

Abstract
The Precision Interventions for Severe and/or Exacerbation-prone Asthma (PrecISE) study is an adaptive platform trial designed to investigate novel interventions to severe asthma. The study is conducted under a master protocol and utilizes a crossover design with each participant receiving up to five interventions and at least one placebo. Treatment assignments are based on the patients’ biomarker profiles and precision health methods are incorporated into the interim and final analyses. We describe key elements of the PrecISE study including the multistage adaptive enrichment strategy, early stopping of an intervention for futility, power calculations, and the primary analysis strategy.
Keywords: Master protocol, platform trial, covariate adaptive randomization, adaptive enrichment, CompEx, severe asthma
1. Introduction
One in 13 persons living in the United States has asthma, and 5 – 10% of these patients have severe asthma, for a total of about 2.5 million (Nanda and Wasan, 2020). Patients with severe asthma experience substantial morbidity and require extensive use of healthcare resources (Israel and Reddel, 2017). While most asthma is controlled by currently available therapy, severe asthma is characterized by poor control, low lung function, and/or increased risk of severe exacerbations (Chung et al., 2014). Optimal treatment for patients with severe asthma is uncertain, because the pathophysiologic underpinnings of severe asthma are heterogeneous (Israel and Reddel, 2017; Siddiqui et al., 2019). A treatment may be effective for only a subset of patients. The newest therapies for severe asthma, for example, appear to be primarily effective in patients with high eosinophils (FitzGerald et al. 2016, Bleecker et al 2016).
This trial was funded by the National Heart, Lung, and Blood Institute (NHLBI) of the National Institutes of Health (NIH). The request for funding called for a Phase 2 proof-of-concept clinical trial to assess a number of novel interventions in biomarker-defined subgroups of patients with severe asthma and obtain information on the utility of the biomarkers in identifying patients with a higher likelihood to respond to a specific treatment. Rather than propose more standard, non-adaptive, biomarker-stratified, parallel group designs, applicants were encouraged to propose adaptive trial designs that made use of accumulating data on biomarker-outcome relationships to modify design features during the trial.
The PrecISE network was formed and immediately began discussions about the various strategies proposed for improving the treatment of severe asthma. The protocol was finalized in early 2019, and the PrecISE study (ct.gov number is NCT04129931) began enrollment in December 2019. An overview of the study design, including the selection of interventions and clinical and other scientific considerations in designing the study, is provided elsewhere (Israel, et al., 2021). In this paper we describe the statistical considerations that were critical to designing the PrecISE study.
The primary objectives of the PrecISE study are to (1) identify novel therapies with activity in biomarker-defined subgroups of participants with severe asthma, and (2) optimize the subgroups targeted for treatment by refining the biomarker and subgroup definitions. Key features of the final study design and their relevance to the PrecISE study objectives include:
Master protocol – allows multiple interventions to be investigated under a single protocol, each evaluated relative to placebo, independently of the other interventions in the study. The focus is on generating evidence about the efficacy of each intervention and not on comparing the interventions to each other.
Platform trial – allows interventions to enter the study at different times, as they become available. A platform trial can run in perpetual fashion, once the master protocol infrastructure and trial governance are established.
Adaptive trial – allows for early discontinuation of interventions due to futility. Additionally, the randomization probabilities for the various interventions are adapted as a result of a prospective enrichment strategy based on baseline biomarkers, as information about biomarker and their relationships to outcomes accrues during the study.
A crossover design was selected to allow each participant to receive multiple interventions during the study and to support the planned precision medicine analyses. Consistent with the phase 2, proof-of-concept objectives, interventions are evaluated to provide preliminary evidence of efficacy while also accumulating information about safety and tolerability. The choice of endpoints and determination of sample size and power are described in Section 2, followed by details of the crossover design in Section 3. The adaptive features and enrichment strategy are described in Section 4. The randomization algorithm based on biomarker profiles of participants is described in Section 5, and the analysis model is described in Section 6. Section 7 provides some practical considerations for implementation of the master protocol. Section 8 includes a discussion of several of the many design decisions made in developing the PrecISE protocol.
2. Endpoints, sample size and statistical power
The PrecISE study is planning to enroll 800 participants. We expect that slightly more than 80% of participants, about 650, will be adults (age ≥ 18 years) and about 150 of participants will be adolescents (age ≥ 12 years and younger than 18 years). The primary endpoints of the PrecISE study capture three important dimensions of severe asthma. They are 1) forced expiratory volume (FEV1) percent predicted, assessed prior to bronchodilator administration; 2) asthma symptom control, assessed via the 6-item Juniper Asthma Control Questionnaire (ACQ-6); and 3) CompEx events (Fuhlbrigge et al., 2017), a composite endpoint that includes exacerbations, assessed over 16 weeks of treatment. All three endpoints are equally important in that an intervention is considered efficacious if significant benefit is shown for any one of the three primary endpoints (FEV1 percent predicted, ACQ-6, and CompEx events). We use the Hochberg method (Hochberg, 1988) to adjust for multiplicity of the three endpoints. Multiple interventions are being tested in PrecISE and no multiplicity adjustment will be made across the interventions during data analysis. The study uses crossover allocation with 16-week treatment periods followed by 8-week washout periods (see Section 3 for more details). Each subject can participate in up to 6 treatment periods to receive multiple interventions. Because of the potential for carryover effects in a crossover design, even with washout periods of sufficiently long duration to account for a drug’s pharmacokinetic and pharmacodynamic properties, we are not assessing efficacy outcomes relative to change from baseline at the start of each period but rather comparing active treatments with placebo based on outcomes observed at the end of the treatment period.
The goal of the study is to investigate the efficacy of each of the six interventions compared to placebo with respect to the three primary endpoints. The efficacy of an intervention will be established if the Hochberg adjusted two-sided p-value with respect to at least one of the three endpoints is less than 0.1. For a given intervention, a sample size of 150 participants was chosen to achieve at least 80% power to detect a treatment effect with respect to one or more of the three primary endpoints, if the true standardized effect size for at least one endpoint is equal to 0.3. This power calculation takes into account the possibility to stop for futility (see Section 4 for details of the futility analysis). Power was estimated using a t-test under the assumption of a within-subject, between-period correlation of responses on placebo and test treatment, τ, of 0.38 assuming an exchangeable correlation structure among responses on the same subject (see Section 3 for more details about our within-subject correlation assumptions). We perform prospective subgroup re-estimation in this trial (Section 4). These power calculations assume that the effect size is 0.3 in each of the three subgroups included in the final data analysis (the initial subgroup and the two estimated subgroups). Table 1 below gives the probability to reject the null hypothesis for two values of within-subject correlation, τ, and for three values of the correlation between each pair of the three endpoints (FEV1 percent predicted, ACQ-6, and CompEx events), ρ. We do not have preliminary data on the value of ρ. Fortunately, power does not vary substantially with the values of ρ. The Type I error rate is controlled at the two-sided 0.1 level assuming the futility rule is non-binding, that is, the study may or may not be stopped when the futility analysis suggests it should. Assuming that the stopping rule for futility is followed, the Type I error rate is equal to about 0.05, two-sided, for τ = 0.38 and a wide range of values for ρ (Table 1).
Table 1.
Power for various effect sizes and values of within-subject (τ) and between-endpoint (ρ) correlation.
| Effect sizes | Correlation τ = 0.38 | Correlation τ = 0.20 | ||||||
|---|---|---|---|---|---|---|---|---|
| FEV1% | ACQ-6 | CompEx | ρ = 0 | ρ = 0.3 | ρ = 0.5 | ρ = 0 | ρ = 0.3 | ρ = 0.5 |
| 0 | 0 | 0 | 0.05 | 0.04 | 0.04 | 0.06 | 0.05 | 0.05 |
| 0.3 | 0 | 0 | 0.82 | 0.80 | 0.80 | 0.74 | 0.75 | 0.73 |
| 0.3 | 0.3 | 0 | 0.97 | 0.94 | 0.92 | 0.94 | 0.91 | 0.87 |
| 0.3 | 0.3 | 0.3 | 1.00 | 0.98 | 0.95 | 0.99 | 0.96 | 0.93 |
With 30 participants enrolled every month to obtain a total of 800 participants, and with at least 38-months study duration, we will accumulate enough participant-periods to study six interventions. Note that we will analyze a combined population of adults and adolescents. The study is not powered to look at adults or adolescents separately. These calculations take into account a drop-out rate of 0.02 every eight weeks (or approximately 12% per year). That is, the probability for a participant to permanently drop-out of the study in an 8-week time period is about 0.02. A similar drop-out rate was observed in recent phase 3 studies in severe asthmatics during one year of follow-up (FitzGerald et al. 2016, Bleecker et al. 2016).
As described above, the study should have sufficient power to detect standardized effect sizes of 0.3, that is, treatment effects equal to 0.3 times the standard deviation. For our three primary endpoints, this translates to a difference of 4.3 percent predicted FEV1 between intervention and placebo with a standard deviation (SD) of 14.5 percent predicted; a difference of 0.3 in average ACQ-6 symptom scores between the intervention and placebo with a SD of 1; and a difference in the CompEx 16-week event rate of 0.66, e.g., a difference between an event rate in the test treatment group of 1.55 (SD = 1.78) over 16 weeks and in the placebo group of 2.17 (SD = 2.50) over 16 weeks. The FEV1 and ACQ-6 treatment effects are thought to be both clinically relevant and able to be observed in a trial with severe asthma patients. The CompEx is a novel endpoint without an established clinically relevant treatment effect.
3. Crossover design
Severe asthma is a chronic disease, and, hence, a crossover trial design was considered a viable option. Crossing patients over from one treatment to another provides higher power than a parallel group design with the same number of subjects. In a crossover study, each participant can serve as his/her own control since for a given endpoint, responses from the same participant on active treatment and placebo are likely positively correlated. This leads to further increases in power. In fact, data from the BARD trial (Wechsler et al., 2019) show that the within-subject correlation can be quite high. The BARD trial was a crossover trial with four 14-week periods. We estimated that the correlation of FEV1 percent predicted values observed at the end of each period in that trial was 0.89. The correlation between period-wise asthma control days, another main endpoint in the BARD study, was 0.77.
The shortcomings of a crossover approach include the potential for carryover effects from a prior treatment and more complex data analysis compared to a traditional parallel group trial. In crossover studies, the power to detect a treatment effect and the operating characteristics of stopping rules for futility and efficacy depend not only on the variability of an outcome but also on the within-subject correlation. The latter is often unknown or with little data available, making the planning for a crossover study somewhat more challenging than for a more traditional, parallel group study. In planning PrecISE, we were able to access the BARD trial data cited above to get an idea about the within-subject correlation for one of our endpoints.
Following an initial screening period, eligible participants are randomized and enter into the multi-period crossover study. Each period is comprised of a 16-week treatment period followed by an 8-week washout period. Washout periods can be longer depending on the half-life of a drug; in general, the length of a washout should be at least five times the known pharmacokinetic half-life of the drug under consideration (Hedaya 2012). We expect that some participants will be able to be in the study for up to six treatment periods, that is, approximately three years including the washouts. As the likelihood of participants dropping out of the study increases with increased duration of participation, we expect that most subjects will participate in 3–4 treatment periods.
The interventions selected for evaluation in PrecISE all have matching placebos. For oral medications, if identical pills/tablets/powders are not available from the manufacturer, over-encapsulation is used to create a matching placebo. For medications administered by injection, normal saline is used, and although the pharmacist preparing the vials as well as the clinical personnel administering the injection are unblinded, the rest of the clinical staff remain blinded to treatment assignment for the study. Each subject participates in multiple treatment periods corresponding to different interventions. Each participant’s involvement in the study consists of three phases: (i) an initial screening phase, (ii) a two-period crossover phase with sequences T:P or P:T for an active treatment (T) and matching placebo (P), and (iii) a multi-period crossover phase during which participants receive up to four different interventions and are re-randomized to a new intervention following washout from the prior intervention. A small percentage of participants will receive placebo a second time during this phase in order to maintain masking and assess sequence effects throughout the study. The probability to receive a placebo is 0.13 in each period for periods 3–6, but no subject will have more than two placebo periods. An example of a crossover sequence for a subject in PrecISE is illustrated in Figure 1, and the randomization algorithm generating this sequence is described further in Section 5.
Figure 1.
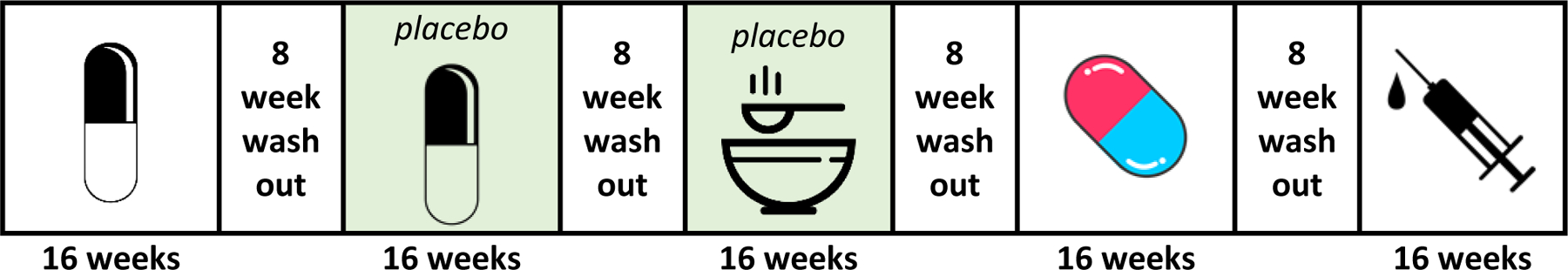
An example of a multiple-period crossover allocation in the PrecISE study. A subject is first randomized to a two-period, T:P or P:T, crossover sequence with an oral intervention and its matching placebo. In period 3, the subject is randomized to receive a placebo matching a dietary supplement intervention. In period 4, the subject is randomized to receive another oral intervention (non-placebo) and in period 5, to an intervention administered as an injection (non-placebo).
The asthma literature indicates that there is placebo response in asthma (Dutile, Kaptchuk, and Wechsler 2014). In chronic diseases other than asthma, mode of administration of an intervention including placebo might augment response to treatment (de Wit et al. 2016). Since interventions in PrecISE have different modes of administration, namely, an injectable, pill, or dietary supplement, we would ideally repeat the two-period crossover phase, phase (ii), allowing each participant to contribute data on several active interventions and their matching placebos. This would, however, require a much longer study than is planned for PrecISE. Our hybrid approach combining phases ii and iii allows reducing the number of placebos and increasing the number of experimental therapies each participant may receive. This makes the study more attractive to potential participants (Israel et al. 2021). Under the hybrid approach, the placebo data are shared across interventions for analysis (see Section 6).
4. Precision medicine and adaptive design features
Six interventions were selected for the PrecISE study (Israel et al. 2021) (see Table 2). It is hypothesized that each intervention studied in PrecISE works best in a biomarker-defined subgroup of patients. Initial enrollment to each therapy targets the a priori best subgroup of patients that was specified for each intervention based on available information before the trial. Table 2 shows the interventions that are studied in the PrecISE trial and their a priori best subgroups. The a priori best subgroup for each intervention is specified based on one or two baseline biomarkers, that is, biomarkers obtained on each participant at the end of the screening phase (i) before the participant’s first randomization. We use biomarkers obtained before the first randomization because some interventions in PrecISE might temporarily change the value of these biomarkers. Although the treatment may alter the biomarker, it is not expected to change the underlying disease pathology. The prevalence of each subgroup (Table 2) was estimated using the data on subjects with severe asthma recruited by the Severe Asthma Research Program (SARP) (Teague et al. 2018).
Table 2.
The list of interventions and their a priori best subgroups defined based on blood eosinophils (Eos), plasma level of IL 6 (IL 6), and fractional exhaled nitric oxide (FeNO) test
| Intervention | A priori best subgroup | Prevalence |
|---|---|---|
| Imatinib | Eos < 300 cells/μl | 62% |
| Clazakizumab | IL 6 > 3.1 pg/|μl | 33% |
| Itacitinib | Eos ≥ 300 cells/μl or FeNO > 20 ppb | 57% |
| Cavosonstat | Genotypes | 64% |
| Broncho-Vaxom | Eos ≥ 300 cells/μl | 38% |
| Medium Chain | FeNO ≥ 15 ppb | 64% |
| Triglycerides (MCT) |
When a sufficient number of patients are enrolled in the a priori best subgroup, we will undertake futility testing of that intervention. All of the proposed interventions are novel in severe asthma; thus, there is a high likelihood that at least one of them will prove ineffective. It was decided in study planning that a fairly aggressive futility rule would be used, to free up the study’s resources for interventions with a high probability of success. A single interim futility analysis is planned for each intervention (independently of the others) after test treatment and placebo (matched or unmatched) data are available on 60 participants in the a priori best subgroup. To set up a futility rule for this trial with crossover allocation we investigated various futility rules and optimal timing to stop for futility (Chang et al. 2020). We use the futility rule from He et al. (2012). For each intervention, the futility analysis is performed after data on 40% of the total sample size of 150 are available. For a single endpoint, the probability to stop for futility under the alternative hypothesis is 0.15. This is when the treatment effect and the SD are equal to those hypothesized. The futility test will be applied to each of the three primary endpoints separately. An intervention must show futility for all three endpoints to be dropped from the study. Table 3 illustrates the probability of stopping for futility for two values of a within-subject, between-period correlation of responses on placebo and test treatment, τ, and the correlation ρ between each pair of the three endpoints, FEV1 percent predicted, ACQ-6, and CompEx events.
Table 3.
Probability of stopping for futility
| Effect sizes | Correlation τ = 0.38 | Correlation τ = 0.20 | ||||||
|---|---|---|---|---|---|---|---|---|
| FEV1% | ACQ-6 | CompEx | ρ = 0 | ρ = 0.3 | ρ = 0.5 | ρ = 0 | ρ = 0.3 | ρ = 0.5 |
| 0 | 0 | 0 | 0.63 | 0.68 | 0.71 | 0.49 | 0.55 | 0.58 |
| 0.3 | 0 | 0 | 0.11 | 0.14 | 0.15 | 0.10 | 0.12 | 0.14 |
| 0.3 | 0.3 | 0 | 0.02 | 0.04 | 0.06 | 0.02 | 0.04 | 0.06 |
| 0.3 | 0.3 | 0 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | 0.01 |
If the interim analysis for an intervention does not indicate futility, we continue enrolling to the intervention until we not only have data from 60 participants who belong to the a priori best subgroup and but also 30 participants outside the a priori best subgroup. For example, for Broncho-Vaxom the a priori best subgroup is defined as subjects with blood eosinophils measured before the first randomization greater than 300 cells/μl, and the subjects outside the a priori best subgroup are those with blood eosinophils less than or equal to 300 cells/μl. The first precision medicine analysis for Broncho-Vaxom will be performed when test treatment and placebo data are available for at least 60 participants with ≥ 300 cells/μl and 30 participants with < 300 cells/μl. The goal of this analysis is to refine the definition of the targeted subgroups through modifications made to the cut points delineating marker positive and negative participants. The purpose of subgroup estimation is to enrich the population prospectively to increase the chances of demonstrating treatment effects at the end of the trial, as testing the efficacy of the intervention in an unenriched population of all participants may result in failure to detect the treatment effect. The best subgroup is defined as the subgroup that maximizes the power of detecting a treatment effect when testing the null hypothesis that the intervention has no effect on any of the three primary endpoints versus the alternative hypothesis that there is an effect on at least one of the primary endpoints. This approach to defining the best subgroup reflects not only the treatment effect in the subgroup but also the size of the subgroup (Lai, Lavori and Liao 2014; Zhang et al. 2017; Joshi et al. 2019, Joshi, Nguyen and Ivanova 2020). That is, an intervention with a large estimated effect in a small targeted subgroup is deemed less desirable than a slightly smaller effect in a broader targeted subgroup with this approach.
After the first precision medicine interim analysis, treatment assignment probabilities for participants are adjusted to reflect the updated target subgroup definitions. The treatment assignment probabilities will be set to achieve an approximate 2:1 ratio of biomarker-positive to biomarker-negative participants for that intervention, using the updated best subgroup definitions, provided the prevalence of the new best subgroup, p, is less than 0.667. Otherwise, the treatment assignment probabilities will be set to achieve an approximate ratio of p:(1 – p). The second precision medicine interim analysis will be performed as soon as test treatment and placebo data are available for 45 additional subjects in the estimated best subgroup and 20 subjects (or less than 20 for p > 0.67) outside the best subgroup (beyond the 60 + 30 participants used in the first precision medicine interim analysis). Some of these participants may come from the cohort of participants enrolled prior to the first precision medicine interim analysis that were still in follow-up during the time of that analysis. It is important that the statistician performing the first precision medicine interim analysis is masked with regards to data of participants still in follow-up.
All available response data will be used in the second precision medicine interim analysis. Up to two additional biomarkers may be considered in determining the updated best subgroup definition. This analysis will provide the final estimate of the best subgroup. The final efficacy analysis will be performed as soon as data on 150 participants from a best subgroup are available, including 60 participants from the a priori best subgroup, 45 participants from the best subgroup estimated in the first precision medicine analysis, and 45 participants from the best subgroup estimated in the second precision medicine analysis. This approach of prospective enrichment increases our chances to establish efficacy of the interventions we investigate (Joshi et al. 2020). As discussed in Section 6, conducting the final efficacy analysis on the union of the a priori and updated subgroups ensures control of the Type I error probability for the analysis. The test of efficacy, however, includes three targeted patient subgroups rather than using a single subgroup.
The PrecISE Data and Safety Monitoring Board (DSMB), established by and advisory to the NHLBI, is tasked with reviewing interim analysis data for purposes of stopping an intervention due to futility, and for potential decrease to sample size, as described above, in addition to performing their other safety oversight responsibilities. A second independent advisory group, the Protocol and Adaptations Review Committee (PARC), also set up by the NHLBI, will advise on study adaptations and on new interventions that may be considered for entry into the study. The PARC will not review any unblinded data or comparative analyses to avoid the potential for bias in making their recommendations.
At the end of the trial, an exploratory precision medicine analysis to estimate individualized treatment rules for severe asthmatics with different biomarker profiles will be performed. Interventions that were found to be significantly better than placebo, and whose estimated best target subgroups overlap, will be analyzed to identify the best intervention in each overlapping region. For example, if the target subgroups for two interventions (found to be effective in the study) both include participants with high IL-6 plasma levels and high blood eosinophils, exploratory analyses will be conducted to determine if either appears to provide superior benefit in that overlapping subgroup region. Potential methods for this exploratory analysis include random forests (Zhu, Zeng and Kosorok 2015), linear regression models with quadratic terms and interactions, and outcome weighted learning models and other individualized treatment rule estimation methods (Zhao et al. 2012; Kosorok and Laber 2019). Equivalence of two treatments for a given value of the vector of baseline biomarkers will be defined as a difference in expected outcome for those two treatments of less than 10% of the overall standard deviation of outcome.
5. Randomization
For the initial two-period crossover phase (phase (ii) defined in Section 3), each participant will first be assigned to an intervention based on his/her biomarker profile using a biased-coin type design that favors treatments targeting the particular profile of that participant. A second randomization then occurs to determine the crossover sequence, i.e., whether participant receives test treatment followed by placebo (T:P) or placebo followed by test treatment (P:T). In subsequent treatment periods (periods 3–6 in phase ii as defined above), the same biased-coin design will be repeated to assign interventions, but restricted to interventions not yet received by a participant.
The a priori best subgroups initially defined for each intervention (Table 2) have some degree of overlap, such that some participants will be targeted by more than one intervention. The randomization probabilities at any point in the trial will reflect the priorities in place at that time. For example, interventions that have not yet reached the required sample size to test for futility will be favored over interventions that have already enrolled enough participants to conduct the futility analysis.
To illustrate the randomization algorithm, consider the example illustrated in Figure 1 (see Section 3). Assume the first subject enrolled is a male with blood eosinophils ≥300 cells/μl, FeNO<15 ppb, and IL-6 ≥3.1 pg/μl, and he has the SNP genotype targeted by Cavosonstat. Based on the intervention-specific exclusions, the subject is eligible for all of the interventions, and based on his biomarker profile, he is in the a priori best subgroup for clazakizumab, itacitinib, Cavosonstat, and Broncho-Vaxom, but not for MCT or imatinib. At the beginning of the trial, there are three treatments ready for enrollment: imatinib, clazakizumab and MCT. Each of the interventions is enrolling in a 2:1 ratio inside vs outside the a priori best subgroup. The odds of the hypothetical subject to be randomized to imatinib, clazakizumab and MCT are 1:2:1, as he is in the a priori best subgroup for clazakizumab only. These odds are approximate, because the randomization algorithm also balances with respect to the three potential prognostic covariates described above. Suppose the subject is randomized to imatinib. Because this is the first randomization, the subject is next randomized to sequence, e.g., T:P or P:T. In this example, the subject receives active imatininb in period 1 and its matching placebo in period 2.
At the beginning of period 3, assume all six interventions have entered the study. The subject is randomized to MCT in period 3, and because he has not yet received a second placebo, he is randomized to active treatment with probability 0.87 and to matching placebo with probability 0.13. In this example, he receives the MCT matching placebo, and as a result, is no longer eligible for placebo assignments in the remaining periods (periods 4–6). In addition, he is no longer eligible for active MCT. This is to prevent unblinding of interventions -- if the subject were to receive both active and placebo MCT in periods 3 and 4, for example, he would know that all subsequent interventions are active, because the maximum number of placebo assignments a subject can receive is two, as indicated in the informed consent. In the next period the subject is randomized to subject Broncho-Vaxom. There is no second randomization to active versus placebo because the subject has received the second placebo already. In period 5 the subject is randomized to clazakizumab.
In the example above only three interventions were available in the beginning of the study. If an intervention enters the study with a targeted subgroup similar to other interventions already in the study, the randomization algorithm is adapted so that priority is given to the interventions closest to reaching the sample size required for the interim futility analysis.
6. Efficacy Analysis
The final analysis to assess the efficacy of an intervention will be performed when accrual and follow-up for that intervention are complete. Treatment effects with respect to each of the first two primary endpoints will be estimated using a mixed model for repeated measurements (MMRM) that appropriately takes into account the within-subject correlations between periods. The MMRM will include treatment, period (1–6), and baseline values (FEV1 and ACQ-6), measured before the first randomization, as fixed effects. A Toeplitz, diagonal-constant, variance-covariance matrix will be used for the MMRM analysis. CompEx event rates will be modeled using a log-linear model for mean event count, with log follow-up time as an offset variable, the same set of covariates as described above for the MMRM, and assuming variance proportional to the mean. Generalized estimating equations with the Toeplitz correlation matrix will be used to estimate regression parameters and compute a robust (sandwich) covariance matrix estimator.
For a given intervention and outcome j, j = 1,2,3, let Zj1 be the test statistic computed based on data from participants in the a priori best subgroup assigned to the intervention before the first precision medicine analysis, Zj2 be the test statistic based on data collected on biomarker-positive subjects after the first precision medicine analysis, and Zj3 be the test statistic based on data collected on biomarker-positive subjects after the second precision medicine analysis. A separate MMRM will be fit to obtain Zj1, Zj2, and Zj3. The intervention subscript is omitted here, for brevity. In the final analysis, the treatment effect of a given intervention with respect to outcome j, j = 1,2,3, is tested by computing the inverse normal combination of the three test statistics (Lehmacher, Wassmer 1999) Zj = sqrt(m1/m)Zj1 + sqrt(m2/m)Zj2 + sqrt(m3/m)Zj3, where mk is the number of participants contributing to the kth test statistic, k = 1,2,3, with m = m1 + m2 + m3 = 60 + 45 + 45 =150.
All available placebo data for a participant will be included in the efficacy analysis of an intervention together with the participant’s data from an active period on that intervention. Sensitivity analyses will be conducted using the same MMRM approach as described above but with the addition of a fixed effect for mode of administration of test treatment or placebo (oral medication as a reference group vs injection vs dietary supplement). For a given outcome, a p-value for at least one of the mode of administration effects of 0.1 or less would suggest that the response to treatment is affected by the mode of administration. If this is the case, we will repeat the primary analysis of that outcome by adding the mode of administration in the model.
Period-specific baseline values of FEV1 percent predicted and ACQ-6 will be analyzed to look for evidence of carryover effects from one treatment period to another due to the crossover nature of the study design. If there is an indication of a carryover effect, an analysis will be performed using the primary analysis models but with the addition of effects corresponding to the treatment a participant received prior to the current treatment to control for the carryover effect.
Sensitivity analyses will be conducted to examine treatment effects estimated only from the first treatment period. First period data from for all participants will form the basis of comparisons of each intervention versus pooled placebo (pooling from first periods only). Data from the first two periods will be analyzed similarly. For both analyses, results will be compared to those from the primary analysis to better understand the impact of the crossover design on analysis results.
To assess the sensitivity of the primary analysis results to the potential for a ceiling effect, i.e. improvement in participants’ asthma symptoms over time due to trial participation, an analysis will be conducted to determine whether participants enrolled in the study improve regardless of the interventions they receive. In this analysis, we will compare response to placebo in periods 1 and 2 to response to placebo in periods 3–6 with adjustment for the previous treatment received. Additionally, we will evaluate the association between the period and subject’s baseline by fitting a model similar to the primary analysis model with baseline as the outcome and period and previous treatment as covariates.
7. Master protocol considerations
The PrecISE study is being conducted under a master protocol that capitalizes on two areas of innovation (see Woodcock and LaVange 2016), infrastructure and trial design. A single master protocol was developed that includes all of the study design elements common across interventions. Separate appendices to the protocol provide intervention-specific information, including additional safety exclusion or monitoring criteria, dose-adjustments, etc. Enrollment began with two of the six planned interventions available. New appendices for the protocol for DSMB, Institutional Review Board (IRB), and FDA review are then made as each intervention enters the study.
Use of shared trial governance, data and randomization systems, oversight boards, and central laboratories and reading centers across all interventions provides efficiencies compared to conducting individual trials for each intervention. Similarly, the use of common protocol elements, case report forms, analysis models, and other features helps reduce study start-up times as new interventions are added to the trial.
Design innovations include the adaptive enrichment strategy described above and the ability to share placebo data across interventions, thereby reducing the time participants spend on placebo. Incorporating a second placebo in a subset of patients enables issues of seasonality, ceiling effect and other time-varying confounders to be examined in the final analysis. Israel, et al. (2021) provides additional discussion of the PrecISE master protocol and trial governance structure.
8. Discussion
Several key decisions were made in designing the PrecISE study. Notable among these was the decision to use a frequentist rather than Bayesian approach to trial design and analysis. Other well-known biomarker-based platform phase 2 clinical trials designed to screen multiple therapies, such as the I-SPY 2 trial in neo-adjuvant breast cancer (Barker, et al. 2009) and BATTLE trials (Liu and Lee 2015) in non-small cell lung cancer, employ Bayesian methods. PrecISE investigators considered whether a Bayesian approach would be a good fit for this study. Although investigators had formulated a priori hypotheses about the biomarker-defined subgroups of patients each intervention should target, there was very little evidence of the effects of the interventions in those subgroups from which to borrow in a formal Bayesian analysis (e.g., as prior distributions about treatment effects). Second, unlike oncology drug development, early endpoints reasonably likely to predict later endpoints that can be used to screen out less effective interventions early in the study are not available in asthma, or at least not well-accepted for that purpose. In the absence of pertinent prior information on the selected interventions’ effects or scientific rationale that suggests two or more interventions may have comparable effects, designs that used Bayesian methods would have essentially identical operating characteristics compared to frequentist designs when held to the equivalent type I error probability control requirements.
Another important design consideration was whether the evaluation of multiple interventions in this single master protocol warranted multiplicity adjustments in efficacy evaluations. Adjustments are needed to control for multiplicity due to the three primary endpoints, each providing a chance for an intervention to demonstrate success. But each intervention’s success is determined without consideration of the other interventions’ results, and there is no concept of the overall success of the study in terms of how many interventions might prove successful. In fact, the master protocol provided a common infrastructure with shared resources and sharing of information (e.g., placebo data) across interventions, but for inferential purposes, it did not differ from six individual trials, each conducted to evaluate a single intervention. Certainly, multiplicity adjustments would not be considered across those six trials, and in the same sense, are not needed for the master protocol.
The PrecISE study design shares control data across interventions, across time, and across modes of drug administration. Each of these decisions was supported by the literature and by simulations to evaluate the potential impact of sharing. Sensitivity analyses are planned to assess their impact on the final analysis results for each intervention. Sharing across time is probably the most controversial, given the sensitivity of asthma symptoms and control to seasonal allergies, coupled with the fact that each treatment period is only 4 months in duration, not enough to span all four seasons of the year. Note, however, that the planned enrollment period for the trial is 30 months in duration, and patients complete their initial two-period crossover, representing a 1:1 randomization ratio of test treatment to placebo, as soon as they complete screening. This implies that we will have placebo data for all interventions across all seasons of the year. In addition, we will have two placebo periods on the same participant in a subset of participants. Because of both design features, we anticipate very little impact of seasonal differences on the ability of the study to demonstrate efficacy of each intervention, and the simulations conducted to date support this.
PrecISE investigators discussed the need to include an active control in the study, selected from among the newer approved therapies for asthma, as a way to determine if the design itself would be able to differentiate between effective and ineffective therapies, often referred to as a study’s assay sensitivity. One advantage to including such a control would be the ability to offer one of the newer asthma treatments to participants for whom it is not otherwise available (e.g., due to lack of insurance coverage). The decision was made, however, to not include such a control, primarily because the study is not designed to compare interventions to each other, and the power for this comparison may be lacking. It was also noted that interpretation of the study’s results could be impaired, or at best, ambiguous, with inclusion of an active control, particularly if one of the interventions demonstrated efficacy, and the active control did not.
In summary, these and numerous other considerations were taken into account in the 1.5-year long design and planning phase for PrecISE, with the net result being a strategic, innovative trial design that, if successfully implemented, should be able to efficiently and effectively evaluate multiple therapies to address a critical unmet public health need in the US population. The design is somewhat complex but attractive to patients in allowing each patient to receive more than one therapy, most targeted specifically for that patient’s biomarker profile, and in minimizing the time a patient is exposed to placebo. Patients are screened once to determine their eligibility match to as many as six interventions, possibly more, offering a stark contrast to sequential trials, where patients are screened for each trial separately, often spanning a very long period of time. This use of a common biomarker screening platform and subsequent biomarker-based randomization assignment is a hallmark of master protocols designed to simultaneously evaluate multiple therapies in a precision medicine context with the most efficient use of patient resources as possible. PrecISE capitalizes on this and other advantages of its master protocol design, in conjunction with the adaptive platform features that ensure information accrued during the trial is used to full advantage through design adaptations. In addition, if any of the originally selected six therapies fail early in the study, there is the ability to add new interventions, and PrecISE investigators identified several candidates early on for that purpose. We believe the PrecISE trial design is fit for purpose in utilizing an innovative design to provide much needed information on how to target severe asthmatic patients with different interventions by recognizing the heterogeneity of the disease.
Acknowledgements
The PrecISE study is funded by the National Heart, Lung, and Blood Institute, National Institutes of Health, grant U24 HL138998. The study also gratefully acknowledges receiving contributed product from Vitaeris, owned and operated by CSL group (clazakizumab), Vitaflo (MCT), Sun Pharma (imatinib), OM Pharma, a Vifor Pharma Group Company (OM-85, Broncho-Vaxom), Incyte (itacitinib), Laurel Venture (cavosonstat) and GlaxoSmithKline (Advair Diskus and Ventolin). The authors thank anonymous reviewers for their helpful comments.
REFERENCES
- Barker AD, C Sigman C, Kelloff GJ, Hylton NM, Berry DA, and Esserman LJ. 2009. I-SPY 2: an adaptive breast cancer trial design in the setting of neoadjuvant chemotherapy. Clinical Pharmacology and Therapeutics 86(1):97–100. doi: 10.1038/clpt.2009.68 [DOI] [PubMed] [Google Scholar]
- Bleecker ER, FitzGerald JM, Chanez P, Papi A, Weinstein SF, Barker P, Sproule S, Gilmartin G, Aurivillius M, Werkström V, and Goldman M. 2016. Efficacy and safety of benralizumab for patients with severe asthma uncontrolled with high-dosage inhaled corticosteroids and long-acting β2-agonists (SIROCCO): a randomised, multicentre, placebo-controlled phase 3 trial. The Lancet 388(10056):2115–2127. [DOI] [PubMed] [Google Scholar]
- Chang Y, Song T, Monaco J, and Ivanova A. 2020. Futility stopping in clinical trials, optimality and practical considerations. Journal of Biopharmaceutical Statistics in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung KF, Wenzel SE, Brozek JL, Bush A, Castro M, Sterk PJ, Adcock IM, Bateman ED, Bel EH, Bleecker ER, Boulet LP, Brightling C, Chanez P, Dahlen SE, Djukanovic R, Frey U, Gaga M, Gibson P, Hamid Q, Jajour NN, and Teague WG. 2014. International ERS/ATS guidelines on definition, evaluation and treatment of severe asthma. The European respiratory journal, 43(2), 343–373. 10.1183/09031936.00202013. [Dosage error in article text] [published correction appears in Eur Respir J. 2018 Jul 27;52(1):]. Eur Respir J. 43(2):343‐373. doi: 10.1183/09031936.00202013 [DOI] [PubMed] [Google Scholar]
- de Wit HM, Te Groen M, Rovers MM, and Tack CJ. 2016. The placebo response of injectable GLP-1 receptor agonists vs. oral DPP-4 inhibitors and SGLT-2 inhibitors: a systematic review and meta-analysis. British journal of clinical pharmacology 82(1):301–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutile S, Kaptchuk TJ, and Wechsler ME. 2014. The placebo effect in asthma. Current allergy and asthma reports 14(8):456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhlbrigge AL, Bengtsson T, Peterson S, Jauhiainen A, Eriksson G, Da Silva CA, Johnson A, Sethi T, Locantore N, Tal-Singer R, and Fagerås M. 2017. A novel endpoint for exacerbations in asthma to accelerate clinical development: a post-hoc analysis of randomised controlled trials. The Lancet. Respiratory medicine 5(7), 577–590. 10.1016/S2213-2600(17)30218-7 [DOI] [PubMed] [Google Scholar]
- FitzGerald JM, Bleecker ER, Nair P, Korn S, Ohta K, Lommatzsch M, Ferguson GT, Busse WW, Barker P, Sproule S, Gilmartin G, Werkström V, Aurivillius M, and Goldman M. 2016. Benralizumab, an anti-interleukin-5 receptor α monoclonal antibody, as add-on treatment for patients with severe, uncontrolled, eosinophilic asthma (CALIMA): a randomised, double-blind, placebo-controlled phase 3 trial. The Lancet 29; 388(10056):2128–2141. [DOI] [PubMed] [Google Scholar]
- He P, Lai TL, and Liao OY. 2012. Futility stopping in clinical trials. Statistics and Its Interface 5 (4):415–423. doi: 10.4310/SII.2012.v5.n4.a4. [DOI] [Google Scholar]
- Hedaya MA 2012. Basic Pharmacokinetics, 2nd edition. CRC Press. [Google Scholar]
- Hochberg Y 1988. A sharper Bonferroni procedure for multiple tests of significance. Biometrika 75:800–802. [Google Scholar]
- Israel et al. , 2021. The Precision Intervention in Severe and/or Exacerbation Prone Asthma (PrecISE) adaptive platform trial with biomarker ascertainment. Journal of allergy and clinical immunology forthcoming. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israel E, and Reddel HK. 2017. Severe and Difficult-to-Treat Asthma in Adults. The New England journal of medicine 377(10):965–976. [DOI] [PubMed] [Google Scholar]
- Joshi N, Fine J, Chu R, and Ivanova A. 2019. Estimating the subgroup and testing for treatment effect in a post-hoc analysis of a clinical trial with a biomarker. Journal of Biopharmaceutical Statistics 29(4), 685–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi N, Nguyen C, and Ivanova A. 2020. Multi-stage adaptive enrichment trial design with subgroup estimation. Journal of Biopharmaceutical Statistics in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosorok MR and Laber EB 2019. Precision medicine. Annual Reviews of Statistics and Its Application 6:263–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai T, Lavori P, and Liao O. 2014. Adaptive choice of patient subgroup for comparing two treatments. Contemporary Clinical Trials 39(2):191–200. doi: 10.1016/j.cct.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SJ, and Lee J. 2015. An overview of the design and conduct of the BATTLE trials. Chinese Clinical Oncology 4(3):33. doi: 10.3978/j.issn.2304-3865.2015.06.07. [DOI] [PubMed] [Google Scholar]
- Nanda A, and Wasan AN. 2020. Asthma in Adults. The Medical clinics of North America, 104(1), 95–108. 10.1016/j.mcna.2019.08.013 [DOI] [PubMed] [Google Scholar]
- Siddiqui S, Denlinger LC, Fowler SJ, Akuthota P, Shaw DE, Heaney LG, Brown L, Castro M, Winders TA, Kraft M, Wagers S, Peters MC, Pavord ID, Walker S, and Jarjour NN. 2019. Unmet needs in severe asthma subtyping and precision medicine trials. Bridging clinical and patient perspectives. American Journal of Respiratory and Critical Care Medicine 199(7), 823–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teague WG, Phillips BR, Fahy JV, Wenzel SE, Fitzpatrick AM, Moore WC, Hastie AT, Bleecker ER, Meyers DA, Peters SP, Castro M, Coverstone AM, Bacharier LB, Ly NP, Peters MC, Denlinger LC, Ramratnam S, Sorkness RL, Gaston BM, Erzurum SC, and Jarjour NN. 2018. Baseline features of the severe asthma research program (SARP III) cohort: differences with age. The journal of allergy and clinical immunology. In practice, 6(2), 545–554.e4. 10.1016/j.jaip.2017.05.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wechsler ME, Szefler SJ, Ortega VE, Pongracic JA, Chinchilli V, Lima JJ, Krishnan JA, Kunselman SJ, Mauger D, Bleecker ER, Bacharier LB, Beigelman A, Benson M, Blake KV, Cabana MD, Cardet JC, Castro M, Chmiel JF, Covar R, hDenlinger L, and NHLBI AsthmaNet. 2019. Step-Up therapy in black children and adults with poorly controlled asthma. The New England journal of medicine 381(13), 1227–1239. 10.1056/NEJMoa1905560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodcock J, and LaVange LM. 2017. Master protocols to study multiple therapies, multiple diseases, or both. The New England Journal of Medicine 377(1):62–70. [DOI] [PubMed] [Google Scholar]
- US Food and Drug Administration. Draft Guidance for Industry: Adaptive Designs for Clinical Trials of Drugs and Biologics. In: (CDER) CfDEaR, (CBER) CfBEaR, eds: U.S. Food and Drug Administration; 2018. [Google Scholar]
- Zhao Y, Zeng D, Rush AJ, and Kosorok MR. 2012. Estimating individualized treatment rules using outcome weighted learning. Journal of the American Statistical Association 107(449):1106–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu R, Zeng D, and Kosorok MR. 2015. Reinforcement Learning Trees. Journal of the American Statistical Association 110(512):1770–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]