

Abstract
Primary sclerosing cholangitis (PSC) is a progressive fibrosing cholestatic liver disease that is strongly associated with inflammatory bowel disease (IBD). PSC-associated IBD (PSC-IBD) displays a unique phenotype characterized by right-side predominant colon inflammation and increased risk of colorectal cancer compared to non-PSC-IBD. The frequent association and unique phenotype of PSC-IBD suggest distinctive underlying disease mechanisms from other chronic liver diseases or IBD alone. Multidrug resistance protein 2 knockout (Mdr2−/−) mice develop spontaneous cholestatic liver injury and fibrosis mirroring human PSC. As a novel model of PSC-IBD, we treated Mdr2−/− mice with dextran sulfate sodium (DSS) to chemically induce colitis (Mdr2−/−/DSS). Mdr2−/− mice demonstrate alterations in fecal bile acid composition and enhanced colitis susceptibility with increased colonic adhesion molecule expression, particularly mucosal addressin cell adhesion molecule 1 (MAdCAM-1). In vitro, ursodeoxycholic acid (UDCA) co-treatment resulted in a dose dependent attenuation of TNF-α-induced endothelial MAdCAM-1 expression. In the combined Mdr2−/−/DSS model, UDCA supplementation attenuated colitis severity and down-regulated intestinal MAdCAM-1 expression. These findings suggest a potential mechanistic role for alterations in bile acid signaling in modulating MAdCAM-1 expression and colitis susceptibility in cholestasis-associated colitis. Together, our findings provide a novel model and new insight into the pathogenesis and potential treatment of PSC-IBD.
Keywords: Primary sclerosing cholangitis, inflammatory bowel disease, dextran sulfate sodium, ursodeoxycholic acid, multidrug resistance protein 2
INTRODUCTION
Primary sclerosing cholangitis (PSC) is a chronic progressive cholestatic liver disease characterized by inflammation and bile duct stricturing leading to hepatobiliary cirrhosis.1 Among PSC patients, approximately 75% will develop coexisting inflammatory bowel disease (IBD), predominantly in the form of ulcerative colitis (UC).1 Compared to UC without PSC, PSC-associated IBD (PSC-IBD) displays a unique phenotype, characterized by right-side predominant colitis with rectal sparing, backwash ileitis, and a markedly increased risk of colorectal carcinoma2.
The strong association between PSC and IBD suggests co-dependent disease mechanisms which are likely distinct from other cholestatic liver diseases or IBD alone. Although the pathogenesis remains unclear, evidence suggests multiple contributing factors including polygenetic risk alleles3, innate and adaptive immune dysregulation4, intestinal dysbiosis5, and alterations in bile acid signaling6, 7 among others. In particular, cholestasis-induced disruptions in the tightly regulated enterohepatic circulation of bile acids offer a compelling link between the concurrent hepatobiliary and intestinal inflammation in PSC-IBD. Indeed, a variety of therapies directed at manipulation of bile acid signaling have demonstrated beneficial effects in various models of cholestasis as well as colitis8-11. Additionally, increasing evidence suggests that alterations in adhesion molecule expression and lymphocyte trafficking may be of particular interest in PSC. Mucosal vascular addressin-cell adhesion molecule 1 (MAdCAM-1) is predominantly expressed within the intestinal vasculature and plays a critical role in gut-specific lymphocyte trafficking in IBD through its interaction with the α4β7 integrin on circulating T lymphocytes12, 13. Interestingly, evidence of abnormal hepatic MAdCAM-1 expression in PSC suggests a potential role for aberrant lymphocyte trafficking along the gut-liver axis, promoting both hepatobiliary and intestinal inflammation in PSC-IBD14-16. However, mechanisms of inflammatory cross-talk along the gut-liver axis require further investigation.17
To date, there are no established murine models of combined cholestasis and colitis that mimic human PSC-IBD. Multidrug resistance protein 2 (Mdr2) is a phospholipid flippase expressed in biliary canaliculi, which mediates hepatocyte secretion of phospholipids into bile18. Mdr2 knockout mice (Mdr2−/−) develop spontaneous cholangitis and periductal liver fibrosis mirroring human PSC and provide the best available model of cholestatic liver disease18-20. Mdr2 heterozygotes (Mdr2+/−) do not develop liver injury at baseline and this appears unchanged with the addition of experimental colitis, however the impact of experimental colitis in Mdr2−/− mice with established liver disease has not been assessed21. Utilizing the Mdr2−/− mouse in combination with chemically induced DSS colitis, we present a novel model of combined cholestatic liver injury and colitis demonstrating similar features to human PSC-IBD. Our murine model demonstrates enhanced colitis susceptibility and increased intestinal adhesion molecule expression in the setting of cholestasis. Moreover, we demonstrate that such enhanced colitis susceptibility is, at least in part, attenuated by dietary ursodeoxycholic acid (UDCA) supplementation. These results suggest a mechanistic role for bile acid signaling in the pathogenesis of cholestasis-associated colitis and provide useful insights into understanding the unique relationship between PSC and IBD.
RESULTS
Cholestatic liver injury does not induce spontaneous colitis in Mdr2−/− mice at baseline.
To confirm both the presence and severity of spontaneous cholestatic liver injury in the Mdr2−/− model, 6 and 10-week old mice were assessed for fibrosis, hepatic injury and changes in bile acid composition. When compared to age matched C57BL/6 (WT) control mice, 6- and 10-week old Mdr2−/− mice demonstrated spontaneous and progressive liver fibrosis as assessed by picrosirius red stain (PSR) under polarized light (Supplementary Fig. 1A, B). Mdr2−/− mice also had higher serum aminotransferase levels when compared to WT mice. (Supplementary Fig. 1C). Similar to human cholestatic liver diseases including PSC, fibrosis in the Mdr2−/− mice was predominantly peri-portal in distribution. Bile acid analysis by mass spectrometry in 10-week old mice demonstrated increased serum bile acids confirming significant cholestasis in Mdr2−/− mice (Supplementary Fig. 1D). Consistent with previously published literature, these data confirm the presence of spontaneous and progressive cholestatic liver injury in the Mdr2−/− mouse model18-20.
We subsequently assessed the presence or absence of spontaneous intestinal inflammation in the setting of progressive cholestatic liver injury in Mdr2−/− mice at baseline. Bile acid analysis by mass spectrometry demonstrated marked differences in fecal bile acid pools between Mdr2−/− and WT mice (Fig. 1A). Detailed histologic evaluation revealed no evidence of colonic inflammation in 6, 10, or 30-week old Mdr2−/− mice with established cholestasis as compared to age matched WT controls (Fig. 1B, C). We further investigated the Mdr2−/− mice for changes in colonic inflammation by measuring colon permeability and proinflammatory cytokine levels. As illustrated in Fig. 1D-E, 10-week old Mdr2−/− mice demonstrated no difference in colon permeability (Fig. 1D) or pro-inflammatory cytokine levels (Fig. 1E) as compared to WT controls. Collectively, this data demonstrates that cholestatic liver injury alone is not sufficient to induce spontaneous colitis in the Mdr2−/− model regardless of age or liver disease severity.
Figure 1. Baseline colonic phenotype in Mdr2−/− mice with active liver disease.

WT and Mdr2−/− mice were euthanized at age 6, 10 and 30 weeks. Feces and colon tissues were collected. (A) Levels of fecal bile acids from 10-week old WT and Mdr2−/− mice were measured using mass spectrometry. (B, C) Representative colon histology (hematoxylin and eosin [H&E]; 200X) and histological scores. Positive control in panel C is WT/DSS from Fig. 2E. (D) Colonic barrier function in 10-week old mice was assessed by FITC-dextran gavage. Serum levels of FITC-dextran were measured four hours post gavage. Positive control is WT/DSS from Fig. 2G. (E) Colon tissue was homogenized and cytokine protein levels were measured. n ≥ 4 mice per group. Data are expressed as means ± SEM, two-way analysis of variance.
Developing a novel murine model of combined hepatic cholestasis and colitis (MDR2−/−/DSS).
To develop and characterize a model of combined cholestatic liver injury and intestinal inflammation, 10- to 12-week-old Mdr2−/− mice with established cholestatic liver injury were administered 1.5% (wt/vol) DSS in DI H2O (Fig. 2A) to induce colitis. There was a significant increase in weight loss and disease activity index (DAI) scores in the Mdr2−/− mice (Fig. 2B, C) compared to WT controls. Histopathologic examination revealed markedly increased histologic colitis severity and colonic shortening in the Mdr2−/− mice (Fig. 2D-F). In addition, Mdr2−/− mice demonstrated significantly increased mucosal permeability as measured by FITC dextran flux assay (Fig. 2G). Colonic tissue cytokine analysis revealed significantly increased levels of the pro-inflammatory cytokines IL-1β, KC, and IL-6 in Mdr2−/− mice as compared to WT controls (Fig. 2H). Tissue levels of IFN-γ, TNF-α, and IL-12p70 proteins were similar between groups (Supplementary Fig. 2). Together this data demonstrates that despite the absence of baseline colonic inflammation, established cholestatic liver disease significantly enhances susceptibility to DSS-induced colitis in the Mdr2−/− mice.
Figure 2. Induction of DSS colitis in Mdr2−/− mice with established cholestatic liver disease.

Ten-week-old WT and Mdr2−/− mice were subjected to 1.5% dextran sodium sulfate (DSS) colitis or water control (diH2O) for 5 days and then allowed to recover. Tissues and sera were collected upon euthanasia on day 9. (A) Scheme of treatment. (B) Body weight curves over the course of DSS treatment and recovery. (C) Disease activity index (DAI) measured by combining weight loss, stool consistency, and bleeding. (D, E) Representative colon histology (H&E; 100X) and histological scores. (F) Colon length at time of euthanasia divided by baseline body weight. (G) Colonic barrier function in 10-week old mice was assessed by FITC-dextran gavage. (H) Colon tissue was homogenized, and cytokine protein levels were measured. n ≥ 10 mice per group. Data are expressed as means ± SEM. *P<0.05, **P<0.01, ***P<0.001, two-way analysis of variance.
Upregulation of adhesion molecule expression in the combined hepatic cholestasis and colitis model (Mdr2−/−/DSS).
Upregulation of adhesion molecule expression has been demonstrated in IBD22 as well as cholestatic liver disease23. In particular, recent evidence suggests a compelling role for abnormal hepatic expression of MAdCAM-1 in facilitating aberrant lymphocyte trafficking along the gut-liver axis in PSC-IBD14-16. Colonic adhesion molecule expression was unchanged at baseline in Mdr2−/− mice (Fig. 3A). DSS-treated Mdr2−/− mice exhibited a marked increase in colonic MAdCAM-1 and vascular cell adhesion protein 1 (VCAM-1) expression as compared to DSS-treated WT controls (Fig. 3A). Intercellular adhesion molecule 1 (ICAM-1) expression did significantly change with DSS treatment. Increased MAdCAM-1 protein expression in DSS-treated Mdr2−/− mice was confirmed by ELISA (Fig. 3B). MAdCAM-1 expression was also measured by tissue immunofluorescence (IFC), normalized to the endothelial marker CD31, demonstrating higher MAdCAM-1 staining in DSS-treated Mdr2−/− mice (Fig. 3C, D). MAdCAM-1 is important in recruiting α4β7+ T cells 12, 13 and the IL17/23 axis has been shown to be important a component of the adaptive immune response in both IBD24 and PSC.25 Colonic IL-17A, IL-17F, and IL-23 levels were measured by qPCR displaying significantly increased IL-17A expression in both C57BL/6 and Mdr2−/− mice following DSS treatment (Supplementary Fig. 3A). While there was a trend toward higher levels in Mdr2−/− mice, this difference did not reach statistical significance (Supplementary Fig. 3A). IL-17F and IL-23 were unchanged with DSS treatment. Infiltrating α4β7+ cells in the colon were evaluated by tissue immunofluorescence. Baseline α4β7+ staining appeared greater in Mdr2−/− mice as compared to WT controls, however this difference did not reach statistical significance. Following DSS treatment, α4β7+ staining increased in both C57BL/6 and Mdr2−/− mice, however this reached statistical significance in C57BL/6 mice only. (Supplementary Fig. 3B,C). These findings suggest an association between MAdCAM-1 expression and cholestasis associated increases in colitis-susceptibility in our combined model of cholestatic liver injury and colitis.
Figure 3. Colonic expression of adhesion molecules in DSS-treated WT and Mdr2−/− mice.

WT and Mdr2−/− mice were subjected to DSS treatment as described in Fig. 2. (A) Real-time quantitative PCR (qPCR) analysis of colon tissue (B) ELISA measurement of colonic MAdCAM-1 protein. (C, D) Representative immunofluorescence double staining of MAdCAM-1 (red; Alexa Fluor 555) and CD31 (green; Alexa Fluor 488) on colon tissue (100X) shown in panel D. Quantitation of MAdCAM-1 density normalized to CD3 is shown in panel C. n ≥ 5 mice per group. Data are expressed as means ± SEM. *P<0.05, ***P<0.001, two-way ANOVA.
Bile acids modulate endothelial adhesion molecule expression in vitro.
Based on our observations of altered serum and fecal bile acid pools that are associated with augmented adhesion molecule expression in the Mdr2−/− mice, we next sought to evaluate the role of bile acids in regulating endothelial adhesion molecule expression in vitro. Following a screen of multiple immortalized endothelial cell lines for preserved MAdCAM-1 expression, we utilized transformed sinusoidal endothelial cells (TSECs) for the induction of MAdCAM-1 using TNF-α. Brain endothelial cells (bEnd.3 cells) were used for confirmation of results in a second endothelial cell line. A dose response assay identified 5ng/mL and 10 ng/mL for TSECs and bEnd.3 cells, respectively, to be the appropriate working concentration for MAdCAM-1 induction by TNF-α (Supplementary Fig. 4A). To determine if bile acids attenuated TNF-α-induced adhesion molecule expression, TSECs and bEnd.3 cells were co-treated with TNF-α and bile acids (100μM ursodeoxycholic acid (UDCA), cholic acid (CA), chenodeoxycholic acid (CDCA), deoxycholic acid (DCA), and lithocholic acid (LCA)) in combination. Bile acid treatment alone revealed no change in adhesion molecule expression (Supplementary Fig. 4B-D). Both UDCA and LCA co-treatments attenuated TNF-α-induced MAdCAM-1 expression across TSEC and bEND.3 cell types (Fig. 4A, Supplementary Fig. 3B). UDCA is known to be metabolized to LCA in the colon and both have demonstrated protective effects in DSS-induced colitis26. The inhibitory effect of the other bile acids on TNFα-induced MAdCAM-1 expression were inconsistent across cell types (Supplementary Fig. 4B). TNF-α-induced ICAM-1 and VCAM-1 were also attenuated by UDCA in TSECs, however this was not seen in bEnd.3 cells (Fig. 4B, C). Other bile acids demonstrated minimal effects on TNF-α-induced ICAM-1 and VCAM-1 expression (Supplementary Fig. 4C, D). These results indicate a role for UDCA and LCA in modulating endothelial MAdCAM-1 expression.
Figure 4. Effects of UDCA on TNF-α-induced MAdCAM-1 expression in vitro.
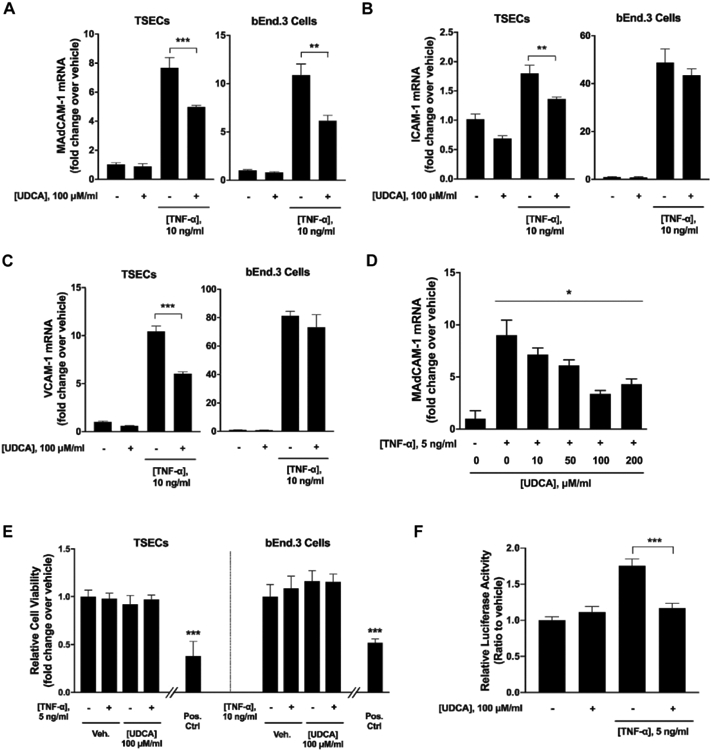
(A-C) TSECs or bEnd.3 cells were co-treated with TNF-α and ursodeoxycholic acid (UDCA) for four hours. Cells were collected and subjected to RT-qPCR analyses of adhesion molecules. (D) Dose titration of UDCA effects on TNF-α induction of MAdCAM-1 in TSECs. (E) TSECs or bEnd.3 cells were co-treated with UDCA and TNF-α and cell viability was assessed. Positive control is treatment of TSECs or bEnd.3 cells with 1% sodium azide (0.76 M). (F) TSECs were transfected with NF-κB luciferase reporter and treated with TNF-α and/or UDCA. NF-κB luciferase activities were measured using a Dual-Luciferase Reporter Assay System. Data are expressed as means ≥ SEM. *P<0.05, **P<0.001, ***P<0.0001, two-way ANOVA and Student’s t-test.
Given the potential protective effects of UDCA in cholestatic liver disease27-29, we next investigated the role of UDCA as a therapeutic agent in attenuating endothelial MAdCAM-1 expression. UDCA dose-titration demonstrated a clear dose response with increasing UDCA concentrations up to 100μM eliciting greater attenuation of MAdCAM-1 expression (Fig. 4D). A cell counting kit-8 (CCK-8) cytotoxicity assay revealed no evidence of bile acid induced cytotoxicity at a 100μM UDCA concentration (Fig. 4E) or 100μM concentration for other bile acids (CA, CDCA, DCA, LCA) (Supplementary Fig. 4E). MAdCAM-1 is known to be induced through the NF-κB pathway30, 31. Assessment of MAdCAM-1 induction utilizing additional NF-κB (IL-1β) and non- NF-κB (IFN-γ) signaling cytokines confirmed induction of MAdCAM-1 with IL-1β but not IFN-γ (Supplementary Fig. 5 A-C). Utilizing an NF-κB luciferase promoter assay, we found that TNF-α markedly increased NF-κB activity which was significantly decreased with UDCA co-treatment (Fig. 4F). Collectively, these findings suggest a direct effect of UDCA in modulating endothelial expression of MAdCAM-1 through the inhibition of NF-κB activity and offer one potential mechanism supporting a mucosal protective role of UDCA in vivo.
UDCA treatment in vivo is protective in a murine model of combined hepatic cholestasis and colitis.
UDCA is a hydrophilic secondary bile acid present at low concentrations in human bile28. UDCA supplementation has demonstrated protective effects in a variety of cholestatic liver diseases including PSC and is FDA approved for the treatment of primary biliary cholangitis (PBC)28, 32. Additionally, UDCA supplementation has demonstrated efficacy in DSS colitis26, 33 and has been associated with a decreased risk of colitis-induced dysplasia in human IBD34. Based on the previous findings, we sought to evaluate the influence of UDCA supplementation on colitis susceptibility and adhesion molecule expression in our combined model of cholestatic liver injury and colitis. Mice were fed normal chow or chow fortified with 0.5% UDCA for 2 weeks prior to induction of colitis and throughout the study period. Mice were then administered 1.5% (wt/vol) DSS in DI H2O to induce colitis for 5 days and allowed 4 days of recovery (Fig. 5A). Focusing specifically on the role of UDCA in modulating DSS colitis severity, and to minimize the number of comparative experimental groups, non-DSS controls were excluded from these experiments. As expected, fecal bile acid analysis confirmed a significant shift in bile acid composition following UDCA supplementation with UDCA comprising 70% of the total fecal bile acid pool (Supplementary Fig. 6A). UDCA supplemented Mdr2−/− and WT mice exhibited a reduced susceptibility to DSS-induced colitis as evidenced by attenuated weight loss and DAI scores (Fig. 5B, C). Although UDCA supplementation proved protective in both Mdr2−/− and WT mice, the magnitude of protection was significantly more pronounced in the Mdr2−/− mice which, after treatment with UDCA, exhibited similar weight loss and DAI scores to DSS-treated WT mice; this was also reflected in histological severity scores (Fig.5D, E). A similarly protective role was observed in analysis of colon length (Fig. 5F) and colon tissue cytokine levels that revealed significantly decreased levels of the pro-inflammatory cytokines KC and IL-6 (Fig. 5G) in DSS-treated Mdr2−/− mice. Despite the beneficial effects of UCDA on colitis severity, liver fibrosis score (Supplementary Fig. 7A, B) and serum ALT (Supplementary Fig. 7C) remained unchanged in DSS treated WT and Mdr2−/− mice. Colonic MAdCAM-1 mRNA and protein levels (Fig. 5H-I) were significantly higher in DSS-treated Mdr2−/− mice as compared to DSS-treated WT controls. Interestingly, although UDCA supplementation resulted in a marked decrease in colonic MAdCAM-1 mRNA and protein levels in the Mdr2−/− mice, this decrease was not observed in WT controls after UDCA treatment as demonstrated in Fig. 5H and I showing no change in MAdCAM-1 mRNA or protein levels in WT mice with UDCA supplementation despite attenuated disease severity by weight loss and DAI score. Colonic adhesion molecule expression profile demonstrated that MAdCAM-1 and VCAM-1 were significantly attenuated by UDCA supplementation in Mdr2−/− mice (Fig. 5H, I). Additionally, we evaluated colonic IL-17 and IL-23 expression as well as α4β7+ immunofluorescence in response to UDCA treatment. Increased IL-17A expression in DSS treated Mdr2−/− mice was significantly attenuated with UDCA supplementation (Supplementary Fig. 8A). Similarly, UDCA supplementation was associated with a trend towards decreased mucosal a4b7+ staining in both DSS treated WT and Mdr2−/− mice, however this did not reach statistical significance (Supplementary Fig. 8 B,C). Taken together, our findings demonstrate successful alteration of fecal bile acid pools through dietary UDCA supplementation resulting in a marked attenuation of cholestasis-associated DSS-susceptibility. Additionally, improvements in intestinal adhesion molecule expression profiles and associated alterations in markers of the adaptive immune response with UDCA supplementation provides important insights into potential mechanisms of inflammatory crosstalk along the gut-liver axis in cholestasis associated colitis.
Figure 5. Dietary UDCA supplementation in DSS-treated WT and Mdr2−/− mice.

Ten-week-old mice were fed 0.5% UDCA supplemented chow for 2 weeks prior to and throughout the DSS treatment and recovery. For DSS treatment, mice were given 1.5% DSS in drinking water for 5 days and then allowed to recover. Tissues and sera were collected upon euthanasia on day 9 after DSS. (A) Scheme of treatment. (B) Body weight curves over the DSS treatment and recovery. (C) Disease activity measured by combining weight loss, stool consistency, and bleeding. (D and E) Representative colon histology (H&E; 100X) and histological scores. (F) Colon length at time of euthanasia divided by the starting body weight. (G) Colon tissue was homogenized, and cytokine protein levels were measured by Meso Scale Discovery analysis. (H) RT-qPCR analysis of adhesion molecules after UDCA and DSS treatment. (I) Colonic MAdCAM-1 protein levels were measured using an ELISA kit. n ≥ 8 mice per group. Data are expressed as means ± SEM. *P<0.05, ***P<0.0001, two-way ANOVA.
DISCUSSION
PSC carries a strong association with IBD, suggesting co-dependent pathogeneses which are likely distinct from those of other cholestatic liver diseases and IBD alone. Despite this unique association, mechanisms of inflammatory crosstalk along the gut-liver axis remain incompletely understood. Our studies establish a novel murine model of combined cholestasis and colitis resembling human PSC-IBD. These findings suggest an important role for bile acids in modulating inflammatory signaling within the intestine and offer useful insights into disease mechanisms in PSC-IBD.
The enterohepatic circulation of bile acids is a tightly regulated and highly efficient system. Bile acids are synthesized by hepatocytes and secreted into the small intestine where approximately 95% are reabsorbed via active transport in the ileum and recycled back to the liver through the portal circulation35. In addition to their roles in cholesterol catabolism and intestinal nutrient absorption, bile acids also act as important signaling molecules. Bile acids have been shown to bind to a variety of cell surface and nuclear receptors with down-stream effects on bile acid homeostasis, energy metabolism, and inflammatory signaling pathways35. As demonstrated in the Mdr2−/− murine model, and as seen in human PSC, cholestasis-induced alterations in bile acid homeostasis are associated with hepatobiliary inflammation and progressive liver injury36. Alterations in fecal bile acid pools have also been implicated in colon carcinogenesis37. These effects are likely mediated through multiple mechanisms including direct cytotoxicity from a relative increase in hydrophobic bile acids, differential effects on bile acid receptor signaling related, and shifts in the fecal microbiota37. Importantly, it is likely that these effects are driven primarily through changes in the relative profile of bile acids in the overall pool as opposed to specific changes in individual bile acids. Consistent with previous studies, our data demonstrate marked alterations of bile acid pools in Mdr2−/− mice with established liver disease20. While the absence of colonic inflammation in Mdr2−/− mice at baseline suggests that cholestasis alone is insufficient to induce colitis, our finding of increased colitis susceptibility in this model offers a compelling association between perturbations in bile acid homeostasis and modulation of inflammatory signaling along the gut-liver axis in PSC-IBD.
In our model of combined cholestasis and colitis, dietary supplementation with UDCA resulted in a marked attenuation of colitis severity, further supporting a role for bile acids in modulating colitis susceptibility. Targeting bile acid signaling as a therapeutic intervention has demonstrated efficacy through a variety of targets. For example, inhibition of the apical sodium-dependent bile acid transporter (ASBT) improves liver injury in Mdr2−/− mice8 and the FXR agonist obeticholic acid (OCA) has demonstrated protective effects in PSC10, PBC9, as well as colitis38. UDCA supplementation has been studied extensively in cholestatic liver disease and is an FDA approved therapy for the treatment of PBC27. Dietary UDCA improved liver histology in Mdr2−/− mice29 and has been associated with improved liver function tests in human PSC. However, studies regarding the benefits on long-term disease progression and transplant free survival in PSC are conflicting with intermediate dosing (17-23 mg/kg/day) suggesting possible benefit, and high doses (28-30mg/kg/day) associated with an increase in serious adverse events11, 32, 39. The potential protective role of UDCA in cholestatic liver injury is mediated through a variety of mechanisms including improved bile acid solubility and hepatobiliary secretion, protection from bile acid and cytokine mediated cytotoxicity, and inhibition of pro-inflammatory cytokine production.28 Additionally, UDCA has demonstrated efficacy in multiple murine models of colitis26, 33 as well as colitis-associated dysplasia in mice and humans34. In this setting, UDCA exhibits protection to the intestinal epithelium through decreases in the relative abundance of the cytotoxic bile acid DCA40, direct anti-secretory influences through regulation of intestinal chloride secretion41, promotion of intestinal barrier integrity42, and inhibition of cellular proliferation43. Similar to human studies, UDCA supplementation in our studies resulted in a significant shift in bile acid composition with UDCA comprising 70% of the total fecal bile acid pool44. Although DSS-colitis susceptibility was attenuated in both Mdr2−/− and WT mice, the magnitude of protection was much more pronounced in the Mdr2−/− mice across all metrics of disease severity. This suggests that alterations in bile acid homeostasis are likely to play a more relevant role in modulating disease severity in the setting of combined cholestasis and colitis as compared to colitis alone.
Bile acids and their metabolites have been shown to modulate innate and adaptive immune responses within the intestines through a variety of pathways. These include direct effects on barrier integrity and macrophage activation45, 46 through bile acid receptor signaling as well as indirect effects on adaptive immunity through their interaction with intestinal microbiota. There exists a complex and inter-dependent relationship between intestinal bile acid and microbial composition in both maintaining intestinal homeostasis as wells as in contributing to intestinal dysbiosis and inflammatory signaling in chronic IBD.47, 48 A recent study by Song et al. demonstrated that bacterial transformed secondary bile acids modulate colonic RORγ+Treg populations and protect against DSS-induced colitis through their interaction with the vitamin D nuclear receptor.49 Similarly, the work by Hang et al. identified a direct effect of two distinct bacterial derived lithocholic acid metabolites on modulating TH17 and Treg homeostasis within the intestinal lamina propria.50 Interestingly, signatures of dysbiosis in PSC appear distinct to those of UC or CD suggesting a unique association between cholestasis and alterations in gut microbial composition5. Adding to the collective understanding, our finding that UDCA attenuates NF-kB induced MAdCAM-1 expression represents an additional novel protective mechanism for bile acids in modulating intestinal immune responses.
Tissue lymphocyte recruitment plays a critical role in disease progression in a variety of autoimmune disorders23, 51, 52. Both PSC and IBD are characterized by a marked lymphoplasmacytic infiltrate facilitated by upregulation of adhesion molecules at local sites of tissue injury23, 51. Increased expression of VCAM-1, ICAM-1, and MAdCAM-1 is associated with active IBD and multiple pharmacologic therapies targeting the integrin-adhesion molecule interaction have demonstrated clinical efficacy, both in humans and in murine models of colitis51, 53. Vedolizumab, a humanized monoclonal antibody against the α4β7-integrin which binds MAdCAM-1, is currently FDA approved for treatment of Crohn’s disease and ulcerative colitis12, 13. Additionally, increased hepatic expression of VCAM-1, ICAM-1, and MAdCAM-1 has been demonstrated in PSC16, 23, 54. Recent studies suggest that aberrant hepatic expression of MAdCAM-1 may be of particular relevance in PSC-IBD by facilitating trafficking of gut-homing lymphocytes between the liver and intestine14-16. MAdCAM-1 is a transmembrane protein that is predominantly expressed on high endothelial venules in the intestine. Unlike other adhesion molecules, MAdCAM-1 exhibits an intriguing homology to the Cα2 constant region immunoglobulin loop of human and gorilla Iga1 that are highly expressed in mucosal tissues, suggesting MAdCAM-1 is relatively gut-specific55. Under inflammatory conditions, MAdCAM-1 binds the α4β7-integrin on circulating T-cells to facilitate recruitment of gut-homing lymphocytes to sites of intestinal injury56. However, available evidence on the use of Vedolizumab in PSC has demonstrated conflicting results on the utility of MAdCAM-1 as a relevant therapeutic target in patients with established liver disease17, 57. Our finding of increased intestinal MAdCAM-1 in colitic Mdr2−/− mice suggests that upregulation of MAdCAM-1 may be similarly important in promoting intestinal inflammation in the setting of cholestasis and that adhesion molecule expression may be driven, in part, by alterations in bile acid signaling. Furthermore, we demonstrate that endothelial expression of MAdCAM-1 as well as other adhesion molecules can be directly attenuated by UDCA through inhibition of NF-κB signaling. In vivo, UDCA supplementation is associated with a significant attenuation of colitis severity, decreased colonic MAdCAM-1 expression, and a trend towards decreased accumulation of α4β7+ lymphocytes within the colon. These findings suggest a direct influence of UDCA on endothelial MAdCAM-1 expression and associated lymphocyte trafficking as a novel potential mechanism for the protection afforded by UCDA.
Recent studies suggest that a new side chain-shortened C23 homologue of UDCA, nor-UDCA,23 may be an effective treatment for cholestasis in PSC with an improved safety profile over UDCA. The unique chemical structure of nor-UDCA confers distinctive protective properties including increased biliary HCO3 excretion, promotion of hepatocellular bile acid detoxification and renal bile acid excretion, as well as anti-fibrotic and anti-inflammatory effects58. Nor-UDCA treatment demonstrated significant improvement in liver function tests and histology as compared to UDCA in Mdr2−/− mice59 and a Phase II study of nor-UDCA in PSC showed significant improvements in liver function tests with no adverse safety signals60. A phase III trial of nor-UDCA is ongoing and will assess both liver function tests as well as histologic outcomes. The potential efficacy benefit of nor-UDCA over UDCA in modulating colitis susceptibility remains unknown; however, this will be of particular relevance to evaluate in our murine model if the Phase III results in PSC remain favorable.
As with all preclinical models, our model carries some limitations regarding its translational relevance to human disease. Although the Mdr2−/− mouse is well-accepted as a model of cholestatic liver disease, it is not a unique model of PSC specifically, and has been used in the study of multiple other chronic cholestatic liver disorders. Additionally, while mutations in the human ortholog gene Mdr3 are associated with a variety of liver disorders,61 genetic variations in Mdr3 have been described in less than 10% of PSC cases arguing against a prominent pathogenic role in PSC62, 63. Notably, although the Mdr2−/− mouse is a whole-body knockout, model concerns regarding the role of Mdr2 in the intestine should be tempered by the fact that colonic expression of Mdr2 appears limited to low level mRNA with no demonstration of protein or a defined functional role for Mdr2 outside of the biliary canaliculi64. While the mucosal predominant injury of DSS colitis provides an accepted “UC-like” model, the acute nature of this model along with distal colonic disease distribution is in contrast to the chronic and typically right-side predominant colitis of PSC-IBD. Likewise, PSC-IBD often follows a mild clinical course in contrast to the increased disease severity noted in the combined Mdr2−/−/DSS model.1
Importantly, although DSS is primarily an innate inflammatory model, our evidence of increased colonic IL-17 expression and alterations in α4β7+ staining demonstrates an additional adaptive immune overlay consistent with prior phenotypic studies of DSS colitis.65 Indeed, disruption of the epithelium associated with DSS colitis can result in a mixed T cell response in animals with an intact adaptive immune system66, which lends credence to our findings with MAdCAM-1. Additionally, given that UDCA has demonstrated efficacy in previous murine models of colitis26, 33, a question remains whether the observed decrease in colonic MAdCAM-1 in vivo is mediated directly through UDCA modulation of intestinal MAdCAM-1 expression or whether this could be a down-stream response to generalized anti-inflammatory influence of UDCA. Acknowledging that the beneficial impact of UDCA are likely multi-factorial, we believe that the combined in vitro and in vivo evidence in our study offers a compelling argument that the presence of cholestasis is associated with an increased susceptibility to DSS colitis and that the beneficial effects of UDCA supplementation may be mediated, in part, through direct modulation of intestinal MAdCAM-1 expression.
Model limitations notwithstanding, given that PSC represents the only cholestatic liver disease exhibiting a clear and strong association with IBD, we believe our combined model of cholestasis and colitis offers a novel tool for evaluating liver-intestine cross-talk mechanisms in PSC-IBD. Acknowledging the limitations, the unique insights gleaned from the current studies offer novel disease markers and therapeutic targets for subsequent application in future models such as chronic DSS or combined genetic models which may more closely resemble the chronic course of PSC-IBD.
In summary, disruptions in bile acid homeostasis are associated with increased colitis susceptibility and upregulation of intestinal adhesion molecule expression in a novel murine model of combined cholestasis and colitis to mimic PSC-IBD. Therapeutic manipulation of bile acid pools through dietary UDCA supplementation results in amelioration of colitis and down-regulation of intestinal adhesion molecules. In vitro, UDCA attenuates TNF-α/NF-κB induced adhesion molecule expression suggesting a potentially unique protective mechanism for UDCA in modulating inflammatory signaling. Taken together, MAdCAM-1 expression is associated with increased disease severity in this model of cholestasis associated colitis and UDCA, by virtue of its ability to inhibit MAdCAM-1 expression, may be useful in attenuating inflammatory crosstalk pathways between the liver and intestine in PSC-IBD.
METHODS
Animal Experiments.
Wild-type C57BL/6J mice were originally purchased from Jackson Laboratories. Breeding pairs of Mdr2−/− mice on a C57BL/6J background were obtained from Dr. Ronald Elferink (Tytgat Institute for Liver and Intestinal Research, Amsterdam). Both WT C57BL/6J and Mdr2−/− mice were bred simultaneously in the same on-site facility and under identical environments. For colitis experiments, mice received control (DI H2O) or 1.5% (wt/vol) DSS in DI H2O for 5 days and were then allowed to recover for 4 days on DI H2O before being euthanized. For UDCA experiments, mice received standard chow (2020X Teklad, Envigo) or 0.5% UDCA (Sigma-Aldrich, St. Louis, MO) supplemented chow compounded through Envigo Teklad laboratory animal diets. Disease Activity Index (DAI) considered stool consistency (0-4), presence of blood (0-4, and weight loss (0-4). The scores were added with a maximum possible score of 12. For colon permeability assessment, mice underwent oral gavage of 10 mg of 4kD FITC-dextran with collection of serum at 4 hours. Serum fluorescence was measured (Promega fluorescent plate reader) and serum FITC concentration determined by standard curve. All animal procedures were approved by the Institutional Animal Care and Use Committee of the University of Colorado Anschutz Medical Campus.
Bile Acid Analysis.
Bile acid analysis was completed by CU School of Pharmacy Mass Spectrometry Facility (M.A.). Fecal and serum bile acid analyses were performed following a protocol previously described by Sarafian et al67, with slight modifications that is described in more detail in the supplementary methods.
Cell Lines.
Mouse transformed endothelial sinusoidal cells (TSECs) were received as a generous gift from Vijay Shah (Mayo Clinic, Rochester, MN) and brain endothelial cells (bEnd.3) were purchased from ATCC. Cells were cultured and treated with cytokines and bile acids as described in detail in the supplemental methods. NF-κB luciferase reporter assays are also described in detail in the supplemental methods.
Cytokine Analysis.
Colon tissue was homogenized, and cytokine protein levels were measured using a Mouse Pro-inflammatory 7-Plex kit (Meso Scale Diagnostics, Rockville, MD) read on a MESO QuickPlex SQ 120 imager (Meso Scale), as described in detail in the supplemental methods.
Histology and Immunohistochemistry.
Colon injury assessment and liver fibrosis were assessed by a trained histologist (D.O.) blinded to mouse strain and treatment group. A previously described semi-quantitative scoring system was used to assess histologic colitis severity68 and is described in more detail in supplementary methods. Paraffin-embedded colon sections were used for immunofluorescence stain. All sections used a heat-induced epitope retrieval in retrieval in Tris-EDTA (pH 9.0). Anti-MAdCAM1 antibody (Cat. #ab80680, Abcam, Cambridge, MA) and anti-CD31 antibody (Cat. #77699, Cell Signaling Technology, Danvers, MA) primaries were visualized with Alexa Fluor 488 and 555 labelled secondary antibodies (Cat. #4412 and 4417, Cell Signaling Technology) were used to visualize the staining, respectively. Anti-α4β7 (Cat#73261, Abcam) primary antibody was visualized with an Alexa Fluor 488 labelled secondary antibodies (Cat#A11006, Invitrogen). Quantification of PSR and IFC is described in more detail in supplementary methods. Histologic images were captured on an Olympus BX51 microscope equipped with a 4MP Macrofire digital camera (Optronics). All images were assembled using Photoshop CS2 (Adobe Systems, Inc.; Mountain View, CA).
Statistical Analysis.
GraphPad Prism 8 (GraphPad Software, La Jolla, CA) was used to generate figures and perform statistical analyses. To compare values obtained from two groups, Student's t-test was performed. Data from three or more groups were compared with one-way ANOVA followed by Tukey’s post hoc test. Probability values of P < 0.05 were considered statistically significant.
Supplementary Material
ACKNOWLEDGEMENTS
We greatly appreciate Michael Armstrong, Dan Koyanagi (University of Colorado), Terrin Manes (University of Colorado), and Dechun Feng (NIH) for excellent technical assistance; Sarah Williams (University of Colorado) for care and maintenance of mouse colonies; Ronald Elferink (Tytgat Institute for Liver and Intestinal Research, Amsterdam) for providing original breeding pairs of Mdr2−/− mice; and Vijay Shah (Mayo Clinic) for providing TSEC cell lines.
This work was supported by a pilot grant from the University of Colorado GI and Liver Innate Immune Program (GALIIP), a grant from the Crohn’s and Colitis Association and by NIH grants DK50189 and DK095491.
Footnotes
Conflict of interest statement: The authors declared that no conflict of interest exists
REFERENCES
- 1.Hirschfield GM, Karlsen TH, Lindor KD, Adams DH. Primary sclerosing cholangitis. Lancet 2013; 382(9904): 1587–1599. [DOI] [PubMed] [Google Scholar]
- 2.Loftus EV Jr., Harewood GC, Loftus CG, Tremaine WJ, Harmsen WS, Zinsmeister AR et al. PSC-IBD: a unique form of inflammatory bowel disease associated with primary sclerosing cholangitis. Gut 2005; 54(1): 91–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Folseraas T, Melum E, Rausch P, Juran BD, Ellinghaus E, Shiryaev A et al. Extended analysis of a genome-wide association study in primary sclerosing cholangitis detects multiple novel risk loci. J Hepatol 2012; 57(2): 366–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aron JH, Bowlus CL. The immunobiology of primary sclerosing cholangitis. Semin Immunopathol 2009; 31(3): 383–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sabino J, Vieira-Silva S, Machiels K, Joossens M, Falony G, Ballet V et al. Primary sclerosing cholangitis is characterised by intestinal dysbiosis independent from IBD. Gut 2016; 65(10): 1681–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vaughn BP, Kaiser T, Staley C, Hamilton MJ, Reich J, Graiziger C et al. A pilot study of fecal bile acid and microbiota profiles in inflammatory bowel disease and primary sclerosing cholangitis. Clin Exp Gastroenterol 2019; 12: 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Torres J, Palmela C, Brito H, Bao X, Ruiqi H, Moura-Santos P et al. The gut microbiota, bile acids and their correlation in primary sclerosing cholangitis associated with inflammatory bowel disease. United European Gastroenterol J 2018; 6(1): 112–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miethke AG, Zhang W, Simmons J, Taylor AE, Shi T, Shanmukhappa SK et al. Pharmacological inhibition of apical sodium-dependent bile acid transporter changes bile composition and blocks progression of sclerosing cholangitis in multidrug resistance 2 knockout mice. Hepatology 2016; 63(2): 512–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nevens F, Andreone P, Mazzella G, Strasser SI, Bowlus C, Invernizzi P et al. A Placebo-Controlled Trial of Obeticholic Acid in Primary Biliary Cholangitis. N Engl J Med 2016; 375(7): 631–643. [DOI] [PubMed] [Google Scholar]
- 10.Larusso NF, Bowlus CL, Levy C, Vuppalanchi R, Floreani A, Andreone P et al. PC.01.8 THE AESOP TRIAL: A RANDOMIZED, DOUBLE-BLIND, PLACEBO-CONTROLLED, PHASE 2 STUDY OF OBETICHOLIC ACID IN PATIENTS WITH PRIMARY SCLEROSING CHOLANGITIS. Digestive and Liver Disease 2018; 50(2): e67. [Google Scholar]
- 11.Tabibian JH, Lindor KD. Ursodeoxycholic acid in primary sclerosing cholangitis: if withdrawal is bad, then administration is good (right?). Hepatology 2014; 60(3): 785–788. [DOI] [PubMed] [Google Scholar]
- 12.Feagan BG, Rutgeerts P, Sands BE, Hanauer S, Colombel JF, Sandborn WJ et al. Vedolizumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med 2013; 369(8): 699–710. [DOI] [PubMed] [Google Scholar]
- 13.Sandborn WJ, Feagan BG, Rutgeerts P, Hanauer S, Colombel JF, Sands BE et al. Vedolizumab as induction and maintenance therapy for Crohn's disease. N Engl J Med 2013; 369(8): 711–721. [DOI] [PubMed] [Google Scholar]
- 14.Eksteen B, Mora JR, Haughton EL, Henderson NC, Lee-Turner L, Villablanca EJ et al. Gut homing receptors on CD8 T cells are retinoic acid dependent and not maintained by liver dendritic or stellate cells. Gastroenterology 2009; 137(1): 320–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eksteen B, Grant AJ, Miles A, Curbishley SM, Lalor PF, Hubscher SG et al. Hepatic endothelial CCL25 mediates the recruitment of CCR9+ gut-homing lymphocytes to the liver in primary sclerosing cholangitis. J Exp Med 2004; 200(11): 1511–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grant AJ, Lalor PF, Hubscher SG, Briskin M, Adams DH. MAdCAM-1 expressed in chronic inflammatory liver disease supports mucosal lymphocyte adhesion to hepatic endothelium (MAdCAM-1 in chronic inflammatory liver disease). Hepatology 2001; 33(5): 1065–1072. [DOI] [PubMed] [Google Scholar]
- 17.de Krijger M, Wildenberg ME, de Jonge WJ, Ponsioen CY. Return to sender: Lymphocyte trafficking mechanisms as contributors to primary sclerosing cholangitis. J Hepatol 2019; 71(3): 603–615. [DOI] [PubMed] [Google Scholar]
- 18.Smit JJ, Schinkel AH, Oude Elferink RP, Groen AK, Wagenaar E, van Deemter L et al. Homozygous disruption of the murine mdr2 P-glycoprotein gene leads to a complete absence of phospholipid from bile and to liver disease. Cell 1993; 75(3): 451–462. [DOI] [PubMed] [Google Scholar]
- 19.Pollheimer MJ, Trauner M, Fickert P. Will we ever model PSC? - "it's hard to be a PSC model!". Clin Res Hepatol Gastroenterol 2011; 35(12): 792–804. [DOI] [PubMed] [Google Scholar]
- 20.Fickert P, Fuchsbichler A, Wagner M, Zollner G, Kaser A, Tilg H et al. Regurgitation of bile acids from leaky bile ducts causes sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology 2004; 127(1): 261–274. [DOI] [PubMed] [Google Scholar]
- 21.Jahnel J, Fickert P, Langner C, Hogenauer C, Silbert D, Gumhold J et al. Impact of experimental colitis on hepatobiliary transporter expression and bile duct injury in mice. Liver Int 2009; 29(9): 1316–1325. [DOI] [PubMed] [Google Scholar]
- 22.Bamias G, Clark DJ, Rivera-Nieves J. Leukocyte traffic blockade as a therapeutic strategy in inflammatory bowel disease. Curr Drug Targets 2013; 14(12): 1490–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bloom S, Fleming K, Chapman R. Adhesion molecule expression in primary sclerosing cholangitis and primary biliary cirrhosis. Gut 1995; 36(4): 604–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fujino S, Andoh A, Bamba S, Ogawa A, Hata K, Araki Y et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut 2003; 52(1): 65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Katt J, Schwinge D, Schoknecht T, Quaas A, Sobottka I, Burandt E et al. Increased T helper type 17 response to pathogen stimulation in patients with primary sclerosing cholangitis. Hepatology 2013; 58(3): 1084–1093. [DOI] [PubMed] [Google Scholar]
- 26.Ward JBJ, Lajczak NK, Kelly OB, O'Dwyer AM, Giddam AK, Ni Gabhann J et al. Ursodeoxycholic acid and lithocholic acid exert anti-inflammatory actions in the colon. Am J Physiol Gastrointest Liver Physiol 2017; 312(6): G550–G558. [DOI] [PubMed] [Google Scholar]
- 27.Poupon RE, Bonnand AM, Chretien Y, Poupon R. Ten-year survival in ursodeoxycholic acid-treated patients with primary biliary cirrhosis. The UDCA-PBC Study Group. Hepatology 1999; 29(6): 1668–1671. [DOI] [PubMed] [Google Scholar]
- 28.Paumgartner G, Beuers U. Ursodeoxycholic acid in cholestatic liver disease: mechanisms of action and therapeutic use revisited. Hepatology 2002; 36(3): 525–531. [DOI] [PubMed] [Google Scholar]
- 29.Van Nieuwkerk CM, Elferink RP, Groen AK, Ottenhoff R, Tytgat GN, Dingemans KP et al. Effects of Ursodeoxycholate and cholate feeding on liver disease in FVB mice with a disrupted mdr2 P-glycoprotein gene. Gastroenterology 1996; 111(1): 165–171. [DOI] [PubMed] [Google Scholar]
- 30.Ando T, Langley RR, Wang Y, Jordan PA, Minagar A, Alexander JS et al. Inflammatory cytokines induce MAdCAM-1 in murine hepatic endothelial cells and mediate alpha-4 beta-7 integrin dependent lymphocyte endothelial adhesion in vitro. BMC Physiol 2007; 7: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ogawa H, Binion DG, Heidemann J, Theriot M, Fisher PJ, Johnson NA et al. Mechanisms of MAdCAM-1 gene expression in human intestinal microvascular endothelial cells. Am J Physiol Cell Physiol 2005; 288(2): C272–281. [DOI] [PubMed] [Google Scholar]
- 32.Cullen SN, Rust C, Fleming K, Edwards C, Beuers U, Chapman RW. High dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis is safe and effective. J Hepatol 2008; 48(5): 792–800. [DOI] [PubMed] [Google Scholar]
- 33.Van den Bossche L, Hindryckx P, Devisscher L, Devriese S, Van Welden S, Holvoet T et al. Ursodeoxycholic Acid and Its Taurine- or Glycine-Conjugated Species Reduce Colitogenic Dysbiosis and Equally Suppress Experimental Colitis in Mice. Appl Environ Microbiol 2017; 83(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Singh S, Khanna S, Pardi DS, Loftus EV, Jr., Talwalkar JA. Effect of ursodeoxycholic acid use on the risk of colorectal neoplasia in patients with primary sclerosing cholangitis and inflammatory bowel disease: a systematic review and meta-analysis. Inflamm Bowel Dis 2013; 19(8): 1631–1638. [DOI] [PubMed] [Google Scholar]
- 35.Chiang JY. Recent advances in understanding bile acid homeostasis. F1000Res 2017; 6: 2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cai SY, Boyer JL. The Role of Inflammation in the Mechanisms of Bile Acid-Induced Liver Damage. Dig Dis 2017; 35(3): 232–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gadaleta RM, Garcia-Irigoyen O, Moschetta A. Bile acids and colon cancer: Is FXR the solution of the conundrum? Mol Aspects Med 2017; 56: 66–74. [DOI] [PubMed] [Google Scholar]
- 38.Gadaleta RM, van Erpecum KJ, Oldenburg B, Willemsen EC, Renooij W, Murzilli S et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut 2011; 60(4): 463–472. [DOI] [PubMed] [Google Scholar]
- 39.Lindor KD, Kowdley KV, Luketic VA, Harrison ME, McCashland T, Befeler AS et al. High-dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Hepatology 2009; 50(3): 808–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Batta AK, Salen G, Holubec H, Brasitus TA, Alberts D, Earnest DL. Enrichment of the more hydrophilic bile acid ursodeoxycholic acid in the fecal water-soluble fraction after feeding to rats with colon polyps. Cancer Res 1998; 58(8): 1684–1687. [PubMed] [Google Scholar]
- 41.Kelly OB, Mroz MS, Ward JB, Colliva C, Scharl M, Pellicciari R et al. Ursodeoxycholic acid attenuates colonic epithelial secretory function. J Physiol 2013; 591(9): 2307–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Golden JM, Escobar OH, Nguyen MVL, Mallicote MU, Kavarian P, Frey MR et al. Ursodeoxycholic acid protects against intestinal barrier breakdown by promoting enterocyte migration via EGFR- and COX-2-dependent mechanisms. Am J Physiol Gastrointest Liver Physiol 2018; 315(2): G259–G271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim EK, Cho JH, Kim E, Kim YJ. Ursodeoxycholic acid inhibits the proliferation of colon cancer cells by regulating oxidative stress and cancer stem-like cell growth. PLoS One 2017; 12(7): e0181183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Colombo C, Setchell KD, Podda M, Crosignani A, Roda A, Curcio L et al. Effects of ursodeoxycholic acid therapy for liver disease associated with cystic fibrosis. J Pediatr 1990; 117(3): 482–489. [DOI] [PubMed] [Google Scholar]
- 45.Vavassori P, Mencarelli A, Renga B, Distrutti E, Fiorucci S. The bile acid receptor FXR is a modulator of intestinal innate immunity. J Immunol 2009; 183(10): 6251–6261. [DOI] [PubMed] [Google Scholar]
- 46.Guo C, Xie S, Chi Z, Zhang J, Liu Y, Zhang L et al. Bile Acids Control Inflammation and Metabolic Disorder through Inhibition of NLRP3 Inflammasome. Immunity 2016; 45(4): 802–816. [DOI] [PubMed] [Google Scholar]
- 47.Duboc H, Rajca S, Rainteau D, Benarous D, Maubert MA, Quervain E et al. Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel diseases. Gut 2013; 62(4): 531–539. [DOI] [PubMed] [Google Scholar]
- 48.Long SL, Gahan CGM, Joyce SA. Interactions between gut bacteria and bile in health and disease. Mol Aspects Med 2017; 56: 54–65. [DOI] [PubMed] [Google Scholar]
- 49.Song X, Sun X, Oh SF, Wu M, Zhang Y, Zheng W et al. Microbial bile acid metabolites modulate gut RORgamma(+) regulatory T cell homeostasis. Nature 2020; 577(7790): 410–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hang S, Paik D, Yao L, Kim E, Trinath J, Lu J et al. Bile acid metabolites control TH17 and Treg cell differentiation. Nature 2019; 576(7785): 143–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ghosh S, Panaccione R. Anti-adhesion molecule therapy for inflammatory bowel disease. Therap Adv Gastroenterol 2010; 3(4): 239–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McMurray RW. Adhesion molecules in autoimmune disease. Semin Arthritis Rheum 1996; 25(4): 215–233. [DOI] [PubMed] [Google Scholar]
- 53.Rivera-Nieves J, Gorfu G, Ley K. Leukocyte adhesion molecules in animal models of inflammatory bowel disease. Inflamm Bowel Dis 2008; 14(12): 1715–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Borchers AT, Shimoda S, Bowlus C, Keen CL, Gershwin ME. Lymphocyte recruitment and homing to the liver in primary biliary cirrhosis and primary sclerosing cholangitis. Semin Immunopathol 2009; 31(3): 309–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Briskin MJ, McEvoy LM, Butcher EC. MAdCAM-1 has homology to immunoglobulin and mucin-like adhesion receptors and to IgA1. Nature 1993; 363(6428): 461–464. [DOI] [PubMed] [Google Scholar]
- 56.Streeter PR, Berg EL, Rouse BT, Bargatze RF, Butcher EC. A tissue-specific endothelial cell molecule involved in lymphocyte homing. Nature 1988; 331(6151): 41–46. [DOI] [PubMed] [Google Scholar]
- 57.Lynch KD, Chapman RW, Keshav S, Montano-Loza AJ, Mason AL, Kremer AE et al. Effects of Vedolizumab in Patients with Primary Sclerosing Cholangitis and Inflammatory Bowel Diseases. Clin Gastroenterol Hepatol 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Steinacher D, Claudel T, Trauner M. Therapeutic Mechanisms of Bile Acids and Nor-Ursodeoxycholic Acid in Non-Alcoholic Fatty Liver Disease. Dig Dis 2017; 35(3): 282–287. [DOI] [PubMed] [Google Scholar]
- 59.Fickert P, Wagner M, Marschall HU, Fuchsbichler A, Zollner G, Tsybrovskyy O et al. 24-norUrsodeoxycholic acid is superior to ursodeoxycholic acid in the treatment of sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology 2006; 130(2): 465–481. [DOI] [PubMed] [Google Scholar]
- 60.Fickert P, Hirschfield GM, Denk G, Marschall HU, Altorjay I, Farkkila M et al. norUrsodeoxycholic acid improves cholestasis in primary sclerosing cholangitis. J Hepatol 2017; 67(3): 549–558. [DOI] [PubMed] [Google Scholar]
- 61.Trauner M, Fickert P, Wagner M. MDR3 (ABCB4) defects: a paradigm for the genetics of adult cholestatic syndromes. Semin Liver Dis 2007; 27(1): 77–98. [DOI] [PubMed] [Google Scholar]
- 62.Pauli-Magnus C, Kerb R, Fattinger K, Lang T, Anwald B, Kullak-Ublick GA et al. BSEP and MDR3 haplotype structure in healthy Caucasians, primary biliary cirrhosis and primary sclerosing cholangitis. Hepatology 2004; 39(3): 779–791. [DOI] [PubMed] [Google Scholar]
- 63.Degiorgio D, Crosignani A, Colombo C, Bordo D, Zuin M, Vassallo E et al. ABCB4 mutations in adult patients with cholestatic liver disease: impact and phenotypic expression. J Gastroenterol 2016; 51(3): 271–280. [DOI] [PubMed] [Google Scholar]
- 64.Oude Elferink RP, Paulusma CC. Function and pathophysiological importance of ABCB4 (MDR3 P-glycoprotein). Pflugers Arch 2007; 453(5): 601–610. . [DOI] [PubMed] [Google Scholar]
- 65.Hall LJ, Faivre E, Quinlan A, Shanahan F, Nally K, Melgar S. Induction and activation of adaptive immune populations during acute and chronic phases of a murine model of experimental colitis. Dig Dis Sci 2011; 56(1): 79–89. [DOI] [PubMed] [Google Scholar]
- 66.Kiesler P, Fuss IJ, Strober W. Experimental Models of Inflammatory Bowel Diseases. Cell Mol Gastroenterol Hepatol 2015; 1(2): 154–170. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sarafian MH, Lewis MR, Pechlivanis A, Ralphs S, McPhail MJ, Patel VC et al. Bile acid profiling and quantification in biofluids using ultra-performance liquid chromatography tandem mass spectrometry. Anal Chem 2015; 87(19): 9662–9670. [DOI] [PubMed] [Google Scholar]
- 68.Dieleman LA, Palmen MJ, Akol H, Bloemena E, Pena AS, Meuwissen SG et al. Chronic experimental colitis induced by dextran sulphate sodium (DSS) is characterized by Th1 and Th2 cytokines. Clin Exp Immunol 1998; 114(3): 385–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.