---|
| Spike glycoprotein | 1WYY | Hyoscyamine Pubchem ID: 154417 Source: Datura stramonium |
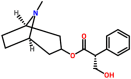 |
−8.14 | 0.0 | Ser924, Thr925, Gly928, Asp932, Gln1161, ILe1164, Asn1168 (Chain A), Ser924, Thr925, Gly928, Gln1161, Ile1164 (Chain B) | Asn1168: 2 |
| Spike glycoprotein-Closed state | 6VXX | −5.7 | 0.0 | Leu335, Pro337, Phe338, Gly339, Asp364, Val367 | 0 | ||
| Spike glycoprotein-Open state | 6VYB | −6.0 | 0.0 | Trp104, Ile119, Val126, Ile128, Tyr170, Ser172, Ile203, Val227 | Ser172: 1 | ||
| Membrane protein | 3I6G | −0.7 | 0.0 | Gly0 | 0 | ||
| Envelope small membrane protein | 2MM4 | −2.2 | 0.0 | Lys63, Asn64 | 0 | ||
| Replicase polyprotein 1 ab | 1Q2W | −6.1 | 0.0 | Glu156, Met165, Asp187, Arg188, Gln189, Gln192 | Gln192 | ||
| Replicase polyprotein 1a | 5RE4 | −6.0 | 0.0 | Trp218, Asn221, Leu271 | Trp218: 1 | ||
| Protein 7a | 1XAK | −4.4 | 0.0 | Tyr3, Tyr5, His47, Gln61 | 0 | ||
| Protein 9b | 2CME | −4.4 | 0.0 | Arg68, Ala69, Phe70 | 0 | ||
| Receptor binding domain of membrane protein | 6M17 | −5.4 | 0.0 | Thr376, Val407, Ala411, Val433, Tyr508 | 0 | ||
| Non-structural protein 7 | 6M71_C | −5.3 | 0.0 | Thr46, Phe49, Val53 | 0 | ||
| Non-structural protein 8 | 6M71_B, D | −5.9 | 0.0 | Leu128, Val130, Thr141, Tyr149 | 0 | ||
| Non-structural protein 12 | 6M71_A | −5.4 | 0.0 | Pro412, Phe415, Phe440, Phe843, | 0 | ||
| Non-structural Protein 6 | Hypothetical model | −5.2 | 0.0 | Asp6, Phe7, Leu15, Leu40, Ile60 | 0 | ||
| Non-structural Protein 10 | Hypothetical model | −6.1 | 0.0 | Ile55, Cys74, Tyr76, His83, Asp91, Leu112, Thr115, Val116 | His83:1 | ||
| Protein 3a | Hypothetical model | −5.3 | 0.0 | Phe56, Ile63, Lys66, Tyr189, Glu191, | 0 | ||
| Spike glycoprotein | 1WYY | Rotiorinol C Pubchem ID: 11703984 Source: Chaetorium cupreum |
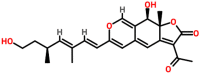 |
−9.82 | 0.0 | Arg4, Lys5, Tyr126, Gln127, Arg131, Asp289 and Glu290 | 5, Lys5, Gln127, Asp 289, Glu290 (A chain) Glu127 (B chain) |
| Spike glycoprotein-Closed state | 6VXX | −6.2 | 0.0 | Arg34, Thr208, Pro209, Leu212, Pro217, Gln218, Phe220 | Phe220: 1 | ||
| Spike glycoprotein-Open state | 6VYB | −6.3 | 0.0 | Thr33, Phe59, Asp287 | 0 | ||
| Membrane protein | 3I6G | −0.6 | 0.0 | Gly0 | 0 | ||
| Envelope small membrane protein | 2MM4 | −2.2 | 0.0 | Lys63, Asn64 | 0 | ||
| Replicase polyprotein 1 ab | 1Q2W | −7.0 | 0.0 | Phe3, Lys5, Arg131, Trp207, Phe291, Ile286, Asp289 | Trp207: 1 | ||
| Replicase polyprotein 1a | 5RE4 | −6.7 | 0.0 | Lys137, Thr199, Tyr239, Leu286, Leu287, Glu288, Asp289 | Tyr239, Asp289: 3 | ||
| Protein 7a | 1XAK | −5.4 | 0.0 | Tyr3, His47, Arg57, Thr59 | 0 | ||
| Protein 9b | 2CME | −5.0 | 0.0 | Ser56, Leu65, Glu66, Ala67, Arg68, Ala69, Phe70, Ser72 | Ser56: 1 | ||
| Receptor binding domain of membrane protein | 6M17 | −6.1 | 0.0 | Cys336, Phe338, Gly339, Ala363, Asp364, Val367, Ser371, Ser373, Phe374, | Ser373: 1 | ||
| Non-structural protein 7 | 6M71_C | −5.9 | 0.0 | Lys2, Asp5, Thr9, Thr46, Phe49, Val53, Met52 | 0 | ||
| Non-structural protein 8 | 6M71_B, D | −6.0 | 0.0 | Pro133, Asp134, Tyr135, Trp182, Pro178 | Tyr135: 1 | ||
| Non-structural protein 12 | 6M71_A | −6.6 | 0.0 | Arg181, Gln184, Asn213 | 0 | ||
| Non-structural protein 6 | Hypothetical model | −5.6 | 0.0 | Gln8, Lys38, Lys42, Leu44 | Lys42: 1 | ||
| Non-structural Protein 10 | Hypothetical model | −6.8 | 0.0 | Ile55, His83, Lys96, Val116 | His83:1 | ||
| Protein 3a | Hypothetical model | −6.3 | 0.0 | Phe28, Val29, Arg68, Ala72, Val90 | 0 | ||
| Spike glycoprotein | 1wyy | Scutifoliamide A Chemspider ID: 23311285 Source: Piper scutifolium |
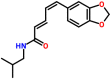 |
−6.4 | 0.0 | Asn910, Ala940, Thr943, Leu944, Gln947, Asn951, Leu1178, Leu1181, Glu1183 | 0 |
| Spike glycoprotein-Closed state | 6VXX | −5.4 | 0.0 | Leu118, Val120, Phe135, Leu141, Leu241 | 0 | ||
| Spike glycoprotein-Open state | 6VYB | −6.6 | 0.0 | Phe823, Asn824, Val826, Pro863, Pro1057, His1058 | Asn824: 1 | ||
| Membrane protein | 3I6G | −0.7 | 0.0 | Gly0 | 0 | ||
| Envelope small membrane protein | 2MM4 | −0.8 | 0.0 | Ly63 | 0 | ||
| Replicase polyprotein 1 ab | 1Q2W | −6.9 | 0.0 | Arg4, Lys5 and Phe291 (A chain), Lys 5 (B chain) | 1; Lys5 (B chain) | ||
| Replicase polyprotein 1a | 5RE4 | −6.8 | 0.0 | Val212, Arg217, Leu220, Gln256, Ile259, Asp263 | 0 | ||
| Protein 7a | 1XAK | −5.6 | 0.0 | Gln6, Cys8, Val9, Thr12, Leu16, Lys17 | Val9, Lys17: 2 | ||
| Protein 9b | 2CME | −4.4 | 0.0 | Ala58, Arg68, Ala69, Phe70, Ser72 | 0 | ||
| Receptor binding domain of membrane protein | 6M17 | −5.9 | 0.0 | Gln493, Tyr495, Gly502, Tyr505 | Gln493, Gly502: 2 | ||
| Non-structural protein 7 | 6M71_C | −5.9 | 0.0 | Asp5, Thr9, Thr46, Phe49, Glu50, Val53 | Thr9: 1 | ||
| Non-structural protein 8 | 6M71_B, D | −6.0 | 0.0 | Leu128, Met129, Pro133, Thr141, Tyr149 | 0 | ||
| Non-structural protein 12 | 6M71_A | −6.1 | 0.0 | Asp484, Ile488, Gln573, Ser578, Ala581 | 0 | ||
| Non-structural Protein 6 | Hypothetical model | −5.5 | 0.0 | Leu16, Ile17, Arg20, Thr21 | Arg20: 1 | ||
| Non-structural Protein 10 | Hypothetical model | −5.6 | 0.0 | His83, Leu92, Asn114, Thr115, Val116 | Val116: 2 | ||
| Protein 3a | Hypothetical model | −5.4 | 0.0 | Cys130, Trp131, His150, His204, His227 | 0 | ||
| Spike glycoprotein | 1wyy | Tamaridone Pubchem ID: 15345466 Source: Tamarix dioica |
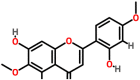 |
−7.2 | 0.0 | Asp931, Asn935, Lys1162, Asp1165, Asn1168, Lys1172 | 1; Asn935 |
| Spike glycoprotein-Closed state | 6VXX | −7.3 | 0.0 | Ala520, Phe559, Phe562, Gln563, Gln564, Phe565, Gly566, Arg567 | Gln564, Phe565, Arg567: 4 | ||
| Spike glycoprotein-Open state | 6VYB | −6.8 | 0.0 | Thr732, Thr778, Ser780, Pro863, His1058 | Ser730: 2 | ||
| Membrane protein | 3I6G | −0.8 | 0.0 | Gly0 | 0 | ||
| Envelope small membrane protein | 2MM4 | −3.1 | 0.0 | Lys63, Asn64 | Asn64: 1 | ||
| Replicase polyprotein 1 ab | 1Q2W | −6.3 | 0.0 | Gly11, Lys12, Glu14, Gly15, | Gly11: 1 | ||
| Replicase polyprotein 1a | 5RE4 | −7.0 | 0.0 | Leu87, Lys137, Thr190, Tyr237, Tyr239, Leu285 | Tyr237: 1 | ||
| Protein 7a | 1XAK | −5.7 | 0.0 | Val9, Thr12, Val14, Arg65 | 0 | ||
| Protein 9b | 2CME | −5.6 | 0.0 | Arg68, Gln71, Ser72 | 0 | ||
| Receptor binding domain of membrane protein | 6M17 | −6.5 | 0.0 | Thr345, Arg346, Ser349, Lys441, Lys444, Asn448, Asn450, Tyr451 | Lys444: 1 | ||
| Non-structural protein 7 | 6M71_C | −5.9 | 0.0 | Lys2, Val6, Thr9, Phe49, Met52 | Thr9: 1 | ||
| Non-structural protein 8 | 6M71_B, D | −5.7 | 0.0 | Leu128, Val130, Val131, Thr141, Tyr149 | 0 | ||
| Non-structural protein 12 | 6M71_A | −6.7 | 0.0 | Lys47, His133, Asn138, Lys780 | 0 | ||
| Non-structural Protein 6 | Hypothetical model | −5.6 | 0.0 | Lys48, Ser50, Leu52, Asp53, Gln56 | Leu52: 1 | ||
| Non-structural Protein 10 | Hypothetical model | −6.7 | 0.0 | Pro37, Asn105, Asp106, Phe110, Lys113, Asn114, Tyr126 | Asn105, Asn114: 2 | ||
| Protein 3a | Hypothetical model | −6.6 | 0.0 | Gln213, Val255, Ile263, Thr270 | 0 |
Hyoscyamine ([(1S,5R)-8-methyl-8-azabicyclo [3.2.1] octan-3-yl] (2S)-3-hydroxy-2-phenyl-propanoate), an alkaloid derivative and anticholinergic, derived from Datura stramonium (Jimson weed) is known to inhibit receptors in smooth and cardiac muscle, the sino-atrial and atrio-ventricular node for a slow ventricular response during atrial fibrillation [74,75]. The molecular docking studies suggested that Hyoscyamine showed good binding potential towards the spike glycoprotein in the post-fusion, closed state, and open state conformations with the binding energies of −8.14, −6.0, and −5.7 kcal/mol, respectively. The interacting residues present in the binding cavity of post-fusion conformation of spike protein were identified to be Ser924, Thr925, Gly928, Asp932, Gln1161, ILe1164, Asn1168 (Chain A) and Ser924, Thr925, Gly928, Gln1161, Ile1164 (Chain B). The interaction stabilized by two hydrogen bonds with Asn1168 of Chain A (Fig. 4 a). It was observed that these residues located at one of the binding sites as predicted by the Castp and Depth servers. Similarly, Trp104, Ile119, Val126, Ile128, Tyr170, Ser172, Ile203, and Val227 were identified to be the major residues in the binding site of the open state conformation of spike protein, and the interaction stabilized by a hydrogen bond (Fig. 4b). The main interacting residues at the binding site of the closed state conformation were identified to be Leu335, Pro337, Phe338, Gly339, Asp364, and Val367 (Fig. 4c). Thus, it is clear that the conformations play a vital role in the binding of the ligand, and maximum interactions were observed between the post-fusion conformation of the spike glycoprotein and the lead molecule in comparison with other conformations. The binding energy of the docked complex of Hyoscyamine and replicase polyprotein 1a was estimated to be −6.1 kcal/mol, and the interacting residues were Glu156, Met165, Asp187, Arg188, Gln189, and Gln192 along with a hydrogen bond (Fig. 4d). Similarly, the lead molecule showed binding energy of −5.4 kcal/mol towards the RBD of the membrane protein. The main interacting residues present in the binding cavity were found to be Thr376, Val407, Ala411, Val433, and Tyr508 (Fig. 4e). Hyoscyamine showed binding energy of −0.7 kcal/mol (interacting residue: Gly) towards membrane protein (Fig. 4f), and the lead molecule exhibited the binding energy of −2.2 kcal/mol towards the envelope protein. Lys63 and Asn64 were found to be the main interacting residues (Fig. 4g). The binding energy of the docked complex of replicase polyprotein1a and Hyoscyamine was estimated to be −6.0 kcal/mol. The main interacting residues were found to be Trp218, Asn221 and Leu271, which formed a hydrogen bond (Fig. 4h). The binding energies of the docked complexes of Protein 3a, 7a, and 9b and ligand were estimated to be −5.3, −4.4, and −4.4 kcal/mol, respectively. The interacting residues of protein 3a included Phe56, Ile63, Lys66, Tyr189, and Glu191 (Fig. 4i). Similarly, interacting residues of protein 7a included Tyr3, Tyr5, His47, Gln61 (Fig. 4j), and the residues present in the binding cavity of protein 9b were found to be Arg68, Ala69, and Phe70 (Fig. 4k). The binding energies of ligand and nsp7, nsp8, and nsp12 of RdRp were estimated to be −5.3, −5.9, and −5.4 kcal/mol, respectively. The nsp7 interacted with the lead molecule via Thr46, Phe49, and Val53 (Fig. 4l). Similarly, the interacting residues present at the binding site of nsp8 were observed to be Leu128, Val130, Thr141, and Tyr149 (Fig. 4m), and the interacting residues present in the binding pocket of nsp12 were found to be Phe412, Phe415, Phe440, and Phe843 (Fig. 4n). The binding energy associated with the interaction between ligand and non-structural protein 6, and 10 were estimated to be −5.2 and −6.1 kcal/mol, respectively. The interacting residues present at the binding site were identified to be Asp6, Phe7, Leu15, Leu40, and Ile60 (Fig. 4o). Similarly, the interacting residues present in the binding pocket of non-structural protein 10 were identified to be Ile55, Cys74, Tyr76, His83, Asp91, Leu112, Thr115, and Val116 along with a hydrogen bond (Fig. 4p). Thus, the interaction modeling of natural lead Hyoscyamine towards the selected targets of SARS-CoV-2 suggested that the highest binding potential observed towards the spike glycoprotein in the post-fusion conformation with a binding energy of −8.14 kcal/mol, and several stabilizing interactions were observed in comparison with other selected molecular targets.
Fig. 4.

Prediction of the binding potential of Hyoscyamine towards the prioritized molecular targets of SARS-CoV by molecular docking visualized in MGL tools of AutoDock. The figure displayed the binding pocket of the ligand-receptor complex. The interacting residues and the ligands are displayed in stick figures. The interacting residues and binding energy are labeled (a) Binding of the ligand and post-fusion conformation of spike glycoprotein (binding energy: −8.14 kcal/mol) (b) Binding of the ligand and open state spike glycoprotein (binding energy: −6.0 kcal/mol) (c) Binding of the ligand and closed state spike glycoprotein (binding energy −5.7 kcal/mol) (d) Binding of the ligand and replicase polyprotein 1 ab (binding energy: −6.1 kcal/mol) (e) Binding of the ligand and receptor binding domain of membrane protein (binding energy: −5.4 kcal/mol) (f) Binding of the ligand and membrane protein (binding energy: −0.7 kcal/mol) (g) Binding of the ligand and small envelope protein (binding energy: −2.2 kcal/mol) (h) Binding of the ligand and replicase polyprotein 1a (binding energy: −6.0 kcal/mol) (i) Binding of the ligand and protein3a (binding energy: −6.1 kcal/mol) (j) Binding of the ligand and protein7a (binding energy: −4.4 kcal/mol) (k) Binding of the ligand and protein 9b (binding energy: −4.4 kcal/mol) (l) Binding of the ligand and non-structural protein7 (binding energy: −5.3 kcal/mol) (m) Binding of the ligand and non-structural protein 8 (binding energy: −5.9 kcal/mol) (n) Binding of the ligand and non-structural protein12 (binding energy: −5.4 kcal/mol) (o) Binding of the ligand and non-structural protein 6 (binding energy: −5.2 kcal/mol) (p) Binding of the ligand and non-structural protein 10 (binding energy: −6.1 kcal/mol).
Rotiorinol-C ((9R,9aR)-3-acetyl-9-hydroxy-6-[(1E,3E,5S)-7-hydroxy-3,5-dimethylhepta-1,3-dienyl]-9a-methyl-9H-furo [3,2-g] isochromen-2-one), an azaphilone derived from the fungus Chaetomium cupreum (Dark-walled mold), which is known to exhibit antimicrobial activities [76]. Rotiorinol-C showed the binding potential to the spike glycoprotein in the post-fusion, closed state, and open state conformations with the binding energies of −6.5, −6.2, and −6.3 kcal/mol, respectively. The main residues present in post-fusion conformation were included Gln931, Asn936, Asn942, Val1157, Asn1159, Gln1161, Asn1168, and Lys1172, and the interaction stabilized by two hydrogen bonds (Fig. 5 a). The interacting residues present in the closed state conformation were observed to be Arg34, Thr208, Pro209, Leu212, Pro217, Gln218, and Phe220. The lead molecule interacted with Phe220 by a hydrogen bond (Fig. 5b). Similarly, the major interacting residues present at the binding site of the open state conformation of spike glycoprotein were identified to be Thr33, Phe59, and Asp287 (Fig. 5c). Rotiorinol-C showed higher interaction with the post-fusion conformation of the spike glycoprotein when compared to the open and closed state conformations. The binding energy of the docked complex of replicase polyprotein 1 ab and the lead molecules were estimated to be −9.82 kcal/mol, and the major residues involved in the binding were observed to be Arg4, Lys5, Tyr126, Gln127, Arg131, Asp289, and Glu290. The interactions were stabilized by five hydrogen bonds (Fig. 5d). The interaction of lead and receptor-binding domain of the membrane protein showed the binding energy of −6.1 kcal/mol, and the major residues involved in the binding were observed to be Cys336, Phe338, Gly339, Ala363, Asp364, Val367, Ser371, Ser373, and Phe374. A hydrogen bond formed between the ligand and Ser373 of the receptor-binding domain (Fig. 5e). The docked complexes were demonstrated the binding energies of −0.6 and −2.2 kcal/mol towards the membrane protein (Fig. 5f) and envelope protein (Fig. 5g), respectively. The binding energy of the docked complex of ligand and replicase polyprotein1a was estimated to be −6.7 kcal/mol. The major interacting residues involved in the interaction were found to be Lys137, Thr199, Tyr239, Leu286, Leu287, Glu288, and Asp289. The interactions were stabilized by three hydrogen bonds (Fig. 5h). The binding energies associated with the interaction of lead molecule and Protein 3a, 7a, and 9b were estimated to be −6.3, −5.4, and −5.0 kcal/mol, respectively. The major interacting residues present at the binding site of protein 3a were identified to be Phe28, Val29, Arg68, Ala72, and Val90 (Fig. 5i). The major interacting residues present in the protein 7a were identified to be Tyr3, His47, Arg57, Thr59 (Fig. 5j), and the main interacting residues present in protein 9b were included Ser56, Leu65, Glu66, Ala67, Arg68, Ala69, Phe70, and Ser72, and the interaction stabilized by a hydrogen bond (Fig. 5k). The binding energies of the docked complexes of lead molecules and nsp7, nsp8, and nsp12 of RdRp were estimated to be −5.9, −6.0, and −6.6 kcal/mol, respectively. The major residues that interacted with nsp7 were identified to be Lys2, Asp5, Thr9, Thr46, Phe49, Met52, and Val53 (Fig. 5l). Similarly, the main residues that interacted with nsp8 were found to be Pro133, Asp134, Tyr135, Trp182, and Pro178. The interaction stabilized by a hydrogen bond (Tyr135) (Fig. 5m). The major interacting residues present at the binding site of nsp12 were found to be Arg181, Gln184, and Asn213 (Fig. 5n). The binding energies associated with the docked complex of lead molecules and non-structural proteins 6 and 10 and were estimated to be −5.6 and −6.8 kcal/mol, respectively. The major interacting residues present in the non-structural protein 6 were observed to be Gln8, Lys38, Lys42, and Leu44. The interaction stabilized by a hydrogen bond (Fig. 5o). Similarly, the main interacting residues present in the non-structural protein10 were identified to be Ile55, His83, Lys96, and Val116. The interaction stabilized by a hydrogen bond (Fig. 5p). From the interaction modeling, it is clear that Rotiorinol-C showed greater binding potential towards the replicase polyprotein (binding energy: −9.82 kcal/mol) when compared to the binding energies of the interaction between the ligand and other prioritized targets of SARS-CoV-2.
Fig. 5.

Prediction of the binding potential of Rotiorinol-C towards the prioritized molecular targets of SARS-CoV by molecular docking visualized in MGL tools of AutoDock. The figure displayed the binding pocket of the ligand-receptor complex. The interacting residues and the ligands are displayed in stick figures. The interacting residues and binding energy are labeled (a) Binding of the ligand and post-fusion conformation of spike glycoprotein (binding energy: −6.5 kcal/mol) (b) Binding of the ligand and open state spike glycoprotein (binding energy: −6.3 kcal/mol) (c) Binding of the ligand and closed state spike glycoprotein (binding energy −6.2 kcal/mol) (d) Binding of the ligand and replicase polyprotein 1 ab (binding energy: −9.8 kcal/mol) (e) Binding of the ligand and receptor binding domain of membrane protein (binding energy: −6.1 kcal/mol) (f) Binding of the ligand and membrane protein (binding energy: −0.6 kcal/mol) (g) Binding of the ligand and small envelope protein (binding energy: −2.2 kcal/mol) (h) Binding of the ligand and replicase polyprotein 1a (binding energy: −6.7 kcal/mol) (i) Binding of the ligand and protein3a (binding energy: −6.3 kcal/mol) (j) Binding of the ligand and protein7a (binding energy: −5.4 kcal/mol) (k) Binding of the ligand and protein 9b (binding energy: −5.0 kcal/mol) (l) Binding of the ligand and non-structural protein7 (binding energy: −5.3 kcal/mol) (m) Binding of the ligand and non-structural protein 8 (binding energy: −5.9 kcal/mol) (n) Binding of the ligand and non-structural protein12 (binding energy: −6.6 kcal/mol) (o) Binding of the ligand and non-structural protein 6 (binding energy: −5.6 kcal/mol) (p) Binding of the ligand and non-structural protein 10 (binding energy: −6.8 kcal/mol).
Scutifoliamide-A ((2E, 4Z)-5-(1, 3-Benzodioxol-5-yl)-N-isobutyl-2, 4-pentadienamide) is commonly present in Piper scutifolium (Pepper) [77]. It showed the binding potential to the spike glycoprotein in the post-fusion, closed state, and open state conformations with the binding energies of −6.4, −5.4, and −6.6 kcal/mol, respectively. The residues present at the binding cavity of the post-fusion conformation were identified to be Asn910, Ala940, Thr943, Leu944, Gln947, Asn951, Leu1178, Leu1181, and Glu1183 (Fig. 6 a). The interacting residues present in the binding site of the closed state conformation were observed to be Leu118, Val120, Phe135, Leu141, and Leu241 (Fig. 6b). Similarly, the main interacting residues present in the binding cavity of open state conformation were found to be Phe823, Asn824, Val826, Pro863, Pro1057, and His1058. The interaction stabilized by a hydrogen bond (Fig. 6c). The binding energy of the docked complex of the lead molecule and replicase polyprotein 1 ab were identified to be −6.9 kcal/mol, and the main interacting residues were identified to be Arg4, Lys5, and Phe291 (A chain), Lys 5 (B chain) (Fig. 6d). The binding energy of the docked complex of the ligand and the receptor-binding domain of the membrane protein was estimated to be −5.9 kcal/mol. The major residues that interacted with the ligand were identified to be Gln493, Tyr495, Gly502, and Tyr505. The interaction stabilized by hydrogen bonds with Gln493 and Gly502 (Fig. 6e). The binding energies associated with the docked complexes of the ligand and membrane protein, and ligand and envelope protein were estimated to be −0.7 (Fig. 6f) and −0.8 kcal/mol (Fig. 6g), respectively. The binding energy associated with the complex of the lead molecule and replicase polyprotein1a was identified to be −6.8 kcal/mol, and the main interacting residues at the binding cavity were identified to be Val212, Arg217, Leu220, Gln256, Ile259, and Asp263 (Fig. 6h). The binding energies associated with the docked complex of ligand and Protein 3a, 7a and 9b were estimated to be −5.4, −5.6, and −4.4 kcal/mol, respectively. The main interacting residues present at the binding site of protein 3a were identified to be Cys130, Trp131, His150, His204, and His227. The interaction stabilized by a hydrogen bond (Fig. 6i). Similarly, the major interacting residues present at the binding site of protein 7a were found to be Gln6, Cys8, Val9, Thr12, Leu16, and Lys17. The interaction stabilized by two hydrogen bonds (Fig. 6j). The major binding residues present in the protein 9b were included Ala58, Arg68, Ala69, Phe70, and Ser72 (Fig. 6k). The binding energies of the docked complex of ligand and nsp7, nsp8, and nsp12 were estimated to be −5.9, −6.0, and −6.1 kcal/mol, respectively. The main residues present at the binding site of nsp7 included Asp5, Thr9, Thr46, Phe49, Glu50, and Val53. The interaction stabilized by a hydrogen bond (Fig. 6l). The residues present at the binding site of nsp8 were found to be Leu128, Met129, Pro133, Thr141, and Tyr149 (Fig. 6m). The main amino acid residues present at the binding site of nsp12 were identified to be Asp484, Ile488, Gln573, Ser578, and Ala581 (Fig. 6n). The binding energies associated with the docked complexes of the ligand and non-structural proteins 6 and 10 were estimated to be −5.5 and −5.6 kcal/mol, respectively. The interacting residues present in the non-structural protein 6 were observed to be Leu16, Ile17, Arg20, and Thr21, and the interaction stabilized by a hydrogen bond with Arg20 (Fig. 6o). Similarly, the major interacting residues present in the non-structural protein10 were included His83, Leu92, Asn114, Thr115, and Val116. The interaction stabilized by two hydrogen bonds (Fig. 6p). The computational modeling suggested that Scutifoliamide-A showed the profound binding potential to several prioritized targets, which showed the highest binding energy to replicase polyprotein 1 ab (−6.9 kcal/mol) when compared to the binding energy of the interaction between Scutifoliamide-A and other selected targets of SARS-CoV-2.
Fig. 6.

Prediction of the binding potential of Scutifoliamide-A towards the prioritized molecular targets of SARS-CoV by molecular docking visualized in MGL tools of AutoDock. The figure displayed the binding pocket of the ligand-receptor complex. The interacting residues and the ligands are displayed in stick figures. The interacting residues and binding energy are labeled (a) Binding of the ligand and post-fusion conformation of spike glycoprotein (binding energy: −6.4 kcal/mol) (b) Binding of the ligand and open state spike glycoprotein (binding energy: −6.6 kcal/mol) (c) Binding of the ligand and closed state spike glycoprotein (binding energy −5.4 kcal/mol) (d) Binding of the ligand and replicase polyprotein 1 ab (binding energy: −6.9 kcal/mol) (e) Binding of the ligand and receptor binding domain of membrane protein (binding energy: −5.9 kcal/mol) (f) Binding of the ligand and membrane protein (binding energy: −0.7 kcal/mol) (g) Binding of the ligand and small envelope protein (binding energy: −0.8 kcal/mol) (h) Binding of the ligand and replicase polyprotein 1a (binding energy: −6.8 kcal/mol) (i) Binding of the ligand and protein3a (binding energy: −5.4 kcal/mol) (j) Binding of the ligand and protein7a (binding energy: −5.6 kcal/mol) (k) Binding of the ligand and protein 9b (binding energy: −4.4 kcal/mol) (l) Binding of the ligand and non-structural protein7 (binding energy: −5.9 kcal/mol) (m) Binding of the ligand and non-structural protein 8 (binding energy: −6.0 kcal/mol) (n) Binding of the ligand and non-structural protein12 (binding energy: −6.1 kcal/mol) (o) Binding of the ligand and non-structural protein 6 (binding energy: −5.5 kcal/mol) (p) Binding of the ligand and non-structural protein 10 (binding energy: −5.6 kcal/mol).
Tamaridone (5,7-dihydroxy-2-(2-hydroxy-4-methoxyphenyl)-6-methoxychromen-4-one), a flavone present in Tamarix dioica (Lal jhau/Ban jhau) showed binding potential towards the spike glycoprotein in the post-fusion, closed state, and open state conformations with the binding energies of −7.2, −7.3 and −6.8 kcal/mol, respectively. The interacting residues present in the binding pocket of the post-fusion conformation of spike protein were found to be Asp931, Asn935, Lys1162, Asp1165, Asn1168, and Lys1172 (Fig. 7 a). The interacting residues at the binding pocket of the closed state were identified to be Ala520, Phe559, Phe562, Gln563, Gln564, Phe565, Gly566, and Arg567. The interaction stabilized by four hydrogen bonds (Fig. 7b). Similarly, the major residues present in the binding site of the open state conformation were identified to be Thr732, Thr778, Ser780, His1058 and Pro863. The interaction stabilized by a hydrogen bond (Fig. 7c). The binding energy of the docked complex of the lead molecule and replicase polyprotein 1 ab was estimated to be −6.3 kcal/mol. The main residues involved in the interactions were predicted to be Gly11, Lys12, Glu14, and Gly15. The interaction stabilized by a hydrogen bond (Fig. 7d). The binding energy associated with the complex of ligand and receptor-binding domain of the membrane protein was estimated to be −6.5 kcal/mol, and the major interacting residues were identified to be Thr345, Arg346, Ser349, Lys441, Lys444, Asn448, Asn450, and Tyr451. The interaction stabilized by a hydrogen bond (Fig. 7e). The docked complexes of ligand-membrane protein and ligand-envelope protein showed binding energies of −0.8 (Fig. 7f) and −3.1 kcal/mol (Fig. 7g), respectively. The binding energy of the docked complex of the lead molecule and replicase polyprotein 1a was estimated to be −7.0 kcal/mol. The major interacting residues present in the binding site of replicase polyprotein 1a were found to be Leu87, Lys137, Thr190, Tyr237, Tyr239, and Leu285. The interaction stabilized by a hydrogen bond (Fig. 7h). The binding energies associated with the interaction of ligand and protein 3a, 7a, and 9b were estimated to be −6.6, −5.7 and −5.6 kcal/mol, respectively. The main interacting residues present in protein3a were identified to be Gln213, Ile263, Val255, and Thr270 (Fig. 7i). Similarly, the major interacting residues present at the binding site of protein 7a were included Val9, Thr12, Val14, and Arg65 (Fig. 7j). The main amino acids that interacted with protein9b were identified to be Arg68, Gln71, and Ser72 (Fig. 7k). The binding energies of the docked complexes of nsp7, nsp8, and nsp12 of RdRp were estimated to be −5.9, −5.7, and −6.7 kcal/mol, respectively. The major residues at the binding site of nsp7 were found to be Lys2, Val6, Thr9, Phe49, Met52 with a hydrogen bond formation (Fig. 7l). Similarly, the main interacting residues present in nsp8 were found to be Leu128, Val130, Val131, Thr141, and Tyr149 (Fig. 7m). The major interacting residues present in nsp12 were found to be Lys47, His133, Asn138, and Lys780 (Fig. 7n). The binding energies associated with the ligand and non-structural proteins 6, and 10 were estimated to be −5.7 and −6.7 kcal/mol, respectively. The major interacting residues present in the non-structural protein6 were observed to be Lys48, Ser50, Leu52, Asp53, and Gln56. The interaction stabilized by two hydrogen bonds (Fig. 7o). Similarly, the major interacting amino acids present in non-structural protein 10 were identified to be Pro37, Asn105, Asp106, Phe110, Lys113, Asn114, and Tyr126. The interaction stabilized by two hydrogen bonds (Fig. 7p). Thus, the interaction modeling of Tamaridone and fourteen prioritized targets of SARS-CoV-2 suggested that the lead showed the highest binding potential to the spike glycoprotein (binding energy: −7.2 kcal/mol) when compared to the binding energies associated with the interaction between ligands and other selected targets.
Fig. 7.

Prediction of the binding potential of Tamaridone towards the prioritized molecular targets of SARS-CoV by molecular docking visualized in MGL tools of AutoDock. The figure displayed the binding pocket of the ligand-receptor complex. The interacting residues and the ligands are displayed in stick figures. The interacting residues and binding energy are labeled (a) Binding of the ligand and post-fusion conformation of spike glycoprotein (binding energy: −7.2 kcal/mol) (b) Binding of the ligand and open state spike glycoprotein (binding energy: −7.3 kcal/mol) (c) Binding of the ligand and closed state spike glycoprotein (binding energy −6.8 kcal/mol) (d) Binding of the ligand and replicase polyprotein 1 ab (binding energy: −6.3 kcal/mol) (e) Binding of the ligand and receptor-binding domain of membrane protein (binding energy: −6.5 kcal/mol) (f) Binding of the ligand and membrane protein (binding energy: −0.8 kcal/mol) (g) Binding of the ligand and small envelope protein (binding energy: −3.1 kcal/mol) (h) Binding of the ligand and replicase polyprotein 1a (binding energy: −7.0 kcal/mol) (i) Binding of the ligand and protein3a (binding energy: −6.6 kcal/mol) (j) Binding of the ligand and protein7a (binding energy: −5.7 kcal/mol) (k) Binding of the ligand and protein 9b (binding energy: −5.6 kcal/mol) (l) Binding of the ligand and non-structural protein7 (binding energy: −5.9 kcal/mol) (m) Binding of the ligand and non-structural protein 8 (binding energy: −6.0 kcal/mol) (n) Binding of the ligand and non-structural protein12 (binding energy: −6.7 kcal/mol) (o) Binding of the ligand and non-structural protein 6 (binding energy: −5.6 kcal/mol) (p) Binding of the ligand and non-structural protein 10 (binding energy: −6.7 kcal/mol.
Similarly, the interaction between two currently suggested drugs against COVID-19 and their usual targets were predicted by molecular docking studies. The binding energies associated with the interaction between Chloroquine and Hydroxychloroquine to their normal drug targets namely glutathione S transferase of Plasmodium falciparum and human angiotensin-converting enzyme 2 (hACE2), respectively, were predicted by molecular docking. The interaction modeling suggested that when Chloroquine docked with glutathione S transferase, the complex showed binding energy of −3.71 kcal/mol. The main interacting residues were identified to be Glu90, Leu91, Glu93, Phe94, and Asp97 (Fig. 8 a and b). The number of interacting residues at the binding pockets was less when compared to the interaction of natural molecules and the selected targets selected. When Hydroxychloroquine docked with human angiotensin-converting enzyme 2 (hACE2), the docked conformation showed binding energy of −1.0 kcal/mol. The complex showed that Gln287 interacted with the ligand (Fig. 8c and d) and no hydrogen bonds were involved in the interaction. From the molecular docking studies, it is clear that the natural lead molecules showed better binding to the selected molecular targets based on binding energy (kcal/mol), the number of hydrogen bonds and other weak interactions, and the number of amino acids involved in the binding when compared to the binding of Chloroquine and Hydroxychloroquine and their usual targets.
Fig. 8.

Prediction of the binding of conventional antiviral drugs towards their usual drug targets by molecular docking studies (a) Interaction of Chloroquine with its usual targets glutathione S transferase of Plasmodium falciparum (b) Binding affinity of Chloroquine towards glutathione S transferase (binding energy: −3.7 kcal/mol) illustrated the major residues involved in the interaction with the ligand (c) Interaction of Hydroxy-chloroquine with usual target human angiotensin-converting enzyme 2 (hACE2) (d) Binding affinity of Hydroxyl-chloroquine towards hACE2 (binding energy:-1.0 kcal/mol) illustrated the major residues involved in the interaction with the ligand.
3.5. Molecular dynamic simulation studies
Four potential docked complexes with the minimum binding energy (kcal/mol), cluster RMS, and a greater number of stabilizing interactions were selected for MD simulation studies. The best-docked conformations of spike glycoprotein and Hyoscyamine, replicase polyprotein 1 ab and Rotiorinol-C, replicase polyprotein 1 ab and Scutifoliamide A, Spike glycoprotein, and Tamaridone were simulated at 100 ns using NVT ensemble to confirm the dynamics and stabilities during the binding. Similarly, the docked complex of Chloroquine and glutathione-S-transferase were simulated, and their binding stabilities were compared with the interaction stabilities of the docked complexes of natural lead molecules and selected targets.
The trajectories obtained during the MD simulation of the best-docked pose of Hyoscyamine and the post-fusion conformation of the spike glycoprotein complex are shown in Fig. 9 . The MD simulation studies suggested that the RMSD value of the protein showed a deviation from 8 to 16 Å at100 ns, which indicated that there were substantial changes in the conformation of the target protein during the simulation period (Fig. 9a). The variation in the RMSD probably due to the binding of Hyoscyamine with the protein, which indicated better interaction and stability when compared to the control. The protein RMSF deviated from 2.0 to 14.0 Å and showed that more than 60% of the residues possessed >10 Å. The fluctuations in the protein with the C- and N-terminals are depicted in Fig. 9b. The ligand RMSF ranged from 4.0 to 6.0 Å, indicated that the fluctuations in their entropic role, and the interactions with the target during the binding are shown in Fig. 9c. The protein-ligand interactions throughout the simulation time showed prominent water bridges, hydrogen bonds, and hydrophobic interactions (Fig. 9d). The molecular interactions that occurred during more than 10% of the simulation period are illustrated in Fig. 9e. The simulation studies revealed that the ligand binds to the protein with hydrophobic interaction with Ile1164 of chain B and a polar interaction with Gln1161 of chain A. The interactions between the ligand and the protein are shown in Fig. 9f. The trajectories of total contacts between the ligand and target such as hydrogen bonds, ionic bonds, water bridges, and hydrophobic interactions were found to be constant throughout the simulation. It was found that several residues interacted with the ligand throughout the trajectory frame in which several residues belonged to chain A and chain B, which indicated binding stabilities during the simulation. The conformational changes of protein and ligand that occurred during the MD simulation are shown in Supplementary Materials Fig. S1. Thus, the MD simulation studies suggested that the stabilities of the interaction between Hyoscyamine and spike glycoprotein of SARS-CoV-2 were better when compared to the stability of the docked complex of chloroquine and its usual target (Fig. 13). Thus, the present study suggested that Hyoscyamine can act as a potential lead molecule against the spike glycoprotein of SARS-CoV-2.
Fig. 9.

The binding and stability of the docked complex of natural lead molecule Hyoscyamine and spike glycoprotein of SARS-CoV-2 were studied by molecular dynamic simulation at 100 ns. The trajectories obtained during the MD simulation is shown in figure (a) Protein-ligand RMSD: Protein RMSD (Å) is shown on the y-axis, and simulation time is given on the x-axis (b) Plot of the protein RMSF: RMSF (Å) is given in the y-axis and atom index is showed in the x-axis (c) Plot of ligand RMSF: RMSF (Å) is shown in y-axis and atom index is given in the x-axis (d) The protein-ligand contact is shown in the form of a histogram. The blue, grey, pink, and green regions in the histogram represent water bridges, hydrophobic interactions, ionic bonds, and hydrogen bonds respectively (e) The interactions between Hyoscyamine and post-fusion conformation of the spike glycoprotein observed during MD simulation (f) The protein-ligand contacts shown in the form of timeline representation. The top panel shows the total number of specific contacts between receptor and ligand, and the bottom panel shows the amino acid residues of the target, which is interacted with the ligand.
Fig. 13.

The binding and stability of the docked complex of conventional antiviral drug Chloroquine and Glutathione-S-transferase studied by molecular dynamic simulation at 100 ns? The trajectories obtained during the MD simulation is shown in figure (a) Protein-ligand RMSD: Protein RMSD (Å) is shown on the y-axis, and simulation time is given on the x-axis (b) Plot of the protein RMSF: RMSF (Å) is given in the y-axis and atom index is showed in the x-axis (c) Plot of ligand RMSF: RMSF (Å) is shown in y-axis and atom index is given in the x-axis (d) The protein-ligand contact is shown in the form of a histogram. The blue, grey, pink, and green regions in the histogram represent water bridges, hydrophobic interactions, ionic bonds, and hydrogen bonds respectively (e) The interactions between Hyoscyamine and post-fusion conformation of the spike glycoprotein observed during MD simulation (f) The protein-ligand contacts shown in the form of timeline representation. The top panel shows the total number of specific contacts between receptor and ligand, and the bottom panel shows the amino acid residues of the target, which is interacted with the ligand.
The MD simulation trajectories of Rotiorinol-C and the replicase polyprotein 1 ab are depicted in Fig. 10 . The simulation studies suggested that the target protein showed RMS deviation from 1.8 to 2.7 Å during the simulation at 100 ns (Fig. 10a). This variation in RMSD probably due to the binding of Rotiorinol-C to the protein target. The protein RMSF deviated from 0.6 to 4.8 Å (the majority of the residues deviated from 1.2 to 2.8 Å, and 40% of the residues deviated at 3.6 Å) indicated the fluctuations in the protein structure during the simulation period (Fig. 10b). Similarly, ligand RMSF observed to be fluctuated between 0.5 and 2.0 Å due to the variation in entropy caused by the receptor-ligand interaction (Fig. 10c). The protein-ligand interaction throughout the simulation is shown in Fig. 10c, which depicted that hydrogen and hydrophobic interactions played major roles in the binding. The interaction that occurred during more than 10% of the simulation time is shown in Fig. 10d. The ligand interacted to the protein with Phe3 of chain B, electrostatic negative interactions with Glu288, Asp289, Glu290 of chain A and Glu288 of chain B, electrostatic interactions with Arg4, Arg131, Lys137 of chain A and Arg4, Lys5 of chain B, polar interactions with Gln127 of both the chains. The interactions between the ligand and the protein are shown in Fig. 10f. The trajectory forces such as hydrogen bonds, ionic bonds, water bridges, and hydrophobic interactions during the simulation were evident. The interaction contacts were observed to be constant up to 80ns with a minor increase, and several amino acids interacted with the ligand throughout the trajectory frame. Arg4, Gln127 of chain A and Lys5, Gln127 of chain B were showed constant interaction with the ligand throughout the simulation. The structural changes associated with the protein and ligand during the MD simulation are shown in Supplementary Materials Fig. S2. Thus, the MD simulation suggested that the stability of the interaction between Rotiorinol-C and replicase polyprotein 1 ab were greater when compared to the interaction of chloroquine and its usual target. Thus, this study revealed that Rotiorinol-C can act as one of the potential lead molecules towards replicase polyprotein 1 ab of SARS-CoV-2.
Fig. 10.

The binding and stability of the docked complex of natural lead molecule Rotiorinol-C and replicase polyprotein 1 ab of SARS-CoV-2 studied by molecular dynamic simulation at 100 ns? The trajectories obtained during the MD simulation is shown in figure (a) Protein-ligand RMSD: Protein RMSD (Å) is shown on the y-axis, and simulation time is given on the x-axis (b) Plot of the protein RMSF: RMSF (Å) is given in the y-axis and atom index is showed in the x-axis (c) Plot of ligand RMSF: RMSF (Å) is shown in y-axis and atom index is given in the x-axis (d) The protein-ligand contact is shown in the form of a histogram. The blue, grey, pink, and green regions in the histogram represent water bridges, hydrophobic interactions, ionic bonds, and hydrogen bonds respectively (e) The interactions between Hyoscyamine and post-fusion conformation of the spike glycoprotein observed during MD simulation (f) The protein-ligand contacts shown in the form of timeline representation. The top panel shows the total number of specific contacts between receptor and ligand, and the bottom panel shows the amino acid residues of the target, which is interacted with the ligand.
The simulation trajectories of the interaction between Scutifoliamide-A and replicase polyprotein 1 ab of SARS-CoV-2 are shown in Fig. 11 . The RMS trajectories suggested that the protein showed a substantial deviation from 5.0 to 45.0 Å at100 ns, which indicated the conformational changes in the target protein (Fig. 11a). The variation in RMSD of the protein was probably due to the interaction of Scutifoliamide-A and the target. The RMSF of the protein was found to be 4.0–28.0 Å, in which more than 40% of the residues showed >20 Å (Fig. 11b). The ligand RMSF ranged between 10.0-25.0 Å indicated that the fluctuations corresponding to their variations in entropy due to the interaction with the target (Fig. 11c). The protein-ligand contacts throughout the simulation are represented in Fig. 11d. Water bridges, hydrogen bonds, and hydrophobic interactions were observed as the major stabilizing forces. The interactions observed during the MD simulation revealed that the ligand binds with the protein by a polar bond at Gln127 (chain A) and positive electrostatic interactions at Arg4 (chain A) and Lys5 (both chain A and B) (Fig. 11e). The interaction between the ligand and the protein is shown in Fig. 11f. The simulation trajectories showed that there were stabilizing interactions such as hydrogen bonds, water bridges, and hydrophobic interactions. It was clear that several residues involved in the interaction in which eight residues belonged to chain A and three residues belonged to chain B. Lys5 (chain B), Arg4, Lys5, and Gln127 (chain A) played a major role in the interaction with the ligand. The protein and ligand conformational changes during the MD simulation studies are shown in Supplementary Material Fig. S3. The MD simulation studies suggested that the Scutifoliamide-A-replicase polyprotein 1 ab complex showed stability during the simulation, however, there were deviations observed during MD simulation. Further, in comparison with the binding of the control and its targets, the Scutifoliamide-A-replicase polyprotein 1 ab complex showed better interaction. Thus, Scutifoliamide-A can be considered to be one of the probable molecules against replicase polyprotein 1 ab of SARS-CoV-2.
Fig. 11.

The binding and stability of the docked complex of natural lead molecule Scutifoliamide-A and replicase polyprotein 1 ab of SARS-CoV-2 were studied by molecular dynamic simulation at 100 ns? The trajectories obtained during the MD simulation is shown in figure (a) Protein-ligand RMSD: Protein RMSD (Å) is shown on the y-axis, and simulation time is given on the x-axis (b) Plot of the protein RMSF: RMSF (Å) is given in the y-axis and atom index is showed in the x-axis (c) Plot of ligand RMSF: RMSF (Å) is shown in y-axis and atom index is given in the x-axis (d) The protein-ligand contact is shown in the form of a histogram. The blue, grey, pink, and green regions in the histogram represent water bridges, hydrophobic interactions, ionic bonds, and hydrogen bonds respectively (e) The interactions between Hyoscyamine and post-fusion conformation of the spike glycoprotein observed during MD simulation (f) The protein-ligand contacts shown in the form of timeline representation. The top panel shows the total number of specific contacts between receptor and ligand, and the bottom panel shows the amino acid residues of the target, which is interacted with the ligand.
The simulation trajectories of Tamaridone and the spike glycoprotein in its post-fusion conformation are shown in Fig. 12 . The MD simulation studies suggested that the protein showed a deviation from 8.0 to 15 Å in the RMSD values at 100 ns (Fig. 12a). This variation was probably due to the interaction of Tamaridone and glycoprotein that showed conformational changes. The protein RMSF deviated from 3.0 to 16.0 Å (Fig. 12b). The ligand RMSF showed a fluctuation between 7.5 and 10.0 Å, due to the changes in the entropy caused by protein-ligand interaction (Fig. 12c). The protein-ligand interactions throughout the simulation are shown in Fig. 12d. The forces that stabilized the interactions between the receptor and ligand were found to be water bridges, hydrogen, and hydrophobic interactions. The molecular interactions that occurred more than 10% of the MD simulation are shown in Fig. 12e. Tamaridone interacted with the protein by hydrophobic interactions at Val1170 (chain A) and Tyr1187 (chain B), and polar interactions at Thr923 (chain A). The contacts between the ligand and protein throughout the simulation are shown in Fig. 12f. The trajectories of total contact such as hydrogen bonds, water bridges, and hydrophobic interactions between the protein and the ligand during the simulation were evident, and several residues were involved in the interaction with the ligand throughout the trajectory frame. The interacting residues of chain A were identified to be Ile916, Ser919, Asp1165, Val1170, and the residues that make contact with chain B were observed to be Asp932, Asn935, and Tyr1187. The structural features of the protein and ligand during the MD simulation are shown in Supplementary Material Fig. S4. The MD simulation studies suggested that the docked complexes of Tamaridone and spike glycoprotein of SARS-CoV-2 showed stability throughout the simulation, and the lead molecule demonstrated better binding and dynamics when compared to the binding of Chloroquine to its usual targets (Fig. 13). Thus, the present study prioritized that Tamaridone can also be used as a potential lead molecule towards spike glycoprotein of SARS-CoV-2.
Fig. 12.

The binding and stability of the docked complex of natural lead molecule Tamaridone and spike glycoprotein of SARS-CoV-2 were studied by molecular dynamic simulation at 100 ns? The trajectories obtained during the MD simulation is shown in figure (a) Protein-ligand RMSD: Protein RMSD (Å) is shown on the y-axis, and simulation time is given on the x-axis (b) Plot of the protein RMSF: RMSF (Å) is given in the y-axis and atom index is showed in the x-axis (c) Plot of ligand RMSF: RMSF (Å) is shown in y-axis and atom index is given in the x-axis (d) The protein-ligand contact is shown in the form of a histogram. The blue, grey, pink, and green regions in the histogram represent water bridges, hydrophobic interactions, ionic bonds, and hydrogen bonds respectively (e) The interactions between Hyoscyamine and post-fusion conformation of the spike glycoprotein observed during MD simulation (f) The protein-ligand contacts shown in the form of timeline representation. The top panel shows the total number of specific contacts between receptor and ligand, and the bottom panel shows the amino acid residues of the target, which is interacted with the ligand.
The trajectories of simulation studies of the docked complex of Chloroquine and glutathione-S-transferase complex are shown in Fig. 13. The simulation results suggested that the RMSD values of the protein showed a deviation from 1.6 to 3.2 Å at 100ns indicated that there were fewer or no conformational changes during the simulation period (Fig. 13a). The variation in the RMSD of the protein was probably due to the binding with Chloroquine. The protein RMSF deviated from 0.8 to 2.3 Å (Fig. 13b). Ligand RMSF deviated from 8.5 to 6.0 Å, which indicated the fluctuations corresponding to their entropy in the binding (Fig. 13c). The interactions that stabilized the docked complex were identified to be water bridges, ionic bonds, hydrogen bonds, and hydrophobic interactions, which are shown in Fig. 13d. The molecular interactions that occurred during >10% of the MD simulation are depicted in Fig. 13e. The analysis revealed that the ligand binds to the protein with hydrophobic interactions with Phe94 and Tyr95 and electrostatic interaction with Lys141. The interactions between ligand and the protein are depicted in Fig. 13f. The major forces that stabilized the binding between the ligand and receptor during the simulation were identified to be hydrogen bonds, ionic bonds, water bridges, and hydrophobic interactions. The number of residues involved in the interaction with the ligand was less in comparison with the interaction of the selected natural molecules and targets. It was observed that Phe94 showed predominant interaction during the first 50 ns, and Tyr95 showed maximum interaction during the next 50ns of the simulation. The conformational changes of the interaction between glutathione-S-transferase and chloroquine during the MD simulation are shown in Supplementary material, Fig. S5.
3.6. Binding potential of natural lead calculated by MM-PBSA
The binding potential of the complexes of natural lead molecules and the prioritized targets were confirmed by the energy calculations by MM-PBSA approaches. Gromacs force field was used to calculate the binding affinities of the molecular targets and ligand complexes. The interaction energy of the Chloroquine and its usual target glutathione-S-transferase was estimated to be −33.12 kcal/mol. The binding energies of the docked complex of Hyoscyamine and spike glycoprotein, Rotiorinol-C and replicase polyprotein 1 ab, Scutifoliamide-A and replicase polyprotein, and Tamaridone and spike glycoprotein were estimated to be −48. 25, −38. 40, −45. 97 and −38. 79 kcal/mol, respectively. Thus, the energy calculations by MMPBSA showed that the interaction of four natural lead molecules towards the prioritized targets of SARS-CoV-2 demonstrated better binding interaction compared to the binding of Chloroquine and Glutathione-S-transferase.
From the molecular docking, MD simulation, and energy calculation studies, it is clear that the four potential natural lead molecules demonstrated better binding energies, stabilities, and dynamics towards the prioritized drug targets of SARS-CoV-2 compared to the binding of Chloroquine and glutathione-S-transferase. Thus, the present study revealed that natural molecules and the prioritized targets can be used as potential candidates for the development of therapeutic agents against COVID-19.
Recent studies emphasized that the scope of computer-aided molecular design towards the lead development and identification of molecular targets of COVID-19. Studies suggested that natural lead compounds screened against the targets of SARS-CoV-2 such as spike glycoprotein (S), non-structural proteins (nsp), an envelope protein (E), membrane protein (M) provided novel insights for drug repurposing approach against SARS-CoV-2 [78]. The interaction studies between SARS-CoV spike protein and cell-surface receptor (GRP78) suggested that the binding favorable occurred between the regions 480–488 of the spike protein and GRP78 [79]. Mpro was identified to be a potential drug target by Jin et al. (2020) suggested a mechanism-based inhibitor, N3, by virtual screening [80]. Beclabuvir showed significant binding towards Mpro with a binding energy of −10.4 kcal/mol and was used to screen novel lead molecules by virtual screening. This drug is presently being suggested for clinical trials [81]. Recent studies showed that the phytochemicals from Psorothamnus arborescens, Myrica cerifera, and Hyptis atrorubens showed binding potential towards Chymotrypsin like protease (CL-pro) [82]. SARS-CoV nsp6 has been shown to activate autophagy, inducing vesicles, and is known to interact with nsp2, nsp8, nsp9, and accessory protein 9b [83]. nsp6 is considered to be one of the potential targets to suppress the virulent genes of the pathogen. Envelope (E) proteins were reported to form ion channels, which is mainly associated with pathogenesis, and the inhibition of these channels provided insights for drug discovery. A recent study suggested that Belachinal, Macaflavanone-E, and Vibsanol-B were reported to be potential inhibitors of envelope protein [84]. Similarly, another study showed that several viral peptides were modeled by computational virtual screening for vaccine development [85]. The current study predicted the binding potential of four natural lead molecules, namely Hyoscyamine, Rotiorinol-C, Scutifoliamide-A, and Tamaridone towards selected molecular targets of COVID-19, and the hypothesis probably provide profound insight for the development of therapeutic agents against putative targets of SARS-CoV-2.
The present study provides insight into the molecular mechanism of the binding potential of natural phytochemical towards fourteen molecular targets of SARS-CoV-2 for future investigation. Since there are no standard drugs available for the treatment of COVID-19 to compare our studies, the interaction models of Chloroquine and Hydroxychloroquine and their usual targets were used as the comparative control, and, therefore, this prediction could have a variation when it implements at the experimental level. Further, the study modeled the binding potential of four lead molecules towards the prioritized targets, and due to the time and resource constraints, the molecular dynamics simulation, and energy calculations were performed only for the best-docked complexes that showed minimum binding energies and maximum stabilizing interactions. The molecular dynamics and simulation studies between selected natural compounds and other targets can also be modeled to obtain significant breakthroughs for the development of potential targets and lead molecules. Thus, the present study provides an ample foundation for future investigation and developing natural lead molecules against multiple targets of SARS-CoV-2.
4. Conclusion
At present, COVID-19 is one of the most critical public health concerns with high mortality and morbidity rate worldwide. There are limited therapeutic agents available to treat the disease. Thus, there is a high emergency to identify potential therapeutic strategies to tackle COVID-19. Computer-aided virtual screening is one of the promising approaches which reduce the time and cost required to develop new candidate drugs. This approach can be used to screen lead molecules with druggish and pharmacokinetic features that can be transformed into experimental levels to develop the therapeutic intervention. The natural molecules are known to have inhibitory potential towards several viral targets. The present study aimed to screen potential natural lead molecules towards multiple prospective molecular targets of SARS-CoV-2. Based on the functional and virulence role, fourteen molecular targets of SARS-CoV-2 were identified, and eleven of them were retrieved from the Protein Data Bank, and three proteins that lack the 3D structures, were computationally predicted. By an extensive literature survey and database search, ninety-two natural compounds were selected. The drug likeliness, pharmacokinetic and toxicity properties of selected lead molecules were predicted by various computational biology tools. Based on the virtual screening, four potential lead molecules were screened, and the binding potentials of these lead molecules towards fourteen targets were modeled by molecular docking. The stabilities of the four best-docked complexes were confirmed by molecular dynamic simulation and energy calculation studies. The present study suggested that the four lead molecules were qualified for the drug likeliness, pharmacokinetics, and toxicity features, and demonstrated profound binding with most of the selected targets. Notably, Hyoscyamine and Tamaridone towards spike glycoprotein and Rotiorinol-C and Scutifoliamide-A towards replicase polyprotein 1 ab of SARS-CoV −2 were showed better and significant binding compared to the binding of Chloroquine and glutathione S transferase. Thus, the present study suggested that these natural lead molecules can be used as potential lead molecules against the prospective molecular targets of SARS-CoV-2, and the computational prediction probably provides a substantial breakthrough in the drug discovery pipeline to develop future therapeutic agents against COVID-19 and the prediction can be transformed into the experiential level.
Funding
No funding was involved in the preparation of this manuscript or in the decision to submit it for publication.
Data availability
Data is presented within the manuscript and the supplemental materials.
Author contribution
Sinosh Skariyachan and Dharshini were involved in the Conceptualization; Data curation; Formal analysis; Investigation; Methodology; Project administration; Resources; Software; Supervision; Validation; Visualization; Roles/Writing - original draft; Writing - review & editing of the manuscript. Aditi G Muddebihalkar, Akshay Uttarkar and Vidya Niranjan involved in Investigation, Resources; Software; Supervision and Visualization; Roles/Writing.
Declaration of competing interest
The authors declare that they have no competing interests.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.compbiomed.2021.104325.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Chan J.F., Kok K.H., Zhu Z., Chu H., To K.K., Yuan S., Yuen K.Y. Genomic characterization of the 2019 novel human pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microb. Infect. 2020;9(1):221–236. doi: 10.1080/22221751.2020.1719902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.World Health Organization WHO coronavirus disease (COVID-19) dashborad. December 2020. https://covid19.who.int
- 3.S. Khan, R. Siddique, M. A. Shereen, A. Ali, J. Liu, Q. Bai, N. Bashir, M. Xue, The emergence of a novel coronavirus (SARS-CoV-2), their biology and therapeutic options, J. Clin. Microbiol. JCM.00187-20. Advance online publication. doi: 10.1128/JCM.00187-20.
- 4.Verity R., Okell L.C., Dorigatti I., Winskill P., Whittaker C., Imai N., Cuomo-Dannenburg G., Thompson H., Walker P., Fu H., Dighe A., Griffin J.T., Baguelin M., Bhatia S., Boonyasiri A., Cori A., Cucunubá Z., FitzJohn R., Gaythorpe K., Green W., Hamlet A., hinsley W., Laydon D., Nedjati-Gilani G., Riley S., van Elsland S., Volz E., Wang H., Wang Y., Xi X., Donnelly C.A., Ghani A.C., Ferguson N.M. Estimates of the severity of coronavirus disease 2019: a model-based analysis. Lancet Infect. Dis. 2020;S1473–3099(20):30243–30247. doi: 10.1016/S1473-3099(20)30243-7. Advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beerkens F., John M., Puliafito B., Corbett V., Edwards C., Tremblay D. COVID‐19 pneumonia as a cause of acute chest syndrome in an adult sickle cell patient. Am. J. Hematol. Accepted Author Manuscript. 2020 doi: 10.1002/ajh.25809. [DOI] [PubMed] [Google Scholar]
- 6.Workman D., Welling D.B., Carter B.S., Curry W.T., Holbrook E.H., Gray S.T., Scangas G.A., Bleier B.S. Endonasal instrumentation and aerosolization risk in the era of COVID-19: simulation, literature review, and proposed mitigation strategies. Int. Forum. Allergy Rh. 2020 doi: 10.1002/alr.22577. Accepted Author Manuscript. [DOI] [PubMed] [Google Scholar]
- 7.Carlos W.G., Dela Cruz C.S., Cao B., Pasnick S., Jamil S. Novel Wuhan (2019-nCoV) coronavirus. Am. J. Respir. Crit. Care Med. 2020;201(4):P7–P8. doi: 10.1164/rccm.2014P7. [DOI] [PubMed] [Google Scholar]
- 8.Guo Y.R., Cao Q.D., Hong Z.S., Tan Y.Y., Chen S.D., Jin H.J., Tan K.S., Wang D.Y., Yan Y. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak - an update on the status. Mil. Med. Res. 2020;7(1):11. doi: 10.1186/s40779-020-00240-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gralinski L.E., Menachery V.D. Return of the coronavirus: 2019-nCoV. Viruses. 2020;12(2):135. doi: 10.3390/v12020135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walls C., Park Y., Tortorici M.A., Wall A., McGuire A.T., Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020 doi: 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Behbahani M. bioRxiv; 2020. In Silico Design of Novel Multi-Epitope Recombinant Vaccine Based on Coronavirus Surface Glycoprotein. [DOI] [Google Scholar]
- 12.Kim M., Cho M., Lee J.H., Kim H., Son H.S. Analysis of coronaviral spike proteins and virus–host interactions. KJPH. 2019;56(1):25–32. [Google Scholar]
- 13.Grifoni J., Sidney Y., Zhang R.H., Scheuermann B., Peters A., Sette A sequence homology and bioinformatic approach can predict candidate targets for immune responses to SARS-CoV-2. Cell Host Microbe. 2020 doi: 10.1016/j.chom.2020.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kong R., Yang G., Xue R., Liu M., Wang F., Hu J., Chang S. 2020. COVID-19 Docking Server: an Interactive Server for Docking Small Molecules, Peptides and Antibodies against Potential Targets of COVID-19. arXiv, preprint arXiv:2003.00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fahmi M., Kubota Y., Ito M. Nonstructural proteins NS7b and NS8 are likely to be phylogenetically associated with evolution of 2019-nCoV. Infect. Genet. Evol. 2020;81:104272. doi: 10.1016/j.meegid.2020.104272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020 doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harrison C. Coronavirus puts drug repurposing on the fast track. Nat. Biotechnol. 2020 doi: 10.1038/d41587-020-00003-1. Advance online publication. [DOI] [PubMed] [Google Scholar]
- 18.Zhou Y., Hou Y., Shen J., Huang Y., Martin W., Cheng F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020;6:14. doi: 10.1038/s41421-020-0153-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang D.H., Wu K.L., Zhang X., Deng S.Q., Peng B. In silico screening of Chinese herbal medicines with the potential to directly inhibit 2019 novel coronavirus. J. Integr. Med. 2020;18(2):152–158. doi: 10.1016/j.joim.2020.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Agostini M.L., Pruijssers A.J., Chappell J.D., Gribble J., Lu X., Andres E.L., Bluemling G.R., Lockwood M.A., Sheahan T.P., Sims A.C., Natchus M.G., Saindane M., Kolykhalov A.A., Painter G.R., Baric R.S., Denison M.R. Small-molecule antiviral β-d-N4-hydroxycytidine inhibits a proofreading-intact coronavirus with a high Genetic barrier to resistance. J. Virol. 2019;93(24) doi: 10.1128/JVI.01348-19. e01348-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duquerroy S., Vigouroux A., Rottier P.J., Rey F.A., Bosch B.J. Central ions and lateral asparagine/glutamine zippers stabilize the post-fusion hairpin conformation of the SARS coronavirus spike glycoprotein. Virology. 2005;335(2):276–285. doi: 10.1016/j.virol.2005.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bonanno J.B., Fowler R., Gupta S., Hendle J., Lorimer D., Romero R., Sauder J.M., Wei C.L., Liu E.T., Burley S.K., Harris T. New York Times; 2003. Company Says it Mapped Part of SARS Virus. 30 July. C2-C2. [Google Scholar]
- 24.D. Fearon, C. D. Owen, A. Douangamath, P. Lukacik, A. J. Powell, C. M. Strain-Damerell, F. Resnick, T. Krojer, P. Gehrtz, C. Wild, A. Aimon, J. Brandao-Neto, A. Carbery, L. Dunnett, R. Skyner, M. Snee, N. London, M. A. Walsh, F. von Delft, PanDDA Analysis Group Deposition of SARS-CoV-2 Mainprotease Fragment Screen. (To be published).
- 25.Liu J., Sun Y., Qi J., Chu F., Wu H., Gao F., Li T., Yan J., Gao G.F. The membrane protein of severe acute respiratory syndrome coronavirus acts as a dominant immunogen revealed by a clustering region of novel functionally and structurally defined cytotoxic T-lymphocyte epitopes. J. Infect. Dis. 2010;202(8):1171–1180. doi: 10.1086/656315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Y., Surya W., Claudine S., Torres J. Structure of a conserved Golgi complex-targeting signal in coronavirus envelope proteins. J. Biol. Chem. 2014;289(18):12535–12549. doi: 10.1074/jbc.M114.560094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nelson C.A., Pekosz A., Lee C.A., Diamond M.S., Fremont D.H. Structure and intracellular targeting of the SARS-coronavirus Orf7a accessory protein. Structure (London, England. 1993;13(1):75–85. doi: 10.1016/j.str.2004.10.010. 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meier C., Aricescu A.R., Assenberg R., Aplin R.T., Gilbert R.J., Grimes J.M., Stuart D.I. The crystal structure of ORF-9b, a lipid binding protein from the SARS coronavirus. Structure (London, England. 1993;14(7):1157–1165. doi: 10.1016/j.str.2006.05.012. 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yan R., Zhang Y., Li Y., Xia L., Guo Y., Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science (New York, N.Y.) 2020;367(6485):1444–1448. doi: 10.1126/science.abb2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gao Y., Yan L., Huang Y., Liu F., Zao Y., Cao L., Wang T., Sun Q., Ming Z., Zhang L., Ge J., Zheng L., Zhang Y., Wang H., Zhu Y., Zhu C., Hu T., Hua T., Hua T., Zhang B., Yang X., Li J., Yang H., Liu Z., Xu W., Guddat L.W., Wang Q., Luo Z., Rao Z. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science. 2020;368(6492):779–782. doi: 10.1126/science.abb7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.UniProt Consortium UniProt: a hub for protein information. Nucleic Acids Res. 2015;43(Database issue):D204–D212. doi: 10.1093/nar/gku989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang C., Zheng W., Huang X., Bell E.W., Zhou X., Zhang Y. Protein structure and sequence re-analysis of 2019-nCoV genome refutes snakes as its intermediate host and the unique similarity between its spike protein insertions and HIV-1. J. Proteome Res. 2020 doi: 10.1021/acs.jproteome.0c00129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu D., Zhang Y. Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization. Biophys. J. 2011;101(10):2525–2534. doi: 10.1016/j.bpj.2011.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heinig M., Frishman D. STRIDE: a web server for secondary structure assignment from known atomic coordinates of proteins. Nucleic Acids Res. 2004;32(Web Server issue):W500–W502. doi: 10.1093/nar/gkh429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wiederstein J., Sippl M. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35(Web Server):W407–W410. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luthy R., Bowie J.U., Eisenberg D. Assessment of protein models with three-dimensional profiles. Nature. 1992;356:83–85. doi: 10.1038/356083a0. [DOI] [PubMed] [Google Scholar]
- 37.Laskowski, MacArthur W., Moss D., Thornton M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993;26(2):283–291. doi: 10.1107/S0021889892009944. [DOI] [Google Scholar]
- 38.Colovos T., Yeates Verification of protein structures: patterns of non-bonded atomic interactions. Protein Sci. 1993;2(9):1511–1519. doi: 10.1002/pro.5560020916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Torsten S., Jurgen K., Nicolas G., Manuel P. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 2003;31:3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim S., Chen J., Cheng T., Gindulyte A., He J., He S., Li Q., Shoemaker B.A., Thiessen P.A., Yu B., Zaslavsky L., Zhang J., Bolton E.E. PubChem 2019 update: improved access to chemical data. Nucleic Acids Res. 2019;47(D1):D1102–D1109. doi: 10.1093/nar/gky1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pence H.E., Williams A. ChemSpider: an online chemical information resource. J. Chem. Educ. 2010;87(11):1123–1124. doi: 10.1021/ed100697w. [DOI] [Google Scholar]
- 42.O'Boyle N.M., Banck M., James C.A., Morley C., Vandermeersch T., Hutchison G.R. Open Babel: An open chemical toolbox. J. Cheminf. 2011;3(1):33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Veber F., Johnson R., Chen H.Y., Smith B.R., Ward K.W., Kopple K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002;45:2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 44.Daina O. Michielin, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lipinski, Lombardo F., Dominy B., Feeney P. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 46.Egan W.J., Walters W.P., Murcko M.A. Guiding molecules towards drug-likeness. Curr. Opin. Drug Discov. Dev. 2002;5(4):540–549. [PubMed] [Google Scholar]
- 47.Frimurer T., Bywater R., Nærum L., Lauritsen L., Brunak S. Improving the odds in discriminating “drug-like” from “nondrug-like” compounds. J. Chem. Inf. Comput. Sci. 2000;40:1315–1324. doi: 10.1021/ci0003810. [DOI] [PubMed] [Google Scholar]
- 48.Ghose K., Viswanadhan V.N., Wendoloski J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999;1(1):55–68. doi: 10.1021/cc9800071. [DOI] [PubMed] [Google Scholar]
- 49.Ajay, Walters P., Murcko A. Can we learn to distinguish between “drug-like” and “nondrug-like” molecules? J. Med. Chem. 1998;41:3314–3332. doi: 10.1021/jm970666c. [DOI] [PubMed] [Google Scholar]
- 50.Martin Y.C. A bioavailability score. J. Med. Chem. 2005;48(9):3164–3170. doi: 10.1021/jm0492002. [DOI] [PubMed] [Google Scholar]
- 51.Ajay, Bemis W., Murcko A. Blood brain barrier: design in libraries with CNS activity. J. Med. Chem. 1999;42:4942–4951. doi: 10.1021/jm990017w. [DOI] [PubMed] [Google Scholar]
- 52.Yazdanian M., Glynn L., Wright L. Hawi, Correlating partitioning and Caco-2 cell permeability of structurally diverse small molecular weight compounds. Pharm. Res. (N. Y.) 1998;15:1490–1494. doi: 10.1023/a:1011930411574. [DOI] [PubMed] [Google Scholar]
- 53.Irvine D., Takahashi L., Lockhart K., Cheong J., Tolan W., Selick E., Grove R. MDCK (Madin-Darby canine kidney) cells: a tool for membrane permeability screening. J. Pharm. Sci. 1999;88:28–33. doi: 10.1021/js9803205. [DOI] [PubMed] [Google Scholar]
- 54.Daina, Michielin O., Zoete V. iLOGP: a simple, robust, and efficient description of n-octanol/water partition coefficient for drug design using the GB/SA approach. J. Chem. Inf. Model. 2014;54(12):3284–3301. doi: 10.1021/ci500467k. [DOI] [PubMed] [Google Scholar]
- 55.Delaney J.S. ESOL: estimating aqueous solubility directly from molecular structure. J. Chem. Inf. Comput. Sci. 2004;44(3):1000–1005. doi: 10.1021/ci034243x. [DOI] [PubMed] [Google Scholar]
- 56.Mortelmans K., Zeiger E. The Ames Salmonella/microsome mutagenicity assay. Mutat. Res. 2020;2:29–60. doi: 10.1016/s0027-5107(00)00064-6. [DOI] [PubMed] [Google Scholar]
- 57.Trott O., Olson A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tan K.P., Varadarajan R., Madhusudhan M.S. DEPTH: a web server to compute depth and predict small-molecule binding cavities in proteins. Nucleic Acids Res. 2011;39(2):W242–W248. doi: 10.1093/nar/gkr356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tian W., Chen C., Lei X., Zhao J., Liang J. CASTp 3.0: computed atlas of surface topography of proteins. Nucleic Acids Res. 2018;46(W1):W363–W367. doi: 10.1093/nar/gky473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schrödinger Release 2019-3 . Schrödinger; New York, NY: 2019. Maestro-Desmond Interoperability Tools. Desmond Molecular Dynamics System, D. E. Shaw Research, New York, NY, 2019. [Google Scholar]
- 61.Baker N.A., Sept D., Joseph S., Holst M.J., McCammon J.A. Electrostatics of nanosystems: application to microtubules and the ribosome. PNAS U.S.A. 2001;98(18):10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kumari R., Kumar R. Open Source Drug Discovery Consortium, A. Lynn, g_mmpbsa--a GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014;54(7):1951–1962. doi: 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- 63.Pronk S., Páll S., Schulz R., Larsson P., Bjelkmar P., Apostolov R., Shirts M.R., Smith J.C., Kasson P.M., van der Spoel D., Hess B., Lindahl E. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29(7):845–854. doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wong S.K., Li W., Moore M.J., Choe H., Farzan M. A 193-amino acid fragment of the SARS coronavirus S protein efficiently binds angiotensin-converting enzyme 2. J. Biol. Chem. 2004;279:3197–3201. doi: 10.1074/jbc.C300520200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jeffers S.A., Tusell S.M., Gillim-Ross L., Hemmila E.M., Achenbach J.E., Babcock G.J., Thomas W.D., Jr., Thackray L.B., Young M.D., Mason R.J., Ambrosino D.M., Wentworth D.E., Demartini J.C., Holmes K.V. CD209L (L-SIGN) is a receptor for severe acute respiratory syndrome coronavirus, PNAS. U.S.A. 2004;101:15748–15753. doi: 10.1073/pnas.0403812101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lokugamage K.G., Narayanan K., Huang C., Makino S. Severe acute respiratory syndrome coronavirus protein nsp1 is a novel eukaryotic translation inhibitor that represses multiple steps of translation initiation. J. Virol. 2012;86:13598–13608. doi: 10.1128/JVI.01958-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nieto-Torres J.L., Verdia-Baguena C., Jimenez-Guardeno J.M., Regla-Nava J.A., Castano-Rodriguez C., Fernandez-Delgado R., Torres J., Aguilella V.M., Enjuanes L. Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology. 2015;485:330–339. doi: 10.1016/j.virol.2015.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Huang, Lokugamage K.G., Rozovics J.M., Narayanan K., Semler B.L., Makino S. SARS coronavirus nsp1 protein induces template-dependent endonucleolytic cleavage of mRNAs: viral mRNAs are resistant to nsp1-induced RNA cleavage. PLoS Pathog. 2011;7 doi: 10.1371/journal.ppat.1002433. E1002433-E1002433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tan Y.J., Tham P.Y., Chan D.Z., Chou C.-F., Shen S., Fielding B.C., Tan T.H., Lim S.G., Hong W. The severe acute respiratory syndrome coronavirus 3a protein up-regulates expression of fibrinogen in lung epithelial cells. J. Virol. 2005;79:10083–10087. doi: 10.1128/JVI.79.15.10083-10087.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tan Y.J., Fielding B.C., Goh P.Y., Shen S., Tan T.H.P., Lim S.G., Hong W. Overexpression of 7a, a protein specifically encoded by the severe acute respiratory syndrome coronavirus, induces apoptosis via a caspase-dependent pathway. J. Virol. 2004;78:14043–14047. doi: 10.1128/JVI.78.24.14043-14047.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cottam E.M., Whelband M.C., Wileman T. Coronavirus NSP6 restricts autophagosome expansion. Autophagy. 2014;10:1426–1441. doi: 10.4161/auto.29309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.te Velthuis J., van den Worm S.H., Snijder E.J. The SARS-coronavirus nsp7+nsp8 complex is a unique multimeric RNA polymerase capable of both de novo initiation and primer extension. Nucleic Acids Res. 2012;40:1737–1747. doi: 10.1093/nar/gkr893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bouvet M., Imbert I., Subissi L., Gluais L., Canard B., Decroly E. RNA 3'-end mismatch excision by the severe acute respiratory syndrome coronavirus nonstructural protein nsp10/nsp14 exoribonuclease complex. PNAS. U.S.A. 2012;109:9372–9377. doi: 10.1073/pnas.1201130109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Helgeson S.A., Mwakyanjala E.J., Parikh P.P., Venkatachalam K.L. Hyoscyamine for a slow ventricular response during atrial fibrillation. Ann. Intern. Med. 2018;169(6):418–419. doi: 10.7326/L18-0037. [DOI] [PubMed] [Google Scholar]
- 75.Gupta N. Hyoscyamine for a slow ventricular response during atrial fibrillation. Ann. Intern. Med. 2019;70(10):735. doi: 10.7326/L19-0092. [DOI] [PubMed] [Google Scholar]
- 76.Kanokmedhakul S., Kanokmedhakul K., Nasomjai P., Louangsysouphanh S., Soytong K., Isobe M., Kongsaeree P., Prabpai S., Suksamrarn A. Antifungal azaphilones from the fungus Chaetomium cupreum CC3003. J. Nat. Prod. 2006;69(6):891–895. doi: 10.1021/np060051v. [DOI] [PubMed] [Google Scholar]
- 77.Marques J.V., Kitamura R.O., Lago J.H., Young M.C., Guimarães E.F., Kato M.J. Antifungal amides from Piper scutifolium and Piper hoffmanseggianum. J. Nat. Prod. 2007;70(12):2036–2039. doi: 10.1021/np070347g. [DOI] [PubMed] [Google Scholar]
- 78.Wu, Liu Y., Yang Y., Zhang P., Zhong W., Wang Y., Wang Q., Xu Y., Li M., Li X., Zheng M., Chen L., Li H. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B. 2020 doi: 10.1016/j.apsb.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ibrahim M., Abdelmalek D.H., Elshahat M.E., Elfiky A.A. COVID-19 spike-host cell receptor GRP78 binding site prediction. J. Infect. 2020 doi: 10.1016/j.jinf.2020.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jin Z., Du X., Xy Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Duan Y., Yu J., Wang L., Yang K., Liu F., You T., Liu X., Yang X., Bai F., Liu H., Liu X., Guddat L.W., Xiao G., Qi C., Shi Z., Jiang H., Rao Z., Yang H. bioRxiv; 2020. Structure-based Drug Design, Virtual Screening and High-Throughput Screening Rapidly Identify Antiviral Leads Targeting COVID-19. [DOI] [Google Scholar]
- 81.Talluri S. 2020. Virtual Screening Based Prediction of Potential Drugs for COVID-19; p. 2020020418. Preprints 2020. [DOI] [PubMed] [Google Scholar]
- 82.ul Qamar M.T., Alqahtani S.M., Alamri M.A., Chen L. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Anal. 2020 doi: 10.1016/j.jpha.2020.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Neuman B.W. Bioinformatics and functional analyses of coronavirus non-structural proteins involved in the formation of replicative organelles. Antivir. Res. 2016;135:97–107. doi: 10.1016/j.antiviral.2016.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gupta M.K., Vemula S., Donde R., Gouda G., Behera L., Vadde R. In-silico approaches to detect inhibitors of the human severe acute respiratory syndrome coronavirus envelope protein ion channel. J. Biomol. Struct. Dyn. 2020:1–11. doi: 10.1080/07391102.2020.1751300. Advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lee Hyun-Jung, Koohy H. In silico identification of vaccine targets for 2019-nCoV. F1000Res. 2020;9:145. doi: 10.12688/f1000research.22507.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data is presented within the manuscript and the supplemental materials.