

Abstract
Dihydroxyacetone (DHA) is a three-carbon sugar that is the active ingredient in sunless tanning products and a by-product of electronic cigarette (e-cigarette) combustion. Increased use of sunless tanning products and e-cigarettes has elevated exposures to DHA through inhalation and absorption. Studies have confirmed that DHA is rapidly absorbed into cells and can enter into metabolic pathways following phosphorylation to dihydroxyacetone phosphate (DHAP), a product of fructose metabolism. Recent reports have suggested metabolic imbalance and cellular stress results from DHA exposures. However, the impact of elevated exposure to DHA on human health is currently under-investigated. We propose that exogenous exposures to DHA increase DHAP levels in cells and mimic fructose exposures to produce oxidative stress, mitochondrial dysfunction, and gene and protein expression changes. Here, we review cell line and animal model exposures to fructose to highlight similarities in the effects produced by exogenous exposures to DHA. Given the long-term health consequences of fructose exposure, this review emphasizes the pressing need to further examine DHA exposures from sunless tanning products and e-cigarettes.
Keywords: dihydroxyacetone, fructose, mitochondria, mitochondrial DNA, metabolic dysfunction, glucose, inflammation, reactive oxygen species, reactive species
Introduction
Dihydroxyacetone (DHA) is the simplest ketone sugar. When applied to the skin surface, it undergoes a Maillard reaction with the proteins to produce pigments called melanoidins. These melanoidins produce the browning effect associated with sunless tanning lotions. Tanning applications of DHA were discovered accidentally when DHA was tested as an alternative carbohydrate source to treat diabetes and other metabolic disorders (Wittgenstein and Berry 1960; Martini 2017). Browning of skin and gums were noted on patients administered DHA. DHA’s topical use was quickly commercialized and received FDA approval for topical applications of up to 20 % w/v in the late 1970s (FDA 2002; FDA 2015). Exposure estimates from a weekly topical application of a self-tanning lotion containing 10% DHA range from 0.91 mg/kg bw/week to 7.68 mg/kg bw/week, depending on absorption (SCCS 2010).
More recently, DHA has also been found in the aerosols generated from electronic cigarettes (e-cigarettes). E-cigarettes use e-liquids containing propylene glycol and vegetable glycerin (glycerol) to mobilize flavorants and nicotine. Free radical oxidation of the e-liquids generates DHA with the amount of DHA generated depending on the device’s combustion temperature (Vreeke et al. 2018). Exposure to DHA from e-cigarette vapor is estimated to be 0.5–2.33 μg/puff (Lee et al. 2018; Vreeke et al. 2018).
Sunless tanning exposures and the popularity of e-cigarettes have increased DHA concentrations to which humans are exposed (Pantini et al. 2007). Beyond skin absorption, DHA can be inhaled and absorbed through mucous membranes from e-cigarettes or the use of home or commercial spray tanning apparatuses. However, the systemic exposure effects of DHA are poorly understood. Several recent studies have demonstrated the cytotoxicity and genotoxicity of DHA, but mechanisms underlying these outcomes are still being investigated.
Endogenously, the phosphorylated form of DHA, dihydroxyacetone phosphate (DHAP), is produced during glycolysis and fructolysis along with its isomer glyceraldehyde-3-phosphate (GAP) (Figure 1) (Burch et al. 1970). When presented exogenously, DHA is rapidly absorbed into cells and is converted to DHAP by Triokinase/Flavin mononucleotide cyclase (TKFC, Figure 1), sometimes known as DHA kinase (DAK) (Cabezas et al. 2005; Moreno et al. 2014; Marco-Rius et al. 2017). DHAP can integrate into nine different metabolic pathways (Burch et al. 1970; Moreno et al. 2014; Marco-Rius et al. 2017), either via glycerol-3-phosphate (G3P) following its reduction or via GAP following a thiamine diphosphate catalyzed isomerization (TPI, Figure 1).
Figure 1.

Fructose is converted into the two 3-carbon metabolites dihydroxyacetone phosphate (DHAP) and glyceraldehyde-3-phosphate (Glyceraldehyde-3-P). Exogenous dihydroxyacetone (DHA) is integrated into 3-carbon metabolism by phosphorylation to DHAP by triose kinase\FMN-cyclase (TKFC). DHAP and Glyceraldehyde-3-P are interconverted by triose phosphate isomerase (TPI). Glyceraldehyde-3-P is converted by Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) to 1,3-Diphospho-glycerate. DHAP can also be directly converted to methylglyoxal by methylglyoxal synthase (MGS). Methylglyoxal and Diacylglycerol are advanced glycation end products (AGE) which can interact with the receptor for advanced glycation end products (RAGE), activate protein kinase c (PKC), or even damage cellular proteins. AGEs and other reactive species like reactive oxygen species (ROS) can induce DNA, RNA, and protein damage which are detrimental to cell health and can induce cell death. Human nomenclature for enzymes is used for this summary figure.
High fructose diets lead to an increase in cellular DHAP and G3P levels, which are linked to increased formation of reactive oxygen and reactive nitrogen species (ROS/RNS), changes in metabolic profile, and reduced mitochondrial function (Gizak et al. 2019; Hernandez-Diazcouder et al. 2019; Taskinen et al. 2019). Elevated DHAP and G3P contribute to the development of insulin resistance associated with type 2 diabetes and advanced glycation end products (AGEs), which form when elevated levels of sugar metabolites react with proteins or lipids (Richard 1993). These harmful compounds can react with amino, nucleic, and fatty acids to cause protein, DNA/RNA, and membrane damage (Yamagishi and Matsui 2010; Fournet et al. 2018). Imbalances in DHAP and G3P due to elevated intracellular fructose levels also alter gene expression and create post-translational modifications that modulate enzyme activity, further hindering the efficiency of cellular metabolic pathways (Moraru et al. 2018).
Given that exogenous DHA exposure can also create imbalances in DHAP and G3P, examining fructose exposures may offer insight into the cellular consequences and mechanism underlying the cytotoxicity and genotoxicity of DHA. Here, we review in vitro and in vivo studies that examine fructose exposures to understand the emerging literature on DHA and contextualize the potential human health consequences of the exogenous DHA exposure.
Fructose exposures generate reactive species
It is well known that elevated cellular levels of reactive species increase oxidative stress within the cell and specific organelles. High levels of ROS/RNS can lead to intracellular damage of DNA and proteins inducing mutations and dysfunction. Within the mitochondria, increased levels of ROS and RNS have been shown to alter electron transport, reduce oxidative phosphorylation, reduce metabolic output, induce mitochondrial DNA (mtDNA) damage, and eventually trigger cell death (Indo et al. 2007).
Fructose exposures induce elevated reactive species formation by disrupting the balance between a cell’s antioxidant defense mechanism and free radical concentration resulting in elevated oxidative stress (Touyz 2012). ROS is produced as a natural consequence of flux through the fructolysis pathway, but increased flux can overwhelm the natural antioxidant balance allowing more reactive species to remain in the cell. ROS is also produced by fructose through the activation of NADPH oxidases (Delbosc et al. 2005). Interestingly, uric acid production also has a role in producing ROS following fructose exposure (Madlala et al. 2016). Increases in dietary fructose consumption promoted uric acid formation, which increased total cellular ROS via activation of transforming growth factor β1 (TGF-β1) and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 4 (Madlala et al. 2016).
ROS and RNS are induced by fructose exposure at both low (≤ 10 mM) and high (> 10 mM) doses (Table 1). At 5 mM doses, the hepatocyte cell lines, rat hepatic parenchymal cells (RHPC), human hepatic (HLO2), and human hepatocellular carcinoma (HepG2) cells show elevated levels of hydrogen peroxide (H2O2), malondialdehyde (MDA), and nitric oxide synthase (iNOS) for up to 48 h (Zhang et al. 2015). Hepatic phagocytic Kupffer cells did not show any increase in total cellular ROS, H2O2, or MDA levels at 5 mM dosing, indicating metabolic differences between cell lines alter the generation of reactive species (Zhang et al. 2015).
TABLE 1.
Fructose exposures in cell line models
| Model-system | Dose | Exposure duration | Methods | Key findings | References |
|---|---|---|---|---|---|
| Reactive species formation | |||||
| Dendritic cells (human) | 15 mM fructose | 24–72 hr | ROS measured using flow cytometry to determine oxidation of dihydrorhodamine 123 to rhodamine 123; AGE formation measured using AGE ELISA kits | Reduced level of ROS production in high fructose treated groups compared to high glucose and control groups | Jaiswal et al., 2019 |
| HEK293 | 30 mM fructose | 4 hr | Total ROS production was quantified by DCFDA | ROS significantly increased in cells exposed to fructose and salt | Dornas et al., 2017 |
| HepG2 | 5 mM fructose | Incubation with 5 mM fructose for 48 hr and incubation with 2 μM allopurinol or 20 μM quercetin for additional 24 hr | Total ROS production was quantified by DCFDA | Elevated ROS, iNOS, and hydrogen peroxide levels in HepG2 cells | Zhang et al., 2015 |
| HLO2 | 5 mM fructose | Incubation with 5 mM fructose for 48 hr and incubation with 2 μM allopurinol or 20 μM quercetin for additional 24 hr | Total ROS production was quantified by DCFDA | Increases in ROS, iNOS, and hydrogen peroxide levels in HLO2 cells | Zhang et al., 2015 |
| Kupffer cells | 5 mM fructose | Incubation with 5 mM fructose for 48 hr and incubation with 2 μM allopurinol or 20 μM quercetin for additional 24 hr | Total ROS production was quantified by DCFDA | No elevation in ROS, hydrogen peroxide, and MDA levels | Zhang et al., 2015 |
| L6 Myotubes | 15 mM fructose | mtROS-1, 3, 6, and 24 hr; superoxide and hydrogen peroxide- 1, 3, and 6 hr | mtROS measured through MitoSOX red fluorescence; superoxide and hydrogen peroxide determined by fluorimetric assay | Increased production of mtROS with 1 hr and NO within 6 hr exposure; NO level reached maximum at 48 hr; non-mitochondrial respiration presented no significant change; iNOS levels peaked at 6 hr of exposure | Jaiswal et al., 2015a |
| L6 Myotubes | 15 mM fructose | 1, 3, and 6 hr | Cytoplasmic ROS measured by DCFDA fluorescence | Time-dependent increase in ROS which peaked at 3 hr; oxidative stress results from increased content of ROS and/or RNS | Jaiswal et al., 2015b |
| Rat hepatic parenchymal cell (RHPC) | 5 mM fructose | Incubation with 5 mM fructose for 48 hr and incubation with 2 μM allopurinol or 20 μM quercetin for additional 24 hr | Total ROS production was quantified by DCFDA | Increased ROS, iNOS, and hydrogen peroxide levels | Zhang et al., 2015 |
| Mitochondrial and metabolic dysfunction | |||||
| Dendritic cells (human) | 15 mM fructose | 24–72 hr | Energetic state measurement- seahorse analyzer | 72 hr exposure induced shift from oxidative phosphorylation toward glycolysis compared to high glucose exposures; decrease in ECAR and OCR at 24 hr; fructose-treated cells exhibited the least OCR compared to all treatment and control groups | Jaiswal et al., 2019 |
| Hepatocytes | 5.5 mM fructose | 72 hr | Apoptosis, lipid content, and lipid peroxidation measured through spectrometry; oxygen consumption monitored using seahorse analyzer; liver steatosis and dysfunction measured through RT-qPCR | Elevated steatosis and liver dysfunction, increased apoptosis, oxidative stress and mitochondrial respiration | Grasselli et al., 2019 |
| HepG2 | 5 mM fructose | 5 mM fructose for 48 hr with 2 μM allopurinol or 20 μM quercetin for additional 24 hr | MMP detection | Altered mitochondrial membrane potential | Zhang et al., 2015 |
| HLO2 | 5 mM fructose | 5 mM fructose for 48 hr with 2 μM allopurinol or 20 μM quercetin for additional 24 hr | MMP detection | Altered mitochondrial membrane potential | Zhang et al., 2015 |
| L6 Myotubes | 15 mM fructose | mtDNA content and mitochondrial membrane potential- 3, 6, 24h, 48, and 72 hr; mitochondrial respiratory function- 3, 6, 24, and 48 hr | mtDNA content- PCR through amplification of a mitochondrial encoded gene; mitochondrial membrane potential- change analyzed with JC-1 absorption; mitochondrial respiratory function- seahorse XF-e24 extracellular flux assay; ATP levels- stay Brite ATP assay; citrate synthase activity monitored by spectrophotometry | Reductions in mtDNA content; loss of mitochondrial membrane potential; impaired mitochondrial energy metabolism; decreased activities of citrate synthase, mitochondrial dehydrogenases, ATP-linked respiration, cellular ATP content, and mitochondrial respiratory complexes | Jaiswal et al., 2015a |
| L6 Myotubes | 15 mM fructose | 1, 3, and 6 hr | 2-DG uptake assay | Reduction in 2-deoxyglucose uptake and an impairment of glucose utilization and insulin signaling through ROS-mediated activation of JNK and ERK1/2 pathways | Jaiswal et al., 2015b |
| RHPC | 5 mM fructose | 5 mM fructose for 48 hr with 2 μM allopurinol or 20 μM quercetin for additional 24 hr | MMP detection | Altered mitochondrial membrane potential | Zhang et al., 2015 |
| Transcriptional changes | |||||
| 3 T3-L1 Preadipocytes | 550 μm | 48 hr | Protein expression measured by immunoblotting | Increase in adipogenesis and overexpression of Pparγ, C/Ebpα and GluT4 | Du and Heaney, 2012 |
| 3 T3-L1 Preadipocytes | 30 mM fructose | Up to 8 days | mRNA expression measured using qPCR; protein expression measured by immunoblotting | FabP4, Pparγ, C/Ebpα and 11β-Hsd1 increased after 4, 6 and 8 days of exposure; C/Ebpβ decreased in same time period; GluT4 increased at days 4 through 6. | Legeza et al., 2014 |
| Dendritic cells (human) | 15 mM fructose | 24–72 hr | CD86 expression measured through mean fluorescence intensity; NF-κB activity determined through Alexa-594 antibody staining; IL-6 and IL-1β determined with corresponding antibody | Increased expression of IL-6, IL-1β, TNF-α, and CD86; exposure caused T-cells to increase secretion of IFN-y; increased production of AGEs led to activation of RAGE and expression of inflammatory cytokines; no significant difference in the expression of pAMPK-α, HIF-1α and p70S6 kinase; GLUT1 expression was 15% higher | Jaiswal et al., 2019 |
| HEK293 | 30 mM fructose | 4 hr | mRNA expression measured using qPCR; protein expression measured by immunoblotting | No significant change in SOD activity; NF-kB subunit p65 was increased when exposed to fructose and salt together | Dornas et al., 2017 |
| Hepatocytes (primary rat) | 5 mM fructose | 5 mM fructose for 48 hr with 2 μM allopurinol or 20 μM quercetin for additional 24 hr | mRNA expression measured using qPCR; protein expression measured by immunoblotting | Increased protein expression levels of Txnip, Nlrp3, Asc, Caspase-1, Il-1β and Il-18; | Zhang et al., 2015 |
| HepG2 | 5 mM fructose | 5 mM fructose for 48 hr with 2 μM allopurinol or 20 μM quercetin for additional 24 hr | mRNA expression measured using qPCR; protein expression measured by immunoblotting | Increased expression of TXNIP and NLRP3 levels; 72 hr exposure led to increased levels of ASC, Caspase-1, STAT3, SOCS3, IL-18, and IL-1β | Zhang et al., 2015 |
| HLO2 | 5 mM fructose | 5 mM fructose for 48 hr with 2 μM allopurinol or 20 μM quercetin for additional 24 hr | mRNA expression measured using qPCR; protein expression measured by immunoblotting | Increased expression of TXNIP and NLRP3 levels; 72 hr exposure led to increased levels of ASC, Caspase-1, STAT3, SOCS3, IL-18, and IL-1β | Zhang et al., 2015 |
| L6 Myotubes | 15 mM fructose | iNOS expression- 3, 6, 24, 48, and 72 hr; Nrf-2 translocation- 24 and 48 hr | Nitrite values determined with differing concentrations of sodium nitrite as a standard; Nrf-2 expression monitored by immunofluorescence; cytochrome C levels determined by immunoblotting | Increased NO production and iNOS expression; iNOS expression highest at 6 hr; elevated localization of Nrf-2 to the nucleus at 24 and 48 hr; apoptosis evident by increased levels of cleaved caspase 3, 9 and 7; Bcl-2 protein expression was significantly reduced and there was an increase in Bax; 48 and 72 hr exposure elevated cytochrome C levels in the cytoplasm from the mitochondria | Jaiswal et al., 2015a |
| L6 Myotubes | 15 mM fructose | 1, 3, and 6 hr | GluT4 translocation measured by the cell surface level of GluT4myc through an antibody-based colorimetric assay; mRNA expression measured using qPCR; protein expression measured by immunoblotting | GluT1 and GluT4 protein and mRNA levels not altered at 3 and 6 hr; no significant change in GluT5 mRNA expression; impaired insulin-stimulated translocation of GluT4myc to the cell surface; lower insulin-stimulated tyrosine phosphorylation of Irs-1; no differences in Irs-1 protein expression; inhibition of insulin-stimulated phosphorylation of Akt without altered total amount of Akt; no altered mRNA expression of Irs-1, Pi3k, Akt, GluT4, and GluT1; elevated ROS activated Jnk, Erk, p38, MapK, and Nf-κb inflammation pathways; increased phosphorylation of Jnk and Erk1/2 without any significant change in the total content; increase in the activity of Nf-κb pathway | Jaiswal et al., 2015b |
| Rat hepatic parenchymal cell (RHPC) | 5 mM fructose | 5 mM fructose for 48 hr with 2 μM allopurinol or 20 μM quercetin for additional 24 hr | mRNA expression measured using qPCR; protein expression measured by immunoblotting | Increased expression of Txnip and Nlrp3 levels; 72 hr exposure led to increased levels of Asc, Caspase-1, Stat3, Socs3, Il-18, and Il-1β | Zhang et al., 2015 |
At high fructose doses, L6 myotubes generated mitochondrial ROS within 1 h and nitric oxide (NO) within 6 h of 15 mM fructose treatment (Jaiswal et al. 2015a; Jaiswal et al. 2015b). iNOS also remained elevated for 48 h indicating the prolonged presence of reactive species (Jaiswal et al. 2015a). While the human embryonic kidney (HEK293) cell line exposed to 30 mM fructose produced elevated total cellular ROS levels (Dornas et al. 2017), 15 mM fructose did not promote the generation of reactive oxygen species in dendritic cells. However, increased production of AGEs did occur (Jaiswal et al. 2019) in dendritic cells, emphasizing that fructose exposures induce reactive species in a cell-specific manner.
Systemic exposures to fructose also increased reactive species and oxidative damage at both low (≤ 10% dietary intake) and high doses (> 10% dietary intake) in animal models (Table 2). Dietary integration of 10% fructose led to the elevated generation of ROS and RNS across animal models. Sprague-Dawley rats exposed to 10% fructose dose for 8 weeks showed elevated total ROS, H2O2, MDA, xanthine oxidase, and iNOS, which all contributed to increases in hepatic oxidative stress (Zhang et al. 2015). A 12-week duration of the same dose in Wistar rats also resulted in high levels of total ROS generation in peripheral blood mononuclear cells but not in bone marrow mononuclear cells (Porto et al. 2015). Even C57BL/6 mice exposed to a 10% fructose dose over 20 weeks showed an 80% increase in total cellular ROS in cardiac tissue along with elevated mitochondrial H2O2 generation (Zhang et al. 2016). These studies suggest that chronic exposure to low dose fructose increases total cellular reactive species in the liver and cardiac tissues.
TABLE 2.
Fructose exposures in animal models
| Model-system | Dose | Exposure duration | Methods | Key findings | References |
|---|---|---|---|---|---|
| Reactive species formation | |||||
| Male Wistar rats | High-fat diet and 5% fructose in drinking water per day | 4 months | ROS in kidney measured DCFDA; MDA measured spectrophotometrically | ROS and malondialdehyde expression were elevated in the kidneys | Rosas-Villegas et al., 2017 |
| Male C57BL/6 mice | 10% fructose in drinking water per day | 20 weeks | Total ROS activity assay kit | 80% increase in ROS production; increased mitochondrial hydrogen peroxide production | Zhang et al., 2016 |
| Male Sprague–Dawley rats | 10% fructose in drinking water per day | 8 weeks | Total ROS measured by DCFDA | Increased ROS, MDA, XO, iNOS, and hydrogen peroxide levels in rat livers | Zhang et al., 2015 |
| Male HTG rats, male Wistar rats, and male Lewis rats | 10% fructose in drinking water per day | 3 weeks | Pentosidine measurement- fluorescence at 332ex/378em nm | Increased post-translational pentosidine levels in rat aorta and skin samples for all three strains of rats | Mikulikova et al., 2008 |
| Male Wistar rats (mononuclear cells) | 10% fructose in drinking water per day | 12 weeks | ROS measured using flow cytometry to quantify the production of *O2−, H2O2, NO and *ONOO−/*OH−, with DHE, DCF, DAF-2 T in peripheral blood and bone-marrow mononuclear cells | Elevated level of ROS generation in peripheral blood mononuclear cells; no ROS increase in bone marrow mononuclear cells | Porto et al., 2015 |
| Male Fischer rats | 20% fructose in drinking water per day and high salt diet (8%) for last 10 weeks | 20 weeks | ROS production quantified using DCFDA; SOD activity measured by autooxidation of pyrogallol; cat activity was measured spectrophotometrically | Salt-dependent hypertension and elevated oxidative stress; decrease in renal enzymatic SOD and catalase activity in the kidney | Dornas et al., 2017 |
| Male and female Sprague–Dawley rats | 20% fructose water solution per day | Dose received during gestation and lactation periods | OxiSelect in vitro ROS/RNS assay kit | 1.5-fold increase in hippocampal LPO which is associated with elevated ROS | Yamada et al., 2019 |
| Male Wistar rats | 250 mL of 25% fructose drinking water per day with 40.6% fats | 6 weeks | ROS levels measured by DCFDA | 8-time increase in lipid peroxidation levels in mitochondria for fructose groups and high fat and fructose groups; no increases in ROS for fructose-treated groups | Garcia-Berumen et al., 2019 |
| Male Sprague–Dawley rats | 30 g of fructose per 100 g of chow (30% fructose) | 8 weeks | DNA/RNA oxidative damage ELISA assay | 8-OHdG levels were increased indicating elevated levels of oxidative stress | Cioffi et al., 2017 |
| Male C57BL/6 mice | 60% fructose diet per day | 6, 10, and 20 weeks | Superoxide production determined through measurement of NADPH activity | Significant increase in superoxide production in mice during 6 weeks of intake; at 10 weeks, there was a 29% increase in superoxide production, and at 20 weeks, there was a 16% increase | Mellor et al., 2010 |
| Male Wistar rats | Fructose content provides 60% of calories in diet per day | 2 weeks | Measured through insulin resistance | Inhibited expression of catalase in the heart and liver; condition leads to a high vulnerability to elevated levels of oxidative stress in insulin resistance | Cavarape et al., 2001 |
| Male spontaneously hypertensive rats | 60% fructose | 3 weeks | ROS by flow cytometry using the fluorescence dye DHE | There were high levels of ROS accumulation and lipid peroxidation in rostral ventrolateral medulla; development of brain oxidative stress can lead to hypertension | Wu et al. 2017 |
| Male Wistar rats | 60% fructose chow in diet per day | 12, 16, 20, and 24 weeks | MDA measured spectrophotometrically | Increase in production of substances such as methylglyoxal, which leads to muscle oxidative stress; no significant increase in the levels of the peroxidation product malondialdehyde in the serum, left ventricular tissue, or in the cardiac level of the nitrooxidative marker 3-nitrotyrosine | Szucs et al. 2019 |
| Mitochondrial and metabolic dysfunction | |||||
| Male C57BL/6 mice | 10% fructose in drinking water per day | 20 weeks | Oxygen consumption rates monitored by a Seahorse XF24 oxygen flux analyzer; complex II enzyme activity measured using microplate assay kit | Cardiac hypertrophy, diastolic dysfunction, decreased complex II activity by 60%, and reduced state III and IV OCR; for complex I and II, rates of ATP synthesis decreased by 37% and 36% respectively | Zhang et al., 2016 |
| Male Sprague–Dawley rats | 10% fructose in drinking water per day | 6 weeks | Respiratory measurements made using Oroboros system for permeabilized muscle fibers | In the extensor digitorum longus (EDL) muscle, there was a significant decrease in complex I respiration; respiratory activity for other complexes were similar; for the soleus muscle, OXPHOS capacity for complex II was much lower | Warren et al., 2014 |
| Male Wistar rats | 250 mL of 25% fructose drinking water per day | 6 weeks | Mitochondrial respiration obtained by determining the oxygen consumption rate of isolated mitochondria using a Clark-type electrode coupled to a YSI 5300A biological oxygen monitor | Mitochondrial respiratory rate in state 3 decreased in the fructose group by 1.6-fold; 1.8-fold increase in state 4 respiration; complex I activity decreased ~2.3-fold in mitochondria for the high fructose group | Garcia-Berumen et al., 2019 |
| Male Sprague–Dawley rats | 30 g of fructose per 100 g of chow (30% fructose) | 8 weeks | mtDNA damage measured using qPCR; changes in mitochondrial biogenesis monitored through the expression of Pcg1α, Nrf1, and Tfam | Increase in mtDNA damage in the liver of rats; 22% decrease in mtDNA fragments; mitochondrial copy number was reduced by approximately 56%; reduced mitochondrial biogenesis | Cioffi et al., 2017 |
| Male and female Sprague–Dawley rats | 20% fructose water solution per day | Dose received during gestation and lactation periods | Mitochondria oxygen consumption rate- MitoXpress oxygen consumption assay; mtDNA damage assessed using qPCR | Reduction mitochondrial OCR; no change in mtDNA amount | Yamada et al., 2019 |
| Transcriptional changes | |||||
| Male Wistar rats | High-fat diet and 5% fructose in drinking water per day | 4 months | Protein expression measured by immunoblotting | Increased protein expression of inflammatory markers Tlr-4, Nf-κb; Tnfα/β, and Mcp1 | Rosas-Villegas et al., 2017 |
| Kupffer cells from male Sprague–Dawley rats | 10% fructose in drinking water per day | 8 weeks | mRNA expression measured using qPCR | Increase in Txnip, Nlrp3 levels in Kupffer cells, and aggravate oxidative stress in livers; in vitro fructose exposure did not change levels of Txnip, Nlrp3, and no ROS overproduction; increased co-localization of Nlrp3; | Zhang et al., 2015 |
| Male Sprague–Dawley rats | 30 g of fructose per 100 g of chow (30% fructose) | 8 weeks | mRNA expression measured using qPCR; protein expression measured by immunoblotting | Pgc1α, Nrf1, and Tfam reduced expression by 19%, 41%, and 43% respectively | Cioffi et al., 2017 |
| Male C57BL/6 mice | 10% fructose in drinking water per day | 20 weeks | mRNA expression measured using qPCR; protein expression measured by immunoblotting | Decreased CFTR expression and induced ROS and oxidative stress | Zhang et al., 2016 |
| Male Sprague–Dawley rats | 10% fructose in drinking water per day | 6 weeks | Protein expression measured by immunoblotting | Decline in Pgc-1α protein levels in both EDL and soleus muscles; increased Sirt3 protein levels in the soleus, and decline in the EDL; no changes in thioredoxin reductase-2 protein levels in the soleus, but there was a decrease in the EDL muscle | Warren et al., 2014 |
| Male Wistar rats | 10% fructose in drinking water per day | 12 weeks | Annexin V-FITC apoptosis detection kit; cytokines measured in the plasma by flow cytometry using a Cytometric bead Array – Mouse inflammation kit | Elevated rates of apoptosis; Il-6 and Il-12p70 were higher | Porto et al., 2015 |
| Male and female Wistar rats | 10% fructose in drinking water per day | 24 weeks | mRNA expression measured using qPCR | Increased expression of Irβ, Irs-1, Irs-2, Akt, Pi3k, eNOS, mTor, Ppary, Nrf2, and eNOS; induction of proinflammatory markers, iNOS, Tnfα, Il-1β, Il-18, MDA, and ALT, as well as anti-inflammatory factors Il-10, and Nrf2 in adipose tissues of male and female rats | Pektas et al., 2016 |
| Male and female Sprague–Dawley rats | 20% fructose water solution per day | Dose received during gestation and lactation periods | mRNA expression measured using qPCR; protein expression measured by immunoblotting | Decreased Ucp5 and Tfam; the amount of genomic Nadh dehydrogenase 1, 2, and 3 did not change; no alterations in mitochondrial complex protein expression | Yamada et al., 2019 |
| Male Wistar rats | Fructose content provides 60% of calories in diet per day | 2 weeks | mRNA expression assessed by northern blot | Decreased expression of Cat and Sod1 in the liver and Cat in the heart; reductions in catalase (cat) and cu-ZnSOD1 activity were detected in skeletal muscle and adipose tissues | Cavarape et al., 2001 |
| Male C57BL/6 mice | 60% fructose diet per day | 6, 10, and 20 weeks | mRNA expression measured using qPCR | No effect on thioredoxin-2 gene expression level; at 6 weeks of exposure, β-myosin gene expression remained unchanged; at 10 weeks, non-significant mRNA expression levels of β-myosin, and NADPH oxidase subunit, Nox2 by 16.8-fold; at 20 weeks, gene expression levels of β-myosin was lowered, and Nox2 was similar for the control and fructose-treated groups | Mellor et al., 2010 |
| Male Wistar rats | 60% fructose chow in diet per day | 24 weeks | mRNA expression measured using qPCR; myosin 6 protein level determined by proteomics | Myosin 6 protein level was increased after fructose intake; Elovl6 and Myh6 also increased; Creatine kinase, creatine kinase-MB, and lactate dehydrogenase levels were not significantly different; 45% decrease in cardiolipin | Szucs et al. 2019 |
| Spontaneously hypertensive rats | 60% fructose | 12 weeks | Protein expression measured by immunoblotting | NADPH oxidase subunit gp91phox and angiotensin II type I receptor levels increased in the RVLM neuron and there was a decrease in extracellular SOD expression | Wu et al. 2017 |
As expected from the low dose fructose results, high fructose doses further increased intracellular ROS and oxidative stress. A 20% fructose diet increased oxidative stress levels in Fischer and Sprague-Dawley animal models for 20 weeks and during lactation/gestation periods, respectively (Dornas et al. 2017; Yamada et al. 2019). Hippocampal analysis of Sprague-Dawley rats demonstrated a 1.5-fold increase in total ROS due to increased fructose consumption (Yamada et al. 2019). Fischer rats treated with 20% fructose combined with 8% saline experienced hypertension, linking dysfunction to AGE formation and contractile changes in the vasculature (Dornas et al. 2017). High-dose fructose also elevated oxidative stress in response to increased 8-OHdG levels in Sprague-Dawley rats (Cioffi et al. 2017). Wistar rats exposed to a 60% fructose dose for 24 weeks showed elevated methylglyoxal and increased oxidative stress (Szucs et al. 2019). 60% fructose also elevated oxidative stress in Spontaneously Hypertensive rats, Wistar rats, and C57BL/6 mice (Cavarape et al. 2001; Mellor et al. 2010; Wu et al. 2017).
Current fructose exposure studies use various animal models and investigate multiple tissue types with little overlap between tissues studied and fructose dosing (Table 2). Of the studies discussed here, dietary supplementation of 7–10% and 60% fructose per day elevated reactive species formation in hepatic tissue of Sprague-Dawley rats and Wistar rats, respectively (Cavarape et al. 2001; Zhang et al. 2015; Cioffi et al. 2017). However, hepatic tissue from Wistar rats with 25% fructose administration exhibited no elevation in ROS levels (Garcia-Berumen et al. 2019; Yamada et al. 2019). Cardiac tissue showed increased ROS for C57BL/6 mice with 10% and 60% dietary fructose and Wistar rats given 60% fructose (Zhang et al. 2015; Szucs et al. 2019). Only limited analysis of kidney tissue was conducted with ROS formation observed after 20% fructose exposure in Fischer rat models (Dornas et al. 2017). Finally, Sprague-Dawley rats given a 20% fructose diet showed elevated ROS formation in hippocampal tissue, and Spontaneously Hypertensive rats administered a 60% fructose diet presented increased ROS in rostral ventrolateral medulla (RVLM) tissue (Wu et al. 2017; Yamada et al. 2019). While the models and dosing vary, these studies demonstrate excess fructose generates reactive species. However, dose-dependence and tissue-specificity cannot be inferred from the current literature.
Fructose exposures induce mitochondrial dysfunction
Fructose exposures also impact mitochondrial health by altering the metabolic output. Animal models show that fructose exposures impair insulin signaling pathways, reducing cellular glucose uptake and establishing a condition of chronic glucose intolerance (Tobey et al. 1982). Reductions in glucose uptake inhibit metabolic productivity exhibited by decreased oxygen consumption rate (OCR) and ATP content (Collins-Nakai et al. 1994). Current literature reveals that both low (≤ 10%) and high fructose (> 10%) supplementation induces mitochondrial dysfunction (Jaiswal et al. 2015a; Jaiswal et al. 2015b; Zhang et al. 2015).
In hepatoma FaO cells, 5.5 mM fructose alters mitochondrial respiration after 72 h (Grasselli et al. 2019). In addition, 5 mM fructose alters mitochondrial membrane potential in RHPC, HLO2, and HepG2 liver cells (Zhang et al. 2015). High-dose fructose exposures increased mitochondrial membrane leakage, lowered mitochondrial membrane potential, and induced mitochondrial DNA damage in L6 myotubes (Jaiswal et al. 2015a). L6 myotubes also showed impaired insulin signaling, inhibited glucose uptake, and insulin resistance following 3 h of the 15 mM fructose (Jaiswal et al. 2015b). Decreased rates of respiration were also observed in both dendritic cells and L6 myotubes after 15 mM fructose supplementation (Jaiswal et al. 2015a; Jaiswal et al. 2019). Moreover, high fructose exposure to L6 myotubes led to reduced rates of mitochondrial biogenesis and total cellular ATP production, contributing to a loss of metabolic function evident with a 30.5% reduction in ATP-related respiration (Jaiswal et al. 2015a).
Low dose fructose exposures to animal models have been linked to reduced rates of mitochondrial respiration, inhibited glucose uptake, and cofactor imbalances (Warren et al. 2014; Zhang et al. 2016). Low dose fructose intake for 20 weeks led to a 60% decrease in complex II respiratory activity and reduced oxygen consumption in States III and IV in C57BL/6 mice (Zhang et al. 2016). Similarly, analysis of Sprague-Dawley revealed reduced respiratory activity of complex I and II in the extensor digitorum longus (EDL) and soleus muscles, respectively; however, the respiratory activity of other complexes remained unchanged (Warren et al. 2014). Findings of reduced mitochondrial respiration correlate with reports of diminished ATP synthesis (Milakovic and Johnson 2005).
Decreases in cellular glucose uptake for high-dose fructose supplementation to animal models leads to a decreased mitochondrial respiration (Table 2). Reduced glucose integration into metabolic pathways contributed to impaired mitochondrial function, as shown with high-dose fructose exposures to Sprague-Dawley and Wistar rats that exhibit decreased rates of respiration (Garcia-Berumen et al. 2019; Yamada et al. 2019).
High-dose fructose exposures in animal model studies have been capable of inducing elevated forms of oxidative damage evident through increased formation of nuclear and mtDNA lesions (Table 2). Fructose concentrations greater than 20% have produced oxidative mtDNA damage in the liver of Sprague-Dawley rats. In male Sprague-Dawley rats administered 30% of fructose per day, mtDNA copy number reduced by half, and there was a significant reduction in mitochondrial biogenesis, which decreased metabolic output (Cioffi et al. 2017). As shown by high-dose exposures to cell line models, fructose can institute mitochondrial impairment through changes in mitochondrial membrane potential (Table 1).
It should be noted that while fructose supplementation induced detrimental metabolic alterations such as reduced glucose uptake and insulin resistance in cell lines and animal models, human clinical trials do not show that fructose has a significant role in reducing metabolic health (Tappy and Le 2012). However, there is evidence showing that diabetic patients exhibit elevated fructose concentrations in blood plasma (Kawasaki et al. 2002). Unfortunately, fructose plasma levels have not been measured in animal studies. More focus has been placed on plasma glucose and leptin measurements. Altogether, there is evidence that low- and high-dose fructose changes metabolism in cell lines and animals (Tables 1 & 2). More work is needed to understand how fructose effects in animal models translate into humans.
Fructose exposure causes transcriptional changes
Fructose exposures also induced unique transcriptional changes in response to elevated cellular stress, ROS, mitochondrial dysfunction, and cell death mechanisms (Figure 2). Oxidation of DNA and RNA by elevated ROS can disrupt replication, transcription and induce mutations within the genome (Poetsch 2020). Oxidative DNA lesions can also act as transcription regulators through epigenetic or enhancer repressor activity to directly regulate gene expression (Ghosh and Mitchell 1999; Pastukh et al. 2015; Fleming et al. 2017).
Figure 2.
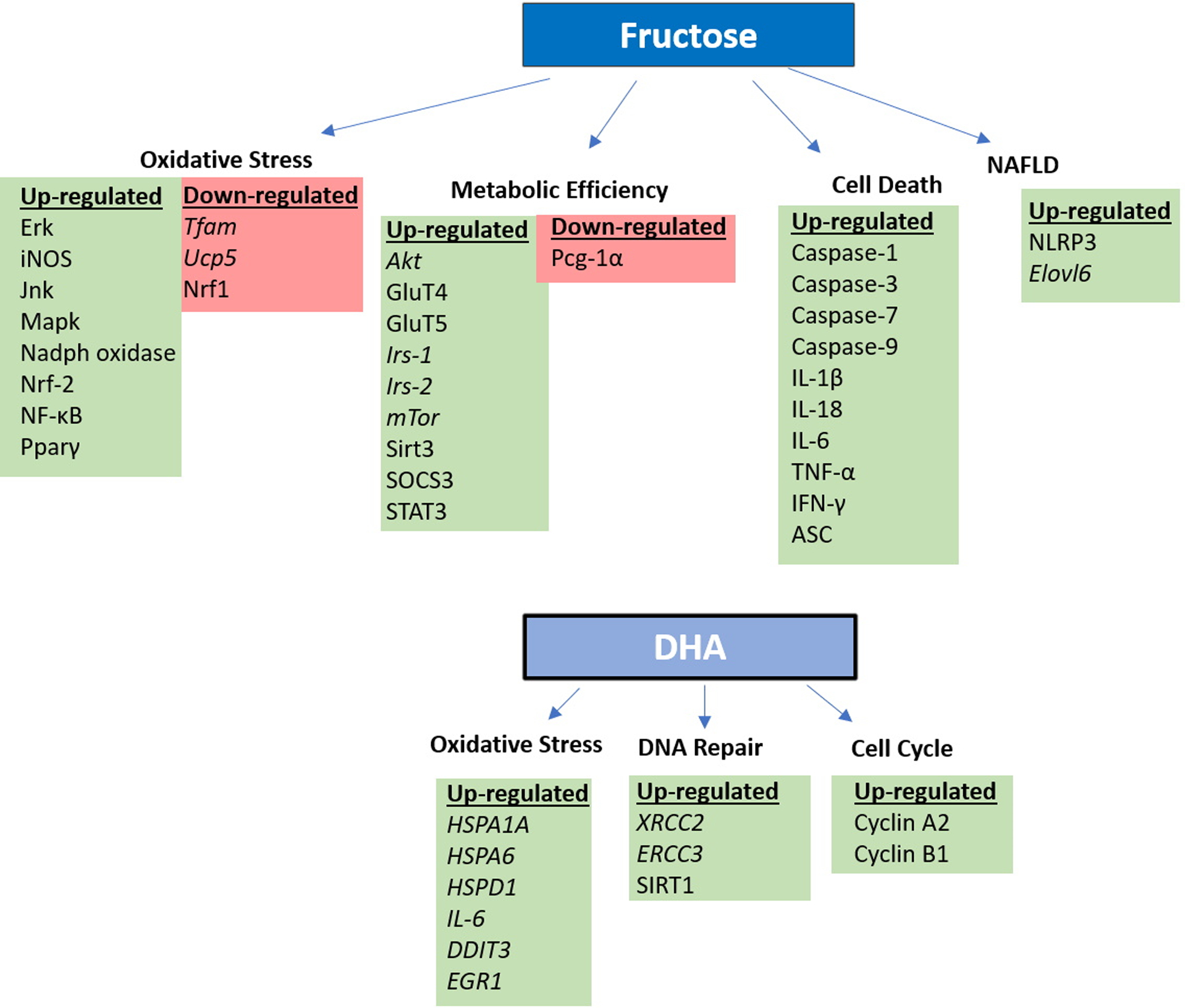
Both fructose and DHA exposures alter the expression of proteins involved in oxidative stress, metabolic efficiency, DNA repair, cell cycle control, and cell death. Changes in gene expression (italics) or protein levels identified in the summarized studies are presented here. Species is denoted by the use or lack of capitalization.
Low dose fructose altered Thioredoxin Interacting Protein (TXNIP) expression, associated with a cellular inflammatory signaling response to oxidative stress, in hepatocytes, RHPCs, HLO2s, and HepG2s cells (Zhang et al. 2015). Low dose fructose exposure also induced NLRP3 expression, which stimulates inflammasome formation and is linked to non-alcoholic fatty liver disease (NAFLD). Cultured hepatocytes (primary rat hepatocytes, RHPCs, HLO2, and HepG2 cells) treated with 5 mM fructose showed significant up-regulation of NLRP3 and inflammasome components Apoptosis-associated speck-like protein containing a CARD (ASC), Caspase-1, interleukin-1 β (IL-1β), and IL-18 (Zhang et al. 2015). Low dose fructose exposures were also able to elevate fatty acid synthase (Fas) activity by 125% and MDA levels by 67% in primary hepatocytes (Grasselli et al. 2019). Elevated protein levels of signal transducer and activator of transcription 3 (STAT3) and suppressor of cytokine signaling-3 (SOCS3), which increase inflammatory cytokines, were also present in cultured HepG2 cells after low dose fructose exposures (Zhang et al. 2015). Even 550 μM fructose caused a 3.5-fold increase in adipogenesis, indicated through overexpression of peroxisome proliferator-activated receptor γ (Pparγ) and Glucose transporter type 4 (GluT4) in 3T3-L1 pre-adipocytes (Du and Heaney 2012).
High-dose fructose exposures elevated iNOS, Nrf2, Jnk, Erk, Nf-κb, and Mapk in response to oxidative stress in L6 myotubes (Jaiswal et al. 2015a; Jaiswal et al. 2015b). Human dendritic cells and HEK293 cells treated with high doses of fructose exhibited elevated expression of NF-κB and its translocation to the nucleus (Dornas et al. 2017; Jaiswal et al. 2019). Human dendritic also showed increased expression of inflammasome proteins IL-1β, IL-6, TNF-α, and IFN-γ at 24, 48, and 72 h after high fructose exposure (Jaiswal et al. 2019). Dendritic cells also showed elevated apoptotic marker proteins and AGEs, which led to the activation of the receptor for advanced glycation end products (RAGE) (Jaiswal et al. 2019), consistent with Caspase-3, Caspase-7, and Caspase-9 activation in L6 myotubes (Jaiswal et al. 2015a).
Glucose exposure to animal models similarly showed elevated oxidative stress and induction of apoptosis (Piro et al. 2002). Glucose exposures disrupt cell metabolic pathways and lead to metabolic syndrome through signs of reduced glucose uptake and reduced OCR (Moreno-Fernández et al. 2018). Given that glucose and fructose can use similar metabolic pathways, it is not surprising that elevation in GLUT proteins was also observed. GLUT1 and GLUT4 participate in glucose uptake, while GLUT5 aids in fructose transport. Although there were no GluT4 and GluT5 mRNA expression changes for L6 myotubes treated with high-dose fructose, GluT4myc translocation to the cell surface was impaired upon fructose exposure (Jaiswal et al. 2019). GLUT1 expression increased in human dendritic cells exposed to 15 mM fructose (Jaiswal et al. 2019). 3T3-L1 pre-adipocytes exhibited elevated levels of GluT5 expression at days 4 and 6 following high fructose incubation, indicating that cellular activity became altered as a function of fructose exposure, which suggests altered metabolic function (Du and Heaney 2012; Legeza et al. 2014).
Low dose fructose exposures in animal models showed similar changes (Table 2). A 5% fructose dose with high-fat administered to Wistar rats produced elevated levels of Il-1β, Il-6, and Nadph oxidase (Rosas-Villegas et al. 2017). Further, there were altered levels of catalase, glutathione peroxidase, glutathione reductase, and Sod1 due to exposure (Rosas-Villegas et al. 2017). Sprague-Dawley rat livers exhibited increased expression of Nlrp3 inflammation, Asc, Caspase-1, Il-1β, and Il-18 after 10% fructose exposure (Zhang et al. 2015). Wistar male and female rats exposed to low dose fructose also showed elevated levels of inflammatory cytokines, Tnf-α, Il-1β, Il-6, Il-10, and Il-18 expression in adipose tissues (Pektas et al. 2016). A 10% fructose dose in Wistar rats also showed increased expression of advanced oxidation protein products (AOPPs), apoptotic marker proteins, and inflammatory cytokines such as Il-12p70 and Il-6 (Porto et al. 2015). Elevated AGEs were also found in Wistar, Lewis, and hypertriglyceridic (HTG) rats administered with 10% fructose dose through increased post-translational pentosidine levels in rat aorta and skin tissue samples (Mikulikova et al. 2008).
Low dose fructose supplementation in C57BL/6 mice led to decreased cystic fibrosis transmembrane conductance regulator (CFTR) levels in cardiac tissue, contributing to heightened mitochondrial oxidative stress, impaired electron transport chain function, and induced myocardial hypertrophy (Zhang et al. 2016). Mitochondrial efficiency of Sprague-Dawley was also monitored through peroxisome proliferator-activated receptor-γ coactivator-1α (Pgc-1α) and Sirt3 protein expression levels after fructose exposure. There was a decrease in Pgc-1α protein expression in both the EDL and soleus muscles of healthy rats with 10% fructose exposure; however, there was an increase in Sirt3 expression in the soleus but a decline in the EDL muscle (Warren et al. 2014). Low dose exposures in Wistar male and female rats found elevated mRNA expression levels of insulin receptor β (Irβ), insulin receptor substrates (Irs-1, Irs-2), Akt, mTor, Pi3k, Ppary, Nrf2, and eNos, suggesting alterations in glucose uptake (Pektas et al. 2016).
High-dose fructose exposure to animal models similarly led to transcriptional changes due to alterations in oxidative stress and metabolic function upon supplementation (Table 2). Spontaneously, hypertensive rats on a 60% fructose diet per day showed increased Nadph oxidase and suppression of extracellular Sod expression, contributing to elevated oxidative stress (Wu et al. 2017). A 20% fructose dose administered to Sprague-Dawley rats induced decreased lipid hydroperoxide (LPO) and increased 8-OHdG levels in the hippocampal region. Moreover, there was reduced expression levels of Ucp5, which is known to inhibit ROS formation with reduced mitochondrial Tfam gene expression levels (Yamada et al. 2019). Fischer rats administered the same high-dose fructose over 20 weeks exhibited decreased renal Sod and catalase enzymatic expression (Dornas et al. 2017). C57BL/6 mice administered a 60% fructose dose experienced a non-significant increase of thioredoxin-2 (Thx2) gene expression levels (Mellor et al. 2010). Wistar rats administered 60% fructose had elevated expression of the elongation of very-long-chain fatty acids protein 6 (Elovl6) enzyme, which has an important role in NAFLD by transforming C16 saturated and monounsaturated fatty acids into C18 compounds (Szucs et al. 2019).
There are indications that high-dose fructose supplementation to animal models has differing effects on apoptosis triggering pathways (Table 2). High fructose exposed Wistar rats had decreased catalase mRNA expression in the liver, heart, skeletal muscle, and adipose tissue, which is pro-apoptotic (Cavarape et al. 2001). There were also increased levels of creatine kinase (CK), CK-MB, and lactate dehydrogenase (LDH), which are indicative of myocardial injury (Szucs et al. 2019). However, a separate Wistar rat study found no change in caspase-7 and Bax expression upon treatment; however, there was an increase in anti-apoptotic 3-ketoacyl-CoA thiolase in mitochondria of cardiac tissue, which diminished Bnip3-induced apoptosis (Szucs et al. 2019).
Both cell line and animal model studies show fructose induces transcriptional changes and apoptosis (Tables 1 & 2). There are clear links between the elevated reactive species and these outcomes, suggesting that fructose exposure does have a significant role in toxicity (Jaiswal et al. 2015a; Porto et al. 2015; Zhang et al. 2015).
DHA exposures show outcomes similar to fructose exposures
Early toxicological investigations found DHA to be mutagenic in Salmonella typhimurium strain TA100 with and without metabolic activation (Table 3) (Pham et al. 1979). Mutagenicity of DHA is not surprising given that glucose, fructose, and galactose are mutagenic (Brands et al. 2000). Interestingly, ketose sugars like fructose are more mutagenic than glucose and galactose, and higher mutagenic activity corresponds with a higher Maillard reactivity (Brands et al. 2000; Laroque et al. 2008). DHA is a ketose, precursor to ribose, the most reactive sugar in modifying proteins by the Maillard reaction.
TABLE 3.
DHA exposures across various models
| Model-system | Dose | Key findings | References |
|---|---|---|---|
| Cell models | |||
| A375P | 5 mM | Inhibited cell growth; increases in ROS; no DNA damage; cell cycle arrest; delayed induction of apoptosis; increased mitochondrial polarization | Smith et al., 2018 |
| HaCaT | 5 to 100 mM | No change in cell proliferation with 25 mM dose after 3 hr; cell cycle arrest (50% increase of cells in the G2/M); doses greater than 25 mM induced cell death; apoptosis; increase in DNA strand breaks | Petersen et al., 2004 |
| HaCaT | Cell viability analysis by flow cytometry and cell proliferation: ≤50 mM; detection of intracellular oxidative stress: ≤40 mM; comet assay (alkaline single cell gel electrophoresis): 20 mM; detection of γH2AX (S139): ≤40 mM | Impaired cell viability, proliferation, and cell cycle progression, increased gene expression of HMOX1 due to oxidative stress, increased XRCC2 and ERCC3 gene expression due to DNA damage, increased AGE formation, increased ROS (only for greater than 40 mM) | Perer et al., 2020 |
| HEK293T | |||
| HEK293T | 5 mM | Cell cycle arrest confirmed by increased cyclin A2 and cyclin B1 levels; no apoptosis as exposure did not lead to cleavage of PARP1 or Caspase 3; autophagy confirmed with increase in LC3BII to LC3BI ratio and SIRT1 expression; reduced mitochondrial membrane potential after 24 hr exposure; initial decline in OCR; decline in ECAR after 24 hr, decreased ATP production and lactate production; NAD+/NADH cofactor imbalances | Smith et al., 2019 |
| Animal model systems, human samples, and bacteria | |||
| C57BL/6 mice | 8 mM | Exogenous DHA readily enter cells and tissues and form metabolites | Moreno et al., 2014 |
| Sprague–Dawley rats | 80 mM of hyperpolarized [2-13C] DHA | Generation of different metabolites in liver and kidneys; higher levels of DHA and DHAS in the kidney than the liver | Marco-Rius et al., 2017 |
| Skin biopsy samples from pig | 5, 10, and 20% DHA | DHA increases free radical production by 180% in UV-exposed skin relative to the control untreated group | Jung et al., 2008 |
| Human serum albumin | Reaction mixtures included 2, 10, or 30 mM of each DHA or DHAP and 35 mg of HAS | HSA is shown to undergo glycation by DHA | Seneviratne et al. 2012 |
| Salmonella typhimurium strain TA100 | 8% DHA | DHA cytotoxicity with and without metabolic activation; DHA is mutagenic and can lead to DNA damage | Pham et al., 1979 |
Low millimolar doses of DHA are cytotoxic and genotoxic in immortalized keratinocytes, melanoma cells, human embryonic kidney cells, and reconstructed epidermis (Table 3) (Petersen et al. 2004; Smith et al. 2018; Smith et al. 2019; Perer et al. 2020). Genotoxicity of fructose has been previously described and attributed to the generation of genotoxic metabolites like reactive species (ROS/RNS) and glyoxal (Table 1) (Hansen et al. 2008). Similarly, the genotoxicity of DHA involves the generation of reactive species and induction of strand breaks. ROS was generated after 5 mM exposures to DHA in A375P cells (Smith et al. 2018). ROS was also observed in the immortalized keratinocyte cell line HaCaT after 25 and 50 mM doses (Perer et al. 2020). Increases in ROS were also observed when ex vivo skin was exposed to DHA then exposed to ultraviolet (UV) light (Jung et al. 2008). Strand breaks and replication stress were also observed in HaCaT and A375P cells through comet assay and γH2AX foci formation (Petersen et al. 2004; Smith et al. 2018; Perer et al. 2020). HEK293T cells did not show increases in ROS, but changes in NAD(P)H and GSH/GSSG levels indicate the presence of reactive species after 5 mM DHA exposure (Smith et al. 2019).
Both fructose and DHA exposures increase reactive species content in cell models. However, fructose investigations infer that the formation of these species can lead to detrimental cellular effects such as metabolic dysfunction, and evidence is only emerging that DHA exposures cause these outcomes as well (Table 1). It is currently unknown if ROS formation following DHA exposure can be driven by uric acid. Elevated ROS formation for both fructose and DHA investigations are similar in that they are dependent on the activation of NADPH oxidases by AGEs, which contribute to elevated oxidative stress (Wautier et al. 2001; Kuroda et al. 2010).
Fructose cytotoxicity is less well described independent of other stressors, but 12 mM fructose was 50% cytotoxic to primary hepatocytes (Lee et al. 2009). Recent characterization of DHA demonstrates higher genotoxicity and cytotoxicity than is associated with fructose exposures, indicating the metabolic mechanisms to deal with excess fructose are more robust than those that deal with an excess of 3-carbon metabolites. The metabolic dependence of DHA cytotoxicity is supported by variations in DHA sensitivity and the mechanisms of cell death between cell lines. Petersen et al. and Perer et al. report cytotoxic doses for the immortalized keratinocyte cell lines HaCaT at 25 mM, while A375P melanoma cells and HEK293T cells show more sensitivity with IC90 doses of 5 mM (Petersen et al. 2004; Smith et al. 2018; Smith et al. 2019; Perer et al. 2020). The different cell line models had varied cell death mechanisms from apoptosis (HaCaT and A375P) to autophagy (HEK293T) (Petersen et al. 2003; Smith et al. 2018; Smith et al. 2019; Perer et al. 2020). A brief period of senescence was even observed in the A375P melanoma cells exposed to 5 mM DHA (Smith et al. 2018), supporting metabolic dependence for the cytotoxic effects and likely the genotoxic effects.
Work with hyperpolarized DHA injected into C57BL/6 mice and Sprague-Dawley rats demonstrated generation of different metabolites after DHA exposures in the liver and kidney (Moreno et al. 2014; Marco-Rius et al. 2017). These works focused on using the hyperpolarized DHA as a metabolic probe, so high millimolar doses and short exposure time points were used (Table 3). These data confirm DHA is rapidly absorbed by tissues and incorporated into metabolic pathways. Critically, the rates of incorporation were dependent on the fed state of the animal (Moreno et al. 2014). Additionally, conversion of DHA to DHAP by TKFC seems to be a rate-limiting step for metabolic incorporation, with higher levels of DHA seen in the kidney than in the liver (Marco-Rius et al. 2017). TKFC expression varies across tissues, and mutations in TKFC are observed in individuals with metabolic disorders (Uhlen et al. 2015; Uhlen et al. 2017; Wortmann et al. 2020).
Once incorporated into metabolic pathways, DHA and DHAP can induce AGE formation, damaging proteins and lipids, similar to fructose exposures. Perer et al. demonstrated that DHA-derived AGEs could be measured in immortalized keratinocytes, human epidermal constructs, and mouse skin (Perer et al. 2020). This is consistent with observed AGE formation in fructose exposure studies; however, DHA is a more efficient glycation agent (Seneviratne et al. 2012). Perer et al. also showed that non-cytotoxic doses of DHA activated stress-related signal transduction pathways (p-p38, p-Hsp27(S15/S78), p-eIF2α) in immortalized keratinocytes (Perer et al. 2020). Elevated levels of stress response genes HSPA1A, HSPA6, HSPD1, IL6, and DDIT3, were also observed in both cell culture and reconstructed skin (Figure 2) (Perer et al. 2020).
Along with AGEs and stress responses, DHA exposures altered mitochondrial function and impacted NAD(P)H pools (Smith et al. 2018; Smith et al. 2019). Similar to low dose fructose exposures, DHA reduced ATP production and respiration rates in HEK293T cells (Smith et al. 2019). Fructose exposures also showed reductions in mtDNA, mitochondrial biogenesis, and respiratory activity (Table 1 and 2). These findings within fructose literature provide insight into the potential metabolic consequences of DHA exposure. The NAD(P)H pool imbalance likely impacts the efficiency of complex I to alter mitochondrial function. Still, a more in-depth analysis of the mitochondrial impact of DHA is needed to clarify the mechanisms underlying mitochondrial dysfunction.
Discussion
While studies to date have focused on more acute DHA exposures (Table 3), the data from these studies indicate the DHA exposures have metabolic, cytotoxic, and genotoxic outcomes that are strikingly similar to fructose exposures but occur at much lower doses than fructose (Gizak et al. 2019; Hernandez-Diazcouder et al. 2019; Taskinen et al. 2019). These similarities are not surprising, given DHAP is generated from the breakdown of fructose. However, DHAP from fructose exposures is generated in equilibrium with G3P. DHA generates DHAP dependent on TKFC activity within each cell or tissue and produces an imbalance in 3-carbon metabolites uniquely in each tissue.
Imbalance in DHAP and G3P has been linked with anemia, neurological disorders, diabetes, and cancer (Orosz et al. 2009; Kitada et al. 2010; Rabbani et al. 2016). Imbalance in this pathway is most often seen when defects in triosephosphate isomerase (TPI), which interconverts G3P and DHAP, results in the accumulation of DHAP. Excess DHAP is proposed to be converted to methylglyoxal and other AGEs, damaging DNA and proteins within the cell and causing neurological disorders and early childhood death, even though energy metabolism is unaffected (Li et al. 2008; Orosz et al. 2009).
Conversion of exogenous DHA to DHAP should cause a brief imbalance of DHAP/G3P, which the cell can re-equilibrate through multiple enzymatic reactions. However, the studies examining DHA exposure reviewed here show more acute genotoxic, metabolic, and cytotoxic outcomes than expected from equivalent fructose exposures (Tables 1 and 2). This suggests excess DHA and TKFC-converted DHAP are not readily metabolically incorporated for equilibration, or if equilibration occurs, it comes at a price to the cell health. DHA that is not readily converted into DHAP may induce unique protein and DNA damage that has yet to be characterized. More work is needed to understand the fate and impact of DHA that is not converted to DHAP in cells.
The DHA converted to DHAP accumulates endogenous toxins like ROS, RNS, and AGEs that damage DNA and proteins within cells. Therefore, the wealth of fructose studies already in the literature provide critical insight into the potential exposure effects of systemic DHA, which could place significant stress on cellular reduction and oxidation pathways, mitochondrial function, and metabolism (Tables 1 and 2). More importantly, it also highlights unexplored areas of systemic DHA exposures where inflammatory cytokines and inflammasome proteins are up-regulated. Chronic, long-term DHA exposure may act similarly to high fructose diets and induce metabolic reprogramming that reduces glucose uptake, changes glucose tolerance, and alters metabolic dependence of tissues. This type of metabolic reprogramming is important in the pathogenesis of obesity and diabetes.
Additionally, DHAP resulting from DHA exposure could impair lipid metabolism, like fructose exposures, and contribute to increased adipose tissue, higher body weights, blood pressures, and plasma triglyceride concentrations in exposed individuals. These types of outcomes from DHA exposure could impact the development of chronic illnesses like insulin resistance and NAFLD. More work is needed with animals exposed to DHA under different dietary conditions to understand the consequences of exposure across different tissue types.
Critically, work characterizing fructose exposures also demonstrates that other underlying metabolic factors, like high-fat diets, can alter the cellular consequences of DHA exposure. Co-exposure of fructose with a high-fat diet showed more elevated ROS generation alongside increased levels of MDA, H2O2, and oxidative damage in adipose, liver, skeletal, and kidney tissue types at lower fructose doses (Rosas-Villegas et al. 2017). The interaction of DHA or TKFC-converted DHAP with these underlying metabolic conditions could further exacerbate the reactive species generated and the genotoxicity and cytotoxicity of DHA.
One other possibility is that low dose exposures to fructose or DHA could stimulate antioxidant mechanisms and transcription factors like NF-κB to reduce reactive species (Figure 2). As noted in some fructose studies, enzymes that scavenge reactive species are up-regulated and could provide a beneficial cellular effect by reducing reactive species and promoting the degradation of damaged proteins. However, there are insufficient studies of low dose DHA exposure to understand if there is a beneficial vs. adverse dose threshold. More studies, including both high and low doses of DHA, would offer new insight into the cell’s ability to use DHA as a carbon source and identify where chronic or acute exposure effects occur.
Evidence that exogenous DHA exposures contribute to disease states is currently lacking. However, there is ample evidence in the literature that imbalances in DHAP and G3P from fructose exposures, diabetes, and other disease states induce protein and DNA damage, metabolic reprogramming and contribute to disease progression. Significantly, more work is needed both in vitro and in vivo to understand the consequences of acute and chronic exposures to exogenous DHA. Given that DHA exposures are following an increasing trend, the wealth of fructose data already in the literature offers a unique opportunity to investigate the differences between fructose and DHA exposures. Fructose studies also provide comparison points for examining the health consequences of DHA and TKFC-converted DHAP exposures.
Conclusions
Exogenous exposures to DHA can occur through the use of sunless tanning products and e-cigarettes. Exposures to millimolar doses of DHA are genotoxic, cytotoxic, and induce metabolic reprogramming. Comparison with existing fructose exposure literature highlights that both fructose and DHA elevate oxidative stress due to the generation of reactive species. Both agents also induce mitochondrial changes that decreased overall function. The fructose data suggests these changes occur through alter electron transport and through the induction of DNA damage. These mechanisms need to be confirmed in DHA exposed models.
Additionally, gene expression changes observed in both fructose and DHA exposure models confirm the induction of cellular stress, even at non-cytotoxic doses that may induce metabolic reprogramming. In summary, DHA exposures are similar to fructose exposure, but they cause detrimental cellular consequences more rapidly and at lower doses. Therefore, fructose exposures can help develop a framework for investigating the health effects of DHA exposures and provide a better understanding of the potential health consequences for exposed individuals.
Acknowledgments
MS, NRG, and MEM are supported by the US Department of Health and Human Services, National Institute of Health (AT009908 and ES032450).
Footnotes
Conflicts of Interest
The authors declare no competing financial interests.
References
- Brands CM, Alink GM, van Boekel MA, Jongen WM. 2000. Mutagenicity of heated sugar-casein systems: effect of the Maillard reaction. J Agric Food Chem 48(6):2271–2275. [DOI] [PubMed] [Google Scholar]
- Burch HB, Lowry OH, Meinhardt L, Max P Jr., Chyu K 1970. Effect of fructose, dihydroxyacetone, glycerol, and glucose on metabolites and related compounds in liver and kidney. J Biol Chem 245(8):2092–2102. [PubMed] [Google Scholar]
- Cabezas A, Costas MJ, Pinto RM, Couto A, Cameselle JC. 2005. Identification of human and rat FAD-AMP lyase (cyclic FMN forming) as ATP-dependent dihydroxyacetone kinases. Biochem Biophys Res Commun 338(4):1682–1689. [DOI] [PubMed] [Google Scholar]
- Cavarape A, Feletto F, Mercuri F, Quagliaro L, Daman G, Ceriello A. 2001. High-fructose diet decreases catalase mRNA levels in rat tissues. J Endocrinol Invest 24(11):838–845. [DOI] [PubMed] [Google Scholar]
- Cioffi F, Senese R, Lasala P, Ziello A, Mazzoli A, Crescenzo R, Liverini G, Lanni A, Goglia F, Iossa S. 2017. Fructose-Rich Diet Affects Mitochondrial DNA Damage and Repair in Rats. Nutrients 9(4):323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins-Nakai RL, Noseworthy D, Lopaschuk GD. 1994. Epinephrine increases ATP production in hearts by preferentially increasing glucose metabolism. Am J Physiol 267(5 Pt 2):H1862–1871. [DOI] [PubMed] [Google Scholar]
- Delbosc S, Paizanis E, Magous R, Araiz C, Dimo T, Cristol JP, Cros G, Azay J. 2005. Involvement of oxidative stress and NADPH oxidase activation in the development of cardiovascular complications in a model of insulin resistance, the fructose-fed rat. Atherosclerosis 179(1):43–49. [DOI] [PubMed] [Google Scholar]
- Dornas WC, Cardoso LM, Silva M, Machado NL, Chianca DA Jr., Alzamora AC, Lima WG, Lagente V, Silva ME. 2017. Oxidative stress causes hypertension and activation of nuclear factor-kappaB after high-fructose and salt treatments. Sci Rep 7:46051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du L, Heaney AP. 2012. Regulation of adipose differentiation by fructose and GluT5. Mol Endocrinol 26(10):1773–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FDA. 2002. Code of Federal Regulations, Title 21. Washington, D.C.: U.S. Food and Drug Administration. 1150 p. [Google Scholar]
- FDA. 2015. Sunless Tanners & Bronzers. Washington, D.C.: U.S. Food and Drug Administration. [Google Scholar]
- Fleming AM, Ding Y, Burrows CJ. 2017. Oxidative DNA damage is epigenetic by regulating gene transcription via base excision repair. Proc Natl Acad Sci U S A 114(10):2604–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournet M, Bonte F, Desmouliere A. 2018. Glycation Damage: A Possible Hub for Major Pathophysiological Disorders and Aging. Aging Dis 9(5):880–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Berumen CI, Ortiz-Avila O, Vargas-Vargas MA, Del Rosario-Tamayo BA, Guajardo-Lopez C, Saavedra-Molina A, Rodriguez-Orozco AR, Cortes-Rojo C. 2019. The severity of rat liver injury by fructose and high fat depends on the degree of respiratory dysfunction and oxidative stress induced in mitochondria. Lipids Health Dis 18(1):78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh R, Mitchell DL. 1999. Effect of oxidative DNA damage in promoter elements on transcription factor binding. Nucleic Acids Res 27(15):3213–3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gizak A, Duda P, Wisniewski J, Rakus D. 2019. Fructose-1,6-bisphosphatase: From a glucose metabolism enzyme to multifaceted regulator of a cell fate. Adv Biol Regul 72:41–50. [DOI] [PubMed] [Google Scholar]
- Grasselli E, Baldini F, Vecchione G, Oliveira PJ, Sardao VA, Voci A, Portincasa P, Vergani L. 2019. Excess fructose and fatty acids trigger a model of nonalcoholic fatty liver disease progression in vitro: Protective effect of the flavonoid silybin. Int J Mol Med 44(2):705–712. [DOI] [PubMed] [Google Scholar]
- Hansen M, Baunsgaard D, Autrup H, Vogel UB, Moller P, Lindecrona R, Wallin H, Poulsen HE, Loft S, Dragsted LO. 2008. Sucrose, glucose and fructose have similar genotoxicity in the rat colon and affect the metabolome. Food Chem Toxicol 46(2):752–760. [DOI] [PubMed] [Google Scholar]
- Hernandez-Diazcouder A, Romero-Nava R, Carbo R, Sanchez-Lozada LG, Sanchez-Munoz F. 2019. High Fructose Intake and Adipogenesis. Int J Mol Sci 20(11):2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indo HP, Davidson M, Yen HC, Suenaga S, Tomita K, Nishii T, Higuchi M, Koga Y, Ozawa T, Majima HJ. 2007. Evidence of ROS generation by mitochondria in cells with impaired electron transport chain and mitochondrial DNA damage. Mitochondrion 7(1–2):106–118. [DOI] [PubMed] [Google Scholar]
- Jaiswal N, Agrawal S, Agrawal A. 2019. High fructose-induced metabolic changes enhance inflammation in human dendritic cells. Clin Exp Immunol 197(2):237–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal N, Maurya CK, Arha D, Avisetti DR, Prathapan A, Raj PS, Raghu KG, Kalivendi SV, Tamrakar AK. 2015a. Fructose induces mitochondrial dysfunction and triggers apoptosis in skeletal muscle cells by provoking oxidative stress. Apoptosis 20(7):930–947. [DOI] [PubMed] [Google Scholar]
- Jaiswal N, Maurya CK, Pandey J, Rai AK, Tamrakar AK. 2015b. Fructose-induced ROS generation impairs glucose utilization in L6 skeletal muscle cells. Free Radic Res 49(9):1055–1068. [DOI] [PubMed] [Google Scholar]
- Jung K, Seifert M, Herrling T, Fuchs J. 2008. UV-generated free radicals (FR) in skin: their prevention by sunscreens and their induction by self-tanning agents. Spectrochim Acta A Mol Biomol Spectrosc 69(5):1423–1428. [DOI] [PubMed] [Google Scholar]
- Kawasaki T, Akanuma H, Yamanouchi T. 2002. Increased fructose concentrations in blood and urine in patients with diabetes. Diabetes Care 25(2):353–357. [DOI] [PubMed] [Google Scholar]
- Kitada M, Zhang Z, Mima A, King GL. 2010. Molecular mechanisms of diabetic vascular complications. J Diabetes Investig 1(3):77–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda J, Ago T, Matsushima S, Zhai P, Schneider MD, Sadoshima J. 2010. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc Natl Acad Sci U S A 107(35):15565–15570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laroque D, Inisan C, Berger C, Vouland É, Dufossé L, Guérard F. 2008. Kinetic study on the Maillard reaction. Consideration of sugar reactivity. Food chemistry 111(4):1032–1042. [Google Scholar]
- Lee O, Bruce WR, Dong Q, Bruce J, Mehta R, O’Brien PJ. 2009. Fructose and carbonyl metabolites as endogenous toxins. Chem Biol Interact 178(1–3):332–339. [DOI] [PubMed] [Google Scholar]
- Lee YO, Nonnemaker JM, Bradfield B, Hensel EC, Robinson RJ. 2018. Examining Daily Electronic Cigarette Puff Topography Among Established and Nonestablished Cigarette Smokers in their Natural Environment. Nicotine Tob Res 20(10):1283–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legeza B, Balazs Z, Odermatt A. 2014. Fructose promotes the differentiation of 3T3-L1 adipocytes and accelerates lipid metabolism. FEBS Lett 588(3):490–496. [DOI] [PubMed] [Google Scholar]
- Li Y, Cohenford MA, Dutta U, Dain JA. 2008. In vitro nonenzymatic glycation of guanosine 5’-triphosphate by dihydroxyacetone phosphate. Anal Bioanal Chem 392(6):1189–1196. [DOI] [PubMed] [Google Scholar]
- Madlala HP, Maarman GJ, Ojuka E. 2016. Uric acid and transforming growth factor in fructose-induced production of reactive oxygen species in skeletal muscle. Nutr Rev 74(4):259–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marco-Rius I, von Morze C, Sriram R, Cao P, Chang GY, Milshteyn E, Bok RA, Ohliger MA, Pearce D, Kurhanewicz J, Larson PE, Vigneron DB, Merritt M. 2017. Monitoring acute metabolic changes in the liver and kidneys induced by fructose and glucose using hyperpolarized [2–13 C]dihydroxyacetone. Magn Reson Med 77(1):65–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini MC. 2017. [Self-tanning and sunless tanning products]. Ann Dermatol Venereol 144(10):638–644. [DOI] [PubMed] [Google Scholar]
- Mellor K, Ritchie RH, Meredith G, Woodman OL, Morris MJ, Delbridge LM. 2010. High-fructose diet elevates myocardial superoxide generation in mice in the absence of cardiac hypertrophy. Nutrition 26(7–8):842–848. [DOI] [PubMed] [Google Scholar]
- Mikulikova K, Eckhardt A, Kunes J, Zicha J, Miksik I. 2008. Advanced glycation end-product pentosidine accumulates in various tissues of rats with high fructose intake. Physiol Res 57(1):89–94. [DOI] [PubMed] [Google Scholar]
- Milakovic T, Johnson GV. 2005. Mitochondrial respiration and ATP production are significantly impaired in striatal cells expressing mutant huntingtin. J Biol Chem 280(35):30773–30782. [DOI] [PubMed] [Google Scholar]
- Moraru A, Wiederstein J, Pfaff D, Fleming T, Miller AK, Nawroth P, Teleman AA. 2018. Elevated Levels of the Reactive Metabolite Methylglyoxal Recapitulate Progression of Type 2 Diabetes. Cell Metab 27(4):926–934 e928. [DOI] [PubMed] [Google Scholar]
- Moreno-Fernández S, Garcés-Rimón M, Vera G, Astier J, Landrier JF, Miguel M. 2018. High Fat/High Glucose Diet Induces Metabolic Syndrome in an Experimental Rat Model. Nutrients 10(10):1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno KX, Satapati S, DeBerardinis RJ, Burgess SC, Malloy CR, Merritt ME. 2014. Real-time detection of hepatic gluconeogenic and glycogenolytic states using hyperpolarized [2–13C]dihydroxyacetone. J Biol Chem 289(52):35859–35867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orosz F, Olah J, Ovadi J. 2009. Triosephosphate isomerase deficiency: new insights into an enigmatic disease. Biochim Biophys Acta 1792(12):1168–1174. [DOI] [PubMed] [Google Scholar]
- Pantini G, Ingoglia R, Brunetta F, Brunetta A. 2007. Sunless tanning products containing dihydroxyacetone in combination with a perfluoropolyether phosphate. Int J Cosmet Sci 29(3):201–209. [DOI] [PubMed] [Google Scholar]
- Pastukh V, Roberts JT, Clark DW, Bardwell GC, Patel M, Al-Mehdi AB, Borchert GM, Gillespie MN. 2015. An oxidative DNA “damage” and repair mechanism localized in the VEGF promoter is important for hypoxia-induced VEGF mRNA expression. Am J Physiol Lung Cell Mol Physiol 309(11):L1367–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pektas MB, Koca HB, Sadi G, Akar F. 2016. Dietary Fructose Activates Insulin Signaling and Inflammation in Adipose Tissue: Modulatory Role of Resveratrol. Biomed Res Int 2016:8014252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perer J, Jandova J, Fimbres J, Jennings EQ, Galligan JJ, Hua A, Wondrak GT. 2020. The sunless tanning agent dihydroxyacetone induces stress response gene expression and signaling in cultured human keratinocytes and reconstructed epidermis. Redox Biol 36:101594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen AB, Na R, Wulf HC. 2003. Sunless skin tanning with dihydroxyacetone delays broad-spectrum ultraviolet photocarcinogenesis in hairless mice. Mutat Res 542(1–2):129–138. [DOI] [PubMed] [Google Scholar]
- Petersen AB, Wulf HC, Gniadecki R, Gajkowska B. 2004. Dihydroxyacetone, the active browning ingredient in sunless tanning lotions, induces DNA damage, cell-cycle block and apoptosis in cultured HaCaT keratinocytes. Mutat Res 560(2):173–186. [DOI] [PubMed] [Google Scholar]
- Pham HN, DeMarini DM, Brockmann HE. 1979. Mutagenicity of skin tanning lotions. J Environ Pathol Toxicol 3(1–2):227–231. [PubMed] [Google Scholar]
- Piro S, Anello M, Di Pietro C, Lizzio MN, Patane G, Rabuazzo AM, Vigneri R, Purrello M, Purrello F. 2002. Chronic exposure to free fatty acids or high glucose induces apoptosis in rat pancreatic islets: possible role of oxidative stress. Metabolism 51(10):1340–1347. [DOI] [PubMed] [Google Scholar]
- Poetsch AR. 2020. The genomics of oxidative DNA damage, repair, and resulting mutagenesis. Comput Struct Biotechnol J 18:207–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porto ML, Lirio LM, Dias AT, Batista AT, Campagnaro BP, Mill JG, Meyrelles SS, Baldo MP. 2015. Increased oxidative stress and apoptosis in peripheral blood mononuclear cells of fructose-fed rats. Toxicol In Vitro 29(8):1977–1981. [DOI] [PubMed] [Google Scholar]
- Rabbani N, Xue M, Thornalley PJ. 2016. Methylglyoxal-induced dicarbonyl stress in aging and disease: first steps towards glyoxalase 1-based treatments. Clin Sci (Lond) 130(19):1677–1696. [DOI] [PubMed] [Google Scholar]
- Richard JP. 1993. Mechanism for the formation of methylglyoxal from triosephosphates. Biochem Soc Trans 21(2):549–553. [DOI] [PubMed] [Google Scholar]
- Rosas-Villegas A, Sanchez-Tapia M, Avila-Nava A, Ramirez V, Tovar AR, Torres N. 2017. Differential Effect of Sucrose and Fructose in Combination with a High Fat Diet on Intestinal Microbiota and Kidney Oxidative Stress. Nutrients 9(4):393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCCS. 2010. Opinion on dihydroxyacetone. Brussels: Scientific Committee on Consumer Safety. [Google Scholar]
- Seneviratne C, Dombi GW, Liu W, Dain JA. 2012. In vitro glycation of human serum albumin by dihydroxyacetone and dihydroxyacetone phosphate. Biochem Biophys Res Commun 417(2):817–823. [DOI] [PubMed] [Google Scholar]
- Smith KR, Granberry M, Tan MCB, Daniel CL, Gassman NR. 2018. Dihydroxyacetone induces G2/M arrest and apoptotic cell death in A375P melanoma cells. Environ Toxicol 33(3):333–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KR, Hayat F, Andrews JF, Migaud ME, Gassman NR. 2019. Dihydroxyacetone Exposure Alters NAD(P)H and Induces Mitochondrial Stress and Autophagy in HEK293T Cells. Chem Res Toxicol 32(8):1722–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szucs G, Soja A, Peter M, Sarkozy M, Bruszel B, Siska A, Foldesi I, Szabo Z, Janaky T, Vigh L, Balogh G, Csont T. 2019. Prediabetes Induced by Fructose-Enriched Diet Influences Cardiac Lipidome and Proteome and Leads to Deterioration of Cardiac Function prior to the Development of Excessive Oxidative Stress and Cell Damage. Oxid Med Cell Longev 2019:3218275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tappy L, Le KA. 2012. Does fructose consumption contribute to non-alcoholic fatty liver disease? Clin Res Hepatol Gastroenterol 36(6):554–560. [DOI] [PubMed] [Google Scholar]
- Taskinen MR, Packard CJ, Boren J. 2019. Dietary Fructose and the Metabolic Syndrome. Nutrients 11(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobey TA, Mondon CE, Zavaroni I, Reaven GM. 1982. Mechanism of insulin resistance in fructose-fed rats. Metabolism 31(6):608–612. [DOI] [PubMed] [Google Scholar]
- Touyz RM. 2012. Chapter 69 - Reactive Oxygen Species and Oxidative Stress. In: Robertson D, Biaggioni I, Burnstock G, Low PA, Paton JFR, editors. Primer on the Autonomic Nervous System (Third Edition). San Diego: Academic Press. p 335–338. [Google Scholar]
- Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, Edqvist PH, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk JM, Hamsten M, von Feilitzen K, Forsberg M, Persson L, Johansson F, Zwahlen M, von Heijne G, Nielsen J, Ponten F. 2015. Proteomics. Tissue-based map of the human proteome. Science 347(6220):1260419. [DOI] [PubMed] [Google Scholar]
- Uhlen M, Zhang C, Lee S, Sjostedt E, Fagerberg L, Bidkhori G, Benfeitas R, Arif M, Liu Z, Edfors F, Sanli K, von Feilitzen K, Oksvold P, Lundberg E, Hober S, Nilsson P, Mattsson J, Schwenk JM, Brunnstrom H, Glimelius B, Sjoblom T, Edqvist PH, Djureinovic D, Micke P, Lindskog C, Mardinoglu A, Ponten F. 2017. A pathology atlas of the human cancer transcriptome. Science 357(6352). [DOI] [PubMed] [Google Scholar]
- Vreeke S, Korzun T, Luo W, Jensen RP, Peyton DH, Strongin RM. 2018. Dihydroxyacetone levels in electronic cigarettes: Wick temperature and toxin formation. Aerosol Science and Technology 52(4):370–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren BE, Lou PH, Lucchinetti E, Zhang L, Clanachan AS, Affolter A, Hersberger M, Zaugg M, Lemieux H. 2014. Early mitochondrial dysfunction in glycolytic muscle, but not oxidative muscle, of the fructose-fed insulin-resistant rat. Am J Physiol Endocrinol Metab 306(6):E658–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wautier MP, Chappey O, Corda S, Stern DM, Schmidt AM, Wautier JL. 2001. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am J Physiol Endocrinol Metab 280(5):E685–694. [DOI] [PubMed] [Google Scholar]
- Wittgenstein E, Berry HK. 1960. Staining of skin with dihydroxyacetone. Science 132(3431):894–895. [DOI] [PubMed] [Google Scholar]
- Wortmann SB, Meunier B, Mestek-Boukhibar L, van den Broek F, Maldonado EM, Clement E, Weghuber D, Spenger J, Jaros Z, Taha F, Yue WW, Heales SJ, Davison JE, Mayr JA, Rahman S. 2020. Bi-allelic Variants in TKFC Encoding Triokinase/FMN Cyclase Are Associated with Cataracts and Multisystem Disease. Am J Hum Genet 106(2):256–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu KL, Wu CW, Tain YL, Chao YM, Hung CY, Tsai PC, Wang WS, Shih CD. 2017. Effects of high fructose intake on the development of hypertension in the spontaneously hypertensive rats: the role of AT1R/gp91(PHOX) signaling in the rostral ventrolateral medulla. J Nutr Biochem 41:73–83. [DOI] [PubMed] [Google Scholar]
- Yamada H, Munetsuna E, Yamazaki M, Mizuno G, Sadamoto N, Ando Y, Fujii R, Shiogama K, Ishikawa H, Suzuki K, Shimono Y, Ohashi K, Hashimoto S. 2019. Maternal fructose-induced oxidative stress occurs via Tfam and Ucp5 epigenetic regulation in offspring hippocampi. FASEB J 33(10):11431–11442. [DOI] [PubMed] [Google Scholar]
- Yamagishi S, Matsui T. 2010. Advanced glycation end products, oxidative stress and diabetic nephropathy. Oxid Med Cell Longev 3(2):101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zhang JH, Chen XY, Hu QH, Wang MX, Jin R, Zhang QY, Wang W, Wang R, Kang LL, Li JS, Li M, Pan Y, Huang JJ, Kong LD. 2015. Reactive oxygen species-induced TXNIP drives fructose-mediated hepatic inflammation and lipid accumulation through NLRP3 inflammasome activation. Antioxid Redox Signal 22(10):848–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YB, Meng YH, Chang S, Zhang RY, Shi C. 2016. High fructose causes cardiac hypertrophy via mitochondrial signaling pathway. Am J Transl Res 8(11):4869–4880. [PMC free article] [PubMed] [Google Scholar]